

Abstract
With the development of next generation sequencing, more and more common inherited diseases have been reported. However, accurate and convenient molecular diagnosis cannot be achieved easily because of the enormous size of disease causing mutations. In this study, we introduced a new single-step method for the genetic analysis of patients and carriers in real clinical settings. All kinds of disease causing mutations can be detected at the same time in patients with Mendelian diseases or carriers. First, we evaluated this technology using YH cell line DNA and 9 samples with known mutations. Accuracy and stability of 99.80% and 99.58% were achieved respectively. Then, a total of 303 patients were tested using our targeted NGS approaches, 50.17% of which were found to have deleterious mutations and molecular confirmation of the clinical diagnosis. We identified 219 disease causing mutations, 43.84% (96/219) of which has never been reported before. Additionally, we developed a new deleteriousness prediction method for nonsynonymous SNVs, and an automating annotation and diagnosis system for Mendelian diseases, thus greatly assisting and enhancing Mendelian diseases diagnosis and helping to make a precise diagnosis for patients with Mendelian diseases.
Introduction
Mendelian diseases are a series of diseases following the principles of Mendelian inheritance. So far, more than 8,000 Mendelian inheritance diseases have been included in Online Mendelian Inheritance in Man (the OMIM database, www.omim.org). Many methods have been performed for the diagnosis of Mendelian diseases in clinical practice, for example, Sanger sequencing of known disease causing genes. As is known, Sanger sequencing is widely considered as the gold standard for sequencing, however, there are some drawbacks in this technology: laborious, expensive, and time consuming.
With the rapid development of next generation sequencing (NGS) technology, all kinds of diseasing causing mutations, such as SNVs (single nucleotide variants), Indel (small insertion/deletion) and CNVs (copy number variants), can be detected at the same time in patients with Mendelian diseases or carriers1. It has been proved that NGS based technology is a powerful tool for the detection of pathogenic mutations, especially in patients with monogenic disorders2. Researchers performed NGS to analyze tens to thousands of genes simultaneously in a single assay1,3,4. This makes NGS more suitable in the diagnosis of Mendelian diseases, and highly improved the diagnostic yield5. Comprehensive characterization of genetic diseases using NGS technology has become an option for researchers and clinicians. However, annotation and interpretation of NGS data is tedious and time-consuming, and require highly specific expertise in this filed. Our new method introduced here can help clinical scientists to efficiently integrate all their data more quickly and form a whole picture of the detected variants.
WGS (whole genome sequencing) and WES (Whole exome sequencing) have been applied in the diagnostic of Mendelian genetic disorders or the screening of carriers6–9. However, it is still a major challenge to detect few disease causing mutations in the vast potential variants in human genome10. Targeted NGS is recognized as a cost effective method for the diagnostic of Mendelian genetic disorders1,3,4. Targeted NGS with high throughput and lower cost can target disease related regions in human genome more efficiently, and detect variants more sensitively. This technology has been widely used in the screening of mutations and effective diagnosis of genetic diseases in clinical setting11–14. What is more, targeted NGS targets a set of genes related to specific disease phenotypes, which could result in a higher sequencing coverage. Variant calling is more accurate for region of interest when performing targeted NGS15.
In this study, we designed a chip (array based) containing 4,689 nuclear genes related to Mendelian diseases, and tested its performance in detecting clinical relevant mutations using HiSeq platform. Then, we applied this panel to 303 clinical cases, and identified disease causing mutations in 152 patients. What is more, we developed a new deleteriousness prediction method for nonsynonymous SNVs, and an automating annotation and diagnosis system for Mendelian diseases. It can greatly assist and enhance Mendelian diseases diagnosis and help to make a precise diagnosis for the patients.
Results
Evaluation of accuracy and stability
Before applying this technology to real clinical patients, we evaluated the accuracy and stability of targeted NGS as reported before1. To assess the accuracy of this technology, we compared the SNPs of a YH cell line sample (C1) detected using targeted NGS with the genotyping results detected using Illumina’s Human Zhonghua-8 Bead Chips (SNP Array). The analysis of C1 using SNP Array was carried out blindly. In C1, there are a total of 4416 SNPs detected in the region of the chip (10,258,198 bp). In the selected loci, 99.80% (4,407/4,416) genotypes detected by targeted NGS in sample C1 were accordant to the results detected by SNP Array, which showed a high accuracy of our method. We also screened the 4,689 nuclear genes in 9 probands with known mutations using targeted NGS to evaluate the technology (Table 1). Sanger sequencing and real-time PCR validation of the known mutations in the 9 probands had been completed prior to this study. A total of 11 known mutations were detected in the 9 probands using targeted NGS technology. The results are exactly the same as obtained using real-time PCR and Sanger sequencing, indicating complete consistency of the methods. These results indicated that targeted NGS technology used in this study provides high accuracy.
Table 1.
Nine probands with known mutations.
| Proband | Disease | Gene | Mutation | Validation |
|---|---|---|---|---|
| P1 | Dent Disease 1 | CLCN5 | c.778_798delACTCTG GTTATCAAAACCATC |
Sanger |
| P2 | X-Linked Ichthyosis | STS | Whole gene deletion | QPCR |
| P3 | Mucolipidosis II/III Alpha&Beta | GNPTAB | c.1090 C > T; c.2404 C > T | Sanger |
| P4 | Charcot-Marie-Tooth disease | PMP22 | c.215 C > T | Sanger |
| P5 | Polycystic kidney disease 1 | PKD1 | c.8835 C > G | Sanger |
| P6 | Duchenne/Becker Muscular Dystrophy | DMD | CDS 10–12 deletion | QPCR |
| P7 | Nephrotic Syndrome Type 2 | NPHS2 | c.593 A > C; c.538 G > A | Sanger |
| P8 | Pseudohypoaldosteronism Type IB | SCNN1B | c.1853C > G | Sanger |
| P9 | Charcot-Marie-Tooth disease | PMP22 | Whole gene duplication | QPCR |
To assess the stability of the technology, YH cell line were sequenced three times in different batches (C1, C2 and C3). There are 10,133,027 genotype can be detected in the 3 YH cell line samples. We then compared all the genotypes detected in C1, C2 and C3. The results showed that the proportion of the same genotype detected is 99.58% (10,090,060/10,133,027) in different batches, which indicated high stability of this technology.
Targeted NGS approach
In this study, we developed a targeted NGS assay to detect disease causing alterations in 303 patients diagnosed with Mendelian diseases using HiSeq platform.
A total of 303 patients were recruited in this study. Using targeted NGS, we obtained high-quality reads of all the samples. After base calling and image analysis using Illumina HiSeq sequencers, primary data was received in the form of FASTQ. Then we performed a new bioinformatics analysis pipeline to analyze the raw sequencing data. The sequencing results of all the patients are as follows: more than 99.55% coverage of the target region was achieved for each sample, and an average sequencing depth of 250.82-fold was achieved for each sample.
Deleteriousness prediction for nonsynonymous SNVs
In order to attain the best classifier for rare missense variants in human genome, we developed a new pathogenicity prediction method (MPPS, Mutation Pathogenicity Prediction Software) in this study. MPPS combined a total of 17 current deleteriousness-scoring methods, including 8 functional prediction scores (Polyphen2_HDIV16, Polyphen2_HVAR16, MutationTaster17, SIFT18, LRT19, FATHMM20, MutationAssessor21 and M-CAP22), 7 new prediction scores (PROVEAN23, VEST324, MetaSVM25, MetaLR25, fathmm-MKL26, CADD27 and DANN28) and 2 conservation scores (GERP++29 and phyloP100way30).
In the present study, we evaluated the performance of the 17 deleteriousness prediction methods and MPPS (Fig. 1). We collected a total of 25,220 known pathogenic variants from ClinVar (www.ncbi.nlm.nih.gov/clinvar), and 22,885 neutral variants from ClinVar (4,308) and our local database (18,577). The best performing methods in this test were MPPS and MutationTaster. The percentage of correct predictions of MutationTaster is 96.56% for pathogenic variants and 86.84% for neutral variants respectively. MPPS was able to correctly predict 97.12% of credibly pathogenic mutations and 95.21% of the credibly benign variants. There was a noticeable problem with several of the deleteriousness prediction programs: the lack of output because of the method’ inability to predict pathogenicity for a specific variant31. So we introduced the concept of Prediction Coverage. Prediction Coverage is defined by dividing the number of predicted variants (correctly or in correctly) by the total number of variants evaluated. Prediction Coverage was used to evaluate if all the variants can be predicted with a precise result or not. As shown in Fig. 1, 100% of variants (48,105/48,105) were successfully covered by MPPS, which is even better than MutationTaster (97.73%). MPPS could also make a great contribution to the automating result interpretation and reporting system in the next step.
Figure 1.

Accuracy and coverage of 17 prediction programs.
Automating result interpretation and reporting system
In this study, we developed a web-based framework named AADSM (Automating Annotation and Diagnosis System for Mendelian disease) for automating analysis of clinical NGS data (Fig. 2). Clinical scientists can use this website to get information directly for diagnostics of Mendelian diseases by uploading the VCF (variant call format) file and the patients’ phenotypes.
Figure 2.

Automating annotation and diagnosis system for Mendelian disease (AADSM). AADSM is a web-based automating annotation and diagnosis system, which can assist and enhance diagnostics of Mendelian diseases.
Before analysis, we build our own gene-phenotype database and a scoring system. We use the gene-phenotype database and the scoring system to allow clinical researchers to carry out analysis of variant data, and help them to make a precise diagnosis for the patients. AADSM accepts variants input in the VCF formats. Patients’ phenotype also need to be upload before analysis (Fig. 2). The server performs a three step analysis process automatically, which mainly includes variant filtering and phenotype analysis, scoring, sorting and reporting. In the first step, all the variants will be filtered and annotated with our databases. At the same time, a gene list will be generated according to the clinical phenotype uploaded. In the second step, all the filtered mutations will be sorted using the scoring system we developed. The scoring system relies on the following rules: mutation frequency, mutation type, the consequence of a variant, and predicted categories from HGMD to determine the rank of a variant. In order to find the most related disease, the gene list generated in the first step will also be sorted using the scoring system. In the last step, a report will be automatically reported based on gene-disease relationship. The diseases and mutations with the highest score will be automated reported. Analysis results were presented in HTML pages, which contain all the details of annotation and the diagnosed diseases. These results can help clinical scientists to efficiently integrate variant related information and form a whole picture of their data more quickly.
To demonstrate the method’s diagnostic efficacy, we tested 56 patients in this study. A total of 85 disease causing mutations in the 56 patients were validated by methods other than NGS technology and identified previously by experienced researchers in this field. All the 56 patients were then diagnosed to be with specific Mendelian diseases according to the mutations identified. We also performed AADSM for automating analysis of clinical NGS data of the 56 patients. All the 85 disease causing mutations identified by experienced researchers were also detected by AADSM. In the 56 patients, the clearly diagnosed diseases in 32 patients (57.14%) ranked first in all the reported diseases, the clearly diagnosed diseases in 45 (80.36%) patients ranked top ten, and the clearly diagnosed diseases in 55 (98.21%) patients ranked top one hundred (Supplementary Material). These results can greatly assist and enhance Mendelian diseases diagnosis for clinical scientists and help to make a precise diagnosis for the patients.
Identification and characterization of disease causing mutations
In this study, a total of 219 disease causing mutations were detected, including 129 missense, 26 nonsense, 33 Indel, 15 splicing and 16 CNV. 43.84% (96/219) of disease causing mutations has never been reported before (Supplementary Material). Of all the 303 random patients, 50.17% (152/303) were found to have deleterious mutations and have molecular confirmation of the clinical diagnosis (Fig. 3). The 152 patients belonged to 9 clinical classifications. It was also observed that Urinary system disease (20.46% of all the 152 positive patients), Eye disease (14.19%), and Neuromuscular disease (8.91%) were the most frequently detected clinical category in our cohort (Fig. 3). Cataract (22 patients), Alport Syndrome (16 patients), and Retinitis pigmentosa (10 patients) were found to contain the most patients (Supplementary Material). The results strongly suggested that targeted NGS technique is an informative and effective approach to make a precise diagnosis for patients with Mendelian diseases, and has great potential to be used in real clinical practice.
Figure 3.
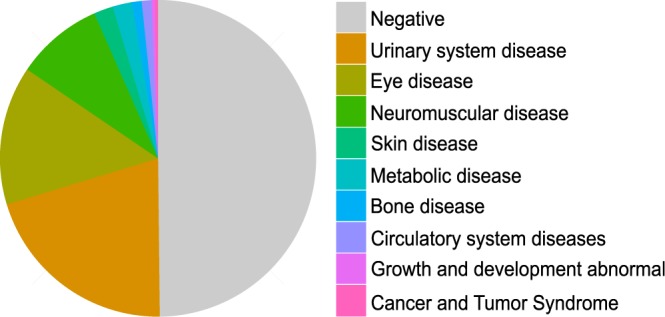
Positive patients found to have deleterious mutations and have molecular confirmation of the clinical diagnosis.
A total of 85 (38.81%) of identified pathogenic mutations were tested and confirmed by Sanger sequencing (Supplementary Material). Co-segregation analysis was also performed among some patients’ family members. The validation further confirmed the clinical diagnosis of the patients.
Discussion
Accurate and timely detection of disease causing mutations in patients with Mendelian diseases plays a pivotal role in the era of personalized medicine. Because the involved genes number is large, traditional methods cannot be used to make a precise genetic diagnosis at a time5. With the rapid development of NGS, comprehensive characterization of genetic diseases using NGS technology has become an option for researchers and clinicians3,4,6,11–14,32,33. It has been proved that NGS based technology is a powerful tool for the detection of pathogenic mutations, especially in patients with Mendelian diseases.
In this study, we designed a new chip (array based) containing 4,689 genes related to Mendelian diseases, and performed targeted region enrichment and NGS for comprehensive mutation detection in 303 clinical cases. We reported disease causing mutations in 152 patients out of 303 random patients using targeted NGS. A total of 219 disease causing mutations (including 96 novel mutations) were identified. Some researchers reported that ~25% random patients were found to have deleterious mutations8. Based on our experience, the detection rate is highly dependent on the experience of the clinical scientist. In this study, all the potential patients were collected by experienced clinical scientists. Comparing to the results of previously publications, we included more genes related to Mendelian diseases, the diagnostic yield was also improved1,3–5,8.
To test the accuracy of this method, we compared the SNPs of a YH cell line sample detected using targeted NGS technology with the genotyping results detected using SNP Array. In the selected loci, 4,407 out of 4,416 genotypes detected by targeted NGS in sample C1 were accordant to the results detected by SNP Array, which means there are 9 genotypes inconsistent using targeted NGS and SNP Array. We did NOT validate these inconsistent genotypes in the present study because in our previous study1, we also found 5 inconsistent genotypes when accessing the accuracy of targeted NGS technology. It turns out that all the genotypes of inconsistent loci detected by targeted NGS were consistent with that of Sanger sequencing1, which suggested that our method could provide high accuracy.
As is known, the most common form of genetic variation is single nucleotide polymorphisms (SNPs) in human. With the development of NGS, the number of detected human genome SNPs grow rapidly. However, it is laborious and time-consuming to attain experimental knowledge about the possible disease association of the variants. Several methods have been developed for deleteriousness prediction of detected SNPs. However, the prediction capabilities of certain programs varied a lot. The correct prediction of the nonsynonymous SNVs pathogenicity of individual program may result in very poor performance. Here we developed a new method named MPPS for pathogenicity prediction of nonsynonymous SNVs. As is known, several prediction programs are unable to predict pathogenicity for specific sets of variants29. Figure 1 showed that the percentage of correct predictions of MPPS was higher than other methods for both pathogenic variants and neutral variants. In the first step of MPPS analysis, we found that a parameter of 3 (3 prediction methods with the most correct predictions for ‘damaging’ SNVs and 3 prediction methods with the most correct predictions for ‘benign’ SNVs) could provide the best performance for pathogenicity prediction. When evaluating the performance of MPPS, we found that a parameter of less than 3 would reduce the accuracy of MPPS because one or two programs would dominate in the analysis process. We also found that a parameter of more than 3 would reduce the accuracy of MPPS because the accuracy will be reduced by some poor-performance programs. That’s the main reason we selected 3 prediction methods for both ‘damaging’ SNVs and ‘benign’ SNVs.
After obtaining the VCF files of the patients, the variants must be annotated and interpreted. This process is tedious and time-consuming, and require highly specific expertise in the domain of the analysis, not to mention the probability of the introduction of human errors. We developed a web-based framework named AADSM for automating analysis of clinical NGS data. AADSM could provide a complete solution in assisting and enhancing Mendelian diseases diagnosis. We could analyze ~20 patients at a time using AADSM. The overall running time of AADSM is related to the variant number of the VCF file. Typically, the whole process took 8 to 20 minutes for 20 samples. The data of the patients are stored and managed over AADSM in a secure manner. Before the submission of this manuscript, AADSM (http://jiyinsou.bgidx.cn/geneM/geneDiagnoseUpload.html) is still under test, and will be online in the near future.
In conclusion, we have developed a novel single test based on targeted NGS approaches for molecular diagnosis of various Mendelian disorders. In this method, we developed a new deleteriousness prediction method for nonsynonymous SNVs, and an automating annotation and diagnosis system for Mendelian diseases. A total of 303 patients were tested using this targeted NGS approaches. And we identified 219 disease causing mutations. The results proved that this method is an informative and effective approach to make a precise diagnosis for patients with Mendelian diseases, and can greatly improve the implementation of targeted NGS in clinical practice.
Methods
Study participants
In the current study, a total of 303 patients were enrolled between April 2015 and June 2017 in Tongji Hospital, Fuyang People’s Hospital and Renmin Hospital of Wuhan University. Clinical information of the patients was also summarized by experienced doctor. To evaluate the accuracy and stability of this technology, 3 samples derived from YH cell line (C1, C2 and C3) and 9 probands (Table 1) with known disease causing mutations were also collected. Written informed consent was obtained from all the participants or their parents before sample collection. This study was approved by the ethics committee of BGI (BGI-IRB15083).
Capture design, library preparation and sequencing
This study was approved by the Institutional Review Board of BGI and was carried out in accordance with the approved guidelines. We designed a new custom human array spanning 10,258,198 bp (~10 M, Roche NimbleGen). The array targets exons and 10 bp flanking intronic sequences of a total of 4,689 Mendelian diseases related genes, which were collected based on 6 well known databases (OMIM (www.omim.org), GeneReviews (www.ncbi.nlm.nih.gov/books/NBK1116), Orphanet (www.orpha.net), GENE Tests (www.genetests.org), CTGT (ctgt.net) and Genetics Home Reference (ghr.nlm.nih.gov)). A total of 4,413 Mendelian diseases can be diagnosed using this array. The 4,413 Mendelian diseases can be classified into 19 clinical categories (Fig. 4), including 870 Neuromuscular system disease, 547 Metabolic disease, 522 Growth and development abnormal, 344 Eye disease, 299 Bone disease, 241 Mental retardation and related syndromes, 238 Skin disease, 224 Circulatory system diseases, 203 Immune and infectious diseases, 173 Deafness and related syndromes, 169 Blood disease, 146 Endocrine system diseases, 111 Urinary system disease, 73 Reproductive system diseases, 72 Digestive system diseases, 51 Psychiatric and behavioral abnormal, 48 Cancer and Tumor Syndrome, 47 Respiratory system diseases, and 35 Oral disease.
Figure 4.

Flow diagram of bioinformatics analysis.
Using the QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany), genomic DNA of all the participants was isolated from the samples’ peripheral blood according to the manufacturer’s standard procedure. Library preparation was conducted at BGI following the guide of the standard protocol1. Briefly, 1 µg of genomic DNA was randomly fragmented to ~250 bp (Covaris S2). Next, end repair and A-tailing were performed. Then, adapters were ligated to both ends of the fragments. After purification, the adaptor-ligated DNA were subsequently amplified, purified, pooled, and hybridized to our ~10 M human array for enrichment. After library quality control, NGS libraries were then sequenced on HiSeq2500 or HiSeq4000 (Illumina) to generate paired-end reads.
Workflow of targeted NGS approaches
In this study, we developed a new bioinformatics analysis pipeline, including data filteration, sequence alignment, variants calling, annotation and automating reporting (Fig. 5).
Figure 5.
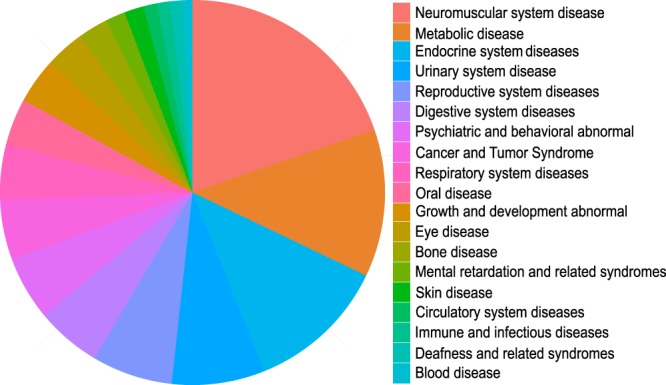
Disease classification of the 10 M chip.
First, raw sequencing reads (FASTQ files) were evaluated using SOAPnuke to generate “clean reads” for further analysis (removing low quality bases, adapters and other technical sequences). After reads filtering, the 100 bp clean reads were then aligned to human reference genome (hg19) with BWA and generated sorted BAM files using Picard. To improve read mapping, we then used GATK and Picard to perform local realignment and removal of PCR duplicates. Next, we used GAKT and in-house scripts to detect sequence variants (SNVs, Indels, CNVs, and microdeletions/microduplications) as reported before1. Variant annotation was performed using ANNOVAR34 with some modifications. In the annotation step, we downloaded 5 well known databases to facilitate annotation, including HGMD (www.hgmd.org), dbSNP (www.ncbi.nlm.nih.gov/SNP), 1000 Genome (www.1000genomes.org), HapMap (www.hapmap.ncbi.nlm.nih.gov), dbNSFP (www.varianttools.sourceforge.net/Annotation/DbNSFP) and a local database of our own11.
For SNVs detection, we developed a new pathogenicity prediction method MPPS (Mutation Pathogenicity Prediction Software) in this study. MPPS combined a total of 17 current deleteriousness-scoring methods and mainly took 3 steps to predict the pathogenicity of nonsynonymous SNVs. In the first step, choose the best 6 prediction methods (3 prediction methods with the most correct predictions for ‘damaging’ SNVs and 3 prediction methods with the most correct predictions for ‘benign’ SNVs) according to the position of a variant. When evaluating the performance of 17 deleteriousness prediction methods, we found that the prediction capabilities of certain programs varied a lot in human genome. For example, the percentage of correct predictions rate for ‘benign’ SNVs of GERP++ is high in some region, but low in other region of the human genome. So we randomly selected subsets of SNPs in human genome to obtain the percentage of correct predictions of the 17 pathogenicity prediction methods. Then we compared the results obtained for the same region to choose the best 6 prediction methods. In the second step, calculate the prediction score of the variant according to the weight of the best prediction methods (3 methods for ‘damaging’ prediction and 3 methods for ‘benign’ prediction). In this step, we assigned the variant a score of 10 when it is classified as ‘damaging’, a score of 5 when it is classified as ‘possibly damaging’, a score of 0 when it could not be classified, a score of −5 when it is classified as ‘possibly benign’, and a score of −10 when it is classified as ‘benign’ by a prediction method. The weight of a prediction method is defined as the percentage of correct predictions in the region. The prediction score of the variant from MPPS is the sum of the scores provided by individual tools according to the weight of the best prediction methods. In the last step, evaluate the pathogenicity of the variant. First, examine whether the variant is located in conserved positions using GERP++ and phyloP100way. If the variant is located in conserved positions, the prediction score of the variant from MPPS will be added one to the original quantity. A final score of −10 (benign) to 10 (damaging) will be generated for the best 3 ‘damaging’ SNV prediction methods and the best 3 ‘benign’ SNV prediction methods respectively. If the results from the best 3 ‘damaging’ SNV prediction methods and the best 3 ‘benign’ SNV prediction methods are the same, the pathogenicity of the variant will be reported, if not, this procedure will be performed again by choosing the second best prediction methods until the results from the 3 ‘damaging’ SNV prediction methods and the 3 ‘benign’ SNV prediction methods are the same. This will be repeated until the results are the same, if not, the variant will be reported mis-classified.
We also developed an automating diagnosis and reporting system (AADSM, Automating Annotation and Diagnosis System for Mendelian disease) for clinical scientists to help themselves in assisting and enhancing diagnostics of Mendelian diseases. Finally, clinical reports of the patients will be generated.
Validation
Clinical disease causing mutations identified using the HiSeq platform in the patients were confirmed by methods other than targeted NGS. If a pathogenic mutation was detected, segregation analysis was also performed for confirmation when the samples were available. For the validation and co-segregation analysis among the proband’s family members, we performed Sanger sequencing. We amplified regions of interest using polymerase chain reaction for sequencing (ABI 3730).
Data Availability
The datasets generated during and/or analyzed during the current study are not publicly available, but are available from the corresponding author on reasonable request.
Electronic supplementary material
Acknowledgements
We thank all the blood donors for their invaluable contribution to this study. This work was supported by National Natural Science Foundation of China (81502593 to Yan Sun). The project is non-profit research projects by government.
Author Contributions
Y.S. designed the research and wrote the first draft of the article. Y.W., L.R.Q., Y.S., Q.G. and H.D.D. provided patient specimens and collected clinical information. L.L., L.W.M., Z.Y.C., C.F., J.Y. and M.F.L. designed and performed the experiments. Y.Y., K.K., Y.W. and X.M.W. contributed to drafting and revising the manuscript. Y.S., J.F.M., G.P. and L.Q.D. performed data analysis. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yin Shen, Email: yinshen@whu.edu.cn.
Xiaoming Wei, Email: weixm@genomics.cn.
References
- 1.Liu Y, et al. Targeted Next-Generation Sequencing for Clinical Diagnosis of 561 Mendelian Diseases. PLoS One. 2015;10:e0133636. doi: 10.1371/journal.pone.0133636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013;14:681–691. doi: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- 3.Wei X, et al. Next-generation sequencing identifies a novel compound heterozygous mutation in MYO7A in a Chinese patient with Usher Syndrome 1B. Clin Chim Acta. 2012;413:1866–1871. doi: 10.1016/j.cca.2012.07.022. [DOI] [PubMed] [Google Scholar]
- 4.Sun Y, et al. Analysis of a Chinese pedigree with Zellweger syndrome reveals a novel PEX1 mutation by next-generation sequencing. Clin Chim Acta. 2013;417:57–61. doi: 10.1016/j.cca.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Neveling K, et al. A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat. 2013;34:1721–1726. doi: 10.1002/humu.22450. [DOI] [PubMed] [Google Scholar]
- 6.Bamshad MJ, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 7.Qing J, et al. Whole-exome sequencing to decipher the genetic heterogeneity of hearing loss in a Chinese family with deaf by deaf mating. PLoS One. 2014;9:e109178. doi: 10.1371/journal.pone.0109178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Y, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bell CJ, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3:65ra64. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooper GM, Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet. 2011;12:628–640. doi: 10.1038/nrg3046. [DOI] [PubMed] [Google Scholar]
- 11.Wei X, et al. Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing. PLoS One. 2011;6:e29500. doi: 10.1371/journal.pone.0029500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alkorta-Aranburu G, et al. Phenotypic heterogeneity in monogenic diabetes: the clinical and diagnostic utility of a gene panel-based next-generation sequencing approach. Mol Genet Metab. 2014;113:315–320. doi: 10.1016/j.ymgme.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weaver JM, Edwards PA. Targeted next-generation sequencing for routine clinical screening of mutations. Genome Med. 2011;3:58. doi: 10.1186/gm274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meder B, et al. Targeted next-generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circ Cardiovasc Genet. 2011;4:110–122. doi: 10.1161/CIRCGENETICS.110.958322. [DOI] [PubMed] [Google Scholar]
- 15.Nijman IJ, et al. Mutation discovery by targeted genomic enrichment of multiplexed barcoded samples. Nat Methods. 2010;7:913–915. doi: 10.1038/nmeth.1516. [DOI] [PubMed] [Google Scholar]
- 16.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 18.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shihab HA, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jagadeesh KA, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581–1586. doi: 10.1038/ng.3703. [DOI] [PubMed] [Google Scholar]
- 23.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter H, Douville C, Stenson PD, Cooper DN, Karchin R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics. 2013;14(Suppl 3):S3. doi: 10.1186/1471-2164-14-S3-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong C, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24:2125–2137. doi: 10.1093/hmg/ddu733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shihab HA, et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015;31:1536–1543. doi: 10.1093/bioinformatics/btv009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015;31:761–763. doi: 10.1093/bioinformatics/btu703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davydov EV, et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooper GM, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bao L, Zhou M, Cui Y. nsSNPAnalyzer: identifying disease-associated nonsynonymous single nucleotide polymorphisms. Nucleic Acids Res. 2005;33:W480–482. doi: 10.1093/nar/gki372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilissen C, Hoischen A, Brunner HG, Veltman JA. Unlocking Mendelian disease using exome sequencing. Genome Biol. 2011;12:228. doi: 10.1186/gb-2011-12-9-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lovric S, et al. Rapid detection of monogenic causes of childhood-onset steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2014;9:1109–1116. doi: 10.2215/CJN.09010813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are not publicly available, but are available from the corresponding author on reasonable request.