

Abstract
Single-molecule (SM) microscopy is a powerful tool capable of visualizing individual molecules and events in real time. SM imaging may rely on proteins or nucleic acids labelled with a fluorophore. Unfortunately photobleaching of fluorophores leads to irreversible loss of signal, impacting the collection of data from SM experiments. Trace amounts of dissolved oxygen (O2) are the main cause of photobleaching. Oxygen scavenging systems (OSS) have been developed that decrease dissolved O2. Commercial OSS enzyme preparations are frequently contaminated with nucleases that damage nucleic acid substrates. In this protocol, we purify highly active Pseudomonas putida protocatechuate 3,4-dioxygenase (PCD) without nuclease contaminations. Quantitation of Cy3 photostability revealed that PCD with its substrate protocatechuic acid (PCA) increased the fluorophore half-life 100-fold. This low cost purification method of recombinant PCD yields an enzyme superior to commercially available OSS that is effectively free of nuclease activity.
Keywords: protein purification, protocatechuate 3, 4 dioxygenase, nuclease contaminants, single-molecule fluorescence, photobleaching, oxygen-scavenging systems
INTRODUCTION
Single biomolecule manipulation and observation techniques play a central role in modern biophysical analysis. Biological processes examined by single-molecule (SM) techniques include the entire central dogma [1–11], DNA damage repair pathways [12–14], RNA structure and function [15, 16], protein structure and function [17, 18], events in viral life cycle [19–24], and conformational transitions of proteins associated with neurobiology [25–27]. A common theme emerging from these studies is that SM techniques are capable of linking biology to the fundamental laws of chemistry and physics. Fluorescence detection is a powerful analytical tool with sensitivity at the SM level that allows visualizing a cohort of individual biomolecules in action. SM fluorescence imaging can generate reaction trajectories of long-range movements such as sliding of an enzyme on DNA [14, 24] as well as short-range movements such as conformational transitions of RNAs or proteins [15–18, 25, 26, 28]. Direct visualization of individual reaction trajectories allows identification of the dynamics, formation of transient intermediates, stochastic and asynchronous events as underlying principles of biology.
Small organic fluorophores or quantum dots attached to biomolecules are usually employed in SM experiments in vitro [29, 30]. These are capable of emitting ~106 − 109 s−1 photons before photobleaching irreversibly [31]. The lifetime and quantum yield of a fluorophore can be severely compromised by premature photobleaching. Imaging fast reactions requires capturing images at a faster rate (smaller detector integration times), which in turn requires stronger illumination of the samples to produce a sufficient signal. In contrast, imaging of slower reactions involves prolonged observation of the samples. Both the stronger and extended illuminations may lead to premature photobleaching of fluorophores resulting in inefficient or incomplete data acquisition [31, 32] during a SM fluorescence experiment.
Trace amounts of molecular oxygen (O2) present in aqueous solutions photochemically convert into singlet-oxygen (SO2) during fluorophore excitation [31]. SO2 may then react with fluorophores and biomolecules leading to photobleaching and phototoxicity. Removing O2 by chemical or enzymatic reactions can prevent this cascade. In SM fluorescence experiments oxygen scavenging systems (OSS) are commonly used for this purpose [33–35]. OSS composed of glucose oxidase and catalase (GODCAT) removes O2 by glucose oxidation albeit with only 50% efficiency due to its O2 reproduction mechanism [36]. In addition, the formation of intermediate H2O2 can in principle lead to more photobleaching and phototoxicity [31]. In contrast, the OSS consisting of the non-heme iron (Fe) metalloenzyme protocatechuate 3,4-dioxygenase and its substrate protocatechuic acid (PCD-PCA) can remove O2 completely and efficiently leading to extended fluorophore lifetimes [36, 37]. Moreover, we have previously shown that widespread nuclease contaminants found in commercial OSS enzymes caused extensive DNA damages such as strand scissions and double-strand breaks (DSB) [37]. We have developed a simple chromatographic purification scheme to remove these contaminants from commercial PCD. Here we extended our previous studies to overexpress recombinant PCD from Pseudomonas putida [38] in E. coli and purify large quantities using two chromatographic steps. Simple methods of testing nuclease and PCD activities are detailed to improve the purification scheme. Furthermore, the methods established in this work allow production of large quantities of highly active PCD that can be frozen for long-term usage without losing the activity upon thawing. Exceptional photostability and the absence of detectable DNA damaging make the recombinant purified PCD superior to any currently available OSS enzyme.
MATERIALS AND METHODS
Expression of PCD
The plasmid pVP91A-pcaHG encodes the α (pcaG) and the β (pcaH) subunits of PCD from Pseudomonas putida (38) (Figure 1) for overexpression of recombinant PCD in E. coli. An octa-Histidine (8xHis) tag is encoded at the N terminus of the β subunit. E. coli strain BL21 was transformed with pVP91A-pcaHG and selected on Luria-Bertani (LB)-Agar plates containing 50 μg/mL Ampicillin. Fresh individual colonies were grown overnight (~18 hours) at 37 °C in LB media containing 50 μg/mL Ampicillin while shaking at 225 rpm. Overnight cultures were then diluted 1:50 into 1 L LB media containing 50 μg/mL Ampicillin and incubated at 37 °C while shaking at 225 rpm. When the optical density of the cells at 600 nm (OD600) reached ~0.5, cultures were transferred to a 17 °C incubator set to 180 rpm shaking. After ~1 hour, when OD600 ~0.7, PCD expression was induced by adding 0.5 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) and 10 mg/L ferrous ammonium sulfate (Fe(NH4)2(SO4)2). Cells were kept at these conditions overnight (~18 hours) for optimal protein production. Cells were harvested by centrifugation at 3000 × g for 20 min. Cell pellets from each 1 L culture were resuspended in 10 mL of Resuspension Buffer (50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 20 mM imidazole, 10% glycerol, 1 μg/mL Leupeptin, 800 ng/mL Pepstatin, and 87.1 μg/mL PMSF). Resuspended cell pellets were flash frozen in liquid nitrogen and stored at −80 °C.
Figure 1. Recombinant expression of P. putida PCD in E. coli.
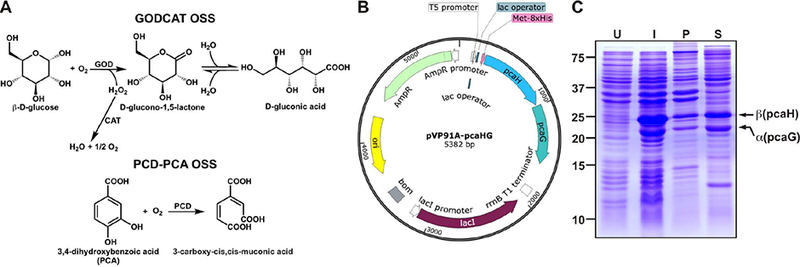
(A) Plasmid map of pVP91A-pcaHG containing the α (pcaG) and β (pcaH) subunits of PCD. The complete sequence of this plasmid is shown in Supplementary Figure 1. Locations of the T5 promoter, lac operator, and 8xHis are also shown. (B) SDS-PAGE analysis showing the whole cell lysate of uninduced (U), and induced (I) BL21 cells. Induction under optimal conditions resulted in a minor insoluble pellet (P) and a major soluble (S) fraction of PCD. The MW of α and β subunits are 22.4 kDa and 28.3 kDa respectively.
Purification of PCD
Cell lysis
Cell pellets were thawed slowly on ice and then sonicated for 1 min using the settings: 1 s pulse on, 1 s pulse off at 30% amplitude (digital sonifier, Branson). To further facilitate the lysis, 0.2 mg/mL lysozyme (Sigma-Aldridge) was added and lysates were incubated for 30 min on ice. The soluble protein fraction containing PCD was separated from the insoluble cell pellet by centrifuging at 120,000 × g for 1 hour at 4 °C.
Ni-NTA affinity column chromatography
All steps were performed at 0.15 mL/min flow rate. Ni-NTA-Buffer A contained 50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 10% glycerol, 1 μg/mL Leupeptin, 800 ng/mL Pepstatin, and 87.1 μg/mL PMSF. Ni-NTA-Buffer B was Ni-NTA-Buffer A supplemented with 250 mM imidazole. The soluble protein fraction was loaded onto a 2 mL Ni-NTA (Qiagen) column pre-equilibrated with 8% Ni-NTA-Buffer B. Non-specifically bound proteins were eluted by washing the column with 20 mL of 8% Ni-NTA-Buffer B. Next, the Ni-NTA-Buffer B was increased to 50% (125 mM imidazole) for 15 mL, and then to 100% (250 mM imidazole) for another 15 mL to elute the protein. Larger (800 μL) fractions were collected for the first elution peak and smaller (200 μL) fractions were collected for the second elution peak. Peak fractions were analyzed on 15% SDS-PAGE gels, and also evaluated for the presence nuclease contaminants as described below.
Nuclease activity of Ni-NTA fractions
The pXba+ plasmid DNA was amplified in XL10 gold E. coli strain and purified using the Qiagen Plasmid Miniprep kit carefully following the protocols provided by the manufacturer. Cell lysates were carefully inverted and maintained on ice during the alkaline lysis step and vortex mixers were avoided during reagent blending to minimize DNA damage that ultimately results in nicked or linear DNA products that could obscure accurate nuclease quantification. The purified supercoiled pXba+ (10 ng/μL) was incubated with 5 μL of each protein fraction in a 50 μL total volume of Ni-NTA Nuclease Buffer (50 mM Tris-HCl, pH 7.5, 130 mM NaCl, 5 mM MgCl2, 0.1 mM DTT, 2–25 mM imidazole) at 37 °C for 1 hour. Reactions were terminated by adding the Stop Buffer (25 mM EDTA, pH 8.0, 0.1% SDS, and 1X gel loading dye), and products were analyzed by a 1% agarose gels supplemented with 0.5 μg/mL ethidium bromide. Gels were imaged on a gel doc system (Bio-Rad), and quantified using ImageQuant software. As a positive control, PCD from Sigma-Aldridge (PCDa) was used at regular 1XSM (~60 nM) concentration, and the reaction buffer for PCDa also contained 25 mM imidazole.
To further evaluate the DNA damaging mechanism, 1XSM PCDa or Ni-NTA fraction #4 (5 μL) was incubated at 37 °C for 1 hour with supercoiled pXba+ DNA (10 ng/μL). Ni-NTA Nuclease Buffer (50 μL total volume) was modified for each reaction by including one of the following components: 10 mM EDTA, 5 mM DTT or 5 mM PCA. Reactions were stopped and analyzed as described above.
Size exclusion column chromatography
All the steps were done at 0.5 mL/min flow rate using a Superose 12 (Sp12) high-resolution gel filtration column (GE Healthcare). The mobile phase consisted of 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 10% glycerol, and 0.1 mM EDTA. Fractions from the second elution peak of the Ni-NTA affinity column were pooled, the volume reduced to 100–200 μL using a 10 kDa molecular weight (MW) cut-off Amicon concentrator (Millipore), and loaded onto the Superose 12 column pre-equilibrated with the mobile phase. Protein fractions (250 μL) were collected during the elution. Peak fractions were analyzed on 15% SDS-PAGE gels and evaluated for PCD enzymatic activity and contaminating nucleases. A molecular weight calibration curve was generated by running a set of globular molecular weight standards (Bio-Rad) and was fit to a line with the equation log10 (MW) = 7.5 − [0.22 × retention volume (mL)].
Nuclease and ensemble level PCD activity following gel filtration
This combined assay was carried out at 37 °C for 1 hour in a flat bottom 96 well plate (Costar) using a multiplex plate reader (Spectra Max M2, Molecular devices). Supercoiled plasmid pXba+ (10 ng/μL) was incubated with 10 μL of each protein fraction (only 1.2 μL for the Load fraction to compensate for the concentration) in a 50 μL total volume of Sp12-Nuclease/PCD Buffer (50 mM Tris-HCl, pH 7.5, 130 mM NaCl, 5 mM MgCl2, 0.1 mM DTT, 5 mM PCA). To minimize the PCD reaction during sample preparation, the plate was kept on ice in a 4 °C cold room. PCD enzymatic activity was measured by monitoring the reduction of PCA absorbance of at 290 nm (A290). Data points were acquired at 20 s intervals while shaking the plate (5 s before readings). After 1 hour, reactions were terminated and the DNA products analyzed by agarose gel electrophoresis as described above. The PCD activity was calculated for each fraction as the change in A290 (ΔA290) during the first 10 min of incubation and normalized to the highest ΔA290 value [37].
PCD concentration measurement
Total PCD concentration in each fraction was determined by UV absorbance at 280 nm (A280) using the extinction coefficient (ε280, Total ) 734,700 M−1cm−1. The online proteomic tool ExPASy-ProtParam (http://web.expasy.org/protparam/) was used to estimate the ε280, Total assuming that the PCD complex consists of 12 αβ dimers [38, 39]. Similarly the Fe(III) concentration in each fraction can be determined by the absorbance at 450 nm (A450). Moreover, the percentage of Fe(III) containing active sites was estimated using the Sp12 chromatograms at 280 nm and 450 nm using the following expression, and successful purification may require this value at least close to 50% [38].
where c is the concentration calculated by Beer-Lambert equation (A = εcl) [40], A450 is the absorbance of the Fe(III)-chromophore at 450 nm, A280 is the absorbance at 280 nm, ε450, Single chromophore = 2,860 M−1 cm−1 [38] and ε280, Single αβ dimer = 61,225 M−1cm−1.
SM photobleaching assay for purified PCD
Cy3-labeled DNA (Supplementary Table 1) was immobilized a on a PEG-passivated quartz surface of an SM-sample chamber via biotin-neutravidin linkage with a surface density of ~0.2 molecules/μm2. Fluorophores were continuously excited by illuminating ~4 mm2 of the sample chamber surface with a green laser (532 nm, 4.5 mW) at 1.1 mW/mm2 utilizing a total internal reflection fluorescence (TIRF) microscope. For each SM movie, emissions from individual Cy3 molecules were recorded on an electron multiplied charge coupled device (EMCCD, Princeton Instruments) camera. Data were collected for 5 min at 250 ms temporal resolution in the absence or presence of increasing concentrations of purified PCD in Imaging Buffer (50 mM Tris-HCl, pH 7.5, 130 mM NaCl, 5 mM MgCl2, 0.1 mM DTT, 5 mM PCA, 2–3 mM Trolox and 0.2 mg/mL BSA). Extended observations for higher PCD concentrations were performed at the same illumination conditions for 20 min at 1 s temporal resolution. Once the activities were confirmed, fractions with the highest PCD activity and no detectable nuclease activity were aliquoted. These aliquots were flash frozen in liquid N2 and stored in −80 °C. SM experiments showed that freezing, thawing and storing at −80 °C did not affect the enzymatic activity.
SM movies were processed using a custom MATLAB (Math Works) particle tracking code [41] as follows. First, the movies were averaged using a 17 frames sliding average, and then a small area (1/10th of the total area) of the averaged-movie was used to generate a coefficient file by manually changing the intensity threshold to select a maximum number of well-defined spots. This file was then used to analyze the entire movie. The percentage of particles that remained fluorescent in each movie was tracked every 40 frames to build survival probability curves. A pseudo-first order kinetic model (y = Ae−kt) was used to fit the data using IGOR Pro (WaveMetrics) and the decay rate (k) was used to estimate the half-life (t1/2) using the expression, t1/2 = ln (2)/k, corresponds to 50% photobleaching of Cy3. The error bars in t1/2 values indicate standard error from the fittings.
RESULTS
PCD expressed in E. coli is mostly soluble
Using the plasmid vector pVP91A-pcaHG (Figure 1A, Supplementary Figure 1), we overexpressed the α and β subunits of Pseudomonas putida PCD in E. coli strain BL21. We did not observe expression of PCD before induction (Figure 1B). When induced with IPTG, a major fraction of PCD was soluble and a small quantity was apparent in the insoluble pellet (Figure 1B). The high yield of soluble protein enabled us to produce ~1–2 mg of purified recombinant PCD per 1 L of cell culture.
Ni-NTA affinity chromatography resulted in nearly pure PCD with undetectable nuclease activity
As the first step of purification we employed a Ni-NTA affinity column to isolate soluble PCD (Figure 2). The 8xHis tag located at the N terminus of the β subunits binds tightly to Ni-NTA. PCD elution observed in flow through and washing steps was due to saturation of the resin (Figure 2A and 2B). The flow through and wash elutions were examined for nuclease activity. These fractions contained the highest DNA damaging activity resulting in complete digestion of a supercoiled plasmid DNA to nicked circular (NC), linear (LN), and small fragments (SF) (Figure 2C and 2D). To separate the DNA damaging contaminants from PCD, we employed a two-step elution scheme (Figure 2A). During the first elution PCD was released with readily apparent protein contaminants (Figure 2B). Fractions from the first elution showed reduced DNA scission and DSB activities compared to Load fraction (Figure 2C and 2D). However, the second elution yielded extremely enriched protein with nearly undetectable nuclease activity (Figure 2B–D). DNA damaging activity of the first elution peak was dependent on Mg2+, but did not change in the presence of DTT or PCA (Supplementary Figure 2). These observations confirmed a contaminating nuclease, rather than Fe(III)-coupled oxidative damage or a PCD OS-coupled DNA damaging mechanism [37].
Figure 2. The first step of purifying PCD by Ni-NTA affinity chromatography, and testing of protein fractions for DNA damaging activities.
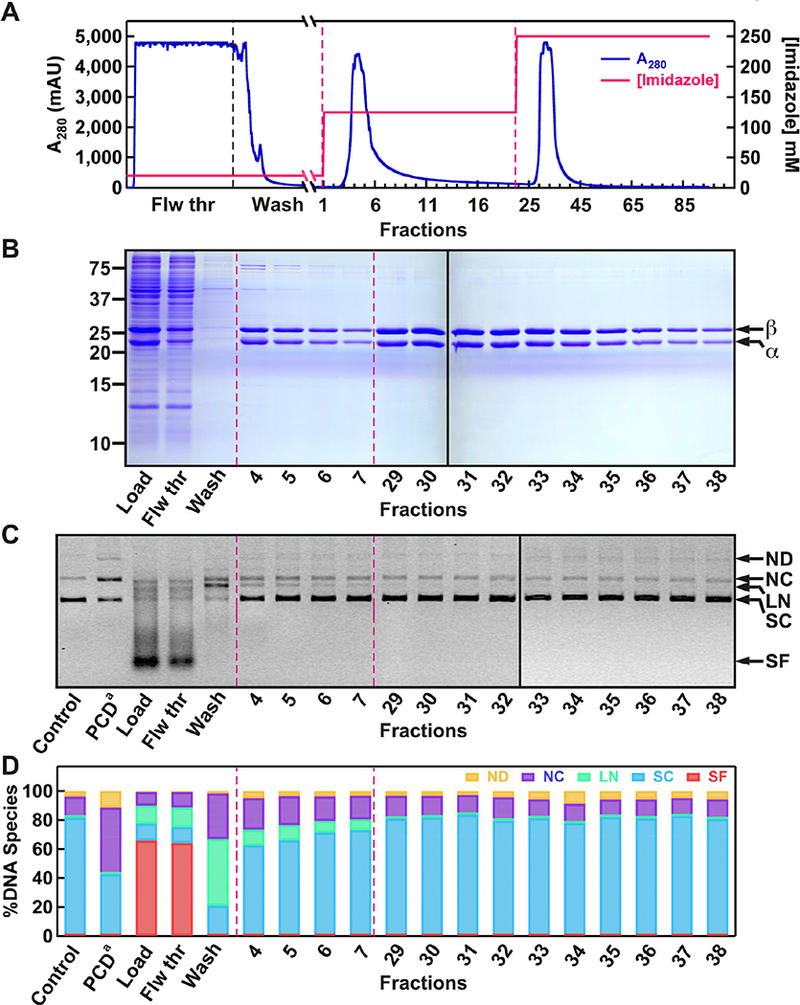
(A) Chromatogram showing the separation of PCD from the rest of soluble protein contaminants using a Ni-NTA column. Different segments of the chromatogram and imidazole concentration profile are also indicated. (B) SDS-PAGE analysis of Ni-NTA protein fractions. (C) Evaluation of protein fractions for any DNA damaging activity using a supercoiled (SC) DNA substrate. (D) Quantification of relative amounts of different DNA species present in each reaction. The resulting products include: nicked dimer (ND), nicked circular monomer (NC), linear DNA (LN) and smaller fragments (SF). PCD from Sigma-Aldrich (PCDa) at 1XSM (~60 nM) was used as a positive control for nuclease activity.
Size exclusion chromatography resulted in near-homogenous highly active PCD
To remove residual nuclease contaminants, we further purified PCD Ni-NTA fractions (#29–38) by Superose 12 size exclusion chromatography (Figure 3) [37]. The PCD complex eluted at ~8–11 mL elution volume, with the peak fractions (#36 and #37) at ~9 mL (Figure 3A). We estimated the MW of the peak PCD complex to be ~458 kDa (Figure 3B). Previous studies have suggested 12 αβ dimers makeup the PCD holoenzyme complex (expected MW of 608 kDa) [39, 42]. The unusual hydrodynamic behaviour associated with gel filtration is consistent with the predictions of previous structural studies [42]. Importantly, fractions 36 and 37 contained stoichiometric amounts of the two subunits (Figure 3C). The plasmid-based nuclease activity assay showed no detectable nuclease in size exclusion fractions #33–49 (Figure 3D and 3E).
Figure 3. The second step of purifying PCD by size exclusion chromatography, and testing of protein fractions for DNA damaging activities.
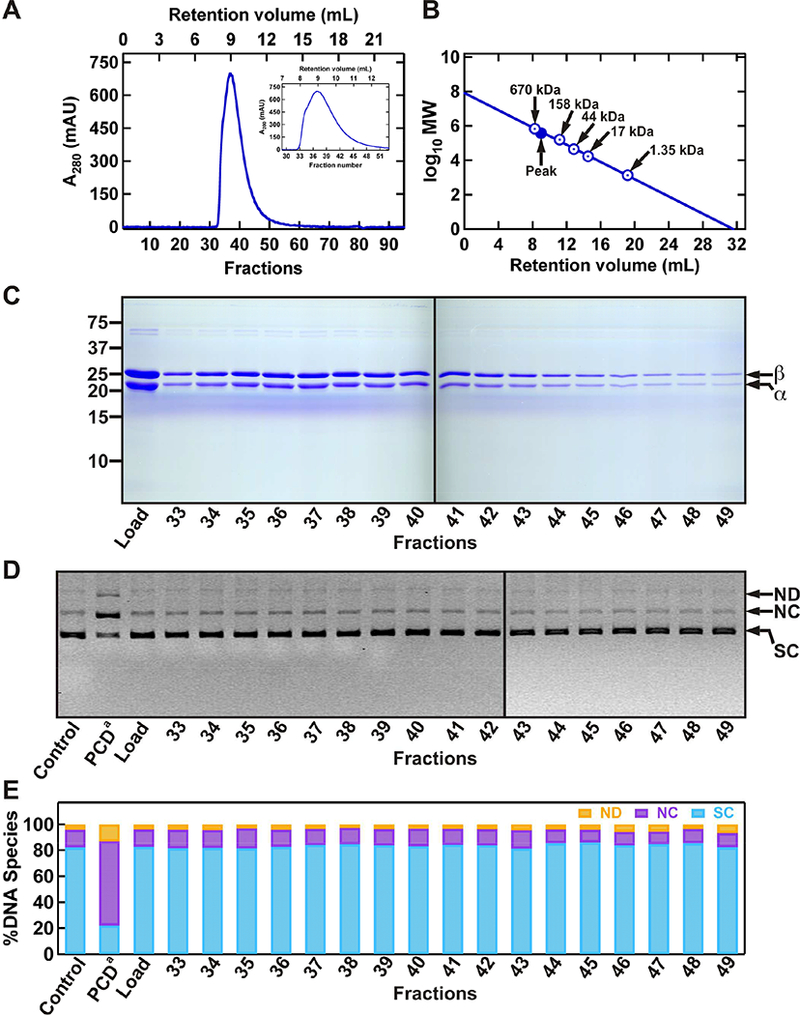
(A) Chromatogram of PCD fractions on the Sp12 gel filtration column. Proteins mainly elute from ~8–11 mL retention volumes (440–670 kDa MW), also shown in zoomed-in view in the inset. (B) MW calibration curve showing the sizes in kDa for the globular molecular weight standards (Bio-Rad), and the position of peak PCD fractions (#36 and #37). (C) Analysis of protein fractions using SDS-PAGE. (D) Evaluation of protein fractions for any DNA damaging activity using a supercoiled (SC) DNA substrate. (E) Quantification of relative amounts of different DNA species present in each reaction. The resulting products include: nicked dimer (ND), nicked circular monomer (NC), linear DNA (LN). PCD from Sigma-Aldrich (PCDa) at 1XSM (~60 nM) was used as a positive control for nuclease activity.
We evaluated the PCD enzymatic activity in several ways. Active sites of the enzyme are located at the interface between each αβ dimer [39]. The percentage of Fe(III) bound to this dimer interface provides a measure of the fraction of active protein [39]. The percentage of bound Fe(III) was determined using Sp12 chromatograms of protein at 280 nm and Fe(III)-center chromophore at 450 nm (Figure 4A, top panel). These data showed that for fractions #34–42 approximately 50% of the active sites were occupied by Fe(III) (Figure 4A, bottom panel).
Figure 4. Testing the ensemble and SM level enzymatic activities of purified PCD.
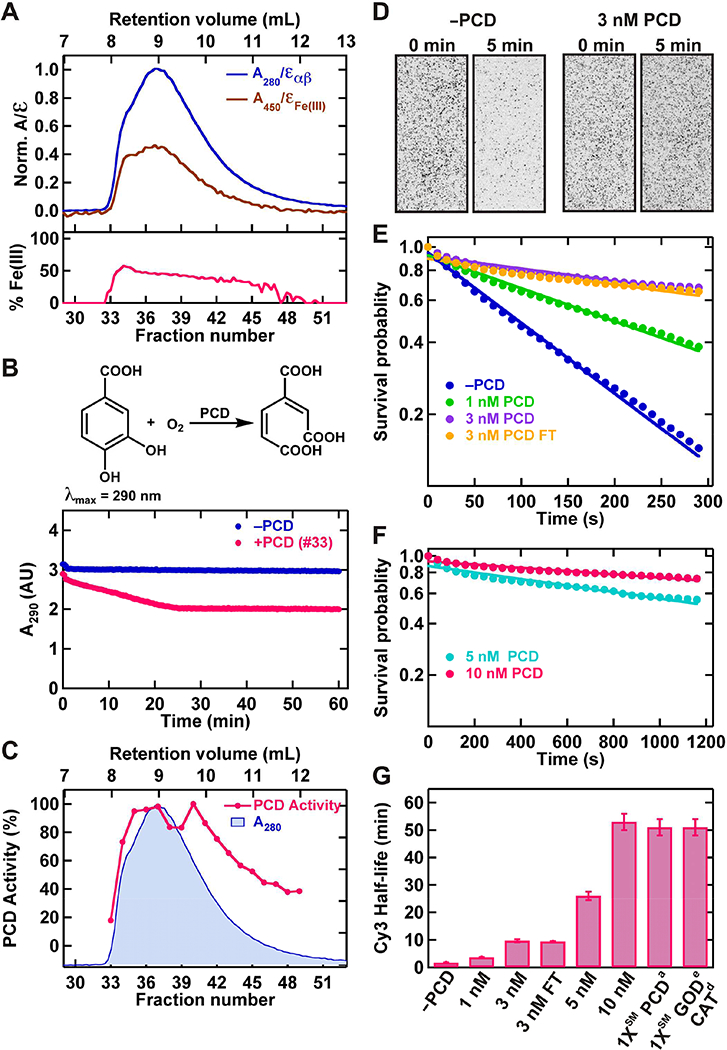
(A) Zoomed-in view of the chromatograms from Sp12 gel filtration column showing relative amounts of αβ dimer, and active site Fe(III) present in each fraction (top panel). The bottom panel shows the corresponding %Fe(III) present in each fraction. (B) PCD catalyzed PCA oxidation to the di-oxygenated product in the presence of dissolved-O2 (top panel). Examples of ensemble level kinetic curves showing: no reduction of A290 of PCA in the absence of PCD, and reduction in A290 in the presence of PCD (bottom panel). (C) Normalized ensemble level PCD activities of gel filtration fractions, overplayed to the normalized protein chromatogram. (D) SM field of view (~34 mm2) showing the fast photobleaching of individual Cy3 molecules (dark spots) in the absence of PCD (left panel), and improved lifetime in the presence of 3 nM PCD OSS (right panel). (E) SM survival probability of Cy3 photobleaching at different concentrations of PCD in the OSS including 3 nM PCD after one cycle of freeze and thaw (3 nM PCD FT). (F) SM survival probability of Cy3 photobleaching at high concentrations (5 nM, 10 nM) of PCD in the OSS as determined by extended observations. (G) Estimated half-life ± error from the fitting of Cy3, which correspond to 50% survival probability, at different concentrations of PCD in the OSS. The data from Sigma-Aldrich PCD (PCDa) at 1XSM (~60 nM) was shown for comparison.
PCD enzymatic activity was also examined by monitoring the oxidation of PCA (Figure 4B, Supplementary Figure 3) [37]. These data indicated an activity distribution nearly equivalent to PCD content presented in SDS-PAGE gel analysis and the Sp12 chromatogram (Figure 3C and 4C). Careful inspection of individual ensemble level kinetic curves (Figure 4B, bottom panel, Supplementary Figure 3) showed that PCD is capable of initiating the reaction at 4 °C during the sample preparation. This reactivity in turn resulted in slight deviation of the bulk kinetic distribution from the expected behavior based on amounts of PCD present (Figure 4C, deviation of fractions #38 and #39).
Finally, we tested the ability of the PCD OSS to minimize photobleaching of fluorophores in SM experiments. Individual surface immobilized Cy3 molecules showed rapid photobleaching in the absence of PCD OSS resulting in a 1.7 ± 0.1 min half-life (Figure 4D–G). PCD OSS prolonged the lifetime of Cy3 in an enzyme concentration-dependent manner (Figure 4D–G). The purified enzyme did not lose this activity during a single freeze-thaw cycle, and storage at −80 °C (compare 3 nM with 3 nM FT in Figure 4E and 4G). Infusion of 5–10 nM PCD in OSS provided exceptional photostability with a half-life of ~25–50 min, albeit with slightly increased background fluorescence. In significant contrast, commercial PCDa showed substantial DNA damaging activity (Figure 2C, 2D and Figure 3D, 3E). At 1XSM (~60 nM) PCDa the photoprotection was comparable to 10 nM purified PCD (Figure 4G) [37].
DISCUSSION
We report a simple, efficient, and cost effective method to produce extremely enriched (>99% purity) oxygen-scavenging enzyme PCD for researchers who apply fluorescence techniques to study nucleic acid-protein interactions. Commercially available PCD preparations are relatively expensive and may be contaminated with nuclease activity (Figures 2 and 3) [37]. Using PCD purified by our method, SM laboratories will be able to acquire data over extended times without confounding nucleic acid damaging contaminants. Importantly, photobleaching of fluorophores may also affect the spatial resolution in super-resolution microscopy. Therefore, the exceptional photoprotective abilities observed with the purified PCD described here may be beneficial in those experiments as well.
Potential drawbacks associated with purified PCD may include increased fluorescence background levels at high concentrations and undesirable interactions with the biological mechanisms under investigation. Therefore, it may be necessary to optimize illumination conditions, data acquisition rates, and biochemical reactions with ensemble assays to confirm that there is minimal or no interference from PCD [37]. Although we employed fast performance protein liquid chromatography (FPLC), it is possible to implement gravity-driven bench-top chromatographic columns to purify PCD if necessary.
Supplementary Material
We present a method to purify nuclease-free oxygen scavenger PCD enzyme.
Purified PCD extends fluorophore Cy3 lifetime two orders of magnitude.
Purified PCD achieves higher fluorophore stability than commerical scavengers.
ACKNOWLEDGMENT
We would like to thank Dr. John Lipscomb and Tim Horn at University of Minnesota for providing the PCD expression plasmid and initial protocols for PCD expression. We would also like to thank Ahmet Ata Akatay and James London for MATLAB code used for Cy3 particle tracking. We thank all our lab members for insights and helpful discussions.
FUNDING
This work was supported by the National Institutes of Health (AI126742 to RF & KEY, GM121284 to KEY and GM080176 to R.F.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLMENTARY DATA
Supplementary Data for this manuscript are provided in a separate document.
Conflict of interest statement: None declared.
REFERENCES
- [1].Wuite GJ, Smith SB, Young M, Keller D, Bustamante C, Single-molecule studies of the effect of template tension on T7 DNA polymerase activity, Nature, 404 (2000) 103–106. [DOI] [PubMed] [Google Scholar]
- [2].Lee JB, Hite RK, Hamdan SM, Xie XS, Richardson CC, van Oijen AM, DNA primase acts as a molecular brake in DNA replication, Nature, 439 (2006) 621–624. [DOI] [PubMed] [Google Scholar]
- [3].Pandey M, Syed S, Donmez I, Patel G, Ha T, Patel SS, Coordinating DNA replication by means of priming loop and differential synthesis rate, Nature, 462 (2009) 940–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Graham JE, Marians KJ, Kowalczykowski SC, Independent and Stochastic Action of DNA Polymerases in the Replisome, Cell, 169 (2017) 1201–1213 e1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Greenleaf WJ, Block SM, Single-molecule, motion-based DNA sequencing using RNA polymerase, Science, 313 (2006) 801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang Z, Revyakin A, Grimm JB, Lavis LD, Tjian R, Single-molecule tracking of the transcription cycle by sub-second RNA detection, Elife, 3 (2014) e01775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fazal FM, Meng CA, Murakami K, Kornberg RD, Block SM, Real-time observation of the initiation of RNA polymerase II transcription, Nature, 525 (2015) 274–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lerner E, Chung S, Allen BL, Wang S, Lee J, Lu SW, Grimaud LW, Ingargiola A, Michalet X, Alhadid Y, Borukhov S, Strick TR, Taatjes DJ, Weiss S, Backtracked and paused transcription initiation intermediate of Escherichia coli RNA polymerase, Proc Natl Acad Sci U S A, 113 (2016) E6562–E6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Blanchard SC, Gonzalez RL, Kim HD, Chu S, Puglisi JD, tRNA selection and kinetic proofreading in translation, Nat Struct Mol Biol, 11 (2004) 1008–1014. [DOI] [PubMed] [Google Scholar]
- [10].Wen JD, Lancaster L, Hodges C, Zeri AC, Yoshimura SH, Noller HF, Bustamante C, Tinoco I, Following translation by single ribosomes one codon at a time, Nature, 452 (2008) 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Uemura S, Aitken CE, Korlach J, Flusberg BA, Turner SW, Puglisi JD, Real-time tRNA transit on single translating ribosomes at codon resolution, Nature, 464 (2010) 1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Qi Z, Redding S, Lee JY, Gibb B, Kwon Y, Niu H, Gaines WA, Sung P, Greene EC, DNA sequence alignment by microhomology sampling during homologous recombination, Cell, 160 (2015) 856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kong M, Liu L, Chen X, Driscoll KI, Mao P, Bohm S, Kad NM, Watkins SC, Bernstein KA, Wyrick JJ, Min JH, Van Houten B, Single-Molecule Imaging Reveals that Rad4 Employs a Dynamic DNA Damage Recognition Process, Mol Cell, 64 (2016) 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu J, Hanne J, Britton BM, Bennett J, Kim D, Lee JB, Fishel R, Cascading MutS and MutL sliding clamps control DNA diffusion to activate mismatch repair, Nature, 539 (2016) 583–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Karunatilaka KS, Solem A, Pyle AM, Rueda D, Single-molecule analysis of Mss116-mediated group II intron folding, Nature, 467 (2010) 935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Haller A, Rieder U, Aigner M, Blanchard SC, Micura R, Conformational capture of the SAM-II riboswitch, Nat Chem Biol, 7 (2011) 393–400. [DOI] [PubMed] [Google Scholar]
- [17].Pirchi M, Ziv G, Riven I, Cohen SS, Zohar N, Barak Y, Haran G, Single-molecule fluorescence spectroscopy maps the folding landscape of a large protein, Nat Commun, 2 (2011) 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ferreon AC, Ferreon JC, Wright PE, Deniz AA, Modulation of allostery by protein intrinsic disorder, Nature, 498 (2013) 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chistol G, Liu S, Hetherington CL, Moffitt JR, Grimes S, Jardine PJ, Bustamante C, High degree of coordination and division of labor among subunits in a homomeric ring ATPase, Cell, 151 (2012) 1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chou YY, Vafabakhsh R, Doganay S, Gao Q, Ha T, Palese P, One influenza virus particle packages eight unique viral RNAs as shown by FISH analysis, Proc Natl Acad Sci U S A, 109 (2012) 9101–9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu S, Chistol G, Hetherington CL, Tafoya S, Aathavan K, Schnitzbauer J, Grimes S, Jardine PJ, Bustamante C, A viral packaging motor varies its DNA rotation and step size to preserve subunit coordination as the capsid fills, Cell, 157 (2014) 702–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Otterstrom JJ, Brandenburg B, Koldijk MH, Juraszek J, Tang C, Mashaghi S, Kwaks T, Goudsmit J, Vogels R, Friesen RH, van Oijen AM, Relating influenza virus membrane fusion kinetics to stoichiometry of neutralizing antibodies at the single-particle level, Proc Natl Acad Sci U S A, 111 (2014) E5143–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vafabakhsh R, Kondabagil K, Earnest T, Lee KS, Zhang Z, Dai L, Dahmen KA, Rao VB, Ha T, Single-molecule packaging initiation in real time by a viral DNA packaging machine from bacteriophage T4, Proc Natl Acad Sci U S A, 111 (2014) 15096–15101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jones ND, Lopez MA Jr., Hanne J, Peake MB, Lee JB, Fishel R, Yoder KE, Retroviral intasomes search for a target DNA by 1D diffusion which rarely results in integration, Nat Commun, 7 (2016) 11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Erkens GB, Hanelt I, Goudsmits JM, Slotboom DJ, van Oijen AM, Unsynchronised subunit motion in single trimeric sodium-coupled aspartate transporters, Nature, 502 (2003) 119–123. [DOI] [PubMed] [Google Scholar]
- [26].Akyuz N, Georgieva ER, Zhou Z, Stolzenberg S, Cuendet MA, Khelashvili G, Altman RB, Terry DS, Freed JH, Weinstein H, Boudker O, Blanchard SC, Transport domain unlocking sets the uptake rate of an aspartate transporter, Nature, 518 (2015) 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang S, Vafabakhsh R, Borschel WF, Ha T, Nichols CG, Structural dynamics of potassium-channel gating revealed by single-molecule FRET, Nat Struct Mol Biol, 23 (2016) 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liu S, Abbondanzieri EA, Rausch JW, Le Grice SF, Zhuang X, Slide into action: dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates, Science, 322 (2008) 1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu J, Hanne J, Britton BM, Shoffner M, Albers AE, Bennett J, Zatezalo R, Barfield R, Rabuka D, Lee JB, Fishel R, An Efficient Site-Specific Method for Irreversible Covalent Labeling of Proteins with a Fluorophore, Sci Rep, 5 (2015) 16883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gorman J, Wang F, Redding S, Plys AJ, Fazio T, Wind S, Alani EE, Greene EC, Single-molecule imaging reveals target-search mechanisms during DNA mismatch repair, Proc Natl Acad Sci U S A, 109 (2012) E3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zheng Q, Jockusch S, Zhou Z, Blanchard SC, The contribution of reactive oxygen species to the photobleaching of organic fluorophores, Photochem Photobiol, 90 448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sabanayagam CR, Eid JS, Meller A, Long time scale blinking kinetics of cyanine fluorophores conjugated to DNA and its effect on Forster resonance energy transfer, J Chem Phys, 123 (2005) 224708. [DOI] [PubMed] [Google Scholar]
- [33].Campos LA, Liu J, Wang X, Ramanathan R, English DS, Munoz V, A photoprotection strategy for microsecond-resolution single-molecule fluorescence spectroscopy, Nature methods, 8 (2011) 143–146. [DOI] [PubMed] [Google Scholar]
- [34].Rasnik I, McKinney SA, Ha T, Nonblinking and long-lasting single-molecule fluorescence imaging, Nature methods, 3 (2006) 891–893. [DOI] [PubMed] [Google Scholar]
- [35].Aitken CE, Marshall RA, Puglisi JD, An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments, Biophysical journal, 94 (2008) 1826–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aitken CE, Marshall RA, Puglisi JD, An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments., iophysical journal, 94 (2008) 1826–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Senavirathne G, Liu J, Lopez MA Jr., Hanne J, Martin-Lopez J, Lee JB, Yoder KE, Fishel R, Widespread nuclease contamination in commonly used oxygen-scavenging systems, Nature methods, 12 (2015) 901–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Frazee RW, Livingston DM, LaPorte DC, Lipscomb JD, Cloning, sequencing, and expression of the Pseudomonas putida protocatechuate 3,4-dioxygenase genes, J Bacteriol, 175 (1993) 6194–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ohlendorf DH, Lipscomb JD, Weber PC, Structure and assembly of protocatechuate 3,4-dioxygenase, Nature, 336 (1988) 403–405. [DOI] [PubMed] [Google Scholar]
- [40].Harris DC, Quantitative chemical analysis, 7th ed, W.H. Freeman and Co., New York, 2007. [Google Scholar]
- [41].Kural C, Tacheva-Grigorova SK, Boulant S, Cocucci E, Baust T, Duarte D, Kirchhausen T, Dynamics of intracellular clathrin/AP1- and clathrin/AP3-containing carriers, Cell Rep, 2 (2012) 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ohlendorf DH, Weber PC, Lipscomb JD, Determination of the quaternary structure of protocatechuate 3,4-dioxygenase from Pseudomonas aeruginosa, J Mol Biol, 195 (1987) 225–227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.