

Abstract
The human transcription elongation factor DSIF is highly conserved throughout all kingdoms of life and plays multiple roles during transcription. DSIF is a heterodimer, consisting of Spt4 and Spt5 that interacts with RNA polymerase II (RNAP II). DSIF binds to the elongation complex and induces promoter-proximal pausing of RNAP II. Human Spt5 consists of a NusG N-terminal (NGN) domain motif, which is followed by several KOW domains. We determined the solution structures of the human Spt5 KOW4 and the C-terminal domain by nuclear magnetic resonance spectroscopy. In addition to the typical KOW fold, the solution structure of KOW4 revealed an N-terminal four-stranded β-sheet, previously designated as the KOW3-KOW4 linker. In solution, the C-terminus of Spt5 consists of two β-barrel folds typical for KOW domains, designated KOW6 and KOW7. We also analysed the nucleic acid and RNAP II binding properties of the KOW domains. KOW4 variants interacted with nucleic acids, preferentially single stranded RNA, whereas no nucleic acid binding could be detected for KOW6-7. Weak binding of KOW4 to the RNAP II stalk, which is comprised of Rpb4/7, was also detected, consistent with transient interactions between Spt5 and these RNAP II subunits.
Introduction
Eukaryotic transcription catalysed by the enzyme RNA polymerase II (RNAP II) is tightly regulated by a variety of mechanisms. An important and widespread regulatory step is promoter proximal transcriptional pausing, which introduces an early block to RNAP II elongation after 20 to 70 transcribed bases. Participation of the general transcription factor DRB (5,6,-dichloro-1-β-D-ribofuranosylbenzimidazole) sensitivity inducing factor (DSIF) in this transcription rate limiting step is required for normal RNA synthesis1–4.
Human DSIF is a heterodimer composed of a 14 kDa (hSpt4) and a 120 kDa (hSpt5) subunit, the human homologues of Spt4 and Spt5 of Saccharomyces cerevisiae2,5. The hSpt5 subunit contains a region that is homologous to the N-terminal domain of the bacterial transcription factor NusG (NGN). NusG/Spt5 proteins are conserved in all three kingdoms of life6. Bacterial NusG and the archaeal Spt5 proteins are composed of the NGN-domain and a flexibly-connected C-terminal Kyrpides-Ouzounis-Woese (KOW) domain. Eukaryotic Spt5 proteins harbour several KOW domain copies whose functions have not been studied in full detail7,8. The function of the NGN is conserved in bacteria, archaea, and eukaryotes. In eukaryotes, the NGN domain binds to the Rpb1 and Rpb2 subunits of RNAP II forming a processivity clamp9,10. It thereby locks the nucleic acids with the RNAP in a closed and pause-resistant state11,12. Spt4, which is not present in bacteria, interacts with Spt5 via the NGN-domain.
Additionally, eukaryotic Spt5 includes an acidic region at its N-terminus and carries two C-terminal repeat (CTR) regions that can be phosphorylated by the positive transcription elongation factor pTEFb. Phosphorylated DSIF activates RNAP II elongation13. Whereas KOW1-5 domains have been detected in all analysed eukaryotes, the region downstream of the CTRs can only be found in metazoan and plant Spt514,15.
Although the structure of the mammalian RNAP II elongation complex has been determined, the precise functions of the different KOW domains of DSIF as well as that of the N-terminal acidic regions remain elusive9,10. hSpt5 is known to interact with RNAP II not only via the NGN but also through the region harbouring the KOW motifs. The KOW1 domain is flexible and binds between the clamp and the RNA exit tunnel of RNAP II9. Crosslinks and protein mass finger prints demonstrated an interaction between the C-terminal region of hSPT5 and Rpb1. The position of DSIF lies over the active centre cleft in the clamp domain of RNAP II9.
A cryo-electron microscopy (EM) structure of the mammalian RNAP II/DSIF complex and cross-linking experiments with Spt4/5 from Saccharomyces cerevisiae show that the Spt5 KOW4 interacts with RNAP II subunits Rpb4 and Rpb710,16. In archaea, RpoE and RpoF are homologous to Rpb7 and Rpb4, respectively, and have similar functions17,18. Rpb4 is suggested to function mainly in mRNA synthesis19. Rpb4/7 is thought to have an additional non-transcriptional role in transcription-coupled DNA repair mechanisms. The Rpb4/7 heterodimer can shuttle between nucleus and cytoplasm bound to mRNA and prevent it from degradation, implicating a role in mRNA export and translation. Furthermore, the RNA-binding protein Nrd1, involved in 3′ end formation of small nucleolar and nuclear RNAs during transcriptional termination, appears to interact with Rpb716,20,21.
The cryo-EM analysis of the RNAP II/DSIF complex and X-ray crystallography revealed that the hSpt5 KOW4 requires a structural element at its N-terminus, KOWx-4, for stability10. Moreover, the C-terminal region of hSpt5 also includes a tandem KOW domain, designated KOW6-7, which is positioned near the exiting RNA, suggesting a function in recruiting factors for RNA capping and in 3′ RNA processing22,23.
To explore in depth the functions of KOW4 and KOW6-7, we determined the solution structures of KOW4 (S522-G647) and KOW6-7 (G961-A1087) of hSpt5 by solution nuclear magnetic resonance (NMR) spectroscopy and performed in vitro interaction studies with nucleic acids and Rpb4/7. Fluorescence and NMR-based titrations showed that only KOW4 variants bind to nucleic acids with micromolar to nanomolar dissociation constants and interact weakly with Rpb4/7. For KOW6-7 no binding to nucleic acids or Rpb4/7 could be detected.
Results and Discussion
Solution structures
During transcription elongation RNAP II associates with DSIF which harbours several KOW domains that have been implicated in different RNA and protein interactions. To probe the functions of KOW4 and KOW6-7, we constructed several variants comprising KOW4 or the C-terminal region of hSpt5 for solution structure determination and interaction studies (Fig. 1). We were able to determine the solution structure of a minimal KOW4 domain spanning the hSpt5 region from amino acid S522 to G647 using multidimensional NMR spectroscopy. N-terminal truncations harbouring only the region of the predicted KOW4 domain did not yield stable proteins (data not shown)24. In addition, we determined the solution structure of the C-terminal KOW6-7 using a construct spanning G961 to A1087 (Fig. 1 and Table 1).
Figure 1.

Schematic representation of Spt4 and Spt5 proteins. Bacterial NusG consists of an N-terminal domain (NTD) followed by one KOW domain. The archaeal dimeric Spt4/5 consists of a NusG N-terminal (NGN) domain in Spt5, which is homologous to NusG and interacts with Spt4, and one KOW domain. Eukaryotic Spt4/5 and the human DSIF (hSpt4/5) harbour an additional N-terminal acidic region that is followed by the NGN domain. KOW1-5 are found in all eukaryotes, however the CTRs and the C-terminal region adjacent to KOW5 are only present in the Spt5 of metazoans and plants. Numbers represent amino acid positions, dotted lines indicate the regions of the KOW4 and KOW6-7 variants used in this study.
Table 1.
Solution structure statistics for KOW4 (S522-G647) and KOW6-7 (G961-A1087).
| Experimentally derived restraints | KOW4 | KOW6-7 | |
|---|---|---|---|
| Distance restraints | |||
| NOE | 628 | 544 | |
| intraresidual | 17 | 14 | |
| sequential | 167 | 135 | |
| medium range | 82 | 85 | |
| long range | 362 | 310 | |
| hydrogen bonds | 2*31 | 2*46 | |
| interdomain NOEs | 52 | 47 | |
| dihedral restraints | 120 | 124 | |
| projection restraints (RDC) | 1D (1H, 15N) | 80 | |
| Restraint violation | |||
| average distance restraint violation (Å) | 0.0063 +/− 0.0007 | 0.0036 +/− 0.001 | |
| maximum distance restraint violation (Å) | <0.10 | 0.11 | |
| average dihedral restraint violation (°) | 0.15 +/− 0.03 | 0.32 +/− 0.10 | |
| maximum dihedral restraint violation (°) | 1.38 | 4.21 | |
| average RDC restraint violation (Hz) | 0.21 +/− 0.03 | ||
| Maximum RDC restraint violation (Hz) | 1.16 | ||
| Deviation from ideal geometry | |||
| bond length (Å) | 0.00062 +/− 0.00003 | 0.00042 +/− 0.00004 | |
| bond angle (°) | 0.11 +/− 0.006 | 0.092 +/− 0.005 | |
| Coordinate precision a,b | |||
| backbone heavy atoms (Å) (all/defined structured region) |
0.91/0.69 | 0.57 | |
| all heavy atoms (Å) | 1.53/1.22 | 1.04 | |
| Ramachandran plot statisticsc (%) | 88.3/11.5/0.3/0.0 | 88.8/10.7/0.5/0.0 | |
aThe precision of the coordinates is defined as the average atomic root mean square difference between the accepted simulated annealing structures and the corresponding mean structure calculated for the given sequence regions.
bCalculated for residues 536–646 (all) or 536–575, 597–646 (defined structured region) (KOW4) and 978–1085 (all, KOW6-7).
cRamachandran plot statistics were determined by PROCHECK and noted by most favored/additionally allowed/generously allowed/disallowed.
The solution structure of KOW4 (S522-G647) shows the β-barrel fold typical for KOW domains (β-strands 7–11) that is covered by a β-hairpin (β-strands 5–6) called lid and stabilized N-terminally by a tilted, convex β-sheet (β-strands 1–4) (Fig. 2a,b). This N-terminal structural feature, formerly described as the connection domain between KOW3 and KOW4, is connected to the lid via a cationic linker and exhibits neither a typical KOW fold nor a KOW motif. It is composed mainly of a four-stranded antiparallel β-sheet (β-strands 1–4) with the strand order found in typical KOW domains. However, in KOW domains a fifth β-strand at the C-terminus adds to the β-sheet by pairing with the first strand, thereby forming the barrel-like domain. As the fifth β-strand is absent the region comprising only β-strands 1–4 should not be regarded as a typical KOW domain. It is required to stabilize the KOW4 domain and is connected to it by a cationic linker spanning K578 to F583.
Figure 2.

Solution structure of KOW4 (S522-G647). (a) Superposition of the 14 lowest energy structures. The disordered loop from K578 – F583 is encircled. The region from S522 to G532 is unstructured and not shown. (b) Ribbon representation depicting the region formerly described as a connection domain (β1–β4) in bright orange, the cationic linker in grey, the lid (β5–β6) in salmon, and the KOW4 domain (β7–β11) in light blue. (c) Stick representation showing relevant amino acids of the domain interface. (d) Distribution of R1/R2 for KOW4 (S522-G647).
The two domains interact at a highly apolar interface of about 700 Å2 in size. The interface mainly involves residues W536, L539, V547, V549, V551, M563 and L561 in the connection domain as well as F618, F621, F623, M635 and V637 in the KOW4 domain. In addition, the interface may be stabilized by a salt bridge between R552 and E631 (Fig. 2c).
To further characterize the domain interaction, we conducted 15N-based spin relaxation experiments. 15N relaxation rates could be determined for 117 residues (Fig. S1a). The heteronuclear {1H}15N steady state nuclear Overhauser effect (hetNOE) at 14 T shows values in the range of 0.7–0.8 for residues, except for the region N581-V585 and the termini. The hetNOE provides information about the motion of individual N-H bond vectors on the sub-ns timescale, significantly faster than the overall rotational tumbling. The values obtained are characteristic for restricted dynamics on the timescale of molecular tumbling for folded proteins. The hetNOE decreases towards the termini, demonstrating the increased flexibility on this timescale as is typical for unstructured termini. The cationic linker region shows slightly reduced values for the hetNOE indicating an enhanced flexibility on the sub-ns timescale. The transverse and longitudinal relaxation rates of 15N spins are very similar throughout the folded region of the protein, and the narrow distribution of the R1/R2 ratios suggests uniform overall tumbling of the protein (Fig. 2d). For proteins with flexibly connected domains, the relative domain motion is reflected in different distributions of the R1/R2 ratios of the two domains25–27. This is not the case for the KOW4 (S522-G647) construct. The fold of the KOW4 (S522-G647) domain deviates from the ideal sphere-like shape suggesting that the overall tumbling cannot be described by an isotropic rotation with a single correlation time. Determination of the rotational diffusion tensor based on the 15N relaxation rates requires a structural model and is a suitable method to validate the determined structure. The overall tumbling of KOW4 (S522-G647) can be well described by a prolate axial symmetric rotational diffusion tensor (Table 2) using 80 residues (36 residues for the connection domain and 44 for the KOW4 domain). A description by an isotropic rotation or an oblate axial symmetric tensor was rejected due to the χ2 statistics. The total asymmetric tensor did not significantly improve the result. The axial rotational diffusion tensor coincides well with the overall shape of the molecule (Fig. S1b) and the determined structure fits well with the relaxation data. Together with the numerous interdomain NOE spectroscopy (NOESY) cross-signals (52), these data confirm tight interdomain interaction.
Table 2.
Rotational diffusion tensor analysis for KOW4 (S522-G647) (80 vectors).
| isotropic | axialsymmetric (prolate) | axialsymmetric (oblate) | asymmetric | ||||
|---|---|---|---|---|---|---|---|
| D⊥ (108 s−1)a | 0.155 | D⊥ (108 s−1)a | 0.180 | Dx (108 s−1) | 0.154 | ||
| D|| (108 s−1)a | 0.202 | D|| (108 s−1)a | 0.158 | Dy (108 s−1) | 0.156 | ||
| Dz (108 s−1) | 0.202 | ||||||
| tc (ns) | 9.68 ± 0.04 | ||||||
| 1.50 · 102 | 8.44 · 101 | 1.32 · 102 | 8.43 · 101 | ||||
| 9.55 · 101 | 9.22 · 101 | 9.18 · 101 | 8.96 · 101 | ||||
| 9.88 · 101 | 9.81 · 101 | 9.75 · 101 | 9.52 · 101 | ||||
aD|| = Dz, D⊥ = Dx = Dy for the axialsymmetric model.
bχ2 = Σ (T1i,exp − T1i,calc)2/σ(T1i) + Σ (T2i,exp − T2i,calc)2/σ(T2i).
cConfidence limits (alpha = 0.1 or 0.05) of 500 Monte Carlo simulations. Models were accepted if χexp < χ0.1.
The C-terminal KOW harbouring region (G961-A1087) is also larger than predicted previously7. The solution structure reveals that it is composed of two domains, both sharing the typical KOW domain β-barrel fold (KOW6-7) (Fig. 3), is in good agreement with the crystal structure of a similar construct determined recently10. The linker between KOW6 and KOW7 consists of only five amino acids and the domain interface of about 500 Å2 is characterized by a small number of hydrophobic residues in the centre (W979, I984 and I1044) that are surrounded by polar residues. In particular, one lysine residue and one arginine residue (K1042, R1049) in KOW7 may form salt bridges with aspartates (D978 and D983) from the linker/KOW6 side (Fig. 3c). This makes this domain interaction distinct from that in KOW4 (S522-G647), which almost exclusively consists of hydrophobic interface residues.
Figure 3.

Solution structure of KOW6-7 (G961-A1087). (a) Superposition of the 20 lowest energy structures from Q974-A1087. The region from G961 to E973 is unstructured and not shown. (b) Ribbon representation highlighting KOW6 β1–β5) in cyan, and KOW7 (β6–β10) in red (c) Stick representation showing relevant amino acids of the domain interface. (d) Distribution of R1/R2 for KOW6-7 (G961-A1087). Colour assignment as in (b).
As described above for KOW4 (S522-G647) we performed 15N-based spin relaxation experiments to characterize the domain movement of KOW6-7 (G961-A1087). 15N relaxation rates were obtained for 112 non-overlapping amide resonances. The hetNOE experiments show the typical values around 0.7–0.8 for the folded part, demonstrating the highly restricted flexibility for the N-H bond vectors of this region. The flexibility on the sub-ns timescale increases towards the termini. For the region linking both domains no increased dynamical behaviour on the sub-ns timescale is observed. Similarly to KOW4 (S522-G647), the narrow, monomodal distribution of R1/R2 ratios suggests that the two KOW domains of KOW6-7 (G961-A1087) interact and the protein moves as one entity (Fig. 3d). Analysis of the rotational diffusion tensor using the 15N relaxation rates (Fig. S2a) of 79 residues (37 residues for KOW6 and 42 for KOW7) and the determined structure show that the overall tumbling can be well described by a prolate axial symmetric tensor (Table 3 and Fig. S2b). Similar to the results obtained with KOW4 (S522-G647), employment of the total asymmetric tensor did not improve the fit, and the isotropic rotation and the oblate axial symmetric tensor were rejected due to the χ2 statistics. Together with 47 interdomain NOE cross signals, the overall tumbling as single entity demonstrates the tight domain interaction of KOW6 and KOW7.
Table 3.
Rotational diffusion tensor analysis for KOW6-7 (G981-A1087) (79 vectors).
| isotropic | axialsymmetric (prolate) | axialsymmetric (oblate) | asymmetric | ||||
|---|---|---|---|---|---|---|---|
| D⊥ (108 s−1)a | 0.145 | D⊥ (108 s−1)a | 0.171 | Dx (108 s−1) | 0.143 | ||
| D|| (108 s−1)a | 0.193 | D|| (108 s−1)a | 0.138 | Dy (108 s−1) | 0.148 | ||
| Dz (108 s−1) | 0.192 | ||||||
| tc (ns) | 10.36 ± 0.04 | ||||||
| 1.96 · 102 | 8.11 · 101 | 1.29 · 102 | 8.06 · 101 | ||||
| 9.56 · 101 | 8.91 · 101 | 9.12 · 101 | 8.76 · 101 | ||||
| 9.99 · 101 | 9.31 · 101 | 9.42 · 101 | 9.11 · 101 | ||||
aD|| = Dz, D⊥ = Dx = Dy for the axialsymmetric model.
bχ2 = Σ(T1i,exp − T1i,calc)2/σ(T1i) + Σ(T2i,exp − T2i,calc)2/σ(T2i).
cConfidence limits (alpha = 0.1 or 0.05) of 500 Monte Carlo simulations. Models were accepted if χexp < χ0.1.
An overlay of all available hSpt5 KOW domain structures discloses that they all share the typical KOW domain β-barrel fold (Fig. 4a). The first of the two KOW domains of KOW6-7 (G961-A1087) (β-strands 1–5), designated as KOW6, had not been identified previously by sequence analysis. KOW6 lacks one highly conserved glycine residue characteristic for a KOW motif. Instead, an insertion between β-strands 1 and 2 results in a larger loop of unknown function between these two β-strands (Fig. 4a,b).
Figure 4.

Structural overlay of hSpt5 KOW domains and sequence comparison with the consensus KOW motif. (a) The structures of hSpt5 KOW2 (PDB: 2E6Z), KOW3 (PDB: 2DO3), KOW4 (S522-G647) (this work, PDB: 6EQY), KOW5 (PDB: 2E70), and KOW6-7 (G961-A1087) (this work, PDB: 6ER0) were used for the overlay. (b) Comparison of the hSpt5 KOW sequences with the consensus KOW motif highlights the conserved residues in yellow and orange and the insertion in KOW6 in bold.
In summary, both KOW4 (S522-G647) and KOW6-7 (G961-A1087) exhibit intramolecular domain-domain interactions and enlarge the structure of the basic KOW fold compared to other KOW domains of hSpt5. The extended structure might be important to present a larger binding surface for additional molecular interactions.
Substrate binding
The cryo-EM structure of the RNAP II/DSIF complex and the crystal structure of the complex from yeast indicated that the linker region between KOW4 and KOW5 is part of the RNA clamp that guides exiting RNA10,28. Thus, we investigated the affinity of several KOW4-5 variants (Fig. 1) and of KOW6-7 (G961-A1087) for nucleic acid substrates. Determination of the KD values by fluorescence anisotropy titrations indicated that KOW4 (S522-G647) has similar sequence-independent micromolar range affinities for DNA and RNA, with some preference for single-stranded substrates (Fig. 5a).
Figure 5.

Determination of nucleic acid binding affinities by fluorescence anisotropy measurements. 50 nM (a,d) or 25 nM (b,c) of fluorescent labelled nucleic acids as indicated were titrated with (a) KOW4 (S522-G647), (b) KOW4 (G531-L705), (c) KOW4 (G531-G754), (d) KOW6-7 (G961-A1087). The curves in (a) represent the best fit to a two-component binding equation to determine the KD values31 for ssDNA1 (12.2 ± 0.6 µM), dsDNA1 (20.1 ± 0.4 µM), ssDNA2 (24.5 ± 3.2 µM), dsDNA2 (41.2 ± 9.0 μM), and ssRNA (39.5 ± 7.4 μM).
We also used KOW4 variants that included the KOW4-KOW5 linker, KOW4 (G531-L705) (Fig. 5b), or, in addition, the KOW5 domain, KOW4-5 (G531-G754) (Fig. 5c), and titrated an ssRNA substrate. Since the data points obtained with both constructs exhibited sigmoidal binding kinetics, implying a complex binding behaviour, we were not able to determine KD values using a two-state binding model. Nevertheless, the data for both constructs indicated similar binding affinities for RNA in the high nanomolar range. We have not defined the mode of RNA binding, however. Compared to KOW4 (G531-L705), the presence of KOW5 in the construct KOW4-5 (G531-G754) did not further enhance RNA binding. These data suggest a substantial contribution of the linker region G648-G705 between KOW4 and KOW5, but not of KOW5 itself, to RNA binding (Fig. 5b,c). These results are in good agreement with the structural data of the RNAP II/DSIF complex, which showed that the KOW4-KOW5 linker is part of the RNA clamp and that KOW 5 primarily interacts with the Rpb1 dock as well as the Rpb2 wall domains to enhance transcription elongation10,28.
Although the twin KOW6-7 domains are positioned near the exiting RNA in the cryo-EM structure of the RNAP II/DSIF complex10, we could not detect nucleic acid binding for the corresponding KOW6-7 (G961-A1087) construct, indicating that it probably interacts exclusively with protein factors necessary for RNA elongation and/or RNA processing during transcription termination (Fig. 5d).
Determination of the nucleic acid binding site of KOW4 by NMR
To determine the nucleic acid binding site on KOW4, we conducted 2D [1H, 15N] heteronuclear single quantum correlation (HSQC)-based titration experiments of 15N-labelled KOW4 (S522-G647) with ssRNA (Fig. 6a), ssDNA and dsDNA (Fig. S3a,b). In each titration experiment, we observed chemical shift changes for some signals, indicating fast chemical exchange on the NMR time scale. Analysis of the chemical shift perturbations revealed the strongest effects on amino acids in the cationic linker and the lid region (around position 585) as well as in the centre of KOW4 (S522-G647) (around position 620) (Figs 6b and S3c,d).
Figure 6.

Determination of the KOW4 (S522-G647) nucleic acid binding interface. (a) Overlay of [1H, 15N] HSQC spectra recorded during titration with different protein (70 µM):ssRNA ratios as indicated. Relevant residues affected by ssRNA addition are labelled by arrows. (b) Normalized chemical shift changes upon ssRNA binding. Changes larger than 0.04 ppm were considered significant, changes from 0.04 to 0.06 ppm were assigned as weak, >0.06–0.08 ppm as medium, and >0.08 ppm as strong. The different regions of KOW4 (S522-G647) are indicated on top of the diagram. (c,d) Mapping of the observed chemical shift changes colour coded as in (b) on the structure of KOW4 (S522-G647) in ribbon (c) and surface representation (d). The amino acids exhibiting significant chemical shift changes are indicated as yellow (weak), orange (medium) and red (strong). (e) Electrostatic surface potential of KOW4 (S522-G647) calculated with the program APBS42, coloured from −3 kT/e to +3 kT/e.
Most residues affected by nucleic acid binding form a well-connected patch centred at the loop between the second and third β-strand of KOW4 (S522-G647). Moreover, most binding site residues carry a polar, positively charged, or aromatic side chain (Fig. 6c,d). This is consistent with their ability to contact the phosphate backbone and bases of the nucleic acid binding partner. KOW4 (S522-G647) includes a positively-charged patch formed by the KOW4 β-barrel and the cationic linker that matches the position of the nucleic acid-binding site (Fig. 6e). The position of the RNA binding region determined here supports the structural data of the RNAP II/DSIF complex in which the KOW4 β-barrel contacts the exiting RNA as part of the RNA clamp10.
Since the fluorescence anisotropy measurements revealed that the binding affinities for nucleic acids of KOW4 (G531-L705), which harbours the KOW4-KOW5 linker region, were higher than of KOW4 (S522-G647), we used an overlay of the 2D [1H, 15N] HSQC spectra of the two 15N labelled proteins to identify the signals corresponding to the KOW4-KOW5 linker (Fig. S4). The spectrum of KOW4 (G531-L705) demonstrates that the linker signals are located in the random coil region and that not all 58 residues of the linker are visible. This is probably due to fast exchange with the solvent and/or line broadening caused by conformational exchange. Titration of KOW4 (G531-L705) with ssRNA did not result in additional chemical shift perturbations in the linker region. Possibly, several linker region arginine residues contribute to binding without forming a defined structure. Indeed, mutagenesis studies suggested that these residues participate in RNA binding and in overall stabilization of the elongation complex10.
These results in combination with the fluorescence titration experiments described above indicate a role of the linker region A584-G705 in RNA binding.
Binding of the RNAP II subunit complex Rpb4/7 to KOW4 S522-G647
In vivo cross-linking experiments suggested an interaction of KOW4 and of the linker between KOW4 and KOW5 with the Rpb4 and Rpb7 subunits of RNAP II16. The 3D structures of the yeast and human RNAP II/DSIF complexes confirmed contacts between the Rpb4/7 stalk and KOW4. However, the precise location and orientation of KOW4 differs in the yeast and human RNAP II/DSIF complexes10,28. To analyse the interaction between Rpb4/7 and the KOW4 domain including the KOW4-KOW5 linker, we expressed and purified the human Rpb4/7 heterodimer and performed in vitro NMR titration experiments with 15N labelled KOW4 (G531-L705) (Fig. 7). The 2D [1H, 15N] HSQC spectra indicate weak interaction of Rpb4/7 with KOW4 (G531-L705) since only small chemical shift changes could be detected for the 1:1 complex (Fig. 7a). However, the 1D [1H, 15N] HSQC spectra of the titration confirmed the interaction of Rpb4/7 with KOW4 (G531-L705) (Fig. 7b). Signals of 15N labelled KOW4 (G531-L705) (19.3 kDa) decreased significantly upon addition of unlabelled Rpb4/7 (36.6 kDa), indicating complex formation (Fig. 7b). The increase in molecular mass upon binding results in faster magnetisation relaxation and thus line broadening.
Figure 7.
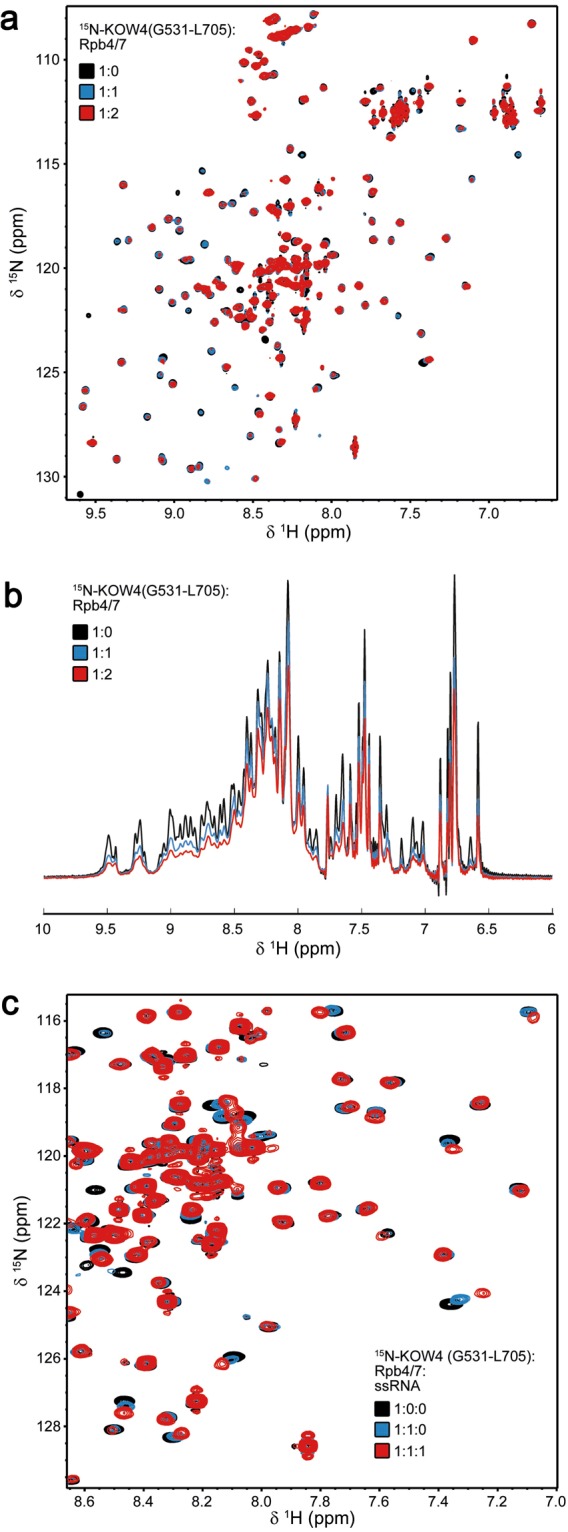
Binding of KOW4 (G531-L705) to Rpb 4/7. Proteins were measured in a buffer containing 50 mM sodium phosphate, pH 7.0, 50 mM NaCl and 1 mM DTT at 298 K. (a) 2D [1H, 15N] HSQC and (b) 1D [1H, 15N] HSQC spectra of 150 µM 15N labelled KOW4 (G531-L705) before (black), and after the addition of Rpb4/7 (molar ratio 1:1, 50 μM each, blue; molar ratio 1:2, KOW4 (G531-L705) 41 μM, red) (c) 2D [1H, 15N] HSQC spectra of 50 μM 15N-labelled KOW4 (G531-L705) in the absence (black) or presence (blue) of equimolar concentrations of Rpb4/7, and after the addition of ssRNA (molar ratio 1:1:1, 45 μM each, red).
Together with the titration experiments of KOW4 (S522-G647) with RNA (Fig. 6) these results support the notion of interactions between KOW4 and Rpb4/7 and/or RNA.
Comparison of the [1H, 15N] HSQC spectra of KOW4 titrated with either Rpb4/7 (Fig. 7a) or RNA (Fig. 6) demonstrated that several of the affected residues were identical, suggesting similar or overlapping binding sites for Rpb4/7 and ssRNA. To confirm this, we carried out a HSQC-based displacement experiment (Fig. 7c). We added ssRNA to the preformed 15N-KOW (G531-L705)/Rpb4/7 complex. The [1H, 15N] HSQC spectrum shows chemical shift changes of the same signals that were already affected upon addition of Rpb4/7 to KOW4 (G531-L705) (Fig. 7c). No additional chemical shift changes could be observed, indicating that ssRNA can displace Rpb4/7 and that the affinity of ssRNA to KOW4 (G531-L705) is higher than that of Rpb4/7.
Conclusion
We postulate that in a transcription initiation complex, the KOW4 domain of DSIF is loosely associated with the Rpb4/7 stalk of RNAP II. However, during elongation, when the transcribed RNA leaves the exit channel, the KOW4 domain and the KOW4-5 linker associate with the exiting RNA, thus stabilizing the elongation complex. Once the transcript is finished, Spt5 no longer binds to Rpb4/7. Without this stabilization, Rpb4/7 is able to dissociate from RNAP II and leave the nucleus together with the RNA, preventing its degradation. Thus Spt5, like NusG, could play a role in coupling transcription with translation, at least indirectly.
The structure of the elongating RNAP II/DSIF complex revealed that KOW4-5 forms an RNA clamp10. Comparison of the RNA binding affinities of KOW4 (S522-G647) lacking the linker and KOW4 (G531-L705) which includes the linker, further indicated that the linker between KOW4 and 5 contributes substantially to RNA binding (Fig. 5).
Interestingly, KOW6-7 (G961-A1087) exhibits a similar spatial arrangement as has been determined for the CTD of human KIN17, which harbours two SH3-like domains (PDB: 2CKK). KIN17 is a 45 kDa DNA and RNA binding protein that plays an important role in nuclear metabolism29. Similar to KOW6-7 (G961-A1087), the dimer interface of KIN17 harbours an Arg and a Lys residue (R351, K391). In addition, the protein also comprises an extended loop between β-strands 1 and 2. In contrast to hSpt5 KOW6-7 (G961-A1087), human KIN17 binds RNA. The negatively charged groove of the domain interface of KIN17 might constitute an additional surface for interaction with other proteins.
In the cryo-EM structure of the RNAP II/DSIF complex, no density was observed beyond the KOW5 domain10. Thus, no function for KOW6-7 could be determined. Since we find no direct interaction between KOW6-7 and nucleic acids, we propose that KOW6-7 might recruit other factors, for example proteins that play a role in RNA elongation, termination and processing, possibly via the positively charged amino acids flanking the groove (residues R1079, R989, K987, K1083, K1017) and the hydrophobic amino acids located within (W979, P1017, I1018).
Materials and Methods
Cloning, expression and protein purifications
KOW domains
The genes coding for KOW4 variants (S522-G647), (G531-L705) and (G531-G754) and KOW6-7 (G961-A1087) of hSpt5 were amplified by PCR using cDNA plasmid pOTB7 huSUPT5H (open biosystems, GE Healthcare) as a template. 5′ and 3′ primers harbouring NcoI and BamH I restriction sites, respectively were used to clone the PCR fragments into the vector pET-GB1a (G. Stier, EMBL, Heidelberg, Germany). The proteins expressed were thus fused to the C-terminus of the B1 domain of streptococcal protein G (GB1) and could be released via a tobacco etch virus (TEV) protease cleavage site located between GB1 and the KOW domain. Gene expression in lysogeny broth or in M9 medium for 15N and 13C labelling was performed in Escherichia coli strain BL21 (DE3) (Invitrogen-Life Technologies, Darmstadt, Germany) as described30. After induction with 100 µM isopropyl-thiogalactoside (IPTG) the temperature was reduced to 20 °C and protein overexpression was performed overnight. Proteins were purified via Ni-affinity chromatography (HisTrap, GE Healthcare, Munich, Germany) and TEV cleavage followed by a second Ni-affinity chromatography which allowed the removal of the 6xHis-GB1 tag. The free KOW proteins were collected in the flow-through and purified further via anion exchange chromatography using a QXL column (GE Healthcare, Munich, Germany). All constructs were flash-frozen with liquid nitrogen and stored at −80 °C.
human Rpb4/7
Genes adapted for E. coli were cloned in tandem into the expression vector pET15b. Rpb4 harboured a sequence coding for an N-terminal 6His tag. E. coli BL21 (DE3) cells (Invitrogen-Life Technologies, Darmstadt, Germany) transformed with the expression plasmid were grown to an optical density at 600 nm of 0.7–0.9 at 37 °C in LB or M9 medium containing 100 µg/ml ampicillin. Gene expression was induced with 1 mM IPTG for 4 h at 37 °C. The cells were then harvested by centrifugation. Cell lysis was performed as described for the KOW constructs in 50 mM sodium phosphate (pH 6.8), 500 mM NaCl, 1 mM dithiothreitol (DTT). The heterodimer was purified via Ni-affinity chromatography (HisTrap, GE Healthcare, Munich, Germany), followed by anion exchange chromatography (5 ml QXL column, GE Healthcare, Munich, Germany) after dialysis against 20 mM Tris/HCl pH 6.8, 20 mM NaCl, 1 mM DTT). The protein complex was eluted with an NaCl step gradient. Rpb4/7 containing fractions were combined, dialyzed against 20 mM Tris/HCl pH 6.8, 20 mM NaCl, 1 mM DTT, concentrated by ultrafiltration, and stored at −80 °C after flash-freezing with liquid nitrogen.
Fluorescence anisotropy measurements
Fluorescence anisotropy measurements were performed at 25 °C on a Synergy 2 microplate reader (biotek) equipped with black, sterile 96-well microtiter plates. The single stranded (ss) or double stranded (ds) DNAs or ssRNA were labelled with 6-FAM at the 5′ ends and contained the following sequences: ssDNA1: 6FAM-CTTATTGAATTA; ssDNA2: 6FAM-GAAAATTGGGTAAG; ssRNA: 6FAM-GGCGGUAGCGUG (metabion, Planegg, Germany). For dsDNA, the corresponding complementary strands without label were hybridized to ssDNA1 and 2 in fluorescence buffer (25 mM Tris/HCl pH 7.0, 50 mM NaCl) at a molar ratio of 1:1.2 (labelled:unlabelled strand) by heating the sample for 3 min at 95 °C, followed by a 10 min incubation step at 34 °C, or 40 °C for dsDNA1, or dsDNA2, respectively. Titrations were performed with individual samples, each containing 25 nM (for KOW4 (G531-L705), and KOW4 (G531-G754)) or 50 nM for KOW4 (G522-G647) and KOW6-7 (G961-A1087) of nucleic acid substrate and increasing amounts of protein in a total volume of 100 μl. The anisotropy of each sample was measured 6 times. Data were analysed by plotting the anisotropy value corrected for the value of the free nucleic acid vs. the protein concentration in the sample. The curves in Fig. 4a represent the best fit to a two-component binding equation describing the binding equilibrium to determine the KD values31. All binding curves were measured in triplicates.
NMR spectroscopy
All NMR experiments were conducted on Bruker Avance 600 MHz, 700 MHz, 900 MHz and 1000 MHz spectrometers, the latter three equipped with cryogenically cooled probes. Standard double and triple resonance experiments32,33 were conducted for backbone and sidechain resonance assignments at 298 K. 15N- and 13C-edited 3D NOESY experiments were recorded with mixing times of 120 ms at 298 K, in a buffer containing 20 mM sodium phosphate, pH 6.4, 20 mM NaCl, and 0.5 mM DTT.
1H, 15N residual dipolar couplings (RDCs) were determined for KOW6-7 (G961-A1087) by in-phase/anti-phase (IPAP) experiments34 using a sample containing 10 mg/ml Pf1 phages (AslaBiotech AB, Latvia)35. Determination of RDCs for KOW4 (S522-G647) were not successful using either Pf1 phages or mixtures of hexa-ethylene glycol monododecyl ether (C6E12), hexanol and water36 due to sample instability.
For characterization of overall and internal motions, 15N longitudinal (R1) and transverse (R2) relaxation rates together with the {1H}15N steady state NOE were determined using standard methods37 at 600.2 MHz (KOW4 (S522-G647)) or 700.2 MHz (KOW6-7 (G961-A1087)) 1H frequency at a calibrated temperature of 298 K.
R1 and R2 relaxation rates were determined by fitting a mono-exponential curve to the signal intensities using the CURVEFIT program (A.G. Palmer, Columbia University, USA). Rotational diffusion tensor analysis was done using the program tensor238. The error of relaxation rates was set to 5% to reflect potential systematic errors due to pulse imperfections, different temperatures in R1 and R2 experiments due to different radio frequency heating, and potential structural noise in the structural models. Residues with {1H},15N steady state values below 0.7 and residues with enhanced R2 rates due to chemical exchange were not included in the analysis. The different models for the rotational diffusion tensor were accepted or rejected based on the χ2 statistics using 500 Monte Carlo simulations.
Solution structure calculation
Distance restraints for structure calculation were derived from 15N-edited NOESY and 13C-edited NOESY spectra. NOESY cross peaks were classified according to their relative intensities and converted to distance restraints with upper limits of 3.0 Å (strong), 4.0 Å (medium), 5.0 Å (weak), and 6.0 Å (very weak). For ambiguous distance restraints the r−6 summation over all assigned possibilities defined the upper limit. Hydrogen bonds were included for backbone amide protons in regular secondary structure, when the amide proton did not show a water exchange cross peak in the 15N-edited NOESY spectrum.
Structure calculations were performed with the program XPLOR-NIH 1.2.139 using a three-step simulated annealing protocol with floating assignment of prochiral groups including a conformational database potential. The 14 (KOW4) and 20 (KOW6-7) structures showing the lowest values of the target function excluding the database potential were further analysed with X-PLOR39, PyMOL, and PROCHECK 3.5.440.
Structure and sequence alignments
Structure alignments were performed using WinCoot’s secondary structure matching algorithm. Sequence alignment of the hSpt5 KOW motifs was performed manually, based on the KOW consensus sequence provided by Pfam41.
Data deposition
The structure coordinates of hSpt5 KOW4 (S522-G647) and KOW6-7 (G961-A1087) were deposited in the Protein Data Bank under the accession codes 6EQY and 6ER0, respectively. Chemical shift assignments were deposited in the BioMagResBank accession numbers 34184 and 34185, respectively.
Electronic supplementary material
Acknowledgements
The authors thank Ulrike Persau, Ramona Heissmann and Andrea Hager for excellent technical assistance. This work was supported by Network Molecular Biosciences of the University of Bayreuth, the Deutsche Forschungsgemeinschaft (DFG) (grants Ro617/21-1, Ro617/17-1) and the Ludwig-Schaefer-prize of Columbia University (PR). This publication was funded by the DFG and the University of Bayreuth in the funding programme Open Access Publishing.
Author Contributions
P.R. and M.E.G. initiated the project and provided conceptual input. B.M.W., K.S. and S.H.K. supervised the project and designed experiments. A.R. carried out the cloning, expression and purification experiments. K.S., P.K.Z. and L.H. performed the NMR experiments and evaluated the data together with K.S. and S.H.K. P.K.Z. carried out the interaction studies. All authors prepared the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Stefan H. Knauer, Email: stefan.knauer@uni-bayreuth.de
Birgitta M. Wöhrl, Email: birgitta.woehrl@uni-bayreuth.de
References
- 1.Kwak H, Lis JT. Control of transcriptional elongation. Annu. Rev. Genet. 2013;47:483–508. doi: 10.1146/annurev-genet-110711-155440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wada T, et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998;12:343–356. doi: 10.1101/gad.12.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartzog GA, Fu J. The Spt4-Spt5 complex: a multi-faceted regulator of transcription elongation. Biochim. Biophys. Acta. 2013;1829:105–115. doi: 10.1016/j.bbagrm.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shetty A, et al. Spt5 Plays Vital Roles in the Control of Sense and Antisense Transcription Elongation. Mol. Cell. 2017;66:77–88.e5. doi: 10.1016/j.molcel.2017.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartzog GA, Wada T, Handa H, Winston F. Evidence that Spt4, Spt5, and Spt6 control transcription elongation by RNA polymerase II in Saccharomyces cerevisiae. Genes Dev. 1998;12:357–369. doi: 10.1101/gad.12.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Werner F. A nexus for gene expression-molecular mechanisms of Spt5 and NusG in the three domains of life. J. Mol. Biol. 2012;417:13–27. doi: 10.1016/j.jmb.2012.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ponting CP. Novel domains and orthologues of eukaryotic transcription elongation factors. Nucleic Acids Res. 2002;30:3643–3652. doi: 10.1093/nar/gkf498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kyrpides NC, Woese CR, Ouzounis CA. KOW: a novel motif linking a bacterial transcription factor with ribosomal proteins. Trends Biochem. Sci. 1996;21:425–426. doi: 10.1016/S0968-0004(96)30036-4. [DOI] [PubMed] [Google Scholar]
- 9.Bernecky C, Herzog F, Baumeister W, Plitzko JM, Cramer P. Structure of transcribing mammalian RNA polymerase II. Nature. 2016;529:551–554. doi: 10.1038/nature16482. [DOI] [PubMed] [Google Scholar]
- 10.Bernecky C, Plitzko JM, Cramer P. Structure of a transcribing RNA polymerase II-DSIF complex reveals a multidentate DNA-RNA clamp. Nat. Struct. Mol. Biol. 2017;24:809–815. doi: 10.1038/nsmb.3465. [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Rucobo FW, Sainsbury S, Cheung AC, Cramer P. Architecture of the RNA polymerase-Spt4/5 complex and basis of universal transcription processivity. EMBO J. 2011;30:1302–1310. doi: 10.1038/emboj.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sevostyanova A, Belogurov GA, Mooney RA, Landick R, Artsimovitch I. The β subunit gate loop is required for RNA polymerase modification by RfaH and NusG. Mol. Cell. 2011;43:253–262. doi: 10.1016/j.molcel.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 14.Jennings BH, et al. Locus-specific requirements for Spt5 in transcriptional activation and repression in Drosophila. Curr. Biol. 2004;14:1680–1684. doi: 10.1016/j.cub.2004.08.066. [DOI] [PubMed] [Google Scholar]
- 15.Guo S, et al. A regulator of transcriptional elongation controls vertebrate neuronal development. Nature. 2000;408:366–369. doi: 10.1038/35042590. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Giles C, Li S. Insights into how Spt5 functions in transcription elongation and repressing transcription coupled DNA repair. Nucleic Acids Res. 2014;42:7069–7083. doi: 10.1093/nar/gku333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Todone F, Brick P, Werner F, Weinzierl RO, Onesti S. Structure of an archaeal homolog of the eukaryotic RNA polymerase II RPB4/RPB7 complex. Mol. Cell. 2001;8:1137–1143. doi: 10.1016/S1097-2765(01)00379-3. [DOI] [PubMed] [Google Scholar]
- 18.Grohmann D, Werner F. Hold on!: RNA polymerase interactions with the nascent RNA modulate transcription elongation and termination. RNA Biol. 2010;7:310–315. doi: 10.4161/rna.7.3.11912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulz D, Pirkl N, Lehmann E, Cramer P. Rpb4 subunit functions mainly in mRNA synthesis by RNA polymerase II. J. Biol. Chem. 2014;289:17446–17452. doi: 10.1074/jbc.M114.568014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choder M. Rpb4 and Rpb7: subunits of RNA polymerase II and beyond. Trends Biochem. Sci. 2004;29:674–681. doi: 10.1016/j.tibs.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Harel-Sharvit L, et al. RNA polymerase II subunits link transcription and mRNA decay to translation. Cell. 2010;143:552–563. doi: 10.1016/j.cell.2010.10.033. [DOI] [PubMed] [Google Scholar]
- 22.Mayer A, et al. The Spt5 C-terminal region recruits yeast 3′ RNA cleavage factor I. Mol. Cell. Biol. 2012;32:1321–1331. doi: 10.1128/MCB.06310-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pei Y, Shuman S. Interactions between fission yeast mRNA capping enzymes and elongation factor Spt5. J. Biol. Chem. 2002;277:19639–19648. doi: 10.1074/jbc.M200015200. [DOI] [PubMed] [Google Scholar]
- 24.Yamaguchi Y, et al. Structure and function of the human transcription elongation factor DSIF. J. Biol. Chem. 1999;274:8085–8092. doi: 10.1074/jbc.274.12.8085. [DOI] [PubMed] [Google Scholar]
- 25.Horstmann M, et al. Domain motions of the Mip protein from Legionella pneumophila. Biochemistry. 2006;45:12303–12311. doi: 10.1021/bi060818i. [DOI] [PubMed] [Google Scholar]
- 26.Burmann BM, Scheckenhofer U, Schweimer K, Rösch P. Domain interactions of the transcription-translation coupling factor Escherichia coli NusG are intermolecular and transient. Biochem. J. 2011;435:783–789. doi: 10.1042/BJ20101679. [DOI] [PubMed] [Google Scholar]
- 27.Drögemüller J, et al. An auto-inhibited state in the structure of Thermotoga maritima NusG. Structure. 2013;21:365–375. doi: 10.1016/j.str.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ehara H, et al. Structure of the complete elongation complex of RNA polymerase II with basal factors. Science. 2017;357:921–924. doi: 10.1126/science.aan8552. [DOI] [PubMed] [Google Scholar]
- 29.le Maire A, et al. A tandem of SH3-like domains participates in RNA binding in KIN17, a human protein activated in response to genotoxics. J. Mol. Biol. 2006;364:764–776. doi: 10.1016/j.jmb.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 30.Leo B, Hartl MJ, Schweimer K, Mayr F, Wöhrl BM. Insights into the structure and activity of prototype foamy virus RNase H. Retrovirology. 2012;9:14. doi: 10.1186/1742-4690-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartl MJ, et al. AZT resistance of simian foamy virus reverse transcriptase is based on the excision of AZTMP in the presence of ATP. Nucleic Acids Res. 2008;36:1009–1016. doi: 10.1093/nar/gkm1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bax A, Grzesiek A. Methodological advances in protein NMR. Acc. Chem. Res. 1993;26:131–138. doi: 10.1021/ar00028a001. [DOI] [Google Scholar]
- 33.Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 1999;34:39–158. doi: 10.1016/S0079-6565(98)00025-9. [DOI] [Google Scholar]
- 34.Ottiger M, Delaglio F, Bax A. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- 35.Hansen MR, Mueller L, Pardi A. Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat. Struct. Biol. 1998;5:1065–1074. doi: 10.1038/4176. [DOI] [PubMed] [Google Scholar]
- 36.Rückert M, Otting G. Alignment of biological macromolecules in novel nonionic Liquid Crystalline Media for NMR Experiments. J. Am. Chem. Soc. 2000;122:7793–7797. doi: 10.1021/ja001068h. [DOI] [Google Scholar]
- 37.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry (N. Y.) 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 38.Dosset P, Hus JC, Blackledge M, Marion D. Efficient analysis of macromolecular rotational diffusion from heteronuclear relaxation data. J. Biomol. NMR. 2000;16:23–28. doi: 10.1023/A:1008305808620. [DOI] [PubMed] [Google Scholar]
- 39.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003;160:65–73. doi: 10.1016/S1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 40.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 41.Finn RD, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–85. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.