

Abstract
Human retinal pigment epithelial (hRPE) cells play important immune-regulatory roles in a variety of retinal pathologic processes, including the production of inflammatory cytokines that are essential mediators of the innate immune response within the ocular microenvironment. The pro-inflammatory “alarmin” cytokine IL-1α has been implicated in both infectious and non-infectious retinal diseases, but its regulation in the retina is poorly understood. The purpose of this study was to elucidate the expression and regulation of IL-1α within hRPE cells. To do this, IL-1α mRNA and protein in hRPE cells was assessed by RT-PCR, qPCR, ELISA, Western blot, and immunofluorescence following treatment with a variety of stimuli and inhibitors. ER stress, LPS, IL-1β, and TLR2 activation all significantly increased intracellular IL-1α protein. Increasing intracellular calcium synergized both LPS- and Pam3CSK4-induced IL-1α protein production. Accordingly, blocking calcium signaling and calpain activity strongly suppressed IL-1α protein expression. Significant but more moderate inhibition occurred following blockage of TLR4, caspase-4, or caspase-1. Neutralizing antibodies to IL-1β and TLR2 partially eliminated LPS- and TLR2 ligand Pam3CSK4-stimulated IL-1α protein production. IFN-β induced caspase-4 expression and activation, and also potentiated LPS-induced IL-1α expression, but IFN-β alone had no effect on IL-1α protein production. Interestingly, all inhibitors targeting the PI3K/Akt pathway, with the exception of Ly294002, strongly increased IL-1α protein expression. This study improves understanding of the complex mechanisms regulating IL-1α protein expression in hRPE cells by demonstrating that TLR4 and TLR2 stimulation and exposure to IL-1β, ER stress and intracellular calcium all induce hRPE cells to produce intracellular IL-1α, which is negatively regulated by the PI3K/Akt pathway. Additionally, the non-canonical inflammasome pathway was shown to be involved in LPS-induced hRPE IL-1α expression through caspase-4 signaling.
Keywords: retinal pigment epithelium, RPE; IL-1α, interleukin-1α, caspase-4, non-canonical inflammasome, TLR2, TLR4
1. Introduction
Human retinal pigment epithelial (hRPE) cells, located at the blood-retina barrier, are important immune-regulatory cells that play key roles in a variety of retinal pathologic processes. hRPE cells and infiltrating leukocytes produce inflammatory cytokines that are essential mediators of the innate immune response within the ocular microenvironment in both infectious and non-infectious retinal diseases (Detrick et al., 2001; Jaffe et al., 1992; Kinnunen et al., 2012; Moyer et al., 2008; Nagineni et al., 2000; Tseng et al., 2013; Vann and Atherton, 1991; Yang et al., 2011). However, inflammation has also been implicated as a pathogenic mechanism in several retinal degenerative diseases including uveitis, diabetic retinopathy and age-related macular degeneration (AMD) (Tseng et al., 2013).
It is known that IL-1α is a pro-inflammatory, pleiotropic cytokine involved in inflammation and immunity and is generated as a central driver of immune responses in tissue damage (Cohen et al., 2010). IL-1α, has been recognized as a pivotal danger signal, exerting its effects on both innate and adaptive immunity. In the eye, enhanced expression of IL-1α has been observed in LPS-induced uveitis (Yoshida et al., 1994) and the tears and conjunctiva of patients with dry-eye disease (Solomon et al., 2001). Moreover, the eye is particularly vulnerable to attack by microbes and has evolved a series of suppression mechanisms to limit damage to the retina (Leung et al., 2009). hRPE cells express TLR4 and TLR2 receptors (Elner et al., 2005; Nazari et al., 2014), which can bind bacterial LPS and lipopeptides, respectively, to initiate an immune response and promote IL-1α production (Agrawal and Gupta, 2011; Bian et al., 2009; Kayagaki et al., 2013).
In addition, IL-1α has been implicated in the pathogenesis of AMD (Brandstetter et al., 2016; Tseng et al., 2013) and promotes necroptosis (Cohen et al., 2010) and necrosis-induced sterile inflammation (Liu et al., 2015), which have been suggested as major causes of cell death in AMD (Hanus et al., 2015; Hanus et al., 2016).
Furthermore, it is well known that caspase-1 cleavage of biologically inactive pro-IL-1β to its active, mature form is required for IL-1β secretion and function (Dinarello, 2009; Uchiyama and Tsutsui, 2015). While it is well known that this pathway does not directly involve IL-1α production or secretion, another inflammasome pathway has recently been discovered in mice that is caspase-11 dependent and induces IL-1α and IL-1β secretion (Hagar et al., 2013; Kayagaki et al., 2013). To distinguish this new signaling pathway from the previously known classical inflammasome pathway, which is caspase-11-independent, the caspase-11-dependent route of inflammasome engagement has been termed non-canonical inflammasome activation (Kayagaki et al., 2011). In humans, caspase-4 and caspase-5 are putative orthologs of rodent caspase-11, and several reports have now demonstrated that caspase-4 mediates non-canonical inflammasome activation and induces secretion of IL-1β and IL-1α (Casson et al., 2015; Schmid-Burgk et al., 2015; Shi et al., 2015; Vigano et al., 2015). We have previously demonstrated that caspase-4 is dually involved in hRPE pro-inflammatory and pro-apoptotic responses and that various pro-inflammatory stimuli and ER stress induce hRPE caspase-4 mRNA synthesis, protein production, and activation (Bian et al., 2009).
It is known that hRPE cells can be induced to express, but not secrete IL-1α (Jaffe et al., 1992; Moyer et al., 2008). However, there have been no follow-up studies regarding hRPE IL-1α expression, regulation, and the signaling pathways necessary for its expression. In this study, we demonstrate that multiple signaling pathways are capable of up-regulating hRPE IL-1α expression, including the caspase-4-mediated non-canonical inflammasome, toll like receptor (TLR) signaling, ER stress, and IL-1β autocrine stimulation.
2. Material and Methods
2.1. Materials
Recombinant human IL-1β was purchased from R&D System (Minneapolis, MN). IL-1α antibody was from Abcam (Cambridge, UK). Caspase-4 inhibitor (Ac-LEVD-CHO), TLR4 inhibitor, PD150606, TLR1/TLR2 Antagonist (Cu-CPT22) were purchased from EMD Millipore (Billerica, MA). Caspase-1 and caspase-4 antibodies, and caspase-1 inhibitor Ac-YVAD-cmk were obtained from BioVision (Mountain View, CA) and Clontech (Mountain View, CA), respectively. The recombinant human interferon-gamma (IFN-γ), human IL-1α ELISA kit, and M-PER Mammalian Protein Extraction Reagent were obtained from Thermo Scientific (Rockford IL). Recombinant IFN-α and -β were obtained from PBL Assay Science (Piscataway, NJ). QIAshredder and RNeasy mini kit were purchased from Qiagen (Valencia, CA.). The TLR1/2 ligand, Pam3CSK4, was purchased from InvivoGen (San Diego, CA). Ionomycin and Wortmannin were from Cell Signaling (Danvers, MA). PI-103 and Mk2206 were from MCE (MedChem Express, Princeton, NJ). Cal-101 was from Selleck Chemicals (Houston, TX). Tunicamycin, LPS, BAPTA-AM, and all other reagents were obtained from Sigma-Aldrich (St. Louis, MO) and Fisher Scientific (Pittsburgh, PA). The main inhibitors and inducers used in this study are listed in Table 1. Concentrations of these compounds were used that have been previously shown to maximally stimulate or inhibit pathways without causing cytotoxic effects (Bian et al., 2009; Elner et al., 1991; Elner et al., 1990), and hRPE cells treated with these chemicals were examined by CCK8 kit and LDH assays to ensure there was no measurable cytotoxicity at the concentrations used.
Table 1.
Inhibitors and inducers used in this study
| Chemical | Major target |
|---|---|
| Wortmannin | PI3K inhibitor |
| CAL-101 | PI3Kδ inhibitor |
| PI-103 | PBKα/β/δ/γ inhibitor |
| MK-2206 | Akt1/2/3 inhibitor |
| Ly294002 | PI3Kα/δ/β inhibitor |
| BAPTA-AM | calcium chelator |
| PD150606 | calpain inhibitor |
| CU CPT22 | TLR1/2 inhibitor |
| TAK-242 | TLR4 inhibitor |
| Ac-LEVD-CHO | caspase-4 antibody |
| Ac-YVAD-cmk | caspase-1 antibody |
| Tunicamycin | ER stress inducer |
| Ionomycin | Calcium ionophore |
| Pam3CSK4 | TLR2 agonist |
| LPS | TLR4 agonist |
2.2. Cell Isolation and Culture
In accordance with the Helsinki agreement, the hRPE cells were isolated within 24 hr of death from donor eyes and cultured as previously described (Elner et al., 1992; Elner et al., 1990). The primary-cultured hRPE cells were propagated for 2-3 passages and were assessed for characteristic RPE marks by immunohistochemistry, including cytokeratin 8/18, fibronectin, laminin, and type IV collagen, to ensure they maintained their differentiated status (Bian et al., 2004; Elner et al., 1991).
2.3. RT-PCR and qPCR for IL-1α
The total cellular RNA was isolated from hRPE cells by QIAshredder and RNeasy mini kit according to manufacturer’s protocol. The cDNA synthesis was set up according to the protocol for a reverse transcription system. Briefly, 5 μg of RNA was added to the reaction mixture with Superscript III reverse transcriptase (200 U/μl) and 1μl Oligod(T)20 (0.5 μg/μl) in a total volume of 20 μl. PCR for each product was performed of the cDNA solution with three different cycles (15, 25 and 35). PCR was accepted as semi-quantitative, when individual amplification was within the mid-linear portion of the response curve. Specific cDNA was amplified by 35, and 20 cycles for IL-1α and b-actin, respectively (Elner et al., 1981). The condition for caspase-4 PCR was as described by Lin et al (Lin et al., 2000) and confirmed by examining three cycles (15, 25 and 35) first and then cycle 32 was selected. The reaction was initiated by adding 0.15 μl of Taq DNA polymerase (5 u/ml) to a final volume of 20 μl. Each PCR product was analyzed by electrophoresis on a 2% agarose gel and stained with ethidium bromide.
β-actin was used as a control with the following forward and reverse primers: 5’-GTGGGGCGCCCCAGGCACCA-3’ and 5’-GCTCGGCCGTGGTGGTGAAGC-3’used in both reverse transcription polymerase chain reaction (RT-PCR) and real-time quantitative PCR (qPCR). qPCR was performed by using CFX96 real-time PCR detection system (Bio-Rad, Hercules, CA) to measure the fluorescence produced by SYBR Green I dye (Molecular Probes, Eugene, OR) that intercalates into PCR product. The PCR reactions were performed in triplicate on each cDNA template along with triplicate reactions of a housekeeping gene, β-actin. A negative control was obtained by performing PCR without cDNA. The primer pair for IL-1α (QT00026075) and caspase-4 (QT00001127) was purchased from QIAGEN. The thermal cycling conditions were: 3 min at 95 °C, followed by 40 cycles at 95 °C for 30 s, 57 °C for 30 s, and 72 °C for 30 s. All PCR reaction products were verified by melting curve analysis and agarose gel electrophoresis. The IL-1β mRNA expression levels were quantified by calculating the average value of triplicate reactions, normalized by the average value of triplicate reactions for the housekeeping β-actin gene.
2.4. ELISA
For IL-α, commercial human IL-1α ELISA kit was used to detect IL-α from 0 to 400 pg/ml in whole cell lysates. Briefly, hRPE cultures were washed by 4°C PBS then added by M-PER Mammalian Protein Extraction Reagent with Halt Protease Inhibitor Cocktail and Phenyl-methane-sulfonyl (PMSF). Next, whole cell lysates were harvested after centrifugation at 4°C, at 14,000rpm for 15 minutes. The plates were added by sample and reconstituted standards. The following steps were sequential incubations with biotinylated antibody reagent and streptavidin–HRP solution. Then, chromogen substrate tetramethylbenzidine (TMB) was added and the plates were incubated to desired extinction and the reaction was terminated with 2M H2SO4. Absorbance for each well at 450 nm was read and corrected with absorbance at 550 nm.
2.5. Western Blotting
Cellular extracts from hRPE cells for Western blots were processed according to the manufacturer’s procedure (Sigma-Aldrich, St. Louis, MO). An amount of 20–50 μg of protein or sample was analyzed by SDS-PAGE. Protein was electro-transferred to nitrocellulose membrane, blocked with a solution of TBS containing 5% of non-fat milk and 0.1% Tween-20 (TBST) for 1 hr, and probed with primary antibodies overnight, followed by washing three times with TBST. Next, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 1 hr at room temperature, and washed three additional times with TBST. The membrane was then visualized using an enhanced chemiluminescent technique (ECL).
2.6. Immunofluorescence Analysis of IL-1α in hRPE Cells
After plated in four-chamber glass slides (Lab-Tek, Polylabo, Strasbourg, France) for 24 h at 37 °C, hRPE cells were stimulated with the indicated condition medium for another 20 hr. Then, the medium was aspirated and the adherent cells were fixed with 4% paraformaldehyde for 15 min. Fixed cells were blocked. After three washes with 1% normal goat serum, the cells were either incubated with or without primary antibody for IL-1α or the corresponding isotype control overnight. The next day the cells were treated with secondary fluorescein isothiocyanate (FITC)-conjugated antibody and diluted in PBS solution containing 2% sheep serum and 1% BSA, at room temperature for 60 min in a humidified dark chamber. This was followed by washes with PBS solution. Finally, the cells were incubated with 1:10,000 bis-benzimide for 2 min and washed. The slides were mounted with prolong anti-fade kit mount (Molecular Probe, Inc) and sealed. The slides were examined under a fluorescence microscope equipped with an argon-krypton laser with blue light for FITC excitation under 400X magnification.
2.7. Statistical Analysis
Each experiment was confirmed by testing samples from three independent primary-cultured hRPE cell lines, thus results were representative of three independent experiments. For ELISA and functional assays the results were representative of three independent experiments that showed similar results with each data point in triplicate. Various assay conditions were compared using ANOVA and t-test by Statview software, and p<0.05 was considered to be statistically significant. In ELISA and functional assays values represent means ± SEM. In Western blots and RT-PCR data, only results from one representative experiment were selected to show in figures.
3. Results
3.1. Proinflammatory Agents and ER Stress Induce Increased IL-1α Expression in hRPE Cells
Pro-inflammatory agents LPS and IL-1β were first used to confirm an initial study that hRPE cells could be induced to express IL-1α mRNA and protein (Fig. 1) (Jaffe et al., 1992). Additionally, as ER stress has been shown to promote IL-1α expression in macrophages (Kandel-Kfir et al., 2015), we tested whether ER stress-inducer tunicamycin could upregulate IL-1β mRNA and protein expression in hRPE cells (Fig. 1). Following 6 hrs of exposure, LPS, tunicamycin, and IL-1α significantly enhanced mRNA synthesis in hRPE cells by 10, 32, and 52-fold, respectively (Fig. 1A). After 24-48 hrs of exposure, whole cell lysate and extracellular media were collected and analyzed separately for IL-1α using ELISA. In the whole cell lysate, LPS, tunicamycin, and IL-1β increased IL-1α protein production from 2 pg/ml in unstimulated hRPE cells to 17, 38 and 72 pg/ml, respectively (Fig. 1B-D). The inductions by LPS and tunicamycin were time-dependent, peaking at 24 hr and decreasing by about half at 48 hr (Fig. 1B-C). Using the same IL-1α ELISA kit, no IL-1α protein was found in the conditioned media.
Fig 1.

IL-1α mRNA synthesis and protein production in hRPE cells. mRNA levels were assessed for cells (A) treated with LPS (1000 ng/ml), tunicamycin (10 μM) or IL-1β (2 ng/ml) for 6 hr. IL-1α protein production was analyzed by ELISA in hRPE cells treated with LPS (B, 1000 ng/ml), tunicamycin (C, 10 μM) or IL-1β (D, 2 ng/ml) for 24 or 48 hr in the presence or absence of caspase-4 inhibitor (LEVD, 4 μM), caspase-1 inhibitor (YVAD, 2 μM), anti-IL-1β antibody (Ab IL-1β) or isotype serium (Ab Ctrl). The p values were calculated by comparing treatment to Ctrl (A), to LPS only (B), to tunicamycin only (C), or to IL-1β only (D). *p<0.05; **p<0.01; ***p<0.001. In order to compare IL-1α mRNA levels under different conditions, expression of the house keeping gene β-action was used to monitor gel loading. Ab, antibody; Ctrl, control; LEVD, Ac-LEVD-CHO; YVAD, Ac-YVAD-cmk.
Next, immunofluorescence was used to assess intracellular localization of the IL-1α protein. Following treatment of hRPE cells with LPS, tunicamycin, or IL-1β, intracellular IL-1α protein expression was enhanced in both the cytosol and nucleus (Fig. 2A-C) compared to weaker basal levels in the control (Fig. 2D). No stain or a very weak background stain was seen without primary antibody (Fig. 2E) or with an isotype control (Fig. 2F) (Bian et al., 2003).
Fig. 2.
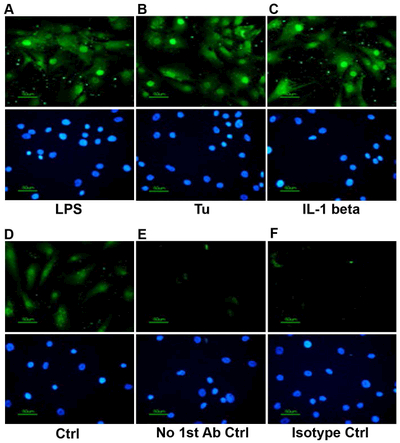
Immunofluorescence analysis of IL-1α expression in hRPE cells. hRPE cells seeded in chamber slides were incubated with LPS (A, 1000 ng/ml), tunicamycin (B, 10 μM) or IL-1β (C, E, F, 2 ng/ml) for 20 hr. Controls include untreated cells (D), IL-1β-treated cells without primary antibody (E), and IL-1β-treated cells with an isotype antibody control (F). The cells were fixed as described in Materials and Methods. The expressed IL-1α is shown in green. The nuclei were stained with 400x bisBenzimide (blue) (A-F, bottom panels). Experiments were repeated twice. Typical images were taken at 400X magnification.
3.2. Non-Canonical Inflammasome Mediates IL-1α Production in hRPE Cells
Several reports have demonstrated that human caspase-4 mediates LPS-induced non-canonical inflammasome activation and IL-1β and IL-1α secretion (Casson et al., 2015; Schmid-Burgk et al., 2015; Shi et al., 2015; Vigano et al., 2015). Our previous study has demonstrated that pro-inflammatory agents, including IL-1β and LPS, as well as ER stress induce caspase-4 expression and activation in hRPE cells (Bian et al., 2009). Thus, we investigated involvement of the non-canonical inflammasome pathway by evaluating caspase-4 and caspase-1, respectively, in inducing IL-1α expression (Vigano et al., 2015). Primary hRPE cells were challenged with LPS, tunicamycin or IL-1β in the presence or absence of caspase-4 inhibitor (Ac-LEVD-CHO) or caspase-1 inhibitor (Ac-YVAD-cmk) for 24 hr. IL-1α protein production in LPS-, tunicamycin-, or IL-1β-treated hRPE cells was reduced by 52, 36 and 27%, respectively, after using the caspase-4 inhibitor (Fig. 1B-D). LPS- and tunicamycin-induced IL-1α protein production was inhibited by the caspase-1 inhibitor by 47 and 34%, (Fig. B and C), respectively. However, the caspase-1 inhibitor had no statistically significant effect on IL-1β-induced IL-1α expression (Fig. 1D).
It is well known that caspase-1 activation is required for IL-1β activation and secretion, but caspase-1 does not directly induce IL-1α production or activation (Vigano et al., 2015). Thus, our finding that caspase-1 inhibition reduces IL-1α production in LPS- or tunicamycin-treated hRPE cells, but not in those treated with IL-1β, suggests that caspase-1 activates IL-1β to promote IL-1α production. Furthermore, as IL-1β is not secreted in hRPE cells in response to LPS, this suggests that the activated IL-1β acts in an autocrine manner in hRPE cells to further enhance LPS-induced hRPE IL-1α production. To examine this possibility, antibody against IL-1β was employed. The result showed that neutralization of IL-1β significantly reduced LPS-stimulated IL-1α production (Fig. 1B) by approximately 20%.
3.3. Type I or II Interferon Priming Increases hRPE Cell Response to LPS
Previous studies have reported that type I IFNs are required for caspase-11 activation in mouse macrophages (Rathinam et al., 2012). However, caspase-4 and caspase-5, the human orthologs of caspase-11, are not activated by type I IFNs in human macrophages and are not required for LPS-induced IL-1α or IL-1β secretion (Vigano et al., 2015). On the other hand, type II IFN, IFN-γ, is known to upregulate the expression and activation of caspase-4 (Bian et al., 2009). So, we evaluated the effects of type I and II IFNs in promoting IL-1α production in hRPE cells with and without LPS stimulation. First, type I IFN-α and IFN-β, but not type II IFN-γ, increased IL-1α mRNA synthesis in hRPE cells (Fig. 3A). Treating hRPE cells with IFN-β plus LPS produced higher IL-1α mRNA levels than that by either alone (Fig. 3B), whereas IFN-γ co-stimulation did not appear to alter LPS-induced IL-1α mRNA levels (Fig. 3C). However, priming with either IFN-β or IFN-γ followed by LPS stimulation increased hRPE IL-1α mRNA synthesis by 4- and 6-fold, respectively compared to LPS alone (Fig. 3D).
Fig. 3.

Effects of IFNs on caspase-4 expression/activation and LPS-induced IL-1α production. mRNA levels of IL-1α and caspase-4 were analyzed by RT-PCR (A-C) and qPCR (D, G). IL-1α protein was analyzed by ELISA (E and F). Caspase-4 protein cleavage in hRPE cells was shown by Western blot analysis (H). hRPE cells were treated with LPS (1000 ng/ml), IFN-α (1000 U/ml), IFN-β (1000 U/ml), IFN-γ (1000 U/ml), IFN-γ priming or in combination for 6 (A-D, G), 24 (E-F) or 0, 16 and 24 hr (H). In E and F, hRPE cells were pre-incubated with IFN-α, -β and -γ at 1000 U/ml for 16 hr and then switched to fresh growth media with or without LPS (1000 ng/ml) for an additional 24 hr. The p values were calculated by comparing treatment with LPS alone (D, E, F), treatment with LPS plus caspase-4 inhibitor (F), or treatment with control (G). *p<0.05; **p<0.01; ***p<0.001. Ctrl, control; IFN, interferon; LEVD, Ac-LEVD-CHO; pre-, pre-incubation with.
Despite our mRNA findings, without LPS-stimulation, ELISA analysis showed that neither type I IFN-α, -β nor type II INF-γ treatment alone induced any statistically significant induction of IL-1α protein production in hRPE cells (Fig. 3E-F). As it is well known that IFN-γ priming induces macrophages to have faster and elevated responses to LPS (Schroder et al., 2004), we hypothesized that LPS may be required for IFNs to promote increased IL-1α protein production (Schroder et al., 2004). Accordingly, priming with IFN-α, -β, or -γ, increased LPS-induced IL-1α protein production by 55, 91 and 122%, respectively (Fig. 3E-F).
3.4. Involvement of Caspase-4 in IL-1α Expression in hRPE Cells
Our previous study has demonstrated that IL-1β, LPS, tunicamycin, and IFN-γ all induced caspase-4 expression (Bian et al., 2009). As with IFN-γ, treating hRPE cells with IFN-α or IFN-β enhanced caspase-4 mRNA synthesis by 3-fold (Fig. 3A and G). Similar to IFN-γ (Bian et al., 2009), IFN-β also promoted caspase-4 activation, as evidenced by peak appearance of cleaved mature caspase-4 after 16 hr of treatment (Fig. 3H). Surprisingly, we found IFN-induced caspase-4 activation (Fig. 3H) without increasing IL-1α protein production (Fig. 3E-F), suggesting that IFNs prime the cells for infection but are not sufficient to induce IL-1α protein production without LPS. Fittingly, priming with either IFN-α, IFN-β or IFN-γ all increased LPS-induced IL-1α protein production (Fig. 3E-F), which was significantly blocked by a caspase-4 inhibitor (Fig. 3F).
3.5. Modulation of IL-1α Protein Production by TLR4 or TLR2 Binding in hRPE Cells
LPS is one of the best-characterized immunostimulatory structural motifs of the outer membrane of Gram-negative bacteria, also known as one type of pathogen-associated molecular pattern (PAMP). PAMP receptors are positioned to act as the first line of host defense in innate and adaptive immune responses within the retina. In addition to the recently discovered finding that intracellular LPS binds directly to caspase-4 (Shi et al., 2014), it has been known for a longtime that LPS is specifically recognized by the extracellular or endosomal TLR4 receptor, which plays a key role in ocular infectious and noninfectious diseases (Lee et al., 2012; Redfern and McDermott, 2010). Complementing this defense mechanism, TLR2 in combination with TLR1 or TLR6, mediates a response to triacylated or diacylated bacterial lipopeptides, respectively and has also been implicated in the defense response against LPS (da Silva Correia et al., 2001). Expression of both TLR4 and TLR2 receptors has been reported in hRPE cells (Elner et al., 2005; Nazari et al., 2014), and activation of both receptors induce and activate caspase-4/11 and produce IL-1α in other cell types (Agrawal and Gupta, 2011; Bian et al., 2009; Kayagaki et al., 2013).
Thus, we examined whether both TLR4 and TLR2 signaling pathways are involved in induction of hRPE IL-1α expression in hRPE cells. First, primary hRPE cells were pre-incubated with or without a TLR4 antagonist (TAK-242) for 0.5 hr. Then hRPE cells were challenged with TLR4-agonist LPS for 24 hr. The whole cell lysates were collected for IL-1α ELISA. The results showed that TAK-242 reduced LPS-induced IL-1α expression by about 55% (Fig. 4A), suggesting that TLR4 receptor signaling participates in LPS-stimulated IL-1α expression. Next, hRPE cells were pre-treated with a TLR2 neutralizing antibody before treatment with LPS or the TLR1/TLR2 agonist Pam3CSK4 for an additional 24 hr. Similar to LPS, in whole cell lysates, Pam3CSK4 induced a significant increase in hRPE IL-1α protein production (Fig. 4B), albeit to a slightly lesser extent than LPS. When Pam3CSK4 was used together with LPS, IL-1α expression was higher than when LPS or Pam3CSK4 was used alone, but less than the sum of both inducers (Fig. 4B). TLR2 neutralizing antibody reduced Pam3CSK4-induced IL-1α protein production by 47% (Fig. 4C), suggesting that the TLR2 signaling pathway is involved in regulating IL-1α expression in hRPE cells. Subsequently, we explored whether the TLR2-induced IL-1α expression may be regulated through caspase-4, in a similar manner to LPS-induced IL-1α expression. Notably, the Pam3CSK4-induced IL-1α production was at least partly caspase-4 dependent, as it was inhibited by 61% following caspase-4 blockage (Fig. 4C).
Fig. 4.

Modulation of IL-1α expression through TLR4 and TLR2 signaling. hRPE cells were cultured with LPS (1000 ng/ml) (A, B) or Pam3CSK4 (Pam 300 ng/ml) or LPS plus 100 ng/ml Pam3CSK4 (B, C) for 24 hr. In some experiments, hRPE cells also were pre-incubated with TAK-242 (75 μM) (A), caspase-4 inhibitor LEVD-CHO (4 μM) (C), TLR2 blocking antibody, or isotype control antibody (C). After stimulations, whole cell lysates were harvested and subjected to ELISA. *p<0.05; **p<0.01; ***p<0.001, as compared with inducers alone. Ab T2, anti-TLR2 antibody; Ab Ctrl, isotype control; LEVD, LEVD-CHO; Pam, Pam3CSK4; TAK, TAK-242.
3.6. Involvement of Calcium Signaling in IL-1α Production in hRPE Cells
Calcium, a ubiquitous intracellular messenger, is intricately involved in a wide spectrum of physiological functions, including signal transduction, gene expression, protein secretion, and ER stress. To investigate the role of calcium signaling in IL-1α expression in hRPE cells, we used BAPTA-AM, a cytosolic calcium chelator, and PD150606, a cell-permeable inhibitor for calpain calcium binding. Treatment with BAPTA-AM reduced LPS-, tunicamycin-, IL-1β- and Pam3CSK4-induced IL-1α protein production by 68% (Fig. 5A), 96% (Fig. 5B), 35% (Fig. 5C) and 30% (Fig. 5D), respectively. As compared to BAPTA-AM, PD150606 only reduced the LPS-induced IL-1α protein production by 13% (Fig. 5A).
Fig. 5.

Ca2+ signaling regulates IL-1α expression. hRPE cells were cultured with or without LPS (1000 ng/ml) (A and F), tunicamycin (Tu, 10μM) (B), IL-1β (2 ng/ml) (C), or Pam3CSK4 (Pam, 100 ng/ml) (D) for 6 (F) or 24 hr (A-D) with or without PD150606 (PD, 100 μM) or BAPTA-AM (BAPTA, 5 μM). hRPE cells were incubated with or without ionomycin (3 μM), LPS, and Pam3CSK4 in presence or absence of caspase-4 inhibitor, LEVD (4 μM), and inhabited with ionomycin plus LPS or ionomycin plus Pam3CSK4 for 24 hr (E). After incubation, the whole cell lysates were harvested and subjected to ELISA. Steady-state IL-1α mRNA was analyzed by qPCR (F). *p<0.05; **p<0.01; ***p<0.001, as compared with inducers alone (A-E), or with control (B).
The almost complete blockade of tunicamycin induction by BAPTA-AM is in agreement with the concept that calcium signaling is a key player in the ER stress-triggered response. To further confirm the involvement of calcium signaling in hRPE IL-1α expression, the calcium ionophore ionomycin was employed. The expected enhancement of intracellular calcium by ionomycin resulted in IL-1α production that was 60% greater than that induced by LPS alone (Fig. 5E). This process was insensitive to caspase-4 inhibitor Ac-LEVD-CHO (Fig. 5E), suggesting that the intracellular calcium-induced IL-1α protein production is caspase-4 independent. The IL-1α protein production by LPS plus ionomycin treatment synergistically enhanced IL-1α protein production by 54% more than the sum by each stimulant alone (Fig. 5E), a result similar to that reported for calcium ionophore in monocytes (Matsushima and Oppenheim, 1985). Ionomycin also synergized TLR2 agonist Pam3CSK4-induced IL-1α protein production by 25% more than the sum by each inducer alone (Fig. 5E). qPCR showed that calcium signaling had no effect on hRPE IL-1α mRNA synthesis (Fig. 5F), indicating that the effect of calcium signaling on IL-1α expression is at the translational level.
3.7. CU-CPT22 Functions to Enhance IL-1α Expression in hRPE Cells
CU-CPT22 is a novel small-molecule antagonist of the TLR1/2 complex, which effectively blocks downstream effectors of Pam3CSK4-TLR1/TLR2 signaling by out-competing Pam3CSK4 binding (Cheng et al., 2012; Daniele et al., 2015). Therefore, we investigated the effects of CU-CPT22 on Pam3CSK4- and LPS-induced hRPE IL-1α expression. Unexpectedly, we found that CU-CPT22 acted in a manner similar to Pam3CSK4, inducing but not inhibiting IL-1α protein production (Fig. 6A). Moreover, CU-CPT22 showed a concentration-dependent synergistic increase in hRPE IL-1α protein production with co-treatment of LPS (Fig. 6A). CU-CPT22 and LPS co-treatment resulted in about 4-fold higher IL-1α expression than the sum of CU-CPT22 and LPS alone and 2-fold higher than the sum of Pam3CSK4 and CU-CPT22 alone (Fig. 6A).
Fig. 6.

The effect of CU-CPT22 on IL-1α expression. hRPE cells were cultured with or without CU-CPT22 or co-cultured with LPS (1000 ng/ml) or Pam3CSK4 (100 ng/ml) for 24 hr (A-C). Unless specified, the concentration of CU-CPT22 was 10 μM, TAK-242 100μM, BAPTA-AM 5 μM, caspase-4 inhibitor, LEVD 4 μM, Ly 294002 75 μM. After incubation, the whole cell lysates were harvested and subjected to ELISA. *p<0.05; **p<0.01; ***p<0.001, as compared with inducers alone (A-C) or control (A, B). Ab T2 anti-TLR2 antibody; Ab Ctrl, isotype control; CU, CU-CPT22; BAPTA, BAPTA-AM; LEVD, LEVD-CHO; Ly, Ly 294002; Pam, Pam3CSK4; TAK, TAK-242.
In contrast to LPS, the CU-CPT22 induced hRPE IL-1α expression was insensitive to caspase-4 or TLR4 inhibition or chelation of intracellular calcium (Fig. 6B). As CU-CPT22 is claimed as a TLR1/TLR2 antagonist, we investigated whether CU-CPT22 induces hRPE IL-1α through TLR2 binding. TLR2 blocking antibody inhibited CU-CPT22-induced IL-1α production by 23% in contrast to 47% of that by Pam3CSK4by, suggesting that the CU-CPT22-induced IL-1α production is, at least in part, mediated by the TLR2 receptor (Fig. 6C). To shed further light on this mechanism, we explored whether the CU-CPT22-induced IL-1α protein production was mediated through the PI3K/Akt pathway, which has previously been reported to promote IL-1α mRNA synthesis and protein production (Bandyopadhaya et al., 2009; Turner et al., 2007). Surprisingly, treatment with the PI3K inhibitor, Ly294002, completely abrogated CU-CPT22-induced IL-1α protein production (Fig. 6B).
3.8. Modulation of PI3K/Akt Signaling Impacts hPRE IL-1α mRNA and Protein Production
Based on our above findings with CU-CPT22, we investigated whether the other agents promoting IL-1α production did so through the PI3K/Akt pathway. In order to do this, and minimize the chance of off-target effects, we selected several different unique PI3K/Akt inhibitors (Table 1), including CAL-101, PI-103, Wortmannin, MK-2206, and Ly294002. . Primary-cultured hRPE cells were pre-incubated with or without each of these compounds for 1 hr and then were challenged with LPS or IL-1β for 6 (mRNA) or 24 hr (ELISA). qPCR showed that Wortmannin potently increased LPS- and IL-1β-induced IL-1α mRNA synthesis by 72- and 67-fold, respectively, while MK-2206 only increased that by 1.3- and 1.7-fold, respectively (Fig. 7A). ELISA showed that LPS-induced IL-1α production was significantly increased by all concentrations tested of Wortmannin, MK-2206, CAL-101, and PI-103 (Fig. 7B). These results suggest negative regulation by PI3K/Akt pathway in hRPE IL-1α expression. In contrast, PI3K-inhibitor Ly294002 completely abolished the LPS-induced IL-1α mRNA synthesis and LPS-, tunicamycin-, IL-1β-, ionomycin-, and Pam3CSK-induced IL-1α protein production (Fig. 7C-D).
Fig. 7.

Effects of PI3K inhibitors on IL-1α expression. hRPE cells were cultured with or without LPS (1000 ng/ml) in the presence or absence of CAL-101, PI-103, Wortmannin and MK 2206 for 6 hr to measure mRNA levels (A, C) or 24 hr to measure protein (B, D). ***p<0.001, as compared with inducers alone.
4. Discussion
Our present study reveals that a variety of proinflammatory agents induce IL-1α expression in hRPE cells. These agents include TLR4- and caspase-4 agonist LPS, TLR2 agonist Pam3CSK4, IL-1β, ER stress inducer tunicamycin, and Ca2+ ionophore ionomycin. These findings suggest that IL-1α is an important danger signal in hRPE cells that responds to both infectious and non-infectious mediators. Notably, we provide the first evidence that the non-canonical inflammasome pathway can be stimulated in hRPE cells, as LPS stimulation led to caspase-4 activation and subsequent IL-1α production. Additionally, the tunicamycin-induced hRPE IL-1α expression was found highly sensitive to the calcium chelator BAPTA-AM with 96% inhibition, indicating that ER stress induction of IL-1α is completely dependent on intracellular [Ca2+]in release.
Mechanistically, IL-1α functions as a membrane receptor agonist and transcription factor-like nuclear protein, and belongs to a unique dual-function cytokine group characterized by having both intracellular and extracellular mechanisms of action (Rider et al., 2013). A distinctive characteristic of pro-IL-1α is the presence of a nuclear localization sequence at its amino-terminal that allows its translocation to the nucleus to exert transcriptional function (Kawaguchi et al., 2006). When synthesized, the intracellular pro-IL-1α may exist in an inactive state by binding to intracellular IL-1 receptor antagonist (IL-1Ra). IL-1Ra is expressed in hRPE cells (Holtkamp et al., 1999; Leung et al., 2009) and both IL-1α and IL-1Ra are present in overlapping peaks (Hammerberg et al., 1992). The binding of IL-1Ra to cytoplasmic pro-IL-1α prevents it from binding to HAX-1 (HS1-associated protein X-1) and translocating into the nucleus (Kawaguchi et al., 2006; Rider et al., 2013). IL-1RII, another cytosolic IL-1 receptor, is also expressed in hRPE cells (Leung et al., 2009), whose binding is thought to inhibit pro-IL-1α activity (Burzynski et al., 2015; Zheng et al., 2013). Once released, IL-1α does not require upregulation or processing to provoke inflammation (Lukens and Kanneganti, 2014), and it is associated with the onset of many inflammatory diseases in the first few hours following pathologic events (Cohen et al., 2010).
In our work, immunofluorescence showed that the induced hRPE IL-1α protein was increased in both the cytosol and nucleus, but there was no detectable secretion of IL-1α in the growth medium. This suggests, as has been reported previously (Di Paolo and Shayakhmetov, 2016) that the IL-1α produced in response to inflammatory mediators may be functioning in multiple roles, such as translocating to the nucleus to induce transcription and also regulating signaling in the cytosol or on the cell-surface. Furthermore, by using neutralizing antibody our study further indicates that about 20% of LPS-induced IL-1α was sensitive to neutralization of IL-1β, suggesting that the low level of IL-1β secretion (< 1 pg, data not shown) was able to strengthen the LPS induction via an autocrine mechanism. This may represent the caspase-1 sensitive portion of IL-1α production by LPS. Moreover, priming hRPE cells with type I IFN-α, -β or type II INF-γ, co-stimulating with ionomycin, and treating with CU-CPT22 all synergized LPS-induced IL-1α production. These synergistic effects imply the existence of cross-reaction and compensatory roles of different signaling pathways for maximal IL-1α expression. Further elucidation of these exact mechanisms is required in future studies.
In addition to binding to cell surface receptor LBP, LPS may also be internalized 61 and directly bind to caspase-4, leading to noncanonical inflammasome activation (Shi et al., 2014; Smith et al., 2015). The present study demonstrates that induction of hRPE IL-1α expression by LPS, Pam3CSKI4, tunicamycin, IFN-primed LPS, and IL-1β, except for ionomycin and CU-CPT22, are all partly caspase-4-dependent with 30 to 60% inhibition by the caspase-4 inhibitor, LEVD-CHO, suggesting that the non-canonical inflammasome can be activated from a variety of stressors in hRPE cells. Our finding that LEVD-CHO blocked induction by Pam3CSK4/TLR2 more than that by LPS/TLR4 (60 vs. 45%), indicates caspase-1 is involved more in TLR2 than TLR4 signaling.
Our findings that IFN-β increased caspase-4 expression and activation and potentiated LPS-induced IL-1α expression, together with the finding of the role of caspase-4 in IL-1α production, indicates that the interferon-α/β receptor (IFNAR) pathway also is involved in hRPE IL-1α production (Vigano et al., 2015). In addition, similar levels of inhibition between caspase-1 inhibitor YVAD-cmk and caspase-4 inhibitor LEVD-CHO were found for LPS- and tunicamycin-induced hRPE IL-1α expression, but for IL-1β-induced IL-1α expression there was no inhibition by YVAD-cmk. This may be due to the fact that recombinant IL-1β is already in its active, cleaved form and does not require caspase-1 for activation, suggesting that extracellular IL-1β acts in an autocrine/paracrine manner to promote IL-1α production. Further study of these exact mechanisms is warranted.
CU-CPT22 is a small molecule inhibitor for TLR1/2 dimerization (Cheng et al., 2012) and has been shown to compete with Pam3CSK4 binding to TLR2 and inhibit Pam3CSK4/TLR2-mediated pro-inflammatory responses in RAW 264.7 cells. Based on this described mechanism, our original goal was to use this compound to investigate blocking of TLR2-mediated IL-1α expression. We unexpectedly found that CU-CPT22 alone was able to induce hRPE IL-1α expression and, more importantly, it greatly enhanced Pam3CSK4/TLR2- and LPS/TLR4-stimulated IL-1α production by 2- and 4-fold, respectively. The partial blockade of CU-CUP22-induced hRPE IL-1α expression by TLR2 receptor blocking antibody indicates that the induction is, at least in part, mediated by TLR2 receptor. Experiments with corresponding inhibitors exclude the involvement of TLR4, intracellular calcium release, and caspase-4 signaling in CU-CPT22-induced IL-1α production. These contradictory results could be due to signaling differences in different cell types. As this compound is currently under development as a promising drug candidate for Parkinson’s disease and likely other diseases (Daniele et al., 2015; Yin and Flynn, 2016) the possible side effects of CU-CPT22 need to be carefully studied and considered.
The inhibition of LPS/TLR4-mediated IL-1α expression by PI3K/Akt inhibitors wortmannin, CAL-101, PI-103 and MK-2206 suggests PI3K/Akt pathway plays a negative regulatory role in TLR4 signaling in hRPE cells, a result consistent with previous reports in other cell types (Aksoy et al., 2012; Fukao and Koyasu, 2003). The negative regulation of TLR signaling as described in this and other studies suggest that PI3K is a key gate-keeping protein in innate immunity (Koyasu, 2003). However, we also found that Ly290004, a well-known and widely used PI3K inhibitor, showed completely opposite results, leading to complete blockage of IL-1α expression. An opposite effect between Ly294002 and Wortmannin has been reported in one previous study (Ueda et al., 2013). A proposed signaling pathway for hRPE IL-1α expression based on our findings and those in the literature is summarized in Fig. 8. Further delineation of the physiologic and pathophysiological engagement of PI3K in balancing immune and inflammatory responses will shed light on the functional roles of PI3K and provide approaches for therapeutic intervention.
Fig. 8.

A schematic view of pathways regulating IL-1α expression in hRPE cells. LPS induced IL-1α expression is mediated by two signal pathways: the TLR4 receptor pathway and the non-canonical inflammasome pathway. After stimulation with LPS, TLR4 binds the sorting adaptor TIRAP, forming an early-acting TIRAP/MyD88 complex, which activates NF-kB, leading to IL-1α expression. An alternative pathway, also involving TIRAP/MyD88, appears to mediate pro-IL-1α expression through TRL2 binding by its agonist, PAM3CSK4. However, the pro-inflammatory response by LPS is subject to negative regulation by PI3K, which stimulates dissociation and degradation of TIRAP and triggers a late-acting anti-inflammatory phase (dotted line), in which TLR4 recruits the TRIF/TRAM complex and induces endosomal-internalization. In this process, LPS may also to be internalized. The intracellular [LPS]in, as with ER-stress and IFNs, mediates non-canonical activation of the inflammasome leading to production of IL-1α. The roles of calcium-dependent calpain in IL-1α production may be due to its stimulation of IκBα degradation, which promotes pro-IL-1α maturation. The inflammasome-mediated release of IL-1β further induces IL-1α expression via an autocrine mechanism. The numbers represent corresponding references: 1 (Aksoy et al., 2012; Siegemund and Sauer, 2012); 2 (Husebye et al., 2006); 3 (Shi et al., 2014); 4 (Bian et al., 2009); 5 (Kayagaki et al., 2015); 6 (Schaecher et al., 2004); 7 (Kavita and Mizel, 1995); 8 (Cui et al., 2013).
Table 2.
Primer sequences used for RT-PCR
| Forward | Reverse | |
|---|---|---|
| IL-1α | 5’- CAGAAGACCTCCTGTCCTATGAG G-3’ |
5’- GTCAGGCATATTGGTGAGGCTGA C-3’ |
| Caspase- 4 |
5’- CAGACTCTATGCAAGAGAAGCAA CGTATGGCAGGA-3’ |
5’- CACCTCTGCAGGCCTGGACAAT GATGAC-3’ |
Highlights:
hRPE cells upregulate IL-1α expression in response to pro-inflammatory signals
IL-1α production is negatively regulated by the PI3K/Akt pathway
LPS induces the non-canonical inflammasome in hRPE cells via caspase-4 signaling
Non-canonical inflammasome activation promotes IL-1α expression
Acknowledgments
Funding: This study was supported by NIH Grants EY-09441, N007361, EY007003, and Research to Prevent Blindness-Senior Scientific Award (VME).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Agrawal S, Gupta S, 2011. TLR1/2, TLR7, and TLR9 signals directly activate human peripheral blood naive and memory B cell subsets to produce cytokines, chemokines, and hematopoietic growth factors. J Clin Immunol 31, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy E, Taboubi S, Torres D, Delbauve S, Hachani A, Whitehead MA, Pearce WP, Berenjeno IM, Nock G, Filloux A, Beyaert R, Flamand V, Vanhaesebroeck B, 2012. The p110delta isoform of the kinase PI(3)K controls the subcellular compartmentalization of TLR4 signaling and protects from endotoxic shock. Nat Immunol 13, 1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhaya A, Bhowmick S, Chaudhuri K, 2009. Activation of proinflammatory response in human intestinal epithelial cells following Vibrio cholerae infection through PI3K/Akt pathway. Can J Microbiol 55, 1310–1318. [DOI] [PubMed] [Google Scholar]
- Bian ZM, Elner SG, Elner VM, 2009. Dual involvement of caspase-4 in inflammatory and ER stress-induced apoptotic responses in human retinal pigment epithelial cells. Investigative ophthalmology & visual science 50, 6006–6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian ZM, Elner SG, Yoshida A, Elner VM, 2003. Human RPE-monocyte co-culture induces chemokine gene expression through activation of MAPK and NIK cascade. Experimental eye research 76, 573–583. [DOI] [PubMed] [Google Scholar]
- Bian ZM, Elner SG, Yoshida A, Elner VM, 2004. Differential involvement of phosphoinositide 3-kinase/Akt in human RPE MCP-1 and IL-8 expression. Investigative ophthalmology & visual science 45, 1887–1896. [DOI] [PubMed] [Google Scholar]
- Brandstetter C, Patt J, Holz FG, Krohne TU, 2016. Inflammasome priming increases retinal pigment epithelial cell susceptibility to lipofuscin phototoxicity by changing the cell death mechanism from apoptosis to pyroptosis. J Photochem Photobiol B 161, 177–183. [DOI] [PubMed] [Google Scholar]
- Burzynski LC, Humphry M, Bennett MR, Clarke MC, 2015. Interleukin-1alpha Activity in Necrotic Endothelial Cells Is Controlled by Caspase-1 Cleavage of Interleukin-1 Receptor-2: IMPLICATIONS FOR ALLOGRAFT REJECTION. The Journal of biological chemistry 290, 25188–25196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casson CN, Yu J, Reyes VM, Taschuk FO, Yadav A, Copenhaver AM, Nguyen HT, Collman RG, Shin S, 2015. Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc Natl Acad Sci U S A 112, 6688–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K, Wang X, Zhang S, Yin H, 2012. Discovery of small-molecule inhibitors of the TLR1/TLR2 complex. Angew Chem Int Ed Engl 51, 12246–12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, Voronov E, Martin MU, Dinarello CA, Apte RN, 2010. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci U S A 107, 2574–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui GM, Zhao YX, Zhang NN, Liu ZS, Sun WC, Peng QS, 2013. Amiloride attenuates lipopolysaccharide-accelerated atherosclerosis via inhibition of NHE1-dependent endothelial cell apoptosis. Acta Pharmacol Sin 34, 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Correia J, Soldau K, Christen U, Tobias PS, Ulevitch RJ, 2001. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex. transfer from CD14 to TLR4 and MD-2. The Journal of biological chemistry 276, 21129–21135. [DOI] [PubMed] [Google Scholar]
- Daniele SG, Beraud D, Davenport C, Cheng K, Yin H, Maguire-Zeiss KA, 2015. Activation of MyD88-dependent TLR1/2 signaling by misfolded alpha-synuclein, a protein linked to neurodegenerative disorders. Sci Signal 8, ra45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detrick B, Nagineni CN, Grillone LR, Anderson KP, Henry SP, Hooks JJ, 2001. Inhibition of human cytomegalovirus replication in a human retinal epithelial cell model by antisense oligonucleotides. Investigative ophthalmology & visual science 42, 163–169. [PubMed] [Google Scholar]
- Di Paolo NC, Shayakhmetov DM, 2016. Interleukin 1alpha and the inflammatory process. Nat Immunol 17, 906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA, 2009. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27, 519–550. [DOI] [PubMed] [Google Scholar]
- Elner SG, Elner VM, Pavilack MA, Todd RF 3rd, Mayo-Bond L, Franklin WA, Strieter RM, Kunkel SL, Huber AR, 1992. Modulation and function of intercellular adhesion molecule-1 (CD54) on human retinal pigment epithelial cells. Laboratory investigation; a journal of technical methods and pathology 66, 200–211. [PubMed] [Google Scholar]
- Elner SG, Petty HR, Elner VM, Yoshida A, Bian ZM, Yang D, Kindezelskii AL, 2005. TLR4 mediates human retinal pigment epithelial endotoxin binding and cytokine expression. Transactions of the American Ophthalmological Society 103, 126–135; discussion 135–127. [PMC free article] [PubMed] [Google Scholar]
- Elner SG, Strieter RM, Elner VM, Rollins BJ, Del Monte MA, Kunkel SL, 1991. Monocyte chemotactic protein gene expression by cytokine-treated human retinal pigment epithelial cells. Laboratory investigation; a journal of technical methods and pathology 64, 819–825. [PubMed] [Google Scholar]
- Elner VM, Schaffner T, Taylor K, Glagov S, 1981. Immunophagocytic properties of retinal pigment epithelium cells. Science 211, 74–76. [DOI] [PubMed] [Google Scholar]
- Elner VM, Strieter RM, Elner SG, Baggiolini M, Lindley I, Kunkel SL, 1990. Neutrophil chemotactic factor (IL-8) gene expression by cytokine-treated retinal pigment epithelial cells. Am J Pathol 136, 745–750. [PMC free article] [PubMed] [Google Scholar]
- Fukao T, Koyasu S, 2003. PI3K and negative regulation of TLR signaling. Trends Immunol 24, 358–363. [DOI] [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA, 2013. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerberg C, Arend WP, Fisher GJ, Chan LS, Berger AE, Haskill JS, Voorhees JJ, Cooper KD, 1992. Interleukin-1 receptor antagonist in normal and psoriatic epidermis. J Clin Invest 90, 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus J, Anderson C, Wang S, 2015. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res Rev 24, 286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus J, Zhao F, Wang S, 2016. Current therapeutic developments in atrophic age-related macular degeneration. The British journal of ophthalmology 100, 122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtkamp GM, de Vos AF, Kijlstra A, Peek R, 1999. Expression of multiple forms of IL-1 receptor antagonist (IL-1ra) by human retinal pigment epithelial cells: identification of a new IL-1ra exon. Eur J Immunol 29, 215–224. [DOI] [PubMed] [Google Scholar]
- Husebye H, Halaas O, Stenmark H, Tunheim G, Sandanger O, Bogen B, Brech A, Latz E, Espevik T, 2006. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J 25, 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe GJ, Van Le L, Valea F, Haskill S, Roberts W, Arend WP, Stuart A, Peters WP, 1992. Expression of interleukin-1 alpha, interleukin-1 beta, and an interleukin-1 receptor antagonist in human retinal pigment epithelial cells. Experimental eye research 55, 325–335. [DOI] [PubMed] [Google Scholar]
- Kandel-Kfir M, Almog T, Shaish A, Shlomai G, Anafi L, Avivi C, Barshack I, Grosskopf I, Harats D, Kamari Y, 2015. Interleukin-1alpha deficiency attenuates endoplasmic reticulum stress-induced liver damage and CHOP expression in mice. J Hepatol 63, 926–933. [DOI] [PubMed] [Google Scholar]
- Kavita U, Mizel SB, 1995. Differential sensitivity of interleukin-1 alpha and -beta precursor proteins to cleavage by calpain, a calcium-dependent protease. The Journal of biological chemistry 270, 27758–27765. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Nishimagi E, Tochimoto A, Kawamoto M, Katsumata Y, Soejima M, Kanno T, Kamatani N, Hara M, 2006. Intracellular IL-1alpha-binding proteins contribute to biological functions of endogenous IL-1alpha in systemic sclerosis fibroblasts. Proc Natl Acad Sci U S A 103, 14501–14506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM, 2015. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM, 2011. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM, 2013. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249. [DOI] [PubMed] [Google Scholar]
- Kinnunen K, Petrovski G, Moe MC, Berta A, Kaarniranta K, 2012. Molecular mechanisms of retinal pigment epithelium damage and development of age-related macular degeneration. Acta Ophthalmol 90, 299–309. [DOI] [PubMed] [Google Scholar]
- Koyasu S, 2003. The role of PI3K in immune cells. Nat Immunol 4, 313–319. [DOI] [PubMed] [Google Scholar]
- Lee HS, Hattori T, Park EY, Stevenson W, Chauhan SK, Dana R, 2012. Expression of toll-like receptor 4 contributes to corneal inflammation in experimental dry eye disease. Investigative ophthalmology & visual science 53, 5632–5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KW, Barnstable CJ, Tombran-Tink J, 2009. Bacterial endotoxin activates retinal pigment epithelial cells and induces their degeneration through IL-6 and IL-8 autocrine signaling. Mol Immunol 46, 1374–1386. [DOI] [PubMed] [Google Scholar]
- Lin XY, Choi MS, Porter AG, 2000. Expression analysis of the human caspase-1 subfamily reveals specific regulation of the CASP5 gene by lipopolysaccharide and interferon-gamma. The Journal of biological chemistry 275, 39920–39926. [DOI] [PubMed] [Google Scholar]
- Liu Y, Kimura K, Orita T, Sonoda KH, 2015. Necrosis-Induced Sterile Inflammation Mediated by Interleukin-1alpha in Retinal Pigment Epithelial Cells. PloS one 10, e0144460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukens JR, Kanneganti TD, 2014. Beyond canonical inflammasomes: emerging pathways in IL-1-mediated autoinflammatory disease. Semin Immunopathol 36, 595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima K, Oppenheim JJ, 1985. Calcium ionophore (A23187) increases interleukin 1 (IL-1) production by human peripheral blood monocytes and interacts synergistically with IL-1 to augment concanavalin A stimulated thymocyte proliferation. Cell Immunol 90, 226–233. [DOI] [PubMed] [Google Scholar]
- Moyer AL, Ramadan RT, Thurman J, Burroughs A, Callegan MC, 2008. Bacillus cereus induces permeability of an in vitro blood-retina barrier. Infect Immun 76, 1358–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagineni CN, Detrick B, Hooks JJ, 2000. Toxoplasma gondii infection induces gene expression and secretion of interleukin 1 (IL-1), IL-6, granulocyte-macrophage colony-stimulating factor, and intercellular adhesion molecule 1 by human retinal pigment epithelial cells. Infect Immun 68, 407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazari H, Karakousis PC, Rao NA, 2014. Replication of Mycobacterium tuberculosis in retinal pigment epithelium. JAMA Ophthalmol 132, 724–729. [DOI] [PubMed] [Google Scholar]
- Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA, 2012. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfern RL, McDermott AM, 2010. Toll-like receptors in ocular surface disease. Experimental eye research 90, 679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider P, Carmi Y, Voronov E, Apte RN, 2013. Interleukin-1alpha. Semin Immunol 25, 430–438. [DOI] [PubMed] [Google Scholar]
- Schaecher K, Goust JM, Banik NL, 2004. The effects of calpain inhibition on IkB alpha degradation after activation of PBMCs: identification of the calpain cleavage sites. Neurochem Res 29, 1443–1451. [DOI] [PubMed] [Google Scholar]
- Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V, 2015. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol 45, 2911–2917. [DOI] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, Hume DA, 2004. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75, 163–189. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F, 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F, 2014. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192. [DOI] [PubMed] [Google Scholar]
- Siegemund S, Sauer K, 2012. Balancing pro- and anti-inflammatory TLR4 signaling. Nat Immunol 13, 1031–1033. [DOI] [PubMed] [Google Scholar]
- Smith C, Wang X, Yin H, 2015. Caspases come together over LPS. Trends Immunol 36, 59–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon A, Dursun D, Liu Z, Xie Y, Macri A, Pflugfelder SC, 2001. Pro- and anti-inflammatory forms of interleukin-1 in the tear fluid and conjunctiva of patients with dry-eye disease. Investigative ophthalmology & visual science 42, 2283–2292. [PubMed] [Google Scholar]
- Tseng WA, Thein T, Kinnunen K, Lashkari K, Gregory MS, D’Amore PA, Ksander BR, 2013. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Investigative ophthalmology & visual science 54, 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner NA, Mughal RS, Warburton P, O’Regan DJ, Ball SG, Porter KE, 2007. Mechanism of TNFalpha-induced IL-1alpha, IL-1beta and IL-6 expression in human cardiac fibroblasts: effects of statins and thiazolidinediones. Cardiovasc Res 76, 81–90. [DOI] [PubMed] [Google Scholar]
- Uchiyama R, Tsutsui H, 2015. Caspases as the key effectors of inflammatory responses against bacterial infection. Arch Immunol Ther Exp (Warsz) 63, 1–13. [DOI] [PubMed] [Google Scholar]
- Ueda K, Nakahara T, Akanuma K, Mori A, Sakamoto K, Ishii K, 2013. Differential effects of LY294002 and wortmannin on neurons and vascular endothelial cells in the rat retina. Pharmacol Rep 65, 854–862. [DOI] [PubMed] [Google Scholar]
- Vann VR, Atherton SS, 1991. Neural spread of herpes simplex virus after anterior chamber inoculation. Investigative ophthalmology & visual science 32, 2462–2472. [PubMed] [Google Scholar]
- Vigano E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A, 2015. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat Commun 6, 8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Elner SG, Chen X, Field MG, Petty HR, Elner VM, 2011. MCP-1-activated monocytes induce apoptosis in human retinal pigment epithelium. Investigative ophthalmology & visual science 52, 6026–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Flynn AD, 2016. Drugging Membrane Protein Interactions. Annu Rev Biomed Eng 18, 51–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Yoshimura N, Hangai M, Tanihara H, Honda Y, 1994. Interleukin-1 alpha, interleukin-1 beta, and tumor necrosis factor gene expression in endotoxin-induced uveitis. Investigative ophthalmology & visual science 35, 1107–1113. [PubMed] [Google Scholar]
- Zheng Y, Humphry M, Maguire JJ, Bennett MR, Clarke MC, 2013. Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity 38, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]