

Abstract
Many of estradiol’s behavioral effects are mediated, at least partially, via extra-nuclear estradiol signaling. Here, we investigated whether two estrogen receptor (ER) agonists, targeting ERα and G protein-coupled ER-1 (GPER-1), can promote rapid anorexigenic effects. Food intake was measured in ovariectomized (OVX) rats at 1, 2, 4, and 22h following subcutaneous (s.c.) injection of an ERα agonist (PPT; 0–200μg/kg), a GPER-1 agonist (G-1; 0–1600μg/kg), and a GPER-1 antagonist (G-36; 0–80μg/kg). To investigate possible cross-talk between ERα and GPER-1, we examined whether GPER-1 blockade affects the anorexigenic effect of PPT. Feeding was monitored in OVX rats that received s.c. injections of vehicle or 40μg/kg G-36 followed 30min later by s.c. injections of vehicle or 200μg/kg PPT. Selective activation of ERα and GPER-1 alone decreased food intake within 1h of drug treatment, and feeding remained suppressed for 22h following PPT treatment and 4h following G-1 treatment. Acute administration of G-36 alone did not suppress feeding at any time point. Blockade of GPER-1 attenuated PPT’s rapid (within 1h) anorexigenic effect, but did not modulate PPT’s ability to suppress food intake at 2, 4 and 22h. These findings demonstrate that selective activation of ERα produces a rapid (within 1h) decrease in food intake that is best explained by a non-genomic signaling pathway and thus implicates the involvement of extra-nuclear ERα. Our findings also provide evidence that activation of GPER-1 is both sufficient to suppress feeding and necessary for PPT’s rapid anorexigenic effect.
Keywords: Estrogen, Rapid effects, Food intake, PPT, G-1
1. Introduction
Estradiol decreases food intake in many species, including humans (Lyons et al., 1989), and loss-of-function studies show that deficits in estradiol signaling promote overeating and weight gain in both sexes (Binh et al., 2011; Chen et al., 2009; Lovejoy et al., 2008). While this provides compelling evidence that estradiol plays a critical role in controlling food intake, the underlying cellular and molecular mechanisms are poorly understood and will remain so until the specific estrogen receptors (ERs) and downstream signaling events are identified.
Estradiol was once thought to exert its diverse effects solely through two members of the nuclear steroid hormone receptor superfamily, ERα and ERβ, which regulate transcription of estradiol-responsive genes (Nilsson et al., 2001). In addition to this genomic signaling pathway, it is now well established that estradiol interacts with extra-nuclear ERs, including cytosolic ERα and ERβ, palmitoylated forms of ERα and ERβ that are trafficked to the plasma membrane (Pedram et al., 2007), and the de novo membrane-associated ER (mER), GPER-1 (originally called GPR30), a G protein-coupled receptor that is structurally unrelated to ERα and ERβ (Carmeci et al., 1997). Ligand-bound, extra-nuclear ERs promote rapid alterations in cell signaling by interacting with effector proteins that activate kinase cascades and other second messenger systems. As a result, extra-nuclear ERs transduce estradiol signals into more rapid changes in cellular activity, and thus behavior, than the canonical nuclear ERs, which require hours to days to manifest a change in behavior (Balthazart et al., 2018). It should be noted, however, that extra-nuclear ER-initiated signaling can also affect gene expression via targeted interactions with downstream transcription factors (Vasudevan et al., 2005). Thus, while extra-nuclear ERs alone transduce rapid cellular responses, including changes in membrane excitability, synaptic plasticity, and cell survival (Levin, 2009), both nuclear and extra-nuclear ERs modulate gene transcription. Taken together, these recent advances in our understanding of rapid estradiol signaling have led to the growing acceptance that extra-nuclear ERs contribute to many of estradiol’s actions that were once believed to be mediated solely by nuclear ERs (Levin, 2009).
Various approaches have been used to investigate the specific ERs that mediate estradiol’s anorexigenic effect. Transgenic studies have shown that a null mutation of ERα, but not ERβ, promotes obesity in mice (Heine et al., 2000; Ohlsson et al., 2000), but it is unclear whether the weight gain is due to changes in energy intake or expenditure (Eckel, 2011). Pharmacological studies provide clearer evidence in support of a role for ERα in the estrogenic control of food intake. Administration of the ERα agonist 4,4′,4″-(4-Propyl-[1H]-pyrazole-1,3,5-triyl) trisphenol (PPT), but not the ERβ agonist 2,3-bis(4-Hydroxyphenyl)-propionitrile (DPN), decreases food intake in ovariectomized (OVX) rats (Santollo et al., 2007; Thammacharoen et al., 2009; Wegorzewska et al., 2008). Unlike the non-specific ER agonist estradiol benzoate (EB), which suppresses feeding with a latency of ~24h (e.g., Asarian and Geary, 2002; Santollo et al., 2007), PPT decreases food intake within 3–6h of treatment (Santollo et al., 2007; Thammacharoen et al., 2009). This suggests that PPT may preferentially target extra-nuclear ERα or increase trafficking of ERα to the membrane. Because PPT’s anorexigenic effect has not been examined until at least 3h post-treatment (Santollo et al., 2007; Thammacharoen et al., 2009), a more detailed time-course analysis, particularly within the first hour after treatment, is needed to determine whether PPT suppresses food intake with a sufficiently short latency that would preclude the involvement of nuclear ERα and thus indirectly implicate extra-nuclear ERα.
The involvement of extra-nuclear ERs in the estrogenic control of food intake is further supported by a study in which central administration of a membrane-delimited form of estradiol (E2-BSA; filtered through a 3-kDA cutoff filter to remove any free estradiol that could trigger intracellular effects) decreased food intake in OVX rats (Santollo et al., 2013). While this provides evidence that mER-initiated signaling is sufficient to decrease food intake, it does not reveal which mERs are involved. One possible candidate is mERα, since activation of ERα by PPT suppresses feeding within 3h (Santollo et al., 2007). Another possible candidate is GPER-1. Imaging studies confirm GPER-1 expression in feeding-related brain areas (Brailoiu et al., 2007; Spary et al., 2013), and some pharmacological studies report an anorexigenic effect of GPER-1. For example, acute administration of the GPER-1 agonist (±)-1-[(3aR*,4S*,9bS*)-4-(6-Bromo-1,3-benzodi-oxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quino-lin-8-yl]-ethanone (G-1) decreased daily food intake in OVX guinea pigs (Washburn et al., 2013), but failed to decrease 24-h food intake in OVX rats (Santollo and Daniels, 2015). These discrepant findings, together with emerging reports of functional cross talk between ERα and GPER-1 in cultured cells (Vivacqua et al., 2009) and dopaminergic neurons in mice (Bourque et al., 2015), highlight the need for further studies investigating both the independent and interactive involvement of ERα and GPER-1 in the estrogenic control of food intake.
The current study investigated the time course over which activation of ERα and GPER-1 suppresses feeding in female rats. First, we tested the hypothesis that PPT, which targets both nuclear and extra-nuclear ERα, decreases food intake with a short latency that is best explained by the more rapid signaling actions of extra-nuclear ERα. We also examined the acute effects of the GPER-1 agonist G-1 and the GPER-1 antagonist (±)-(3aR*,4S*,9bS*)-4-(6-Bromo-1,3-benzo-dioxol-5-yl)-3a,4,5,9b-tetrahydro-8-(1-methylethyl)-3H-cy-clopenta[c]quinolone (G-36) on food intake. Because some ER antagonists can act as selective estrogen receptor modulators (SERMs) with mixed agonist/antagonist effects (Kuiper et al., 1999), the latter experiment was conducted to rule out the possibility that the GPER-1 antagonist G-36 might suppress feeding, similar to that observed following treatment with the GPER-1 agonist G-1. To investigate whether cross talk between ERα and GPER-1 contributes to the estrogenic control of food intake, we investigated whether GPER-1 blockade attenuates PPT’s anorexigenic effect.
2. Methods
2.1. Animals and housing
Female Long-Evans rats (Charles River Breeding Laboratory, Raleigh, NC), weighing 225–250g at study onset, were housed individually in custom plastic tub cages that provided access to spill-resistant food cups. Throughout the study, rats were given ad libitum access to powdered chow (Purina 5001, St. Louis, MO) and tap water unless otherwise specified. Animal rooms were maintained at 20±2°C with a 12:12h reverse light-dark cycle (dark onset=1300h). Animal usage and all procedures were approved by the Florida State University Institutional Animal Care and Use Committee.
2.2. Surgery
Animals were anesthetized with 3% isoflurane (Butler Schein Animal Health, Dublin, Ohio), delivered at a rate of 1L/min, and bilaterally OVX using an intra-abdominal approach. Following surgery, animals received intraperitoneal (i.p.) injections of butorphanol (0.5mg/kg; Fort Dodge Animal Health, Fort Dodge, IA) to minimize postoperative pain, and subcutaneous (s.c.) injections of gentamicin (10mg/kg; Pro Labs Ltd., St. Joseph, MO) to minimize risk of infection. Behavioral testing commenced after two weeks of postoperative recovery.
2.3. Experiment 1: acute effect of the ERα agonist PPT on food intake
A within-subject design was used to assess the anorexigenic effect of varying doses of the ERα agonist PPT (Tocris Bioscience, Minneapolis, MN) in OVX animals (N=9). We choose PPT because it has a 410-fold selectivity for ERα over ERβ (Harris et al., 2002) and has been used extensively to examine the contribution of ERα to the estrogenic control of food intake. PPT was dissolved in 50% dimethyl sulfoxide (DMSO) vehicle (Sigma-Aldrich, St. Louis, MO; diluted in physiological saline (Teknova, Hollister, CA)), to yield the following doses: 0, 10, 20, 40, 100, or 200μg/kg PPT. On test days, food was removed from the animals’ cages during the last 2h of the light phase to prevent the consumption of a meal just prior to drug treatment. Within 5min of dark onset, animals received s.c. injections of a single dose of PPT, administered in random order. Food cups were returned at dark onset and food intake was monitored at 1, 2, 4, and 22h. Test days were spaced at least 3days apart (range=3–5days), and daily food intake was monitored on non-test days. This 3–5day wash-out period was based on a previous study that provided a detailed examination of the time course of PPT’s anorexigenic effect in female rats (Santollo et al., 2007). In this study, acute administration of PPT (300μg/kg) decreased food intake for 15h, with no further decrease in food intake during the 7days following PPT treatment (Santollo et al., 2007). This suggests that PPT’s anorexigenic effect is restricted to the day of injection, as has been reported in other studies (Roesch, 2006; Santollo and Eckel, 2009; Thammacharoen et al., 2009), with no further anorexigenic effect that could reflect a delayed genomic effect. Thus, while our within-subjects design does not allow us to completely rule out the possibility that a delayed genomic effect of one dose of PPT could contribute to the anorexia observed in response to a subsequent dose of PPT, the 24-h restricted time course of PPT’s anorexigenic effect suggests that such an outcome is unlikely. The doses of PPT and the time course over which food intake was monitored were chosen to extend previous studies that utilized higher doses of PPT (300–2000μg/kg) and limited assessment of food intake to the 3–24h period following drug treatment (Roesch, 2006; Santollo et al., 2007; Thammacharoen et al., 2009).
2.4. Experiment 2: acute effects of the GPER-1 agonist (G-1) and GPER-1 antagonist (G-36) on food intake
A within-subject, randomized design was used to assess the anorexi-genic effect of varying doses of the GPER-1 agonist G-1 (Cayman Chemical Company, Ann Arbor, MI) in OVX animals (N=8). G-1 is a racemic but diastereomerically pure compound with high (nanomolar) affinity for GPER-1, no detectable biological activity at ERα at concentrations up to 10μM, and no transcriptional activity in cells lacking GPER-1 (Albanito et al., 2007; Bologa et al., 2006). On test days, animals received s.c. injections of G-1 (in 50% DMSO vehicle) administered in two separate dose ranges: 0, 800 and 1600μg/kg G-1, followed by 0, 0.4, 2, and 4μg/kg G-1. All other aspects of the study design were the same as described for Experiment 1. The higher range of G-1 doses was chosen to be consistent with a previous study investigating the effect of G-1 on food intake in guinea pigs (Washburn et al., 2013). The lower range of G-1 doses was based on preliminary results from our lab and a previous study examining the effects of G-1 on social learning in mice (Ervin et al., 2015).
Pharmacological compounds designed to block ERs can sometimes act like SERMs with mixed, tissue-specific, agonist/antagonist properties (Kuiper et al., 1999). As such, food intake was also assessed in OVX animals (N=8) following acute administration of the cell-permeable, non-steroidal GPER-1 antagonist G-36 (Cayman Chemical Company, Ann Arbor, MI), an isosteric derivative of G-1 with high (nM) affinity for GPER-1 (Dennis et al., 2011). This study was deemed necessary to rule out the possibility that G-36 may possess SERM-like activity with respect to feeding behavior. We chose G-36 over the first-generation GPER-1 antagonist G-15, which has been shown to bind to and activate ERα when administered in the 10μM range. Unlike G-15, G-36 exerts no detectable activity at ERα in cell-based, competitive binding assays (Dennis et al., 2011). On test days, animals received s.c. injections of 0, 20, 40, or 80μg/kg G-36, in 50% DMSO vehicle, administered according to a within-subject, randomized design. We selected our range of doses to be consistent with previous studies (Dennis et al., 2009; Dennis et al., 2011). All other aspects of the study design were the same as described for Experiment 1.
2.5. Experiment 3: effect of GPER-1 blockade on PPT’s anorexigenic effect
Our final study investigated the anorexigenic effect of the ERα agonist PPT both in the presence and absence of the GPER-1 antagonist G-36. On test days, OVX animals (N=9) received s.c. injections of vehicle (50% DMSO) or 40μg/kg of G-36, followed 30min later by s.c. injections of vehicle or 200μg/kg of PPT. This yielded four drug treatments (vehicle/vehicle, G-36/vehicle, vehicle/PPT, and G-36/PPT), which were administered at 3-day intervals according to a within-subject, randomized design. As in the previous experiments, drug treatment was completed just prior to dark onset, food was returned to the animals’ cages at dark onset, and food intake was monitored at 1, 2, 4 and 22-h. Daily food intake was monitored on non-test days.
2.6. Data analysis
Data are presented as means±SEM throughout. Repeated-measures ANOVAs (drug dose) were used to assess the effects of PPT, G-1, and G-36 on cumulative food intake at 1, 2, 4, and 22-h post drug treatment (Experiments 1 and 2). In Experiment 3, the effects of GPER blockade on PPT’s anorexigenic effect at 1, 2, 4 and 22-h were analyzed via two-factor (GPER-1 antagonist × ERα agonist) repeated-measure ANOVAs. Significant ANOVA effects (p<0.05) were followed up using Tukey’s honestly significant difference post-hoc test.
3. Results
3.1. Experiment 1: acute effects of the ERα agonist PPT on food intake
An examination of the time course of PPT’s anorexigenic effect during the first 4h following drug treatment revealed an inhibitory effect of PPT on cumulative food intake at 1, 2 and 4h, F(5,40)=3.12–6.42, p<0.05–0.01, η2=0.28–0.45 (Fig. 1). Post-hoc tests revealed that the duration of PPT’s anorexigenic effect during this period was dose-dependent, with low doses producing a more transient effect than higher doses. At 1h, all but the lowest dose of PPT (20–200μg/kg) decreased food intake, relative to that consumed following vehicle treatment (p<0.05, d=1.33–1.75). At 2h, 40 and 100μg/kg PPT decreased food intake relative to vehicle (p<0.05, d=1.20 and 1.36, respectively), whereas the highest (200μg/kg) dose of PPT decreased food intake relative to vehicle and the two lowest doses of PPT (p<0.05, d=1.04–2.11). At 4h, the two highest doses of PPT (100 and 200μg/kg) decreased food intake relative to vehicle and the two lowest doses of PPT (p<0.05, d=1.00–3.30).
Fig. 1.
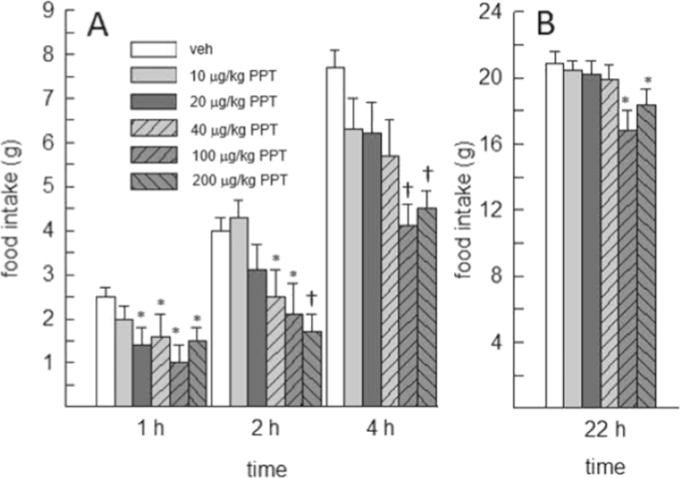
Effect of the ERα agonist PPT on short-term (up to 4h) and daily (22-h) food intake in OVX rats (N=9). (A) At 1h, food intake was decreased by all but the lowest dose of PPT. At 2 and 4h, PPT produced a dose-dependent decrease in food intake. (B) Daily (22-h) food intake was decreased by the two highest doses of PPT (100 and 200μg/kg). *Less than vehicle, p<0.05. †Less than vehicle, 0.01, and 200μg/kg PPT.
Cumulative daily (22-h) food intake was also decreased by PPT, F(5,40)=5.93, p<0.05, η2=0.43. Only the two highest doses of PPT (100 and 200μg/kg) decreased 22-h food intake, relative to vehicle treatment (p<0.05, d=1.58 and 0.76, respectively) (Fig. 1). PPT had no effect on food intake on non-test days.
3.2. Experiment 2: acute effects of the GPER-1 agonist G-1 and the GPER-1 antagonist G-36 on food intake
Administration of G-1 at high doses (800 and 1600μg/kg) failed to affect food intake at 1, 4, and 22h, F(2,14)=0.072–2.139, p=0.08–0.931, η2=0.01–0.23. However, at the 2h time point, food intake was influenced by G-1, F(2,14)=4.628, p<0.05, η2=0.39. Post-hoc analysis revealed that the 800μg/kg dose of G-1 significantly increased food intake relative to vehicle (p<0.05, d=1.07) (Fig. 2).
Fig. 2.
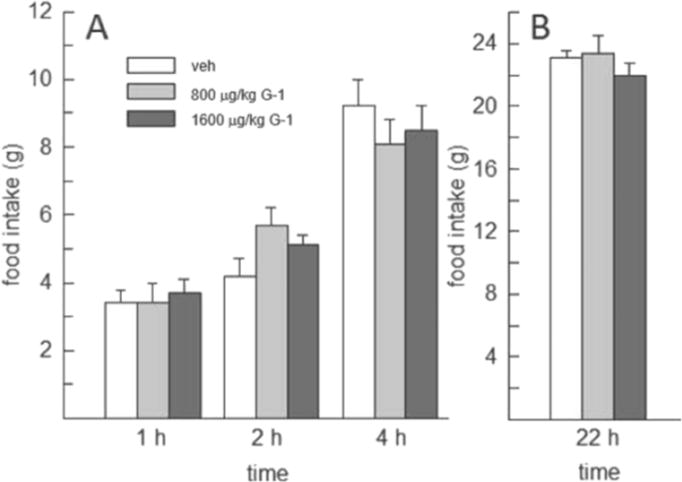
Effect of high doses of the GPER-1 agonist G-1 on food intake in OVX rats (N=8). G-1, at 800 and 1600μg/kg, failed to decrease food intake at any of the time points examined. Food intake was increased, however, by 800μg/kg G-1, relative to vehicle, at 2h.
In comparison, administration of G-1 at lower doses (0.4–4μg/kg) decreased food intake throughout the first 4h following drug treatment, F(3,21)=3.36–3.96, p<0.05, η2=0.25–0.36 (Fig. 3). Post-hoc analysis revealed that all doses of G-1 decreased food intake within the first h of drug treatment, relative to that consumed following vehicle treatment (p<0.05, d=0.85–1.11). At 2 and 4h, only the 2μg/kg G-1 continued to decrease food intake, relative to vehicle (p<0.05, d=0.81 and 1.24, respectively).
Fig. 3.
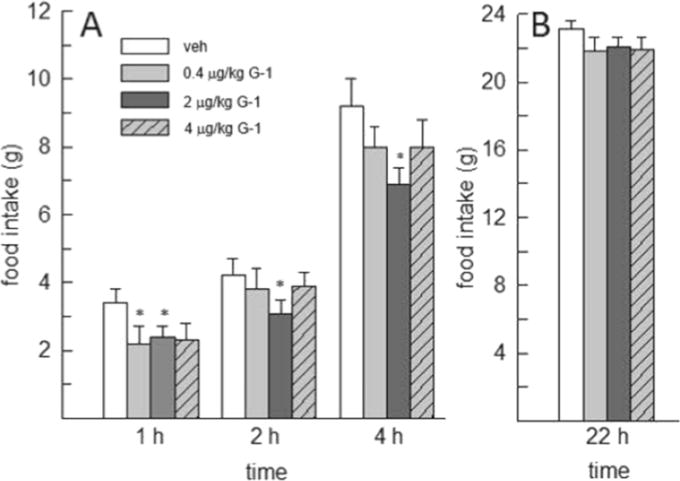
Effect of lower doses of the GPER-1 agonist G-1 on food intake in OVX rats (N=8). (A) At 1h, food intake was similarly decreased by all doses of G-1, relative to vehicle. At 2 and 4h, only the 2μg/kg dose of G-1 had a sustained anorexigenic effect. (B) There was no effect of G-1 at the 22h time point. *Less than vehicle, p<0.05.
The anorexigenic effect of G-1 was limited to the first 4h following drug treatment as G-1 failed to alter cumulative daily (22-h) food intake, F(3,21)=0.575, p=0.638, η2=0.08 (Fig. 3). Daily food intake was also unaffected by G-1 on non-test days.
Administration of the GPER-1 antagonist (G-36) alone had no effect on food intake at any time point examined, F(3,21)=0.704–2.317, p=0.1–0.436, η2=0.09–0.24 (Fig. 4). Food intake on non-test days was also unaffected by G-36 treatment.
Fig. 4.
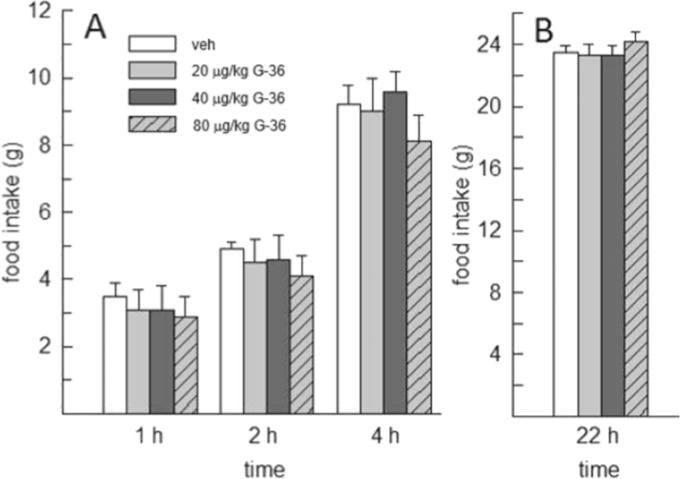
Effect of the GPER-1 antagonist G-36 on food intake in OVX rats (N=8). G-36 failed to influence food intake at any of the time points examined.
3.3. Experiment 3: effect of GPER-1 blockade on PPT’s anorexigenic effect
Pretreatment with the GPER-1 antagonist G-36 influenced PPT’s ability to suppress food intake at 1h, F(1,8)=20.863, p<0.005, η2=0.72 (Fig. 5). Post-hoc tests revealed that PPT decreased 1-h food intake in rats pretreated with vehicle (p<0.05, d=1.03), but this anorexigenic effect of PPT was blocked when animals were pretreated with G-36 (Fig. 5A). This interactive effect of GPER-1 blockade and ERα activation was transient, however, as food intake at 2, 4, and 22h was influenced only by a main effect of PPT, F(1,8)=12.61–34.95, p<0.01–0.001, η2=0.61–0.81. At each of these time points, PPT decreased food intake both in the presence and absence of GPER-1 blockade (p<0.05, d=1.00–2.04) (Fig. 5B–D).
Fig. 5.
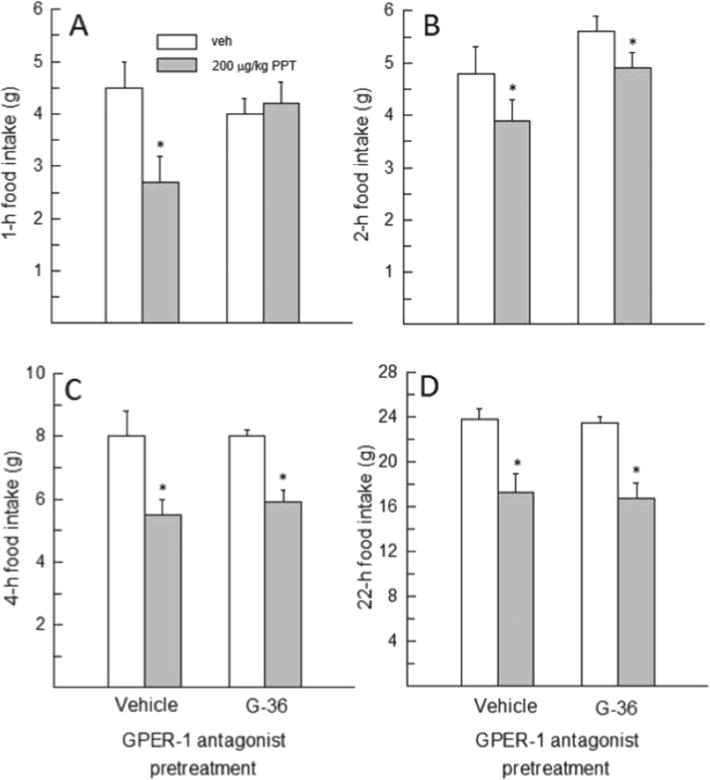
Effect of GPER-1 blockade on PPT’s anorexigenic effect in OVX rats (N=9). (A) Pretreatment with G-36 blocked PPT’s anorexigenic effect at 1h. (B–D) Pretreatment with G-36 did not affect PPT’s inhibitory effect on food intake at 2, 4 and 22h. *Less than vehicle, p<0.05.
4. Discussion
Previous work has shown that selective activation of mERs (via E2-BSA) is sufficient to decrease feeding in female rats (Santollo et al., 2013), but little is known about the specific ERs underlying this response. Here, we found that acute administration of ERα and GPER-1 agonists (PPT and G-1, respectively) suppress food intake within 1h of drug treatment, a time course that is best explained by non-genomic signaling events evoked through activation of extra-nuclear ERs. Our time-course analysis also revealed that PPT suppressed food intake for up to 22h, whereas G-1 suppressed food intake for up to 4h. In a final experiment, we demonstrated that GPER-1 blockade attenuates PPT’s anorexigenic effect at 1h, but not at 2, 4, or 22h, suggesting a rapid but transient interaction between ERα and GPER-1 in the estrogenic control of food intake.
In the current study, acute administration of 20–200μg/kg PPT decreased food intake within 1h, whereas the lowest dose of PPT had no effect. This extends previous reports that PPT decreases food intake within 3–6h in rats (Santollo et al., 2007) and 8h in mice (Ervin et al., 2015). Our findings also demonstrate that lower doses of PPT than used previously (Roesch, 2006; Santollo et al., 2007; Thammacharoen et al., 2009) promote robust decreases in food intake that occur with minimal delay, as female rats typically consume their first nocturnal meal within 1h of dark onset (Eckel et al., 2000). While PPT targets both nuclear and extra-nuclear ERα, which are derived from the same transcript (Razandi et al., 1999), its rapid anorexigenic effect is best explained by non-genomic signaling events involving mERα and/or cy-tosolic ERα. This interpretation is supported by the earlier demonstration that non-selective activation of ERs by membrane-delimited E2-BSA is sufficient to decrease feeding in OVX rats (Santollo et al., 2013). Our findings extend this report by providing indirect evidence that mERα may be involved in mediating E2-BSA’s anorexigenic effect.
The minimally effective anorexigenic dose of PPT (20μg/kg) suppressed feeding for 1h, whereas higher doses of PPT (100 and 200μg/ kg) decreased feeding for up to 22h with no further anorexigenic effect on non-test days. This suggests that low, near-threshold doses of PPT may target extra-nuclear ERα exclusively, resulting in a rapid but transient decrease in food intake. While the more prolonged anorexigenic effect of higher doses of PPT may simply reflect sustained activation of extra-nuclear ERα, it could also reflect a genomic signaling mechanism, resulting in the transcriptional regulation of estradiol-responsive genes. In this context, it is interesting that membrane-delimited E2-BSA has been shown to decrease food intake for up to 24h (Santollo et al., 2013), similar to the time course of PPT’s anorexigenic effect reported here. Thus, activation of mERs alone appears sufficient to produce the kind of cellular response (i.e., transcriptional regulation of estradiol-responsive genes) necessary to promote a sustained decrease in feeding. Thus, PPT’s prolonged anorexigenic effect could be mediated by nuclear and/or extra-nuclear ERα as both can activate signaling cascades that result in a change in gene transcription. Further studies are needed to delineate the relative contributions of nuclear and extra-nuclear ERα to PPT’s prolonged anorexigenic effect.
Activation of GPER-1 by high doses of G-1 (800 and 1600μg/kg) did not decrease food intake at any time point in the current study. However, we did observe an increase in food intake following 800μg/ kg G-1 that was observed only at the 2-h time point. Because activation of GPER-1 was not expected to increase food intake, and there is no obvious physiological mechanism underlying such an isolated response, we believe that this is not a biologically meaningful effect. Previously, these same high doses of G-1 were shown to decrease 24-h food intake in female guinea pigs (Washburn et al., 2013), but we found no similar anorexigenic effect in OVX rats. The reason for the discrepant findings is unclear, but may be related to a species difference in G-1 metabolism, GPER-1 expression, or GPER-1 binding affinity. To the best of our knowledge, similarly high doses of G-1 have not been examined in previous studies involving rats or mice. Moreover, the use of such high doses of G-1 is potentially problematic as a high concentration of ligand can overstimulate its cognate receptor, resulting in rapid receptor internalization via intracellular proteins like arrestin (Lefkowitz, 1998). Indeed, internalization of mERα has been observed in hypothalamic cell lines following high dose PPT treatment (Wong et al., 2015). Thus, high doses of G-1 may have caused rapid internalization of GPER-1 in our rats but failed to exert a similar effect in guinea pigs. This could have been the result of lower circulating levels of G-1 in guinea pigs resulting from their higher body weight relative to the rats in our study, and/or a species difference in G-1 metabolism.
Previous studies have reported behavioral changes in various paradigms following much lower doses of G-1 than those used in the feeding study involving guinea pigs. For example, a 6μg/kg dose of G-1 enhanced social learning in a social transmission of food preference test in female mice at 30min and 2h, but not at 8h (Ervin et al., 2015). In light of these findings, we examined whether similarly low doses of G-1 would be effective in our feeding paradigm. We found that all doses of G-1 decreased food intake within 1h of administration, and one dose of G-1 (2μg/kg) suppressed feeding for up to 4h (Fig. 3). This time course of G-1′s anorexigenic effect is similar to the behavioral changes reported in the mouse study of social learning. It is also interesting that, in this mouse study, higher doses of G-1 (10 and 30μg/kg) failed to alter social learning (Ervin et al., 2015). Thus, low doses of G-1 were optimal to promote rapid but transient changes in social and feeding behavior, whereas higher doses of G-1 were ineffective in both behavioral paradigms. This suggests that G-1 may affect behavior over a fairly narrow dose range in rats and mice. It also suggests that the difference in the duration of G-1′s and PPT’s anorexigenic effect (4h and 22h, respectively) may be best explained by different mechanisms of action (i.e., recruitment of genomic effects by PPT, but not G-1), rather than a difference in dose, since large doses of G-1 failed to decrease food intake here. That said, we cannot rule out the possibility that differences in dosing contributed to the differential time courses of PPT’s and G-1′s anorexigenic effect.
Three studies have examined the effect of G-1 on feeding in rodents. In male mice, lateral ventricular infusion of G-1 decreased food intake at 24h, but not at 2, 4, or 8h post administration (Kwon et al., 2014). The lack of effect at the earlier time points is likely due to a floor effect as the mice had only consumed ~1g of chow in 8h. Nevertheless, this study in mice supports our finding that G-1 decreases food intake in rats, but the time course may be dose- sex-, and/or species-specific. The mouse study reporting G-1-dependent changes in social learning at 30min and 2h did not report a change in total food intake at either time point (Ervin et al., 2015), but a floor effect may have obscured the ability to detect an anorexigenic effect of G-1 in this paradigm as the mice had only consumed ~0.7g by the 2-h time point. It will be important, therefore, to assess G-1′s effect on short-term food intake in mice that are motivated to eat >1g of chow (i.e., when given a highly palatable food or following a period of food restriction). Finally, our findings are not consistent with a study in which acute administration of G-1 (25–400μg/kg) failed to decrease 24-h food intake in OVX rats (Santollo and Daniels, 2015). However, it should be noted that administration of the same doses of G-1 that failed to alter feeding produced a reliable decrease in angiotensin II-stimulated water intake during a brief (30-min) drinking test (Santollo and Daniels, 2015). Because a much higher dose of estradiol (at least ten-fold) is required to decrease water intake compared to that required to decrease food intake (Asarian and Geary, 2002; Findlay et al., 1979) it is possible that an inhibitory effect of G-1 on feeding may have been observed in the previous study if lower doses of G-1, similar to those used in our and previous studies (Ervin et al., 2015; Kwon et al., 2014) had been administered.
Mouse models of GPER-1 deficiency have also been used to assess the involvement of GPER-1 in the regulation of food intake and body weight. Deletion of the GPER-1 gene in C57BL/6J mice produced an increase in visceral adiposity, relative to wildtype controls (Haas et al., 2009). A similar finding was reported by another group but food intake and locomotor activity were unaffected, suggesting that the obesity phenotype manifested via another mechanism (Sharma et al., 2013). A more recent study provided the first comprehensive examination of feeding behavior in GPER-1 null mice (Davis et al., 2014). In this study, daily food intake was unaffected by GPER-1 deletion in both sexes; however, female, but not male, GPER-1 null mice displayed decreased sensitivity to the anorexigenic effects of estradiol, cholecystokinin and lep-tin (Davis et al., 2014). While these findings offer some support for a role of GPER-1 in mediating estradiol’s inhibitory effect on feeding, and are thus consistent with our findings, the overall literature involving GPER-1 null mice is largely equivocal, with discrepant findings often attributed to differences in the techniques used to silence the GPER-1 gene.
Previous studies have shown that compounds classified as ER antagonists can sometimes function as SERMs with tissue-specific, mixed agonist/antagonist properties (Bryant and Dere, 1998). For example, methyl-piperidino-pyrazole (MPP) is a non-steroidal compound that is synthesized via the addition of a basic side chain (BSC) to a member of the pyrazole-triol family that includes PPT (Zhou et al., 2009). While MPP was originally classified as an ERα antagonist based on in vitro assays (Sun et al., 2002), it was subsequently shown to mimic estradiol’s effects on feeding and uterine tissue in vivo (Davis et al., 2008; Santollo and Eckel, 2009), presumably through metabolic cleavage of the BSC that served to reveal the latent agonist activity of MPP in vivo (Zhou et al., 2009). Given that the GPER-1 antagonist G-36 is an isosteric derivative of the GPER-1 agonist G-1 (Dennis et al., 2011), it was important to confirm that G-36 exerts no agonist activity in vivo with regard to food intake. Here, acute administration of G-36 alone failed to decrease food intake across a wide range of doses. This confirms a lack of agonist effect, at least with respect to feeding behavior, with this compound.
There is considerable interest in determining whether specific mERs function alone or interact with other extra-nuclear or nuclear ERs to mediate estradiol’s diverse effects. With the caveat that some degree of cross-reactivity may exist between estrogenic compounds (i.e., G-15 can activate ERα at high doses (Dennis et al., 2011)), multiple reports provide evidence that ERα/GPER-1 cross-talk underlies estradiol-dependent changes in multiple cellular processes. For example, both ERα and GPER-1 are required for estradiol-dependent proliferation of BG-1 ovarian cancer cells (Albanito et al., 2007). Additionally, blockade of both ERα and ERβ in an isolated perfused heart attenuated G-1-mediated changes in heart rate and left ventricular pressure (Filice et al., 2009). In another study, blockade of GPER-1 attenuated the ability of the ERα-derived peptide, ERα17p, to activate ERK1/2 in leiomyoma cells (Leiber et al., 2015). Even fewer studies have investigated the potential cross talk between GPER-1 and ERα in the brain. To the best of our knowledge, only a single report exists in which GPER-1 blockade was shown to prevent PPT’s neuroprotective effect of dopaminergic neurons against MPTP insult in mice (Bourque et al., 2015). While these studies provide evidence of GPER-1/ERα cross talk in cell culture systems and isolated brain tissue, whether a similar interaction mediates any of estradiol’s diverse behavioral effects is currently unknown. As such, we utilized the GPER-1 antagonist G-36 to determine whether GPER-1 blockade would alter the magnitude or time course of PPT’s anorexigenic effect. We found that pretreatment with G-36 blocked PPT’s ability to decrease food intake during the first hour following drug treatment (Fig. 5A). This suggests that GPER-1 is necessary for PPT’s rapid (within 1h) anorexigenic effect, and supports the notion that GPER-1/ERα cross talk may be involved in the estrogenic control of food intake. While the nature of the potential interaction between GPER-1 and ERα is unknown, it has been hypothesized that GPER-1 may aid in the assembly of ERα at the outer plasma membrane (Levin, 2009). It is now well established that post-translational lipid modification of ERα via palmitoylation facilitates ERα conjugation to anchor proteins such as caveolin-1, which can then form a signaling complex with classic GPCRs such as metabotropic glutamate receptors or, possibly, GPER-1 (Micevych and Mermelstein, 2008). Furthermore, co-immunoprecipitation studies have confirmed a close physical interaction between GPER-1 and ERα (Vivacqua et al., 2009). Taken together, these studies suggest that GPER-1/ERα cross talk may be the result of a physical interaction at the outer plasma membrane, rather than a common related downstream mechanism.
While pretreatment with G-36 blocked PPT’s rapid anorexigenic effect at 1h, it had no effect on PPT-induced decreases in food intake at 2, 4, and 22h. This suggests that GPER-1 is not required for PPT’s prolonged anorexigenic effect. The suppression of food intake at these later time points may reflect a genomic signaling mechanism involving nuclear and/or extra-nuclear ERs that activate a downstream transcription factor. However, one cannot rule out the possibility that a rapid half-life of G-36 may have prohibited its ability to provide prolonged blockade of GPER-1. Further experiments are necessary to test this hypothesis.
In conclusion, we have shown that PPT and G-1 exert a rapid (within 1h) inhibitory effect on feeding behavior, and that the duration of PPT’s anorexigenic effect exceeds that of G-1. While this differing time-course may be mediated, at least in part, by dose, it is difficult to equate relative doses of PPT and G-1 as multiple biological and pharmacological factors affect their activity, including drug half-life, mechanism of drug metabolism, and potential differential receptor expression of ERα and GPER-1. Another possibility is that PPT’s more prolonged anorexi-genic effect may be mediated, at least in part, through a genomic signaling mechanism that may not be similarly invoked by G-1. Our findings also provide the first evidence that the rapid (within 1h) inhibitory effect of PPT on feeding may be mediated via cross talk between mERα and GPER-1. Given the almost exclusive focus on the genomic signaling events that underlie estradiol’s ability to suppress feeding, our findings provide an important new direction for further research. It will be particularly important to investigate the cellular and molecular mechanisms underlying the mER-dependent decreases in feeding behavior reported here.
Acknowledgments
This research was supported by a Council on Research and Creativity grant from the Florida State University (LAE), a Program in Neuroscience Fellowship (MJB), and NIH T32 grant MH093311 (MJB).
Footnotes
Conflict of interest
The authors have no conflicts to disclose.
References
- Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, Oprea TI, Prossnitz ER, Musti AM, Andò S, Maggiolini M. G protein–coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17β-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007;67:1859–1866. doi: 10.1158/0008-5472.CAN-06-2909. [DOI] [PubMed] [Google Scholar]
- Asarian L, Geary N. Cyclic estradiol treatment normalizes body weight and restores physiological patterns of spontaneous feeding and sexual receptivity in ovariectomized rats. Horm Behav. 2002;42:461–471. doi: 10.1006/hbeh.2002.1835. [DOI] [PubMed] [Google Scholar]
- Balthazart J, Choleris E, Remage-Healey L. Steroids and the brain: 50years of research, conceptual shifts and the ascent of non-classical and membrane-initiated actions. Horm Behav. 2018;99:1–8. doi: 10.1016/j.yhbeh.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binh TQ, Nakahori Y, Hien VTT, Khan NC, Lam NT, Mai LB, Yamamoto S. Correlations between genetic variance and adiposity measures, and gene-gene interactions for obesity in postmenopausal Vietnamese women. J Genet. 2011;90:1–9. [PubMed] [Google Scholar]
- Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- Bourque M, Morissette M, Di Paolo T. Neuroprotection in Parkinsonian-treated mice via estrogen receptor activation requires G protein-coupled estrogen receptor 1. Neuropharmacology. 2015;95:343–352. doi: 10.1016/j.neuropharm.2015.04.006. [DOI] [PubMed] [Google Scholar]
- Brailoiu E, Dun SL, Brailoiu GC, Mizuo K, Sklar LA, Oprea TI, Prossnitz ER, Dun NJ. Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J Endocrinol. 2007;193:311–321. doi: 10.1677/JOE-07-0017. [DOI] [PubMed] [Google Scholar]
- Bryant HU, Dere WH. Selective estrogen receptor modulators: an alternative to hormone replacement therapy. Proc Soc Exp Biol Med. 1998;217:45–52. doi: 10.3181/00379727-217-44204. [DOI] [PubMed] [Google Scholar]
- Carmeci C, Thompson D, Ring HZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. 1997;45:607–617. doi: 10.1006/geno.1997.4972. [DOI] [PubMed] [Google Scholar]
- Chen HH, Lee WJ, Fann CSJ, Bouchard C, Pan WH. Severe obesity is associated with novel single nucleotide polymorphisms of the ESR1 and PPARgamma locus in Han Chinese. Am J Clin Nutr. 2009;90:255–262. doi: 10.3945/ajcn.2009.25914. [DOI] [PubMed] [Google Scholar]
- Davis AM, Mao J, Naz B, Kohl JA, Rosenfeld CS. Comparative effects of estradiol, methyl-piperidino-pyrazole, raloxifene, and ICI 182 780 on gene expression in the murine uterus. J Mol Endocrinol. 2008;41:205–217. doi: 10.1677/JME-08-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KE, Carstens EJ, Irani BG, Gent LM, Hahner LM, Clegg DJ. Sexually dimorphic role of G protein-coupled estrogen receptor (GPER) in modulating energy homeostasis. Horm Behav. 2014;66:196–207. doi: 10.1016/j.yhbeh.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, Oprea TI, Yamaguchi Y, Hayashi SI, Sklar LA, Hathaway HJ, Arterburn JB, Prossnitz ER. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol. 2011;127:358–366. doi: 10.1016/j.jsbmb.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel LA. The ovarian hormone estradiol plays a crucial role in the control of food intake in females. Physiol Behav. 2011;104:517–524. doi: 10.1016/j.physbeh.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel LA, Houpt TA, Geary N. Spontaneous meal patterns in female rats with and without access to running wheels. Physiol Behav. 2000;70:397–405. doi: 10.1016/s0031-9384(00)00278-x. [DOI] [PubMed] [Google Scholar]
- Ervin KSJ, Mulvale E, Gallagher N, Roussel V, Choleris E. Activation of the G protein-coupled estrogen receptor, but not estrogen receptoralpha orbeta, rapidly enhances social learning. Psychoneuroendocrinology. 2015;58:51–66. doi: 10.1016/j.psyneuen.2015.04.002. [DOI] [PubMed] [Google Scholar]
- Filice E, Recchia AG, Pellegrino D, Angelone T, Maggiolini M, Cerra MC. A new membrane G protein-coupled receptor (GPR30) is involved in the cardiac effects of 17beta-estradiol in the male rat. J Physiol Pharmacol. 2009;60:3–10. [PubMed] [Google Scholar]
- Findlay AL, Fitzsimons JT, Kucharczyk J. Dependence of spontaneous and an-giotensin-induced drinking in the rat upon the oestrous cycle and ovarian hormones. J Endocrinol. 1979;82:215–225. doi: 10.1677/joe.0.0820215. [DOI] [PubMed] [Google Scholar]
- Harris HA, Katzenellenbogen JA, Katzenellenbogen BS. Characterization of the biological roles of the estrogen receptors, ERα and ERβ, in estrogen target tissues in vivo through the use of an ERα-selective ligand. Endocrinology. 2002;143:4172–4177. doi: 10.1210/en.2002-220403. [DOI] [PubMed] [Google Scholar]
- Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-α knockout mice. Proc Natl Acad Sci U S A. 2000;97:12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper GGJM, van den Bemd GJCM, van Leeuwen JPTM. Estrogen receptor and the SERM concept. J Endocrinol Investig. 1999;22:594–603. doi: 10.1007/bf03343616. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ. G protein-coupled receptors: III. New roles for receptor kinases and β-arrestins in receptor signaling and desensitization. J Biol Chem. 1998;273:18677–18680. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- Leiber D, Burlina F, Byrne C, Robin P, Piesse C, Gonzalez L, Leclercq G, Tanfin Z, Jacquot Y. The sequence Pro295–Thr311 of the hinge region of oestrogen receptor α is involved in ERK1/2 activation via GPR30 in leiomyoma cells. Biochem J. 2015;472:97–109. doi: 10.1042/BJ20150744. [DOI] [PubMed] [Google Scholar]
- Levin ER. Plasma membrane estrogen receptors. Trends Endocrinol Metab. 2009;20:477–482. doi: 10.1016/j.tem.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovejoy JC, Champagne CM, de Jonge L, Xie H, Smith SR. Increased visceral fat and decreased energy expenditure during the menopausal transition. Int J Obes. 2008;32:949–958. doi: 10.1038/ijo.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons PM, Truswell AS, Mira M, Vizzard J, Abraham SF. Reduction of food intake in the ovulatory phase of the menstrual cycle. Am J Clin Nutr. 1989;49:1164–1168. doi: 10.1093/ajcn/49.6.1164. [DOI] [PubMed] [Google Scholar]
- Micevych PE, Mermelstein PG. Membrane estrogen receptors acting through metabotropic glutamate receptors: an emerging mechanism of estrogen action in brain. Mol Neurobiol. 2008;38:66–77. doi: 10.1007/s12035-008-8034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- Ohlsson C, Hellberg N, Parini P, Vidal O, Bohlooly M, Rudling M, Lindberg MK, Warner M, Angelin B, Gustafsson Obesity and disturbed lipoprotein profile in estrogen receptor-α-deficient male mice. Biochem Biophys Res Commun. 2000;278:640–645. doi: 10.1006/bbrc.2000.3827. [DOI] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Sainson RCA, Kim JK, Hughes CC, Levin ER. A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem. 2007;282:22278–22288. doi: 10.1074/jbc.M611877200. [DOI] [PubMed] [Google Scholar]
- Roesch DM. Effects of selective estrogen receptor agonists on food intake and body weight gain in rats. Physiol Behav. 2006;87:39–44. doi: 10.1016/j.physbeh.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Santollo J, Daniels D. Activation of G protein-coupled estrogen receptor 1 (GPER-1) decreases fluid intake in female rats. Horm Behav. 2015;73:39–46. doi: 10.1016/j.yhbeh.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santollo J, Eckel LA. Effect of a putative ERα antagonist, MPP, on food intake in cycling and ovariectomized rats. Physiol Behav. 2009;97:193–198. doi: 10.1016/j.physbeh.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santollo J, Wiley MD, Eckel LA. Acute activation of ER alpha decreases food intake, meal size, and body weight in ovariectomized rats. Am J Phys Regul Integr Comp Phys. 2007;293:R2194–R2201. doi: 10.1152/ajpregu.00385.2007. [DOI] [PubMed] [Google Scholar]
- Santollo J, Marshall A, Daniels D. Activation of membrane-associated estrogen receptors decreases food and water intake in ovariectomized rats. Endocrinology. 2013;154:320–329. doi: 10.1210/en.2012-1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Hu C, Brigman JL, Zhu G, Hathaway HJ, Prossnitz ER. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology. 2013;154:4136–4145. doi: 10.1210/en.2013-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spary EJ, Chapman SE, Sinfield JK, Maqbool A, Kaye J, Batten TFC. Novel G protein-coupled oestrogen receptor GPR30 shows changes in mRNA expression in the rat brain over the oestrous cycle. Neurosignals. 2013;21:14–27. doi: 10.1159/000333296. [DOI] [PubMed] [Google Scholar]
- Sun J, Huang YR, Harrington WR, Sheng S, Katzenellenbogen JA, Katzenellenbo-gen BS. Antagonists selective for estrogen receptor alpha. Endocrinology. 2002;143:941–947. doi: 10.1210/endo.143.3.8704. [DOI] [PubMed] [Google Scholar]
- Thammacharoen S, Geary N, Lutz TA, Ogawa S, Asarian L. Divergent effects of estradiol and the estrogen receptor-α agonist PPT on eating and activation of PVN CRH neurons in ovariectomized rats and mice. Brain Res. 2009;1268:88–96. doi: 10.1016/j.brainres.2009.02.067. [DOI] [PubMed] [Google Scholar]
- Vasudevan N, Lee MK, Pfaff D. Steroids. Elsevier; 2005. Integration of steroid hormone initiated membrane action to genomic function in the brain; pp. 388–396. [DOI] [PubMed] [Google Scholar]
- Vivacqua A, Lappano R, De Marco P, Sisci D, Aquila S, De Amicis F, Fuqua SAW, Andò S, Maggiolini M. G protein-coupled receptor 30 expression is up-regulated by EGF and TGFα in estrogen receptor α-positive cancer cells. Mol Endocrinol. 2009;23:1815–1826. doi: 10.1210/me.2009-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn N, Borgquist A, Wang K, Jeffery GS, Kelly MJ, Wagner EJ. Receptor subtypes and signal transduction mechanisms contributing to the estrogenic attenuation of cannabinoid-induced changes in energy homeostasis. Neuroendocrinology. 2013;97:160–175. doi: 10.1159/000338669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AM, Abrams MC, Micevych PE. β-Arrestin regulates estradiol membrane-initiated signaling in hypothalamic neurons. PLoS One. 2015;10:e0120530. doi: 10.1371/journal.pone.0120530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HB, Carlson KE, Stossi F, Katzenellenbogen BS, Katzenellenbogen JA. Analogs of methyl-piperidinopyrazole (MPP): antiestrogens with estrogen receptor α selective activity. Bioorg Med Chem Lett. 2009;19:108–110. doi: 10.1016/j.bmcl.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]