

Abstract
Carbamoyl phosphate synthetase 1 (CPS1) is a urea cycle enzyme that forms carbamoyl phosphate from bicarbonate, ammonia and ATP. Bi-allelic mutations of the CPS1 gene result in a urea cycle disorder presenting with hyperammonemia, often with reduced citrulline, and without orotic aciduria. CPS1 deficiency is particularly challenging to treat and lack of early recognition typically results in early neonatal death. Therapeutic interventions have limited efficacy and most patients develop long-term neurologic sequelae. Using transgenic techniques, we generated a conditional Cps1 knockout mouse. By loxP/Cre recombinase technology, deletion of the Cps1 locus was achieved in adult transgenic animals using a Cre recombinase-expressing adeno-associated viral vector. Within four weeks from vector injection, all animals developed hyperammonemia without orotic aciduria and died. Minimal CPS1 protein was detectable in livers. To investigate the efficacy of gene therapy for CPS deficiency following knock-down of hepatic endogenous CPS1 expression, we injected these mice with a helper-dependent adenoviral vector (HDAd) expressing the large murine CPS1 cDNA under control of the phosphoenolpyruvate carboxykinase promoter. Liver-directed HDAd-mediated gene therapy resulted in survival, normalization of plasma ammonia and glutamine, and 13% of normal Cps1 expression. A gender difference in survival suggests that female mice may require higher hepatic CPS1 expression. We conclude that this conditional murine model recapitulates the clinical and biochemical phenotype detected in human patients with CPS1 deficiency and will be useful to investigate ammonia-mediated neurotoxicity and for the development of cell- and gene-based therapeutic approaches.
Keywords: Carbamoyl Phosphate Synthetase Deficiency, Gene Therapy
1.1 Introduction
Ammonia is readily diffusible and toxic to the brain and thus needs to be maintained at relatively low levels in the plasma. Mammals rely on the urea cycle, consisting of six enzymes, two amino acid transporters, and a cofactor, to detoxify ammonia by incorporating excess nitrogen into urea which is then excreted in the urine [1]. The initial and rate-limiting enzyme of the cycle is carbamoyl phosphate synthetase 1 (CPS1) (EC 6.3.4.16), located primarily in the liver, as 20% of mitochondrial matrix protein [2], but also in enterocytes of the intestinal mucosa of the small intestine [3]. CPS1, a monomeric protein of 1462 amino acids, performs the first committed step of the cycle, catalyzing the formation of carbamoyl phosphate from ammonia, bicarbonate and two molecules of ATP [4]. The reaction requires the allosteric activator N-acetylglutamate. Deficiency of CPS1 (OMIM #237300), an autosomal recessively inherited mutation (2q34), has an estimated incidence of 1 in 1.3 million births [5].
As the urea cycle begins with this step, deficiency of CPS1 results in pure hyperammonemia, which if not treated early, can lead to encephalopathy and death [6]. CPS1 patients do not demonstrate orotic aciduria; this distinguishes this disorder from ornithine transcarbamylase deficiency. Citrulline and arginine levels are typically reduced (but not always) [7] in neonatal patients while it is often normal or low normal in those with late onset. Management is based on a low protein diet with scrupulous monitoring to limit nitrogen intake, arginine supplementation, and the need to provide alternate routes for waste nitrogen disposal [8, 9] including administration of ammonia scavengers, such as sodium benzoate, sodium phenylbutyrate, and glycerol phenylbutyrate. Together these therapies are marginally effective for this deficiency that has been described as the most severe of the urea cycle disorders [10], leaving patients at risk for having recurrent hyperammonemic episodes and further brain injury. At present, the only definitive treatment is liver transplantation [11], which is limited by organ availability, and there remains an unmet need in the form of definitive effective non-surgical therapies.
Here, we describe the successful generation of a conditional Cps1 knockout mouse and illustrate proof in principal of its value for both the study of the disorder and for the investigation of experimental interventions. We demonstrate that the conditional Cps1tm1c/tm1c animals have normal nitrogen metabolism until Cps1 deletion that results in hyperammonemia and elevated plasma glutamine without orotic aciduria, followed by death within 4 weeks after delivery of the Cre recombinase-expressing vector. We also demonstrate that this CPS1-deficient mouse can be corrected by liver-directed gene therapy, showing this model’s potential for use in the development of new clinically-appropriate therapies for this difficult-to-treat disorder. Our findings demonstrate that 13% of native hepatic CPS1 gene expression is necessary to obtain survival in male animals, whereas female mice may require higher levels of CPS1 expression, and suggests a minimum target that likely will be necessary to achieve for treatment of patients with this disorder.
2.1 Materials and Methods
2.1.1 Embryonic Stem Cell Targeting
The strategy for homologous recombination and post allelic modification in germline mice is outlined in Figure 1.
Figure 1. Development of the Cps1cKO (tm1c) mouse.
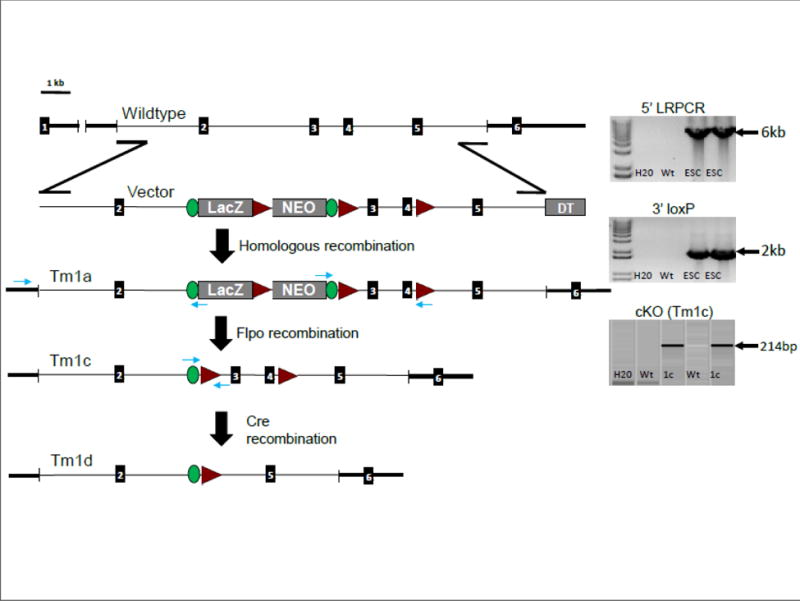
The strategy for successful development of the mouse is diagrammed. 5′ LRPCR image and targeting confirmation of the F1 (tm1a) mice produced from the chimeric backcrossing: This incorporates a gene specific forward primer in the endogenous region upstream of the 5′ homology arm paired with a reverse primer in the lacZ cassette (in the en2/Frt junction). 3′ loxP image: This demonstrates the loxP confirmation of the targeted F1 mice produced from the chimeric backcrossing to ensure that the intact targeted allele has passed through the germline and proves it is the conditionally ready allele (tm1a). cKO (Tm1c) image: Screening of animals that are positive for the Flpo transgene demonstrates that successful recombination at the Frt sites has occurred, generating the conditional knockout (tm1c). This utilized a forward primer in the en2/Frt portion of the lacZ cassette and a reverse primer in the floxed region. With administration of AAV8-Cre recombinase, successful recombination at the loxP sites occurs, generating the knockout (tm1d). (LRPCR, long range polymerase chain reaction; DTA, Diphtheria Toxin A gene; Green ovals, FRT sites; red triangles, loxP sites; numbered black rectangles, exons)
2.1.2 Targeting Vector
The Cps1 conditional ready “KO first” (tm1a) targeting vector PG00187_Z_8_A08 was purchased from the European Conditional Mouse Mutagenesis - EuMMCR at www.eummcr.org[12, 13]. The vector plasmid was transformed into commercial DH10B cells and grown overnight in 2ml LB broth with Spectinomycin (50μg/ml) and Kanamycin (3mg/ul) selection. Plasmid DNA was isolated using Qiagen Mini-prep Kit (Valencia, CA) according to the manufacturer’s protocol. Plasmid identity and purity was assessed using a 5 unique restriction digest analysis.
2.1.3 Vector preparation for ES cell transformation
The Cps1 vector was prepared for electroporation into ES cells as follows. A 50ml overnight culture was grown in LB broth with Spectinomycin (50ug/ml) and Kanamycin (3mg/ul) selection. Plasmid DNA was isolated from the 50ml culture using a Qiagen Midi-prep Kit according to the manufacturer’s protocol. Plasmid DNA was linearized by digestion with the restriction enzyme AsiSI. Following linearization, the plasmid DNA was purified by ethanol precipitation, and the DNA pellet was re-suspended into sterile PBS. An aliquot of the linear vector was visualized on a 1% agarose gel, and the vector was quantified using a NanoDrop spectrophotometer (Thermo Scientific, Grand Island, NY). Prior to electroporation into ES cells, the vector was diluted in sterile PBS to a concentration of 50 ng/μl.
2.1.4 Electroporation
Approximately 5μg of the linearized targeting vector was electroporated into JM8.N4 (C57BL/6N) [14] derived ES cells and cells were subject to positive selection using 200μg/μl G418 selection media for 7 to 8 days. 288 G418 selected clones were picked and expanded in 96-well plates.
2.1.5 Genetic Screening
DNA was extracted from G418 selected clones via proteinase K digestion at 75°C for 30 minutes in lysis buffer with final concentrations of 10mM Tris/HCL pH8, 1mM EDTA, 50mM KCl, 2mM MgCl2, 3mg/ml proteinase K. DNA was then screened using quantitative Taqman® loss of allele (LOA)[15, 16] via relative Ct method multiplexed in quadruplicate using Qiagen QuantiTech qPCR kit with the following oligos: CPS1 gene primers/probe include a forward primer CATGTCGTTATTACTTGGTTACTCTTCA, reverse primer GCTTCTCTAGCCTACCAGATAGTG, probe 6Fam-TGTGAAACTTCCAGCCCTCCACAG-NFQ and were quantified next to a Y chromosome endogenous reference primers/probe including a forward primer CAGCCTGCAGTTGCCTCAA, reverse primer TGCCTGTATGTGATGGCATGT, probe VIC-TACAACCTTCTGCAGTGGGACAGGAACC-TAMRA (0.42μM each primer, 0.14μM each probe). Twelve passing clones were then expanded and DNA extracted via Qiagen blood and tissue kit following manufacturer’s protocol. Approximately 50ng of DNA was tested by long range PCR across the 5′ arm as well as the 3′ distal loxP using Invitrogen SequalPrep long PCR Kit following manufacturer’s protocol with the following thermal cycling: 94°C for 2min; 10 cycles of 94°C for 15 sec, 65°C for 15 sec (↓1C/cycle), 68°C for 10 min; 25 cycles of 94°C for 15 sec, 55°C for 30 sec, 68°C for 10 min (+20 sec/cycle), hold at 4°C. 5′ long range PCR included a genomic forward primer TTTCAGCTTTTAAGGGACTAGCGTGC paired with a cassette reverse primer GGTGGTGTGGGAAAGGGTTCGAAG for an expected 6,007bp targeted band. 3′ distal loxP PCR included a genetrap cassette forward primer CACACCTCCCCCTGAACCTGAAAC paired with a distal loxP cassette reverse primer CATTAATTGCGTTGCGCCATCT for an expected 2,010bp loxP band. PCR fragments were separated on a 0.8% agarose gel matrix at 120 volts for 90 minutes and analyzed on a Kodak (Rochester, NY) gel logic 200. Additionally, selection cassette copy number was confirmed as one copy/cell in a similar fashion as the Taqman® LOA screening above using the same reference primers/probe with the following LacZ target primers/probe: forward primer ATCAGGATATGTGGCGGATGA, reverse primer TGATTTGTGTAGTCGGTTTATGCA, probe 6Fam-CGGCATTTTCCGTGACGTCTCGTT- TAMRA. All qPCR assays were processed on an AB7900HT (Applied Biosystems, Grand Island, NY) in 384 well barcoded plates.
2.1.6 Germline Transmission (GLT) and Allele Modification Testing
At least 20 spreads were chromosome counted from each of three targeted clones. Two genotype confirmed euploid, clones were injected into Balb/c embryos by morula injection and 30 embryos were implanted into each of 2 individual CD1 recipients. 4 male chimeras derived from one recipient were selected and ranked by black coat color and mated to C57BL/6N wildtype females to identify mice with germline transmission.
DNA was extracted on an automated Hamilton STAR robot (Reno, NV) from approximately 3mm tail snips using Agencourt DNAdvance magnetic beads (Beckman-Coulter, Fullerton, CA) according to manufacturer′s protocol. DNA was then screened via LacZ target primers/probe: forward primer ATCAGGATATGTGGCGGATGA, reverse primer TGATTTGTGTAGTCGGTTTATGCA, probe 6Fam-CGGCATTTTCCGTGACGTCTCGTT-TAMRA and quantified next to an endogenous Tcrd reference primers/probe: forward primer CAGACTGGTTATCTGCAAAGCAA, reverse primer TCTATGCCAGTTCCAAAAAACATC, probe VIC-ATTATAACGTGCTCCTGGGACACCC-TAMRA [17, 18]. LacZ positive DNA was then confirmed for both correct targeting and as distal loxP positive using same assays as the ES cell confirmation. PCR reactions included a non-template control (NTC), negative wildtype control (B6), and targeted stem cell DNA as a positive control.
2.1.7 Allelic Modifications
Heterozygous conditional readyCps1 tm1a mice were crossed to C57BL/6N-Tg(CAG-Flpo)1Afst/Mmucd (MMRRC stock number 036512-UCD) in order to remove the genetrap cassette to create the conditional tm1c allele. Briefly, approximately 50ng of DNA was PCR tested for the presence of the Flpo transgene using forward primer GCCACCTTCATGAGCTACAACACC paired with a reverse primer AACAGGAACTGGTACAGGGTCTTGG producing a 383bp Flpo transgene specific amplicon. The flipped Cps1 tm1c allele was PCR screened using forward primer CGCATAACGATACCACGATATCAACAAG paired with reverse primer CCGCCTACTGCGACTATAGAGATATC producing a 214bp post Flpo excised specific amplicon [19] DNA was confirmed quantitatively as lacZ negative using same screening method as utilized at GLT screening. To account for potential mosaic mutants as well as breeding out the Flpo transgene, Cps1 tm1c positive animals were back crossed with C57BL/6N wildtype animals and subsequent progeny screened as above.
For the Flpo crosses: 15μl PCR reactions included 0.4uM of each primer, 1X PCR buffer, 1.7mM MgCl2, 0.2mM each dNTPs, 1 Unit Amplitaq polymerase (Applied Biosystem), and 1.3M Betaine, 1.3% DMSO (Sigma) with approximately 50ng of template DNA. Thermal cycling included an initial denaturing at 94°C for 5 min; 10 cycles of 94°C for 15sec, 65°C to 55°C for 30sec (↓1oC/cycle), 72°C for 40sec; 30 cycles of 94°C for 15sec, 55°C for 30sec, 72°C for 40sec; final extension of 72°C for 5 minutes and maintained at 4°C. PCR reactions included a non-template control (NTC), negative wildtype control (B6), and positive controls when applicable. Amplicons were detected and analyzed on a capillary based QIAxcel (Qiagen) instrument using screening cassette 2400 with AM320 method specifications.
Heterozygous Cps1 tm1c mice were inbred to maintain on the C57BL/6N background and to develop a homozygous floxed line (Cps1tm1c/tm1c).
2.1.8 PCR Screening of the CPS1 conditional Knockout (KO)
Genomic DNA was prepared from tail tip or ear tip by standard methods (5-PRIME ArchivePure™ DNA Purification Kit, 5 Prime Inc, Gaithersburg, MD) and subjected to PCR for genotyping. Gene specific primers for Cps1 wild type and tm1c conditional KO (floxed): 3′gsp reverse primer 5′-AGAAAAGCTGGTGCTCATACAAAGG-3′ and 5′gsp forward primer: 5′-AGGGTTGTATGCTTCATCTTGTATGC-3′. Cycle parameters: Denaturation: 98°C for 15sec; annealing: 60°C for 30sec; elongation: 72°C for 40sec for 35 cycles using DNA polymerase (Takara, Mountain View, CA, Catalog #RR006A). The wild type band is 409bp and the Post-floxed band is 563bp.
2.1.9 Adeno-Associated Viral Vectors
AAV8-TBG-Cre and AAV8-TBG-Luciferase were purchased from the University of Pennsylvania Vector Core (Philadelphia, PA).
2.1.10 Mouse Procedures
All mice were housed under specific pathogen-free conditions; food and water were provided ad libitum. All mice were kept according to the National Institutes of Health guidelines and all experimental procedures were conducted in accordance with guidelines for the care and use of research animals at our institution. All intravenous injections were performed by a retroorbital approach under isoflurane anesthesia. Animals were administered helper-dependent adenovirus expressing murine CPS1 at day -3 followed by injection with 2.0 × 1011genome copies (gc) of AAV8-thyroxine binding globulin (TBG)-Cre. All vectors were prepared in sterile pharmaceutical grade saline. The injections were performed in a total volume of 100 μl. Male and female mice were equally represented throughout the study. After the vector injection, scheduled blood sampling was taken from retro-orbital plexus under isoflurane anesthesia. Plasma was frozen immediately and stored at −80°C until analysis. Urine was collected by holding the mouse over a Petri dish or placing an Eppendorf tube by the urethral meatus and gently applying pressure to the abdominal area. Mice were euthanized if they showed symptoms of lethargy, seizures, or lying on side, as symptoms of hyperammonemia.
2.1.11 Biochemical Analysis
Ammonia
Ammonia determination was performed from plasma samples per manufacturer’s instructions (ab83360, Abcam, Cambridge, MA). Briefly, after equilibrating all reagents at room temperature, standards were prepared. Samples were set up using 5 μl of plasma added to 45 μl of assay buffer in duplicate in a 96-well microplate. The reaction mix was prepared as a master mix with each reaction consisting of 42 μl ammonia assay buffer, 2 μl OxiRed Probe, 2 μl Enzyme Mix, 2 μl developer, and 2 μl converting enzyme. 50 μL of reaction mix was added to samples and standard wells, mixed, and incubated for 1 hour at 37°C in a dark container. Optical density was measured at 570 nm using a microplate reader (iMark 16704, Bio-Rad, Hercules, CA). From the standards a line was generated and sample results in μM were calculated.
Biochemical Analysis of Serum
The concentration of amino acids was determined by HPLC utilizing pre-column derivatization with o-phthalaldehyde as previously described [20].
Urinary Orotic Acid
Urine samples were prepared by mixing 200 μl of sample with isotopic internal standard 15N2-orotic acid (Cambridge Isotope Laboratories, MA). Orotic acid and orotidine were assayed on a Micromass Quattro mass spectrometer (Waters, MA). HPLC was performed on a Waters ODS-AQ analytical column [150 ×2.0mm (i.d.), 5-μm bead size]. Mobile phase was isocratic 0.05 M ammonium formate (pH 4.0). The MS/MS system was set at a flow rate of 0.2 ml/min. Mass spectrometer was operated in the Electrospry ionization (ESI) negative multiple-reaction-monitoring (MRM) mode. Nitrogen was used as nebulizer gas at flow rate of 60-90 l/hr and desolvation gas 500 l/hr. Other optimized mass spectrometer parameters were cone voltage −15V, capillary −3250V and collision voltage −10V.
Alanine Amino Transferase
Plasma samples were analyzed for alanine aminotransferase level (ALT). Forty μl of plasma per sample was pipetted into cups and placed into a Vet Excel Clinical Chemistry Analyzer (Alfa Wassermann Diagnostic Technologies, West Caldwell, NJ) per the manufacturer’s instructions.
Biochemical Analysis of Liver
Amino acids profile in the liver was determined as in [21]. Briefly, the frozen liver tissue was ground into a fine powder, and then, extracted with 4% perchloric acid, neutralized with KOH, and used for measurement of amino acids. The concentration of amino acids was determined with an Agilent 1260 Infinity LC system, utilizing pre-column derivatization with o-phthalaldehyde [21].
2.1.12 Immunohistochemistry
Livers were removed from euthanized animals and fixed in 10% neutral buffered formalin in histology cassettes for 48 hours and then stored in 70% ethanol followed by paraffin embedding using standard procedures. For each embedded tissue, 4 μm thick cross sections were collected on microscope slides, which were subsequently used for immunohistochemistry staining procedures. The sections were deparaffinized in xylene and rehydrated in serial washes of ethanol. Antigen retrieval was performed in10 mM sodium citrate buffer pH 6.0 in a steamer for 30 min followed by cooling at RT for 20 min. Following antigen retrieval, tissue sections were permeabilized with 0.1% Triton X-100 in PBS for 10 min prior to blocking with 10% normal goat serum in PBS for one hour. Later, sections were incubated with CPS1 antibody (Abcam ab45956) at 1 μg/ml in the blocking buffer overnight at 4°C. After overnight incubation, the sections were stained with Goat anti-Rabbit Secondary Antibody (Invitrogen A-11012) for 1 hour at RT. The cell nuclei were counterstained with DAPI using VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA). The stained tissue sections were visualised with LSM 880 with Airyscan Confocal Microscope (Carl Zeiss, Oberkochen, Germany).
2.1.13 Western Blot
Tissue samples were homogenized in RIPA buffer (Sigma, St. Louis, MO) in the presence of Protease inhibitor cocktail kit (ThermoFisher Scientific, Waltham, MA). 50 μg of centrifuge-clarified total protein was used in western analysis and probed with rabbit polyclonal antibody to CPS1 (Abcam ab45956) at 1 μg/ml. β-Actin (1:5000) or β-tubulin antibody (1:500) (HRP Conjugated; Santa Cruz Biotechnology) was used as a loading control. HRP-conjugated goat anti-rabbit IgG (Santa Cruz, Biotechnology, Inc., catalog number sc-2004) was used as the secondary antibody (1:5000) and was visualized by SuperSignal™ West Pico PLUS Chemiluminescent Substrate detection (ThermoFisher Scientific).
2.1.14 Quantitative Real-Time RT-PCR
Total RNA was extracted from the liver using High Pure RNA Isolation Kit (Roche Molecular Systems) according to the manufacturer’s instructions. 1 μg of total RNA was subjected to reverse transcription as described in the Transcriptor First Strand cDNA Synthesis Kit (Roche Molecular Systems, Basel, Switzerland). Following cDNA synthesis, expression level of the genes was quantified by real-time PCR (qPCR) using SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad, Hercules, CA) on MyiQ™2 Two-Color Real-Time PCR Detection System (Bio-Rad).The quantification was analysed using the CT (threshold cycle) values. The expression of the target genes was normalized to that of the endogenous β-Actin and the fold enrichment was measured using the 2−ΔΔCt method.
2.1.15 Viral Copy Number
Viral copy number determination was performed as described previously [22]. In brief, Genomic DNA from the liver tissues was extracted using DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA). Quantification of the viral vector in the genomic DNA was determined with HGH polyadenylation signal(Forward primer: 5′AATCTTGGCTCACTGCAATCTCCG3′, Reverse primer: 5′CATGCATGCCTGGAATCCCAACAA3′) and quantified using SsoAdvanced™ Universal SYBR® Green Supermix on MyiQ™2 Two-Color Real-Time PCR Detection System. Livers from wild-type mice were used for non-template controls and demonstrated no amplification. Determination of copy numbers was based on known of dilutions of plasmid containing HGH polyadenylation signal.
2.1.16 Gene Delivery with Helper-Dependent Adenovirus
The HDAd-CPS1 vector bears the PEPCK-WL-CPS1 expression cassette including the liver-specific promoter of phosphoenolpyruvate carboxykinase (PEPCK) and other regulatory elements like the woodchuck hepatitis virus post regulatory element (WPRE), the Locus Control Region (LCR) from the apoE locus, the bovine growth hormone polyadenylation signal as described elsewhere.[23] Mouse CPS1 cDNA was synthesized by Life Technologies (Carlsbad, CA). HDAds were produced in 116 cells [24] using the helper virus AdNG163 [25] as described elsewhere [24]. HDAd titers were determined by absorbance at 260 nm as described elsewhere [26].
Female (n=5) and male (n=5) adult conditional CPS1 knockout mouse were administered 5×1012 vector particles (vp)/kg of a helper-dependent adenoviral vector expressing murine CPS1 under control of the PEPCK promoter and administered on day −3; control animals (female n=5 and males n=5) were administered pharmaceutical grade saline. To determine the minimum dose for survival beyond that of the vehicle-administered controls, pilot studies were performed followed by confirmation with two groups of mice: 5×1011 vp/kg and 1×1012 vp/kg (N=5 per group, male mice). Blood was sampled from both groups before administration, at time of death (typically at day 21 for the lower dose group), and at day 28 (for surviving experimental animals). All administrations were performed in pharmaceutical grade saline of 100 μls. On day 0, 2×1011 genome copies of AAV8-TBG-Cre was administered by retroorbital injection. Animals were weighed and observed daily.
2.1.17 Statistical Analyses
The data are presented as the arithmetic mean ± standard deviation. Statistical analyses including Kaplan-Meier survival curve (with Log-rank test), one-way ANOVA, two-way ANOVA and unpaired Student’s t-test were performed using GraphPad Prism Software 6.0 (La Jolla, CA). For one-way ANOVA, Tukey’s multiple comparison test and for two-ANOVA Sidak’s multiple comparison test were used. Statistical significance was assigned for a value of p<0.05.
3.1 Results
Deletion of CPS1 expression in adult mice results in weight loss and death
Adult homozygous Cps1 conditional knockout mice (Cps1tm1c) were developed by homologous recombination into embryonic stem cells (ESC), blastocyst microinjection, and germline allelic modification and were used for these studies (Figure 1). To conditionally delete CPS1 expression, we injected intravenously (IV) Cps1tm1c/tm1c mice (n=10) with an adeno-associated viral vector (AAV) serotype 8 expressing Cre recombinase under the control of the hepatocyte-specific promoter thyroxine binding globulin (TBG). As a control, adult animals (n=6) were administered AAV serotype 8 with an irrelevant gene (luciferase) also under the control of the TBG promoter. Whereas control mice exhibited little weight loss over the period of observation (6.5%) (Figure 2A), the experimental mice developed severe weight loss, up to 40.1% weight loss compared to baseline weights measured prior to the injection of the AAV vectors (Figure 2B, C). We also closely examined survival. All experimental mice (males and females, n = 12) died by day 24 (range: 15-24 days; 20.9 ± 3.1 [avg. ± SD]) after injection of AAV Cre recombinase while all controls animals (n = 6) remained alive to at least day 32 (end of study) (p = 0.0002 by log-rank) (Figure 2D).
Figure 2. Characterization of CPS1 Conditional Knockout.
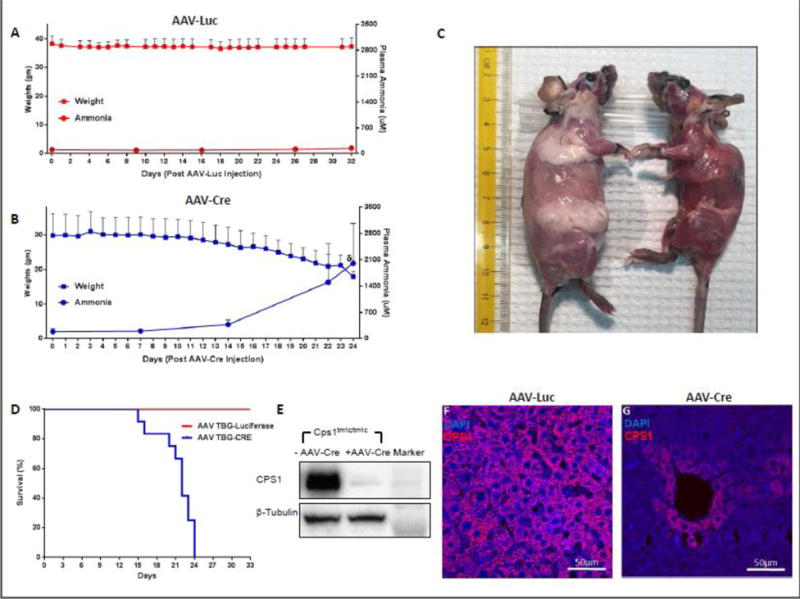
(A) Weights of control mice (n = 6) and mice with Cre-mediated disruption of Cps1 (n = 12) (B) were assessed daily, and plasma ammonia was assessed at assigned intervals. (C) Subcutaneous fat pads in CPS1 disrupted mice (right) become greatly reduced within three weeks after AAV-Cre administration (control mouse for comparison is at left). (D) Kaplan-Meier survival curve of mice undergoing Cre-mediated Cps1 disruption compared to an irrelevant AAV control vector (AAV-Luciferase), both with a liver-specific promoter (p = 0.0002 by log-rank). (E) Western blot analysis of hepatic CPS1 expression in control and CPS1 conditionally deleted mice along with immunostaining of wild type (F) and conditionally deleted liver (G) All values are mean ± S.D. (TBG, thyroxine binding globulin) (δ refers to ammonia measured at death or when euthanized for ethical endpoint; this was not at day 26 in all animals.)
Loss of hepatic CPS1 expression in adult mice results in hyperammonemia and elevated glutamine levels without orotic aciduria
Deletion of CPS1 activity in the Cps1tm1c/tm1c mice resulted in plasma ammonia levels that averaged 2002.7 ± 1065.9 μM (n = 9) (Figure 2B), 13 times that of undeleted controls (146.7 ± 11.4 μM, n = 6) (Figure 2A), confirming the development of severe hyperammonemia with CPS1 deletion. Hyperammonemic CPS1−/− mice (Cps1tm1d/tm1d) also exhibit increased plasma glutamine (1133.7 ± 323.4 μM) compared to controls (795.5 ± 93.7 μM) (p=0.03), and while citrulline was reduced, it was not statistically different (40.8 ± 16.5 μM with AAV-Cre, 50.7 ± 14.3 μM with AAV-Luciferase’ p = 0.27). With deletion of CPS1 mice also showed statistically significant increases (p < 0.05) in plasma taurine, serine, asparagine, glycine, phenylalanine, tryptophan, and lysine, while methionine and serine were decreased (see Supplementary Table 1). Plasma homoalanine, also known as α-amino butyric acid, was markedly increased (3.13 ± 0.95 μM for controls and 103.45 ± 32.71 μM in CPS1−/−) (p = 0.00001). Widespread derangement of hepatic amino acids also occurs with disruption of hepatic CPS1 activity (see Supplementary Table 3 comparing day 0 wild type and day 21 AAV-Cre + saline groups). Finally, plasma alanine aminotransferase (ALT), a measure of general hepatocellular injury, while found to be increased, was not statistically significant (p = 0.37) with disruption of CPS1 (wild type: 75.2 ± 80.4 U/L [n = 5]; CPS1-deleted: 266.3 ± 315.0 U/L [n = 4]; male mice).
A hallmark of human CPS1 deficiency is a lack of elevated urinary orotic acid, in contrast to ornithine transcarbamylase deficiency that typically present with orotic aciduria. Consistent with the human disease, urine samples were collected and urinary orotic acid was found to be no different than control mice: 13.6 ± 8.3 mmoles of orotic acid/mole creatinine in CPS1flox/flox mice with deleted Cps1 expression (n=5) and 10.4 ± 8.1 mmoles of orotic acid/mole creatinine in control mice with normal hepatic Cps1 expression (n=7) (p = 0.54).
Analysis of CPS1 protein in the liver was performed. These studies confirmed that the disruption of Cps1 abolished expression as there was markedly reduced hepatic CPS1 protein by both Western blot (Figure 2E) and by immunohistochemistry (Figures 2F and G).
Helper-dependent adenovirus expressing murine Cps1 rescues deficient animals from lethality
To evaluate the utility of this mouse model for investigation of experimental therapies for CPS1 deficiency, we generated a helper-dependent adenoviral (HDAd) vector expressing murine CPS1 protein under control of a liver-specific promoter (phosphoenolpyruvate carboxykinase [PEPCK]) for administration to this mouse model (Figure 3A). HDAd vector was chosen in part because of the hepatic tropism of this virus and to accommodate the 4.5 kb CPS1 cDNA that has a size (when adding ITRs and regulatory elements) exceeds the AAV packaging capacity. An IV dose of 5×1012 vector particles (vp)/kg was chosen as our prior studies demonstrated this dose to be effective in therapeutic protein expression in the liver [22].
Figure 3. Expression of CPS1 by hepatic gene therapy results in animal rescue.
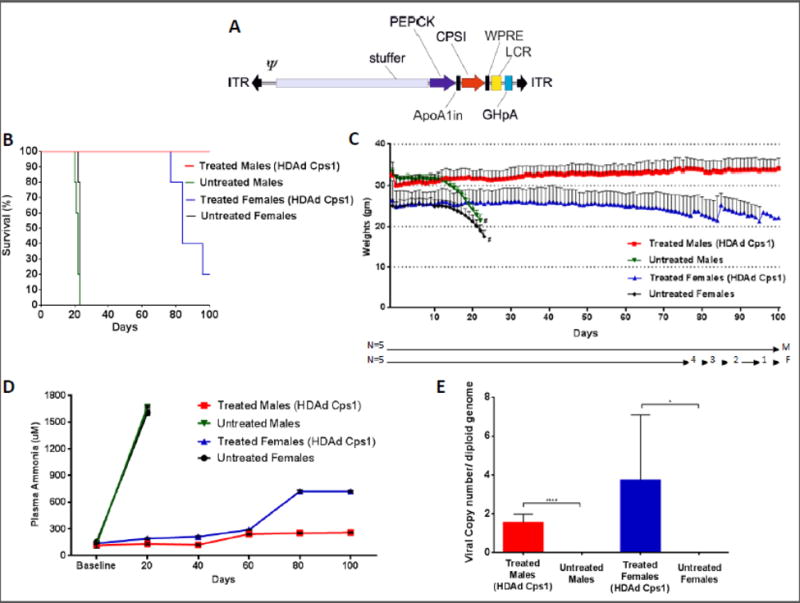
(A) A helper dependent adenovirus was produced to express murine CPS1 under control of the PEPCK promoter. It also contains inverted terminal repeats (ITR), a packaging signal (Ψ), the ApoA1 intron, the hepatic locus control region (LCR), and a growth hormone polyadenylation signal (GHpA). (B) Kaplan-Meier survival curve after conditional knockout of hepatic CPS1 expression in untreated male and female mice (green and black lines) and in those after the administration of a helper-dependent adenovirus expressing CPS1 (red and blue lines) (p < 0.0001 by log-rank). (C) Mouse weights were assessed daily with loss of hepatic CPS1 expression, with or without viral vector-mediated hepatic expression of CPS1. (Number of surviving mice to each time point is shown on the X-axis.) (D) Plasma ammonia levels were assessed at 20-day intervals in mice with conditional deleted CPS1 expression with (red and blue lines) or without (green and black lines) administration of a helper-dependent adenovirus expressing CPS1. (E) Copy number per diploid hepatocyte genome was determined at day 100 for both male and female mice administered helper-dependent adenoviral vector expressing CPS1 (* = p ≤ 0.05, **** = p ≤ 0.0001). All values are mean ± S.D. (# = death or termination for ethical time point)
We administered HDAd intravenously to adult Cps1tm1c/tm1c mice followed by administration of 2×1011 genome copies (gc) of AAV8-TBG-Cre recombinase three days later (n=5 males, n=5 females) (subsequently referred to as ‘treated’ mice). Control animals (subsequently referred to as ‘untreated’ mice) received IV pharmaceutical grade saline (vehicle) only, followed by AAV8-TBG-Cre 3 days later (n=5 males, n=5 females). All untreated animals died by day 23 (males: 21.6 ± 1.1 days, range 20-23 days; females: 22.8 ± 0.4 days, range 22-23 days) while all male experimental mice lived to day 100 (end of study) (Figure 3B). Unexpectedly, only 1 treated female mouse lived to day 100, the rest having expired or were euthanized for humane endpoints between days 77 and 95.
Animals were weighed daily from the time of HDAd helper dependent adenovirus administration until day 100 or their death (Figure 3C). Untreated male and female mice began to lose weight at about 2 weeks after AAV-Cre administration, both groups developing severe weight loss before death or being euthanized for humane endpoints: untreated males lost on average 31.6% of their original weight while untreated females lost 27.9%. With the administration of helper-dependent adenovirus expressing CPS1, male mice gained weight (13.3%, measured at day 100). However, treated females lost weight, declining 26.5% (measured at date of death or, if survival, at day 100).
Plasma ammonia in untreated males and females was markedly elevated by day 20 (males [n=5]: 1672.1 ± 10.5 μM; females [n=5]: 1607.3 ± 28.1 μM) (Figure 3D). In treated males, plasma ammonia was well controlled to day 100 (257.2 ±5.1 μM), having only mildly increased from baseline day (114.5 ± 31.0); plasma ALT was not statistically different from the wild type mice (treated males: 101.4 ± 164.7 U/L [n = 5]; p = 0.77). However, in treated females, plasma ammonia, while well-controlled to day 60 (day 0: 136.9 ± 7.9 μM; day 60: 289.3 ± 3.0 μM), began to increase: by day 80 it was moderately elevated (722.3 ± 14.8 μM) and remained as such until day 100 (or euthanasia) (722.2 ± 9.0 μM) (end of study).
Plasma amino acids were compared in the untreated and treated mice by gender (see Supplementary Table 2). While untreated males had marked abnormalities in multiple amino acids by day 21 (p < 0.05: glutamine, taurine, serine, glycine, phenylalanine, lysine, histidine, aspartic acid, threonine; p < 0.10: methionine [0.06], tyrosine [0.06]), treated mice demonstrated little change at 3 weeks in plasma amino acids. Glutamine was relatively stable during the first three weeks after AAV-Cre administration (from 313.6 ± 27.0 at day 0 to 385.6 ± 27.1 μM at day 21), while by day 100, it increased further (to 523.8 ± 59.5 μM). While mild amino acid abnormalities developed when measured at day 100, none were markedly abnormal (see Supplementary Table 2A). Furthermore, many of the hepatic (i.e. tissue) amino acid abnormalities detected in male mice after CPS1 activity was disrupted improved with CPS1 reexpression by the helper-dependent adenoviral vector (see Supplementary Table 3).
In female untreated mice, similar marked abnormalities occurred in many plasma amino acids by 3 weeks after AAV-Cre administration. Comparable to the males, statistically significant alterations (p < 0.05) were detected in glutamine, taurine, serine, glycine, phenylalanine, lysine, and threonine; however, such statistically significant changes were also detected in the branched chain amino acids, tyrosine, citrulline and asparagine that were not found in the males, suggesting that the severity of the disorder, and associated metabolic derangements, may be greater in female mice. Unlike the treated male mice, by day 100, progressive abnormalities were detected in many of the plasma amino acids of the treated females, including glutamine (560.5 ± 32.9 μM) (see Supplementary Table 2B).
The viral copy number per hepatocyte diploid genome was determined. Livers were collected at terminal time points: days 21-23 for untreated males and females (dead or euthanized for ethical reasons); day 100 for treated males; and at the terminal time point for females (dead or euthanized for ethical reasons) (Figure 3E). While, as expected, untreated animals had essentially undetectable copies in hepatocytes (males: 0.0008 ± 0.001 viral copies/diploid genome; females: 0.00095 ± 000015 copies/diploid genome), treated female mice, demonstrating overall poorer long-term survival, paradoxically had higher copy numbers per hepatocyte than males: 3.72 ± 3.3 viral copies/diploid genome vs. 1.54 ± 0.43 viral copies/diploid genome (p = 0.19).
Further studies revealed the level of expression of Cps1 mRNA and protein in the liver. Helper-dependent adenoviral-mediated Cps1 mRNA (utilizing gender-specific wild type controls [n = 2 for each gender] was greater in males at 100 days (0.47 ± 0.14; n = 5) when compared to females (0.34 ± 0.14; n = 5) (Figure 4A, B); however, this was not statistically significant (p = 0.20). In examining the expression of CPS1 protein, as expected untreated Cps1tm1c/tm1c mice showed minimal hepatic CPS1 protein expression by both Western blot (Figure 4C) and immunohistochemistry (Figure 4D left, 4B left). Conversely, HDAd-mediated CPS1 protein expression by Western blot, as noted by band density, demonstrated that expression in males was more modest compared to females (Figure 4C) and CPS1 protein expression was more homogenously detected in hepatocytes in the long-term treated males compared to female survivors (representative images, Figure 4D right, 4E right).
Figure 4. Expression by helper-dependent adenovirus results in hepatic CPS1 expression.
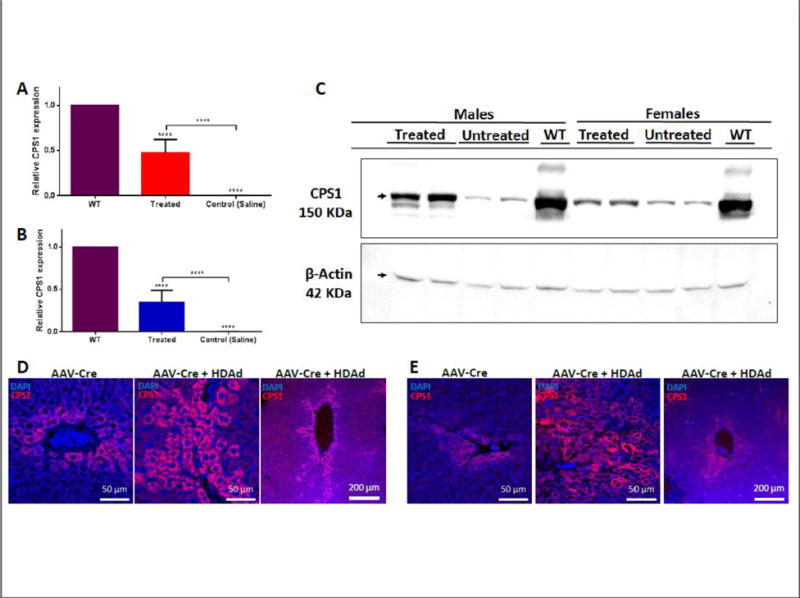
Determination of long-term relative male (A) and female (B) Cps1 mRNA expression in hepatocytes after administration of helper-dependent adenovirus (treated and untreated male and female mice, n=5 per gender; wild type males and females, n=2 per gender). Male vector- administered animals were studied at 100 days after vector administration while females were studied at the terminal time point: death or euthanized for ethical reasons. (C) Western blot of CPS1 expression in untreated mice compared to wild type mouse liver and helper-dependent adenovirus-treated mice. (Each lane represents a separate animal.) Liver immunostaining for long-term CPS1 expression after vector administration (100 days) for both male (D) and female (E) mice: left panels show remaining endogenous hepatic CPS1 expression after administration of AAV8-TBG-Cre recombinase; middle panels show expression at terminal time point (males: 100 days; females 100 days or death) after helper-dependent adenovirus expressing CPS1 administration; right panels show perivenous CPS1 from helper-dependent adenoviral expression. (All values are mean ± S.D.; ****, p ≤ 0.0001).
Studies were next performed to determine the minimum vector dose/body weight and Cps1 mRNA expression necessary for survival of male mice up to 30 days to define a minimum level of Cps1 expression needed for survival. Similar to untreated mice (saline injected), administration of 1×1011 vp/kg led to all animals expiring by day 20 (Figure 5A) accompanied by marked weight loss (24.0%) (Figure 5B). At this dose, vector genome copies detected per diploid hepatocyte were minimal (0.0063 ± 0.005) (Figure 5C) as was the relative expression of hepatic Cps1 (0.0062 ±0.003) (Figure 5D). At sacrifice, blood ammonia (917.5 ± 314.6 μM) was markedly elevated over baseline (95.6 ± 39.7 μM) (Figure 5E). At the dose of 5×1011 vp/kg, 80% of the mice (4 of 5) survived to day 30 post AAV8-TBG-Cre administration and they showed reduced weight loss (weight decline decreased to 17.1%), greater viral copy number per diploid hepatocyte (0.56 ± 0.25 vector copies/diploid genome), and higher relative Cps1 mRNA expression (0.13 ± 0.035) compared to wild type. Plasma ammonia control was also improved (506.6 ± 66.1 μM). Finally, with the administration of 1×1012 vp/kg, 100% of the mice survived, there was less weight loss (10.6% weight decline) and, as expected, greater vector copies per diploid hepatocyte (0.93 ± 0.64), higher relative Cps1 expression (0.27 ± 0.085) to wild type and improved plasma ammonia control (446.7 ± 164.1 μM).
Figure 5. A minimum hepatic CPS1 expression is needed to obtain survival.
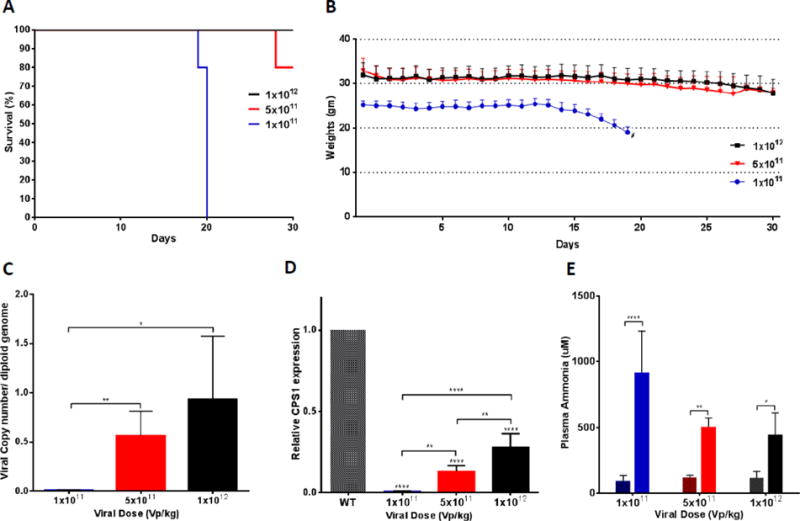
(A) Kaplan-Meier survival curve to 30 days after different doses (male, n=5 per dose group) of helper-dependent adenovirus expressing CPS1 was administered (p = 0.0006 by log-rank). (B) Mice were weighed daily after administration of AAV-Cre and the different helper-dependent adenoviral doses (n=5 per group). (C) Viral copy number per hepatocyte diploid genome was determined at day 30 comparing to dose of helper-dependent adenoviral vector administered (1×1012 vp/kg: 0.93 ± 0.64, n=5; 5×1011 vp/kg: 0.56 ± 0.25, n=5; and 1×1011 vp/kg: 0.0063 ± 0.005, n=5). (D) Relative expression of Cps1 mRNA was assessed compared to dose of vector administered (0.27 ± 0.085 for 1×1012 [n = 5]; 0.13 ± 0.035 for 5×1011 [n = 4]; 0.0062 ±0.003 for 1×1011 [n = 5]; wild type, n=2) (E) Plasma ammonia was assessed before and after conditional Cps1 deletion after administration of different doses of helper-dependent adenovirus expressing Cps1 (at 30 days or death) (1×1011 vp/kg: 917.5 ± 314.6 vs. 95.6 ± 39.7 μM; 5×1011 vp/kg: 506.6 ± 66.1 vs. 121.85 ± 15.1 μM; 1×1012 vp/kg: 446.7 ± 164.1 vs. 117.3 ± 48.9 μM; n=5 per group). (In E, Sidak’s multiple comparisons test was used to calculate P values between baseline and terminal point at each concentration of helper-dependent adenovirus.) (*, p ≤ 0.05; **, p ≤ 0.01; ****, p ≤ 0.0001) (WT = wild type). All values are mean ± S.D.
4.1 Discussion
Depending on the age of onset of CPS1 deficiency, and whether there is any residual enzyme activity, two phenotypes are observed: 1) severe neonatal onset disease presenting shortly after birth and 2) less severe late onset disease seen in both children and adults [7, 27, 28]. Neonatal onset patients typically are healthy at birth and tend to do well for a few days after delivery. However, they suddenly become lethargic, refuse oral intake and develop vomiting, hypothermia, seizures, and coma. As neonatal CPS1 deficiency is the most severe of the urea cycle disorders [10], the prognosis is generally poor with patients suffering irreversible brain injury from hyperammonemia and with many dying from brain edema even with the best of care [29].
A constitutive CPS1-deficient mouse was previously developed by another group by gene targeting of exon 17 with insertion of a PGK-neo selection cassette into an area encoding an ATP binding domain [30]. These neonatal mice, with homozygous disruption of Cps1, died within 36 hours of birth from overwhelming hyperammonemia but without significant liver pathology, consistent with the neonatal onset phenotype. Liver extracts showed no enzyme activity and an RNA transcript was not detected by RT-PCR. This mouse model was not submitted to a repository and was lost [31]. In the present studies reported herein, we developed a novel Cre-lox conditional Cps1 knockout mouse, this time by targeting exons 3-4 and with introduction of a predicted early stop codon in exon 5. We characterized this mouse, defining its response to hepatic CPS1 loss and the subsequent effect on plasma ammonia, glutamine and other amino acids, along with urinary orotic acid. Furthermore, we have been able to demonstrate that this model, more consistent with the latent onset of this deficiency, can be rescued by episomal Cps1 hepatic expression as low as 13% of native Cps1 mRNA. Unexpectedly we also detected a potential gender difference in the model, not previously reported with this disorder.
In this conditional knockout, animals were of normal size and activity until the initiation of gene deletion by Cre recombinase expression. After about 14 days, animals began to develop weight loss as plasma ammonia began to rise; sarcopenia, a common feature of chronic liver disease in humans, is mediated by hyperammonemia, an important effector of the liver-muscle axis [32]. Otherwise, animals did not show any evidence of clinical deterioration until several days to a week prior to death, when they developed a hunched appearance with mild dishevelment of fur late in their course. When ill, animals became ataxic progressing to difficulty righting and then becoming nonambulatory. Evaluating the plasma, mice demonstrated the hallmark of CPS1 deficiency, severe hyperammonemia. Citrulline was reduced but this was not statistically significant; this is likely due to intact CPS1 activity in enterocytes of the small intestine as the administered AAV-Cre would only be expected to disrupt hepatic CPS1 activity due to limitations based on use of a promoter specific for hepatocytes. Finally, urinary orotic acid was normal consistent with the typical presentation in CPS1 deficiency.
Differences in plasma and hepatic amino acids were revealed by comparing to the plasma of untreated animals. Over the 3-4 weeks from initiation of Cre expression to death, there was a slow and eventually marked rise in plasma glutamine; along with alanine, this is the major nontoxic interorgan carrier of nitrogen in the body [33]. In addition, statistically significant alterations of other amino acids occurred along with the marked appearance α-amino butyric acid during the period of terminal illness. While it is conceivable that the broad changes in the plasma and hepatic amino acid profile in part occurs from the derangement in metabolism, the marked elevation of plasma α-amino butyric acid is likely a nonspecific marker of liver dysfunction generated from the breakdown of methionine and threonine [34], both of which were decreased in the plasma of CPS1 deficient mice. While its clinical significance is not completely known, the absolute value of α-amino butyric acid, and its ratio to leucine (to account for nutritional status), has been reported to be elevated in certain inborn errors of metabolism; this has been described most notably for ornithine transcarbamylase deficiency [35]. To our knowledge, plasma α-amino butyric acid values for CPS1 deficiency do not exist in the literature. Potential mechanisms for this elevation in both humans with urea cycle disorders, and in this case in mice with CPS1 deficiency, include simultaneously enhanced production [i.e. tissue catabolism [36], amination of α-ketobutyric acid in hyperammonemia [35] and decreased oxidation (due to underlying liver disease) [35].
While at present there is much pre-clinical and clinical interest in AAV, CPS1 deficiency will likely be a difficult disorder to treat with this vector system. As the wild type viral genome of AAV is 4.7 kb, the CPS1 cDNA alone with only the inverted terminal repeats exceeds that size. Our lab had previously developed a CPS1 AAV with a minimal promoter and a small synthetic polyadenylation signal [37] to keep the construct size reduced: the smallest DNA construct that could be developed was 5.1 kb. However, this vector was not functional and CPS1 protein was not detected (unpublished results). Furthermore, it is likely that gene therapy for this disorder would be initiated early after birth. Recent data suggesting, at least in mice, an association between AAV and hepatocellular carcinoma does raise some concern about genotoxicity and safety for AAV hepatic gene therapy in neonates and children [38]. In addition, the substantial loss of vector from dividing hepatocytes would likely result in loss of efficacy as episomal AAV would be rapidly lost [39, 40].
Importantly, as a model for developing new gene-based therapies, we were able to demonstrate that the Cps1tm1c/tm1c model is amenable to hepatic-based gene therapy and to define a minimum target threshold of hepatic expression, at least in males, necessary for survival and some control of plasma ammonia. With a helper-dependent adenoviral vector expressing murine Cps1, expression was found in individual hepatic lobules and also in a perivenous pattern. While we did not definitively see periportal expression of CPS1 in the sections examined, expression in hepatocytes intermediate along the porto-central axis was likely contributing to urea cycle function. While glutamine synthetase is localized in the 1 to 3 layers of cells of the most distal perivenous hepatocytes [41] in the mouse [42], endogenous CPS1 is expressed in the remainder (>90%) of the parenchyma [43], overlapping with glutamine synthetase only in 2-3% of total hepatocytes [44, 45]. Thus obtaining periportal CPS1 expression is likely not necessary in order to have adequate nitrogen metabolism by the urea cycle in developing hepatic gene therapy for CPS1 deficiency.
A helper-dependent adenovirus was developed based on its hepatotropic qualities and large capacity for DNA inserts that remains episomal, reducing the concern for risk of malignancy. Similar to AAV, helper-dependent adenoviral vectors are precluded from readministration by neutralizing anti-adenoviral antibodies generated following the first administration [46]. In our studies, administration of a helper-dependent adenovirus expressing CPS1 (5×1012 vp/kg), led to long-term survival (100 days, end of study) in all male mice, preventing weight loss and establishing good control of both plasma ammonia and glutamine. Importantly, we were able to determine that at least 13% of wild type hepatic Cps1 mRNA expression was necessary to produce survival to 30 days in male mice.
However, unexpectedly, survival, viral copy number, mRNA, and protein data in aggregate suggest that female mice may require a higher level of hepatic CPS1 expression. While in part this may be due to a more rapid gradual decline of the therapeutic effect from episomal vector loss over time with hepatocellular division, the data do suggest that while higher transduction is achieved in female mice (as demonstrated by vector copy number per diploid hepatocyte nucleus at 100 days), hepatic CPS1 expression is in fact reduced at the same time points compared to males. These results cannot be explained based upon gender and hepatic transduction differences with helper-dependent adenovirus as no known difference has been reported previously. Neither can this be explained on AAV transduction (and thus greater Cre recombinase expression) as it has been shown that AAV hepatic transduction is in fact less efficient in female mice [47]. While inconclusive at this time and requiring further inquiry, the potential for a gender difference and the model itself will be important in the development of clinically applicable gene-and cell-based therapies for this disorder and as a model to improve not only the understanding of the biochemical abnormalities that occur in CPS1 deficiency, but also in developing measures to blunt the deleterious effects of hyperammonemia on the central nervous system.
Supplementary Material
Acknowledgments
We are grateful for the technical support, products, and/or services provided to our research by the Mouse Biology Program (MBP) at the University of California Davis. The mouse strain used for this research project, C57BL/6N-Tg(CAG-Flpo)1Afst/Mmucd, RRID:MMRRC_036512-UCD (MMRRC FLP line) was obtained from the Mutant Mouse Regional Resource Center, a NIH funded strain repository, and was donated by the MMRRC at UC Davis. The original transgenic was donated by Dr. Konstantinos Anastassiadis from Technische Universitaet Dresden. Amino acid profiles and isotopomer enrichment analyses were performed in the Metabolomics Core facility, The Children’s Hospital of Philadelphia (https://metabolomic.research.chop.edu/).
Funding
This work was supported by a gift from Jim and Ada Horwich to generate the CPS1 deficient mouse. Funding for the characterization of the mice and development of gene therapy was provided by a grant from the National Institute of Neurological Disorders and Stroke/National Institutes of Health [grant number 1R21HD076197-01, -02 to G.S.L.].
Abbreviations
- CPS1
Carbamoyl phosphate synthetase 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors claim no conflict of interest.
References
- 1.Nettesheim S, Kolker S, Karall D, Haberle J, Posset R, Hoffmann GF, Heinrich B, Gleich F, Garbade SF, S. Arbeitsgemeinschaft fur Padiatrische, r. European, D. network for Intoxication type Metabolic, D. Erhebungseinheit fur Seltene Padiatrische Erkrankungen in, G. Austrian Metabolic, U. Swiss Paediatric Surveillance Incidence, disease onset and short-term outcome in urea cycle disorders -cross-border surveillance in Germany, Austria and Switzerland Orphanet. J Rare Dis. 2017;12:111. doi: 10.1186/s13023-017-0661-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clarke S. A major polypeptide component of rat liver mitochondria: carbamyl phosphate synthetase. J Biol Chem. 1976;251:950–961. [PubMed] [Google Scholar]
- 3.Christoffels VM, Habets PE, Das AT, Clout DE, van Roon MA, Moorman AF, Lamers WH. A single regulatory module of the carbamoylphosphate synthetase I gene executes its hepatic program of expression. J Biol Chem. 2000;275:40020–40027. doi: 10.1074/jbc.M007001200. [DOI] [PubMed] [Google Scholar]
- 4.Diez-Fernandez C, Haberle J. Targeting CPS1 in the treatment of Carbamoyl phosphate synthetase 1 (CPS1) deficiency, a urea cycle disorder. Expert Opin Ther Targets. 2017;21:391–399. doi: 10.1080/14728222.2017.1294685. [DOI] [PubMed] [Google Scholar]
- 5.Summar ML, Koelker S, Freedenberg D, Le Mons C, Haberle J, Lee HS, Kirmse B, R. European, h.w.e.-i.o.e.i.p. Network for Intoxication Type Metabolic Diseases. Electronic address, h.r.e.u.e.u. Members of the Urea Cycle Disorders Consortium. Electronic address The incidence of urea cycle disorders. Mol Genet Metab. 2013;110:179–180. doi: 10.1016/j.ymgme.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maestri NE, Hauser ER, Bartholomew D, Brusilow SW. Prospective treatment of urea cycle disorders. J Pediatr. 1991;119:923–928. doi: 10.1016/s0022-3476(05)83044-6. [DOI] [PubMed] [Google Scholar]
- 7.Funghini S, Thusberg J, Spada M, Gasperini S, Parini R, Ventura L, Meli C, De Cosmo L, Sibilio M, Mooney SD, Guerrini R, Donati MA, Morrone A. Carbamoyl phosphate synthetase 1 deficiency in Italy: clinical and genetic findings in a heterogeneous cohort. Gene. 2012;493:228–234. doi: 10.1016/j.gene.2011.11.052. [DOI] [PubMed] [Google Scholar]
- 8.Batshaw ML, Brusilow S, Waber L, Blom W, Brubakk AM, Burton BK, Cann HM, Kerr D, Mamunes P, Matalon R, Myerberg D, Schafer IA. Treatment of inborn errors of urea synthesis: activation of alternative pathways of waste nitrogen synthesis and excretion. N Engl J Med. 1982;306:1387–1392. doi: 10.1056/NEJM198206103062303. [DOI] [PubMed] [Google Scholar]
- 9.Batshaw ML, MacArthur RB, Tuchman M. Alternative pathway therapy for urea cycle disorders: twenty years later. J Pediatr. 2001;138:S46–54. doi: 10.1067/mpd.2001.111836. discussion S54-45. [DOI] [PubMed] [Google Scholar]
- 10.Mew NA, Lanpher B, Gropman AL, Chapman KA, Simpson KL, Summar ML. Urea Cycle Disorders Overview. In: RA P, Adam MP, Ardinger HH, SE W, A A, LJH B, TD B, C-T F, Mefford HC, RJH S, K S, editors. GeneReviews. University of Washington; Seattle, WA: 2003–2015. [PubMed] [Google Scholar]
- 11.Kasahara M, Sakamoto S, Shigeta T, Fukuda A, Kosaki R, Nakazawa A, Uemoto S, Noda M, Naiki Y, Horikawa R. Living-donor liver transplantation for carbamoyl phosphate synthetase 1 deficiency. Pediatr Transplant. 2010;14:1036–1040. doi: 10.1111/j.1399-3046.2010.01402.x. [DOI] [PubMed] [Google Scholar]
- 12.Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan W, Costantino N, Li R, Lee SC, Su Q, Melvin D, Court DL, Liu P. A recombineering based approach for high-throughput conditional knockout targeting vector construction. Nucleic Acids Res. 2007;35:e64. doi: 10.1093/nar/gkm163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pettitt SJ, Liang Q, Rairdan XY, Moran JL, Prosser HM, Beier DR, Lloyd KC, Bradley A, Skarnes WC. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat Methods. 2009;6:493–495. doi: 10.1038/nmeth.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valenzuela DM, Murphy AJ, Frendewey D, Gale NW, Economides AN, Auerbach W, Poueymirou WT, Adams NC, Rojas J, Yasenchak J, Chernomorsky R, Boucher M, Elsasser AL, Esau L, Zheng J, Griffiths JA, Wang X, Su H, Xue Y, Dominguez MG, Noguera I, Torres R, Macdonald LE, Stewart AF, DeChiara TM, Yancopoulos GD. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat Biotechnol. 2003;21:652–659. doi: 10.1038/nbt822. [DOI] [PubMed] [Google Scholar]
- 16.PE Applied Biosystems. Foster City, CA: 1997. ABI PRISM 7700 User Bulletin. [Google Scholar]
- 17.Tesson L, Remy S, Menoret S, Usal C, Anegon I. Analysis by quantitative PCR of zygosity in genetically modified organisms. Methods Mol Biol. 2010;597:277–285. doi: 10.1007/978-1-60327-389-3_19. [DOI] [PubMed] [Google Scholar]
- 18.Tesson L, Heslan JM, Menoret S, Anegon I. Rapid and accurate determination of zygosity in transgenic animals by real-time quantitative. PCR Transgenic Res. 2002;11:43–48. doi: 10.1023/a:1013928600442. [DOI] [PubMed] [Google Scholar]
- 19.Horton RM. PCR-mediated recombination and mutagenesis. SOEing together tailor-made genes. Mol Biotechnol. 1995;3:93–99. doi: 10.1007/BF02789105. [DOI] [PubMed] [Google Scholar]
- 20.Jones BN, Gilligan JP. o-Phthaldialdehyde precolumn derivatization and reversed-phase high-performance liquid chromatography of polypeptide hydrolysates and physiological fluids. J Chromatogr. 1983;266:471–482. doi: 10.1016/s0021-9673(01)90918-5. [DOI] [PubMed] [Google Scholar]
- 21.Nissim I, Horyn O, Nissim I, Daikhin Y, Wehrli SL, Yudkoff M, Matschinsky FM. Effects of a glucokinase activator on hepatic intermediary metabolism: study with 13C-isotopomer-based metabolomics. Biochem J. 2012;444:537–551. doi: 10.1042/BJ20120163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu C, Cela RG, Suzuki M, Lee B, Lipshutz GS. Neonatal helper-dependent adenoviral vector gene therapy mediates correction of hemophilia A and tolerance to human factor VIII. Proc Natl Acad Sci U S A. 2011;108:2082–2087. doi: 10.1073/pnas.1015571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brunetti-Pierri N, Ng T, Iannitti DA, Palmer DJ, Beaudet AL, Finegold MJ, Carey KD, Cioffi WG, Ng P. Improved hepatic transduction, reduced systemic vector dissemination, and long-term transgene expression by delivering helper-dependent adenoviral vectors into the surgically isolated liver of nonhuman primates. Hum Gene Ther. 2006;17:391–404. doi: 10.1089/hum.2006.17.391. [DOI] [PubMed] [Google Scholar]
- 24.Palmer D, Ng P. Improved system for helper-dependent adenoviral vector production. Mol Ther. 2003;8:846–852. doi: 10.1016/j.ymthe.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 25.Palmer DJ, Ng P. Physical and infectious titers of helper-dependent adenoviral vectors: a method of direct comparison to the adenovirus reference material. Mol Ther. 2004;10:792–798. doi: 10.1016/j.ymthe.2004.06.1013. [DOI] [PubMed] [Google Scholar]
- 26.Palmer DJ, Ng P. Methods for the production of helper-dependent adenoviral vectors. Methods Mol Biol. 2008;433:33–53. doi: 10.1007/978-1-59745-237-3_3. [DOI] [PubMed] [Google Scholar]
- 27.Ono H, Suto T, Kinoshita Y, Sakano T, Furue T, Ohta T. A case of carbamoyl phosphate synthetase 1 deficiency presenting symptoms at one month of age. Brain Dev. 2009;31:779–781. doi: 10.1016/j.braindev.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 28.Klaus V, Vermeulen T, Minassian B, Israelian N, Engel K, Lund AM, Roebrock K, Christensen E, Haberle J. Highly variable clinical phenotype of carbamylphosphate synthetase 1 deficiency in one family: an effect of allelic variation in gene expression? Clin Genet. 2009;76:263–269. doi: 10.1111/j.1399-0004.2009.01216.x. [DOI] [PubMed] [Google Scholar]
- 29.Kurokawa K, Yorifuji T, Kawai M, Momoi T, Nagasaka H, Takayanagi M, Kobayashi K, Yoshino M, Kosho T, Adachi M, Otsuka H, Yamamoto S, Murata T, Suenaga A, Ishii T, Terada K, Shimura N, Kiwaki K, Shintaku H, Yamakawa M, Nakabayashi H, Wakutani Y, Nakahata T. Molecular and clinical analyses of Japanese patients with carbamoylphosphate synthetase 1 (CPS1) deficiency. J Hum Genet. 2007;52:349–354. doi: 10.1007/s10038-007-0122-9. [DOI] [PubMed] [Google Scholar]
- 30.Schofield JP, Cox TM, Caskey CT, Wakamiya M. Mice deficient in the urea-cycle enzyme, carbamoyl phosphate synthetase I, die during the early neonatal period from hyperammonemia. Hepatology. 1999;29:181–185. doi: 10.1002/hep.510290112. [DOI] [PubMed] [Google Scholar]
- 31.Deignan JL, Cederbaum SD, Grody WW. Contrasting features of urea cycle disorders in human patients and knockout mouse models. Mol Genet Metab. 2008;93:7–14. doi: 10.1016/j.ymgme.2007.08.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dasarathy S, Merli M. Sarcopenia from mechanism to diagnosis and treatment in liver disease. J Hepatol. 2016;65:1232–1244. doi: 10.1016/j.jhep.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nurjhan N, Bucci A, Perriello G, Stumvoll M, Dailey G, Bier DM, Toft I, Jenssen TG, Gerich JE. Glutamine: a major gluconeogenic precursor and vehicle for interorgan carbon transport in man. J Clin Invest. 1995;95:272–277. doi: 10.1172/JCI117651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newsholme EA, Leech AR. Amino acid metabolism, Biochemistry for the Medical Sciences. John Wiley & Sons; New York: 1983. pp. 382–441. [Google Scholar]
- 35.Yudkoff M, Blazer-Yost B, Cohn R, Segal S. On the clinical significance of the plasma alpha-amino-n-butyric acid:leucine ratio. Am J Clin Nutr. 1979;32:282–285. doi: 10.1093/ajcn/32.2.282. [DOI] [PubMed] [Google Scholar]
- 36.Swendseid ME, Yamada C, Vinyard E, Figueroa WG, Drenick EJ. Plasma amino acid levels in subjects fed isonitrogenous diets containing different proportions of fat and carbohydrate. Am J Clin Nutr. 1967;20:52–55. doi: 10.1093/ajcn/20.1.52. [DOI] [PubMed] [Google Scholar]
- 37.Levitt N, Briggs D, Gil A, Proudfoot NJ. Definition of an efficient synthetic poly(A) site. Genes Dev. 1989;3:1019–1025. doi: 10.1101/gad.3.7.1019. [DOI] [PubMed] [Google Scholar]
- 38.Chandler RJ, LaFave MC, Varshney GK, Trivedi NS, Carrillo-Carrasco N, Senac JS, Wu W, Hoffmann V, Elkahloun AG, Burgess SM, Venditti CP. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest. 2015;125:870–880. doi: 10.1172/JCI79213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee EK, Hu C, Bhargava R, Rozengurt N, Stout D, Grody WW, Cederbaum SD, Lipshutz GS. Long-term survival of the juvenile lethal arginase-deficient mouse with AAV gene therapy. Mol Ther. 2012;20:1844–1851. doi: 10.1038/mt.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tai DS, Hu C, Kim EH, Lipshutz GS. Augmentation of transgene-encoded protein after neonatal injection of adeno-associated virus improves hepatic copy number without immune responses. Pediatr Res. 2015;78:239–246. doi: 10.1038/pr.2015.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elias H. A re-examination of the structure of the mammalian liver; the hepatic lobule and its relation to the vascular and biliary systems. Am J Anat. 1949;85:379–456. doi: 10.1002/aja.1000850303. 315 pl. [DOI] [PubMed] [Google Scholar]
- 42.Bennett AL, Paulson KE, Miller RE, Darnell JE., Jr Acquisition of antigens characteristic of adult pericentral hepatocytes by differentiating fetal hepatoblasts in vitro. J Cell Biol. 1987;105:1073–1085. doi: 10.1083/jcb.105.3.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaasbeek Janzen JW, Lamers WH, Moorman AF, de Graaf A, Los JA, Charles R. Immunohistochemical localization of carbamoyl-phosphate synthetase (ammonia) in adult rat liver; evidence for a heterogeneous distribution. J Histochem Cytochem. 1984;32:557–564. doi: 10.1177/32.6.6373912. [DOI] [PubMed] [Google Scholar]
- 44.Gebhardt R, Lindros K, Lamers WH, Moorman AF. Hepatocellular heterogeneity in ammonia metabolism: demonstration of limited colocalization of carbamoylphosphate synthetase and glutamine synthetase. Eur J Cell Biol. 1991;56:464–467. [PubMed] [Google Scholar]
- 45.Gebhardt R. Metabolic zonation of the liver: regulation and implications for liver function. Pharmacol Ther. 1992;53:275–354. doi: 10.1016/0163-7258(92)90055-5. [DOI] [PubMed] [Google Scholar]
- 46.Vetrini F, Ng P. Gene therapy with helper-dependent adenoviral vectors: current advances and future perspectives. Viruses. 2010;2:1886–1917. doi: 10.3390/v2091886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davidoff AM, Ng CY, Zhou J, Spence Y, Nathwani AC. Sex significantly influences transduction of murine liver by recombinant adeno-associated viral vectors through an androgen-dependent pathway. Blood. 2003;102:480–488. doi: 10.1182/blood-2002-09-2889. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.