

Abstract
Genomic instability in disease and its fidelity in health depend on the DNA damage response (DDR), regulated in part from the complex of meiotic recombination 11 homolog 1 (MRE11), ATP-binding cassette–ATPase (RAD50), and phosphopeptide-binding Nijmegen breakage syndrome protein 1 (NBS1). The MRE11–RAD50–NBS1 (MRN) complex forms a multifunctional DDR machine. Within its network assemblies, MRN is the core conductor for the initial and sustained responses to DNA double-strand breaks, stalled replication forks, dysfunctional telomeres, and viral DNA infection. MRN can interfere with cancer therapy and is an attractive target for precision medicine. Its conformations change the paradigm whereby kinases initiate damage sensing. Delineated results reveal kinase activation, posttranslational targeting, functional scaffolding, conformations storing binding energy and en-abling access, interactions with hub proteins such as replication protein A (RPA), and distinct networks at DNA breaks and forks. MRN biochemistry provides prototypic insights into how it initiates, implements, and regulates multifunctional responses to genomic stress.
Keywords: ATPase, nuclease, FHA domain, forkhead-associated domain, BRCT domains, BRCA1 C-terminal domains, coiled-coil domain, CtIP tetramerization, CtBP-interacting protein tetramerization
INTRODUCTION
At the cellular level, life is constantly challenged by genotoxic stress originating from endogenous and exogenous sources, with stalled replication forks (RFs) and double-strand breaks (DSBs) being among the most deleterious and extreme forms of DNA damage. Failure to properly respond to genomic distress can be mutagenic and toxic to cells as well as fatal to the organism. Cells have therefore evolved to counteract genotoxic stress by integrating DNA repair mechanisms and cell cycle regulation through checkpoint signaling (1). In this network of DNA damage responses (DDR), the meiotic recombination 11 homolog 1 (MRE11)–DNA repair protein RAD50–Nijmegen breakage syndrome protein 1 (NBS1) (MRN) complex (Figure 1a,b) holds a key position. It is among the first responders and regulates both signaling and damage responses to at least four types of extreme cellular stress: DNA damage at DSBs, stalled RFs, dysfunctional telomeres, and viral invasion. MRN also has a prime role in kinase [ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and RAD3-related (ATR)] signaling that helps orchestrate cell cycle progression and damage responses (2).
Figure 1.
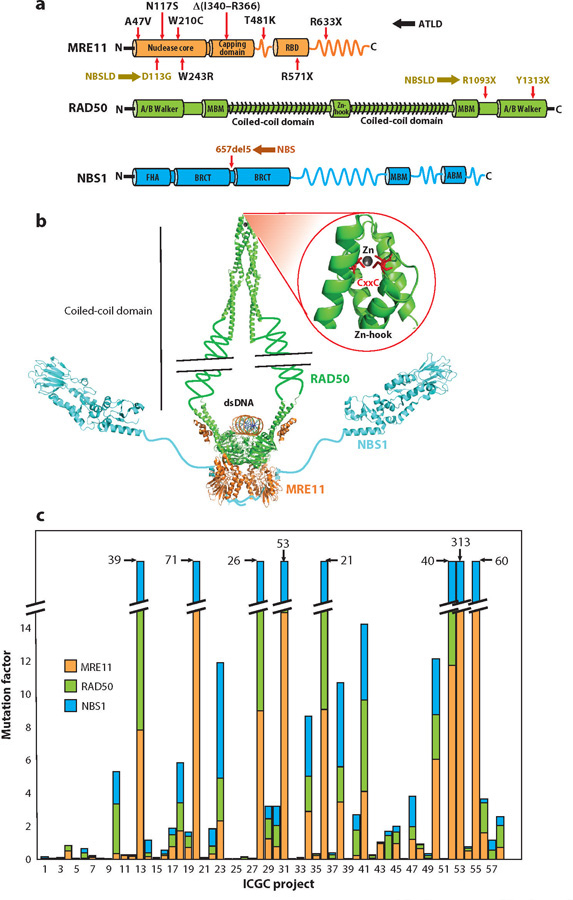
MRN domains, complex assembly, and mutations. (a) Key domains of MRN subunits MRE11 (orange), RAD50 ( green), and NBS1 (cyan). Mutated sites (red arrows) for human inherited diseases (color coded for ATLD, NBS, and NBSLD; mutations corresponding to each disease are color coded with respective abbreviations) are shown on each subunit with respective substitutions, where X represents the stop codon. (b) Modeled assembly of MRN complex and dsDNA based on experimentally derived partial crystal structures (PDB IDs: 5F3W, 5GOX, and 3HUE; color coding as in panel a) showing a dimer of a heterotrimer. The allosterically linked Zn-hook is highlighted in the inset along with CxxC motifs (red ) on two RAD50 monomers. (c) The extent of mutations (reported as mutation factor) in subunits of the MRN complex in cancers documented in the International Cancer Genome Consortium (ICGC) projects. A mutation factor is defined as a number of observed mutations multiplied by the fraction of mutation-positive cases. ICGC projects with high mutation factors (>15 in total) include the following: #13, Breast Cancer, ER+ and HER2−, European Union/United Kingdom; #20, Esophageal Adenocarcinoma, United Kingdom; #28 Liver Cancer, China; #31, Liver Cancer, RIKEN, Japan; #36, Malignant Lymphoma, Germany; #52, Skin Adenocarcinoma, Brazil; #53, Skin Cancer, Australia; and #55, Soft Tissue Cancer, Leiomyosarcoma, France. A complete list of the 58 projects is provided in Supplemental Table 1. Abbreviations: ABM, ATM binding motif; ATLD, ataxia-telangiectasia-like disorder; BRCT, BRCA1 C-terminal domain; dsDNA, double-stranded DNA; FHA, forkhead-associated domain; MBM, MRE11 binding motif; MRE11, meiotic recombination 11 homolog 1; NBS, Nijmegen breakage syndrome; NBSLD, NBS-like disorder; PDB, Protein Data Bank; RAD50, DNA repair protein RAD50; RBD, RAD50 binding domain.
MRN functional significance in both signaling and repair is underscored by the observation that the core Mre11–Rad50 (MR) catalytic subcomplex is conserved across all domains of life, encompassing archaea (e.g., Pyrococcus furiosus), eubacteria (Escherichia coli SbcC/D), some phage (T4 phage gp46/47), and eukaryotes including homologs in Saccharomyces cerevisiae and Schizosac-charomyces pombe (3). The NBS1 (Xrs2 in S. cerevisiae) component is conserved only in eukaryotes. However, mutations in any of the genes encoding MRN components cause severe consequences in humans. Germline mutations (Figure 1a) in MRE11, NBS1, or RAD50 cause ataxia-telangiectasia (A-T)–like disorder (ATLD), Nijmegen breakage syndrome (NBS), or NBS-like disorder (4–10). A-T is caused by inherited mutations in ATM kinase. Aside from germline mutations, all 3 MRN complex components are mutated in >50 cancers, as assessed from the International Cancer Genome Consortium projects (Figure 1c and Supplemental Table 1). Furthermore, the MRN complex is frequently found absent in patients with epithelial ovarian cancer (11). MRN functional disruption may be a synthetic lethal combination with poly(ADP-ribose) glycohydrolase (PARG) inhibition (12) providing a possible strategy to target MRN-defective cancers by inhibition of PARG, whose structure is defined (13). However, upregulation of RAD50 increases cellular radioresistance, and knockdown sensitizes some cancer cells to radiation, suggesting MRN may be a target for precision medicine (14). Overall, its conservation across all domains of life coupled to the catastrophic consequences of its mutations affirms the role of the MRN complex in maintaining essential aspects of genomic stability for all cells.
In this review, we consider exemplary recent developments and paradigm-shifting results linking MRN structural biochemistry to its biology to complement and build upon prior excellent reviews (2, 4, 15, 16). We integrate recent advances to delineate how MRN’s multidomain three protein composition enables its central enzymatic, sensing, signaling, architectural, and scaffolding functions in damage responses. MRN conformations are key to damage signaling through the ATM and ATR kinases, regulating the MRE11 nuclease and opening DNA for nuclease incision. By activating ATM, the MRN complex regulates phosphorylation of >900 sites on >700 proteins, including MRE11, RAD50, and NBS1 (17, 18). NBS1 interacts with ATM kinase plus its N-terminal phosphopeptide-interacting domains that link MRN to other partners including the C-terminal binding protein (CtBP)-interacting protein (CtIP) (19, 20). As it directly binds NBS1, DNA, and RAD50, the MRE11 nuclease dimer is the core of the MRN complex (21–23). We therefore update the two-step MRE11 nuclease incision model to explain its endo- and exonuclease properties in light of recent structures, including the eukaryotic RAD50 Zn-hook and CtIP N-terminal domain. We describe factors affecting MRN functions at DSBs, stalled RFs, dysfunctional telomeres, and complexes combating exogenous viral DNA infection. We discuss how MRN acts more as a dynamic molecular machine for the expeditious formation of protein–nucleic acid functional scaffolds at DSBs, RFs, dysfunctional telomeres, and viral DNA than as part of linear pathways, as also proposed for nonhomologous end joining (NHEJ) complexes (24). As a multifunctional macromolecular machine, the structural biochemistry of the MRN complex initiates sensing of stalled forks and DSBs, cell cycle checkpoint signaling cascades, incision for and commitment to break repair pathways, reestablishment of posttranslational phosphorylations via ATM kinase, and functional regulation of chromatin remodeling. The collective results outlined here suggest how MRN conformations and interactions enable both its specificity and multiple functions in cells with implications for science and biomedicine.
MRN COMPLEX: STRUCTURAL BIOCHEMISTRY AND BIOLOGY
The MRN complex is an intriguing chemo-mechanical molecular machine that converts energy from binding to DNA, ATP, and protein partners into conformational changes that control nuclease activities and network assemblies (2). MRN binds DNA and acts as a sensor of DNA damage (25). It is furthermore the activator for ATM and ATR kinase cascades (26), a tethering scaffold for recognition and stabilization of protein interactions, a processor of RFs, a licensing factor for homologous recombination (HR), and a facilitator of fork restart and DNA repair processes (2, 27, 28). At the basic level, MRN consists of two RAD50 ATP-binding cassettes (ABC)–ATPase sub-units, two MRE11 subunits forming a DNA structure-specific endo/exo/hairpin nuclease, and the NBS1 regulatory docking protein with phosphopeptide-interacting forkhead associated (FHA) and BRCA1 C-terminal (BRCT) domains flexibly linked to an MRE11 interface and adjacent C-terminal ATM kinase interaction motif (16, 29) (Figure 1a,b).
The core MR complex exists as a hetero-tetrameric assembly (M2R2), whose dynamic morphology emerges from shifts in its modular head, coil, and hook domains (Figure 1b). The globular DNA binding head is formed by the two Rad50–ATPase domains and the Mre11 nuclease that bind to the base of the Rad50 helical coiled-coil domains (Figure 2a). The heart of the MR (in prokaryotes) and MRN (in eukaryotes) assembly originates from the Mre11 dimerization interface (29). Yet, RAD50 is the largest subunit: It belongs to the structural maintenance of chromosomes (SMC) protein family (30). Each Rad50 polypeptide assembles into a globular ATPase domain despite a large intramolecular antiparallel coiled-coil sequence insertion. This coiled-coil domain extends far from the head domain (∼500 Å for eukaryotic Rad50 homologs) with a central CxxC motif forming a Zn-hook and reverse turn that caps its distal end and mediates Zn-dependent Rad50 subunit interactions (31–33). The resulting Rad50 bipartite ATPase dimerizes in a Mg2+and ATP-dependent manner (30). Notably, Rad50 conformation at the dimer interface is controlled by binding two ATP molecules: Each is sandwiched between signature motifs conserved in ABC–ATPases on one subunit and the Walker A, Walker B, Q-loop, and D-loop motifs from the other subunit (34, 35).
Figure 2.
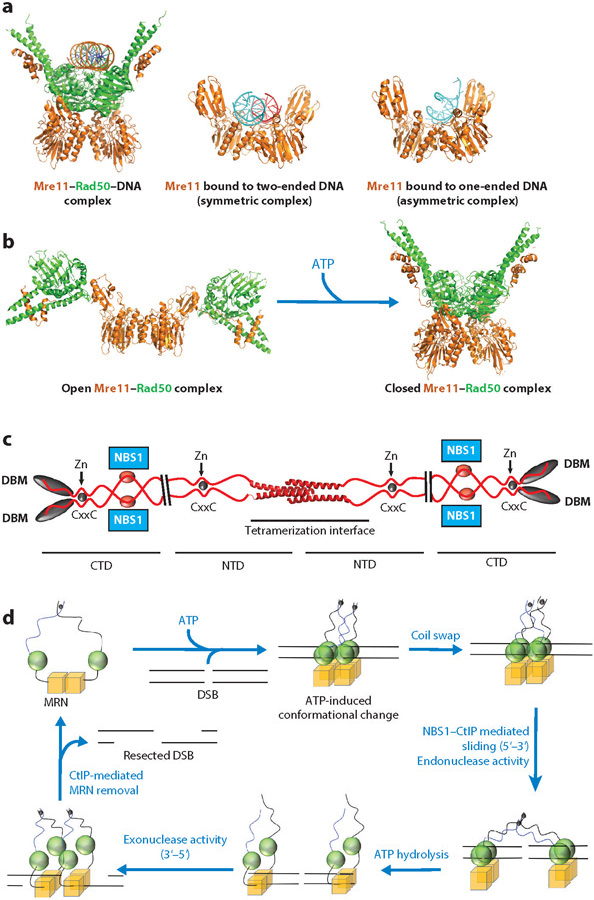
The Mre11–Rad50 (MR) heterotetramer: DNA-binding interface, ATP-mediated conformational changes, and a unified model for MRN’s functions in DSB resection (color coding as in Figure 1) are shown. (a) Crystal structures of ATPγS-bound Methanococcus jannaschii (Mj) Mre11—Rad50–DNA complex (PDB ID: 5F3W) and the symmetric (PDB ID: 3DSC) and asymmetric (PDB ID: 3DSD) Pyrococcus furiosus Mre11 DNA-bound states, highlighting the DNA-binding interface at the Rad50 and Mre11 dimerization interface. (b) Crystal structures of the MR complex in the nucleotide-free state (PDB ID: 3QG5, Thermotoga maritima MR) and in the ATPγS-bound state (PDB ID: 3AV0, MjMR), highlighting the conformational switch in the MRN core complex. (c) Human CtIP model from the recent crystal structure of CtIP N-terminal tetramerization domain (PDB ID: 4D2H). Plausible NBS1 binding sites on four CtIP and C-terminal DBMs, along with CxxC motifs and Zn coordination, are shown. (d ) A testable pathway for DSB resection by the MRN complex. For clarity, only MRE11 (M) and RAD50 (R) subunits are shown. First, two molecules of MRN bind to DSB ends in an ATP-bound state, with MRE11 nuclease activity inhibited by the closed RAD50 conformation. Then, MRN forms an interlinked complex through a RAD50 coil swap. At this point, CtIP–NBS1 interaction allows MRN dimers to slide away from the break, and MRE11 makes the initial licensing cut through its endonuclease activity. Upon complete ATP hydrolysis, the RAD50 dimerization interface opens and dsDNA becomes accessible to the MRE11 exonuclease. DNA resection begins in the 3΄–5΄ direction and proceeds toward the break. Upon reaching the break, the MRN complex may be dislodged from DSB with the help of CtIP and NBS1 (NBS1 not shown for clarity). Because only M and R are shown in the model, NBS1 (N) is colored gray in MRN notation. Abbreviations: CTD, C-terminal domain; CtIP, C-terminal binding protein–interacting protein; DBMs, DNA binding motifs; DSBs, double-strand breaks; dsDNA, double-stranded DNA; MRE11, meiotic recombination 11 homolog 1; MRN, MRE11–RAD50–NBS1 complex; NBS1, Nijmegen breakage syndrome protein 1; NTD, N-terminal domain; PDB, Protein Data Bank; RAD50, DNA repair protein RAD50.
MRE11 key features are an N-terminal core nuclease domain followed by a cap domain that restricts access to the nuclease active site and a flexibly linked RAD50 binding motif (21, 22, 36–39). X-ray structures of P. furiosus Mre11 revealed the two-domain architecture for the catalytic core (40). The N-terminal calcineurin-like phosphoesterase domain bears the Mn2+-coordination and nuclease catalytic motifs. The phosphoesterase is further decorated by an alpha-beta fold C-terminal domain positioned to enforce DNA binding specificities (Figure 1a) (40). The ternary complex of P. furiosus Mre11 bound to Mn2+ and a 5΄҆-dAMP nucleotide hydrolysis product supports the biochemically observed 3΄–5΄ directionality of the Mre11 exonuclease activity. Yet, as noted below, Mre11 nuclease activity also enables 5΄–3΄ resection of DNA ends to generate 3΄ single-stranded DNA (ssDNA) overhangs for template-mediated HR and homologous repair (27). The partly disordered MRE11 C terminus is regulated by posttranslational modifications (Figure 1a).
The NBS1 subunit is specific to eukaryotes. It has one FHA and two BRCT domains in its N terminus (Figure 1a). These FHA and BRCT domains bind multiple phosphorylated proteins to regulate MRN interactions (29). The rest of NBS1 is unstructured except for the functionally critical C-terminal MRE11 and ATM-binding subdomains and motifs (23, 41). The NBS1 ATM-binding motif resembles the C-terminal binding motifs of Ku80 and ATR-interacting protein (ATRIP), which interact with and recruit the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and ATR, respectively (42), suggesting an interaction conserved across the three kinases (43). A recent DNA-PKcs crystal structure with bound Ku80 C terminus showed how Ku80 binds near DNA-PKcs autophosphorylation sites (the PQR cluster) (44). Interestingly, the NBS1 C terminus may also bind near ATM autophosphorylation sites (S367, S1893, and S1891) (42), suggesting that analogous modes of phosphorylation regulate DNA-PKcs and ATM activities. Yet, this NBS1 C-terminal region may not be critical for ATM-mediated checkpoint activation but helps in proper phosphorylation of some of the ATM substrates such as SMC1 and proapoptotic factor BID (45, 46).
The combination of at least two dimer states for structure-specific nuclease (MRE11) and ATPase enzyme (RAD50) activities, long-range allostery from NBS1 and the Rad50 Zn-hook binding domains, and regulatory phosphorylation plus flexible interface linkages enables multiple MRN conformational states. Together, these functional characteristics make MRN a dynamic machine that not only can sense and process DNA damage but also can participate in different pathways and networks, such as NHEJ, alternative nonhomologous end joining (A-NHEJ), or HR, for DNA damage repair. Owing to its dynamic and extended nature, structural analyses of MRN have employed multiple combined methods and biophysical techniques, including X-ray crystallography, small-angle X-ray scattering, electron microscopy (EM), and atomic or scanning force microscopy. Furthermore, structural information on MR has emerged from prokaryotes that can provide more accessible structures than have been analyzed for their eukaryotic counterparts. In fact, prokaryotic Mre11 is used as a molecular avatar (an embodiment of human MRE11–essential features) for design of inhibitors against human MRE11 (47). Excitingly, high-resolution structures of eukaryotic MRN protein domains are also being solved (33, 38, 39). For eukaryotes, CtIP is also emerging as a key player in MRN functions in HR on the basis of structural analyses of the CtIP N-terminal domain (48–50). From these collective MRN structural and biochemical results, we propose a testable integrated model for MRN structure–function relationships (Figure 2).
RAD50 forms a unique SMC-type structure in which subunits interact through a Zn-hook at the tip of antiparallel coiled-coil arms to form an M2R2N2 intralinked complex (Figure 1b) (30). In the ATP-free or hydrolyzed state seen in the Thermotoga maritima MR complex,the Rad50 ATPase subunits are flexible and relatively open with the arms relaxed (51, 52) (Figure 2b). In contrast, upon ATP binding, Rad50 closes into a single, more rigid conformation in which both head domains (N- and C-terminal) are interacting with each other in trans and form a central groove that can accommodate double-stranded DNA (dsDNA) (Figure 2a) (21, 38, 51). Notably, an added DNA binding site at the coiled-coil domain on Rad50 subunits is seen for the complex of Rad50 (nucleotide binding domain) and Mre11 (helix-loop-helix domain) with AMPPNP and dsDNA (53). Single-molecule imaging of the human MRN complex provided additional evidence for this DNA-binding mode. Fluorescence imaging of MRN on nucleosome coated DNA suggested that this binding mode facilitates an efficient search for DNA lesions via facilitated diffusion along chromatin (34). Also, as seen in EM images for human and P. furiosus MRN complexes, the coiled-coil domains can form interlinked complexes (two intralinked MRN complexes bridged through RAD50 Zn-hooks), where they may tether two distant DNA sites, and in this configuration, the arms can extend to 1,200 Å (32). This observation was supported by the crystal structure of the P. furiosus Rad50 Zn-hook domain with a small coiled-coil region, where the Zn2+-coordinating cysteines are arranged such that either intra- or interlinked complex coiled-coil domains can form (32). Recently, however, crystal structure and X-ray scattering analyses of human RAD50 Zn-hook with a portion of coiled-coil domain suggest that RAD50 predominantly forms an intralinked complex (Figure 1b) (33) having a novel interaction interface within the coiled-coil domain that supports a rigid rod-shaped RAD50 dimer.
The Zn-hook and coiled-coil domain are critical for functional RAD50 DNA tethering and ATM activation (54–56). Yet, two Rad50 subunits are also held flexibly by the Mre11 dimer, whose subunits form a flexibly linked helix-bundle interface at the base of two Rad50 subunits that allow the Rad50 dimer to open upon ATP hydrolysis and to expose the Mre11 DNA-binding channel for nuclease excision. Although limited coiled-coil domain–length deletions are tolerated, mutations in the Zn-hook domain share the phenotype of a null mutation in yeast (32, 57), suggesting that long-range allostery connecting the MR core enzymatic activity with the Zn-hook is essential for MRN function in vivo.
Importantly, the closed-state (ATP-bound) Rad50 renders dsDNA inaccessible to the Mre11 nuclease active site (Figure 2a) (21). However, ATP hydrolysis opens dsDNA so that flexible ssDNA may access the Mre11 active site (21). In another ATPase machine, FlaI (58), a structurally defined intermediate with bound phosphate ion (PO43–) released from ADP partially opens subunit contacts: This structure suggests that RAD50 conformational intermediates may allow MRE11 ssDNA endonuclease activity while restricting dsDNA and MRE11 exonuclease excision. By analogy, this as yet unseen RAD50 state would involve binding of arginine on the ABC–ATPase signature helix to the PO43– released by ATP hydrolysis (35). After ATP hydrolysis, Rad50 forms a structurally defined and more open state that makes DNA accessible to Mre11 for 3΄–5΄ exonuclease activity. The switch between closed and open Rad50 states orchestrates conformational changes to the MRN complex and its bound DNA (Figure 2b) that result in end protection or processing, as supported by structure-guided mutations (52). Furthermore, functional analyses of the Rad50 mutants show that the correct Rad50 interface with ATP and Mre11 are required for appropriate DSB processing (35, 59). Moreover, RAD50 mutation L1237F resulted in an exceptional patient response to an otherwise failed clinical trial of a checkpoint kinase-1 (CHK1) inhibitor combined with the DNA-damaging agent irinotecan (60). Our analyses suggest that this mutation promotes an open RAD50 that could activate MRE11 nuclease. We argue that matching the biochemistry of this mutation with a structure-based small-molecule inhibitor suggests a strategy to generalize the exceptional clinical response to other patients for precision medicine.
Crystal structures of Mre11 core domain are solved for both the Mre11 alone and in complex with Rad50 from different prokaryotes and eukaryotes (21, 29, 38, 39). All crystal structures, irrespective of source, show dimer formation, yet angles for the protomer arrangement vary. The dimerization surface is in the N terminus of T4 phage Mre11, followed by a capping domain and an acidic linker to the Rad50 binding domain, where the linker can act as an auto-inhibitor when Mre11 is not bound to Rad50 (61). From the X-ray crystal structures of P. furiosus Mre11 bound to a two-ended dsDNA and to a one-ended DNA fork (36), MR rearrangements can reshape DNA to access the nuclease active site (62). Existing DNA complex structures suggest that the Mre11 interface helps distinguish two-ended dsDNA breaks (symmetric dimer) from one-ended DNA forks (asymmetric dimer) (36) (Figure 2a). In fact, structure-guided small-molecule inhibitors (27), which bind adjacent to the dimer interface, show that blocking MRE11 endonuclease activity promotes DSB repair through NHEJ. An endonuclease nick distal to the DSB end licenses HR by initiating 3΄–5΄ MRE11 exonuclease activity back toward the DSB. Furthermore, the MRE11 endonuclease provides an entry point for long-range EXO1- and DNA2-dependent 5΄–3΄ resection activity away from the DSB. Consequently, inhibition of MRE11 exonuclease activity led to a DNA repair defect owing to a DSB repair defect that could be partially rescued by inhibiting EXO1/BLM resection needed to form extended ssDNA as the committed step for HR (27). Thus, MRN enzyme activities can help determine pathway choice between NHEJ and HR.
Nbs1 structural analyses define the phosphoprotein binding sites and uncover their flexible linkage to the Nbs1 C terminus (19). Furthermore, the crystal structure of Mre11 fused with the Nbs1 C terminus indicates that the Nbs1 C terminus interacts across the Mre11 dimer interface and on the face opposite to the DNA binding groove (23). As the Nbs1 N terminus interacts with several phosphorylated proteins, its flexible connection to its C-terminal interface across the Mre11 dimer extends the reach and network of the MRN complex for orchestrating DDRs (16, 29). For example, comparison of Nbs1-bound versus free structures unveils a long-range conformational change transmitted from the FHA domain bound to the phospho-CtIP-peptide through the BRCT domains, which may impact their binding to their phospho-serine protein targets, to drive an ∼30◦ domain rotation. These changes, which pull the linker to the Mre11 interface by ∼30 Å (19), are postulated to alter the Mre11 dimer symmetry thought to distinguish nuclease binding and activity for two-ended dsDNA breaks (symmetric dimer) and one-ended DNA forks (asymmetric dimer) (19). Overall, NBS1 (Xrs2 in S. cerevisiae) acts a chaperone and adaptor coordinating interactions of the MR complex with other proteins (63, 64) and as a regulated activator of Mre11 nuclease (65).
Consistent with the implications from structures and biochemistry, CtIP binds to the NBS1 FHA domains and evidently promotes resection in HR (50, 66–70) as one mode of conformational control for MRN. Recently, structure analyses of the N terminus for human CtIP and S. pombe Ctp1 reveal its assembly (48, 49). The full length and N terminus of the CtIP predominantly form a tetramer in solution. Furthermore, an extended Zn-binding motif in the coiled-coil domain adjacent to the N terminus extends the dimeric interface in both directions (Figure 2c). This CtIP interface resembles the MRN architecture and may tune the MRN complex in the interlinked complex that joins two sets of MRN–DNA complexes.
Using MRN’s structural biology, we propose a testable model for MRN endo- and exonuclease activity during HR (Figure 2d). Other nucleases, proteins, and spatial constraints likely impact this mechanism. Following initial recruitment to the damaged region by NBS1 N-terminal interactions (in eukaryotes) (71), ATP-bound MRN complexes load onto both sides of the DSB with RAD50 coiled-coil domains as intralinked complexes. Owing to their proximity, the Zn-hook and coiled-coil domains may switch into an interlinked complex, as seen in EM images (32). The ring-like conformation is suitable to permit MRN to move back from the break to allow access to other proteins, as the coiled-coil domain spans ∼1,200 Å , or ∼300 base pairs (bp) (32). Alternatively, a recent single-molecule imaging study (34) shows that the MRN complex can diffuse along the DNA, even in the presence of nucleosomes, to find the DNA end and carry out resection in association with EXO1. This study shows that Rad50 is critical for promoting facilitated diffusion along the nucleosomes-coated DNA, whereas Mre11 recognizes the DNA ends. This separation of function suggests one possible reason for the coevolution of Mre11 and Rad50 in all domains of life. Nbs1 is dispensable for both activities, allowing recruitment of the MR complex to the break in lower organisms for which Nbs1 is absent.
Through its interactions with both MRN and DNA (72, 73), the tetrameric CtIP configuration suggests a mechanical role in geometrically activating an MRE11 endonuclease cut in the 5΄ strand of the duplex with possible associated or sequential ATP hydrolysis. Upon ATP hydrolysis, RAD50 opens to allow MRE11 to carry out 3΄–5΄ resection toward the break, and the arms may be freed from the interlinked complex. At the nick, other 5΄–3΄ enzymes, such as EXO1/DNA2, resect away from the break to form extended ssDNA as the committed step and 3΄ single-strand substrate for HR. Upon reaching the DSB end, the CtIP dissociation upon a posttranslational modification of MRN may regulate MRN off rate and avoid release of unprotected DNA ends. To gain sequence-level information to test this model and other possible MRN mechanisms, it will likely be necessary to employ cryo–electron microscopy (cryo-EM), as enabled by advances in sample preparation for large complexes, plus EM detection technology and data analysis (74, 75). Integrated models of NHEJ from component structures have proved useful to guide both biochemical and cellular experiments (76), so we hope this may be the case for MRN.
MRN AND DOUBLE-STRAND BREAK REPAIR
MRN’s role in DDR is extensively studied for DSB repair. Unrepaired DSBs are toxic and can lead to chromosomal rearrangements (cancer hallmarks) and cell death through apoptosis (77). DSBs are caused by endogenous events (such as RF stalling; see Section 4) and severe damage from oxidation or ionizing radiation (IR) (78). MRN functions in both DSB sensing and repair: It is recruited to DSBs through interaction between NBS1 and cell cycle checkpoint protein RAD17 (71). RAD17 is phosphorylated by ATM and recruited to DSBs independent of the MRN complex (ATM-activation mechanisms are thoroughly reviewed in 79). Once at DSBs, the MRN complex recruits and activates ATM through its NBS1 interaction (Figure 3), with the likely participation of other unidentified interfaces. Activated ATM phosphorylates mediator of DNA damage checkpoint 1 (MDC1), which in turn recruits more MRN to DSBs and thereby amplifies MRN/ATM signaling (80).
Figure 3.
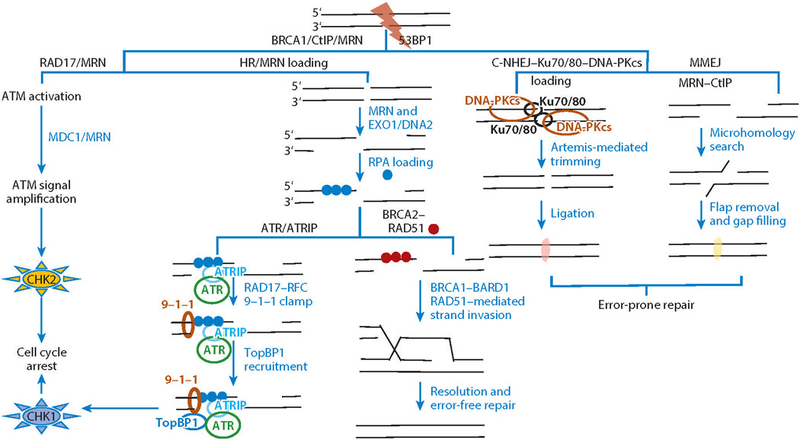
The MRN complex activates possible (alternative) pathways at DSBs. The MRN complex can initially be recruited to a DSB by RAD17 and activates the ATM kinase. Once activated, ATM phosphorylates downstream substrates including MDC1, which in turn recruits MRN, and the ATM signal is further amplified at the DSB. ATM checkpoint signaling leads to cell cycle regulation through CHK2 activation ( far left). On the basis of the cell cycle phase and nature of the DSB ends, DSBs are channeled through either HR or NHEJ for repair (central paths). In HR, the MRN complex initiates the 5΄ resection followed by long-range resection by EXO1 or DNA2. The resulting 3΄ ssDNA overhangs are loaded with ssDNA-binding protein RPA. The RPA-coated ssDNA can activate a second checkpoint pathway: ATR signaling. ATR is recruited to the RPA-coated ssDNA through ATRIP and RPA interaction. Then the RAD17–RFC clamp loading complex loads the 9–1–1 (RAD9–HUS1–RAD1) checkpoint clamp at the ssDNA and dsDNA junction on the RPA-coated ssDNA arms. The ATR activator TopBP1 is loaded by RAD9, leading to activation of CHK1 kinase and cell cycle regulation. For high-fidelity DSB repair, the BRCA2–RAD51 complex nucleates the formation of RAD51 nucleofilaments and displaces RPA from ssDNA. RAD51 nucleofilaments promote homology search and strand invasion in association with BRCA1–BARD1 for error-free HR repair through the resolution of repair intermediates. The two NHEJ pathways (right) depend upon DNA-PKcs and MRN–CtIP. In C-NHEJ, DSB ends are bluntly fused through Ku70/80–DNA-PKcs complex tethering, Artemis-mediated end processing, ligase IV, and XLF-XRCC4 scaffolding factor–mediated ligation. The C-NHEJ is independent of the MRN complex; however, MMEJ operates through MRN–CtIP-mediated resection followed by priming based on microhomology, flap removal, and gap filling. Abbreviations: ATM, ataxia-telangiectasia mutated; ATR, ataxia-telangiectasia and RAD3-related; ATRIP, ATR-interacting protein; C-NHEJ, canonical nonhomologous end joining; DNA-PKcs, DNA-dependent protein kinase catalytic subunit; DSB, double-strand break; HR, homologous recombination; MMEJ, microhomology-mediated end joining; MRE11, meiotic recombination 11 homolog 1; MRN, MRE11–RAD50–NBS1 complex; NBS1, Nijmegen breakage syndrome protein 1; NHEJ, nonhomologous end joining; RAD50, DNA repair protein RAD50; RFC, replication factor C; RPA, replication protein A; ssDNA, single-stranded DNA.
MRN/ATM signaling is regulated in part by factors that influence the stability and loading of the complex to DSBs. Interestingly, ATM and NBS1 are protected by the molecular chaperone heat shock protein 90 (Hsp90) alpha from polyubiquitination-mediated proteasomal degradation (81). Similarly, PIH1D1, a subunit of Hsp90 cochaperone R2TP complex, stabilizes MRE11 through its C-terminal interactions (82). Ubiquitination of NBS1 by the ring finger protein RNF8 promotes optimal binding to DSBs (83), whereas MRE11 phosphorylation by Polo-like kinase 1 (Plk1) inhibits recruitment of the MRN complex to DSBs (84). In addition, ATM-interacting protein (ATMIN) competes with NBS1 for ATM binding, therefore decreasing DSB-related ATM signaling (85, 86). In contrast, the recently discovered MRN-interacting protein (MRNIP) promotes MRN loading onto DSBs, thus contributing positively to MRN/ATM signaling (87). Consequently, activated ATM phosphorylates many downstream substrates to regulate checkpoint signaling and allow cells time to repair damage (88).
There are two major pathways through which cells repair DSBs (89) (Figure 3): the error-prone canonical nonhomologous end joining (C-NHEJ) and the error-free HR. NHEJ mends DSBs by end joining with minimal end processing. This process is initiated by Ku heterodimer (Ku70/Ku80) binding at DSB ends and recruitment of DNA-PKcs to phosphorylate various substrates and aid end processing and ligation by the DNA ligase IV complex (90). Two added pathways that repair DSBs with minimum end processing are resection-dependent NHEJ (rd-NHEJ) and microhomology-mediated end joining (MMEJ) (91, 92). In rd-NHEJ, an initial resection step involves MRE11 exonuclease, EXD2, Plk3-phosphorylated CtIP/BRCA1, EXO1, and Artemis endonuclease before repair through C-NHEJ occurs. MRE11 endonuclease activity may be dispensable in rd-NHEJ (92), whereas in MMEJ, initial resection involves both endo- and exonuclease activity of the MRN complex plus other enzymes, such as CtIP, PARP1, FEN1, and DNA ligases I and III (93, 94). The minimum homology for ligation is ∼5 bp (94). However, Werner syndrome helicase (WRN) and Bloom syndrome helicase (BLM) regulate these alternative pathways by blocking MRE11 and CtIP recruitment to DSB ends to promote repair through C-NHEJ (95, 96).
In HR, DSB ends are extensively resected to generate 3 ssDNA overhangs that inhibit NHEJ repair (Figure 3). As noted above, the evolving general model suggests that the MRN complex can initiate and license resection through an MRE11 endonuclease cut (nicking several hundred base pairs back from the DSB). Subsequent MRE11 exonuclease (3΄–5΄ direction toward the DSB) acts in association with phosphorylated CtIP (Sae2 in yeast). At the nick, 5΄–3΄ nucleases, such as EXO1 or DNA2 in conjunction with BLM or WRN, promote extended resection away from the DSB to generate 3΄ ssDNA overhangs more than 1,000 bp in length that constitute the committed step for HR (29). EXO1 catalyzes long-range resection of MRN-processed DNA ends in human cells (97, 98). However, EXO1 is strongly inhibited by replication protein A (RPA) binding to the resulting ssDNA (99, 100). Single-molecule imaging identified an additional, noncatalytic role for MRN in long-range resection. Here, MRN physically interacts with EXO1, traveling with the resection machinery as a processivity factor that prevents RPA inhibition (34).
Via interaction with the MRN complex or CtIP, extended DNA resection is stimulated by additional enzymes, including RECQL4 helicase and possibly by EXD2 exonuclease (101, 102), which has alternatively been localized to the cytoplasm and regulates translation in mitochondria (103). Regardless, the biochemistry for these regulating partners suggests changes in protein–DNA interaction stability and off rates. As described in the proposed model (Figure 2d), Sae2 (CtIP in humans) may dislodge the yeast MRX complex (Mre11–Rad50–Xrs2, MRN in humans) from ssDNA ends after resection (104, 105). In the absence of Sae2, DSB repair is impaired due to prolonged MRX complex binding. This defect is rescued by depleting the yeast 53BP1-ortholog RAD9, as 53BP1 promotes NHEJ by inhibiting dsDNA resection for HR (106, 107). In humans, the tumor suppressor BRCA1 inhibits 53BP1 at breaks in association with MRN and CtIP to promote HR over NHEJ (107–110). Extended RPA-coated ssDNA can activate ATR signaling (discussed in Section 4). For HR, RPA is displaced from ssDNA by RAD51 that forms nucleofilaments after loading by BRCA2 (111). RAD51 filaments enable a homology search, subsequent strand invasion, and template-dependent HR repair, as reviewed previously (111–113). Besides contributing to HR pathway choice, the BRCA1–BARD1 complex also plays an indispensable role in stimulating RAD51 recombinase activity (114). On the basis of the in vitro EM imaging data, the MRN complex may support HR by clamping the invading strand and HR template independent of its nuclease properties (32). Also, Mre11 nuclease-deficient or null TK46 cells show delays in Rad51 foci resolution after IR-induced breaks compared with wild-type cells. This delay suggests that the MRN complex may function downstream (as well as upstream) of RAD51-filament formation for proper resolution of HR intermediates (115).
Key factors regulating pathway choice for DSB repair are the cell cycle (116) and the nature of DNA ends (69). Whereas NHEJ-based repair pathways are active throughout the cell cycle, HR requires a DNA template for the repair and is active in only the S and G2 phases, during which the homologous sister chromosome is present. The nature of the DSB ends also regulates the repair pathway choice (69). If the DSB ends are free of protein adducts and damaged nucleotides, they are likely channeled through NHEJ for repair in human cells. Otherwise, they require processing through resection and the phosphatase activity of polynucleotide phosphatase/kinase (PNKP) (117). MRN biochemical endo- and exonuclease activities act on both 5΄ and 3΄ strands of DNA with protein adducts, and its endonuclease activity is stimulated by CtIP (72). In contrast, NBS1 inhibits the MR 3΄–5΄ exonuclease activity on clean DSB ends (72). The MRN complex can thus remove covalently linked topoisomerase complexes from genomic DNA in a mechanism requiring CtIP and BRCA1 (115, 118, 119). The MRN complex in association with CtIP also protects genomic loci at common fragile sites and palindromic repeats (120). Indeed, DSBs occurring at these sites often lead to the formation of secondary structures that can attract structure-specific nucleases and result in chromosomal rearrangements (120). At such sites, however, MRE11 nuclease activity promotes HR for error-free repair.
MRN AND REPLICATION FORK DYNAMICS
Accurate replication of genetic information is a prime genetic selection, and stress during replication can slow, stall, or collapse the RF (121). Sources for replication-induced stress include damaged DNA, chromatin compaction, non-B-DNA (i.e., DNA structures that do not conform to the canonical Watson-Crick double helix) forming sequences (G-quadruplex, small inverted repeats, trinucleotides repeats), covalent protein-DNA adducts, and DNA/RNA hybrids (121). Cells can overcome these barriers through RF stabilization, protection, remodeling, and restart (121–124) along with dormant origin firing in higher organisms (125–128). HR is implicated in fork transactions during fork recovery; however, HR protein responses at forks can be independent of dsDNA break repair (124). Although the HR response restores the replication robustness under stress, it can also contribute to genomic instability as a result of its intrinsic reliance on a homologous sequence (reviewed in 124). Given its HR-initiating roles, the MRN complex helps orchestrate RF stress responses (Figure 4). Its role at forks can be both beneficial and detrimental and thus requires regulation.
Figure 4.
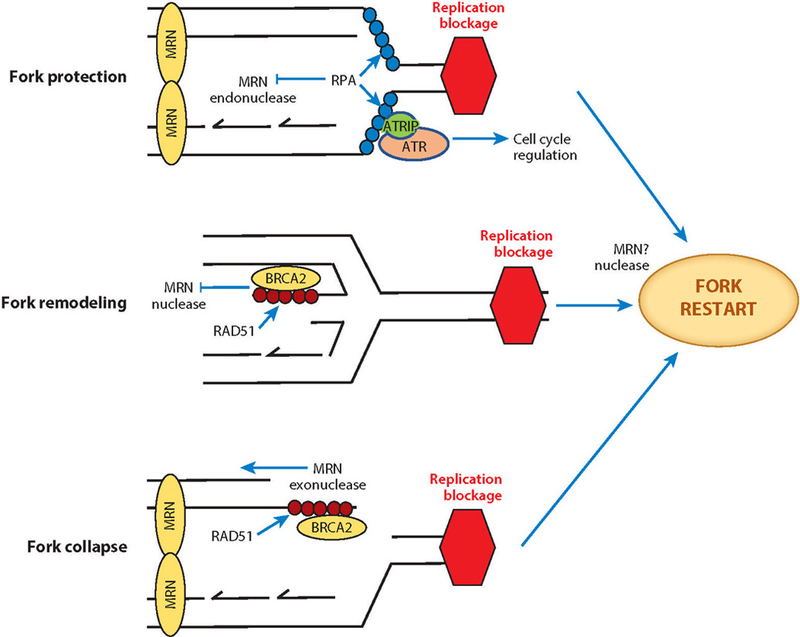
The MRN complex with RPA and ATR orchestrates replication fork protection, remodeling, and restart as well as HR rescue of collapsed forks. At stalled forks, the MRN complex participates in fork protection through non-nuclease functions (e.g., DNA tethering), and its ssDNA endonuclease activity is inhibited by RPA. RPA-coated ssDNA initiates ATR checkpoint signaling that provides cells time for repair and restart. Replication barriers are removed by remodeling through fork reversal that must then be processed for fork restart. The nascent ssDNA at reversed forks is protected against MRN exonuclease activity by RAD51 loading in association with BRCA2. For restart, MRE11 excision of one strand may facilitate fork reversal and restart. Unrepaired collapsed forks can become DSBs where the MRN complex initiates homologous recombination by initiating resection. Abbreviations: ATR, ataxia-telangiectasia and RAD3-related; ATRIP, ATR-interacting protein; MRE11, meiotic recombination 11 homolog 1; MRN, MRE11–RAD50–NBS1 complex; NBS1, Nijmegen breakage syndrome protein 1; RAD50, DNA repair protein RAD50; RPA, replication protein A; ssDNA, single-stranded DNA.
At forks, protein-coated ssDNA is a platform for binding by MRN and other DNA repair proteins that initiate damage signaling, fork protection, and repair (129). An encounter between the replication machine (or replisome) and a replication barrier can uncouple the replicative helicase and polymerase. This uncoupling results in long stretches of ssDNA, leading to an ATR checkpoint response that helps to stabilize the fork and allows time to resolve the replication barrier by arresting cell cycle progression (Figure 4) (121). Stretches of ssDNA are protected by ssDNA-binding proteins such as RPA or human single-stranded DNA binding protein 1 (hSSB1) (130, 131). RPA binding to ssDNA inhibits annealing of short homologous sequences (as in short repeats) through microhomology-mediated repair and reduces secondary structures recognized by nucleases (132). The resulting protein-coated ssDNA not only is protected but also promotes binding of MRN and other DNA repair proteins (133). ATR, along with ATRIP, is recruited to ssDNA-RPA regions through ATRIP and RPA interactions (134). At the damaged forks, RAD9–RAD1–HUS1 (also known as the 9–1–1 clamp), is loaded onto the DNA end adjacent to RPA-bound ssDNA by the clamp loader RAD17–RFC complex (134). The 9–1–1 clamp recruits the ATR-activator protein topoisomerase-binding protein 1 (TopBP1) that assists ATR in phosphorylating its downstream substrates, including CHK1 and RPA (Figure 3) (134). Notably, TopBP1 was recruited independently of the 9–1–1 complex to the ATR-activating sites at ssDNA-dsDNA junctions at the RFs through the MRN complex in Xenopus egg extracts (135). This result is consistent with the observation that the MRN-depleted Xenopus extracts show a decrease but not complete loss of CHK1 phosphorylation (136).
In fact, ATR activation is a critical function for MRN in damage signaling. MRE11 (in addition to ATM) is required for ATR activation upon IR-induced damage (137, 138). Furthermore, NBS1 activates ATR in vitro independently of MRE11 (139). Notably, for replication-associated DSBs, a substantial amount of RPA phosphorylation by ATR is dependent on the NBS1–RPA interaction and is independent of RAD17 (140). These functions are likely conserved in yeast, in which RPA recruits the MRX complex to stalled forks and DSBs (141). Upon recruitment, MRX-mediated sister-chromatid tethering is essential for survival of genomic stress at both forks and breaks.
ATR activation is also regulated by RAD50 (142). Once activated, ATR phosphorylates RAD50 at S635, which is required for cohesin complex (that holds together newly replicated chromatids) loading at the forks. Aside from phosphorylation, the neighboring Zn-hook sequence influences cohesin loading at replication sites to promote fork restart (143). These collective data suggest that multiple MRN components can orchestrate ATR signaling events that may themselves regulate the temporal sequence for MRN biochemical activities at RFs.
At forks, MRN-mediated resection plays a major functional role. MRN-mediated resection of nascent DNA likely frees the replisome from the stalled forks by promoting strand removal, fork inversion, and restart (123). MRE11 can generate 3΄ overhangs to initiate HR in association with DNA2/EXO1. For example, in a recent study in a Werner syndrome–derived fibroblast cell line, the phosphorylation status of the S1133 of the WRN helicase influences the resection at breaks generated at the RF (144). Inhibition of S1133 phosphorylation in WRN results in impaired interaction with MRE11, defective MRN foci formation, and resection at the forks. Consequently, breaks at the fork can be repaired through DNA fusion by NHEJ with associated genome instability.
Although the MRN nuclease function can help resolve stalled RFs, unregulated resection can be catastrophic and lead to fork degradation through MRE11 exonuclease activity (145, 146). Thus, other proteins help regulate MRN-mediated nascent DNA degradation (147–150). The Fanconi anemia and breast cancer protein BRCA2 blocks MRE11-mediated resection of nascent ssDNA by promoting formation of stable nucleofilaments of RAD51 on nascent DNA (147, 151). However, cyclin-dependent kinase 2 (CDK2)–dependent phosphorylation of the BRCA2 C terminus results in disassembly of RAD51 filaments (152). This CDK2 activity is controlled by a complex formation with large tumor suppressor homolog 1 (LATS1) as part of the regulation of BRCA2 phosphorylation. Similarly, WRN helicase interacting protein 1 (WRNIP1) can stabilize RAD51 and thereby inhibit MRN-mediated degradation, a function independent of WRNIP1 ATPase activity (153). Like MRE11, WRN has an exonuclease domain with an associated coiled-coil region allowing assembly and partner interactions (154–156). Interestingly, the WRN exonuclease domain appears essential to inhibit MRE11/EXO1-dependent fork degradation under replication stress induced with low doses of the TOP1 inhibitor camptothecin (157). MRN regulation at forks is thus enforced by protein interactions that block productive MRE11 nuclease binding.
By inhibiting Mre11 using endonuclease- and exonuclease-specific inhibitors, BRCA2 deficiency is rescued in tumor cells lacking functional BRCA2. Although BRCA-defective cancer cells can be killed by PARP inhibitors (PARPi), resistance to PARPi arises in cancer cells from the inactivation of proteins (such as PTIP, CHD4, and PARP1) that recruit MRE11 protein for nuclease processing at stalled forks (158). This unraveled resistance mechanism underscores the critical role for MRE11 in the regulated and error-free fork restart and how loss of MRE11 or its regulation at forks can rebalance damage responses in cancer cells. In fact, this may be the basis for genome instability from p53 mutations (159).
MRN AND DYSFUNCTIONAL TELOMERES
MRN responds to the disfunction of telomeres, which protect linear eukaryotic chromosome ends from DSB-related DDR and suppress replication instability (160). Telomeres consist of long (thousands of base pairs in length) duplex-DNA repeats (-TTAGGG-in humans) with shorter (up to several hundred base pairs) single-stranded 3΄ overhangs and associated telomere protection–related proteins collectively termed the shelterin complex (161). Owing to the strict polarity restrictions of DNA polymerase, the 3΄-DNA end sequence from the lagging strand is not duplicated at telomere ends, leading to shortening of the net telomere length with each replication cycle (162). Conversely, duplication of the leading strand always results in a blunt-ended dsDNA owing to correct polarity (Figure 5). Both leading and lagging strand telomeres are processed by nucleases Apollo and EXO1 to generate functional 3΄ overhangs (163).
Figure 5.
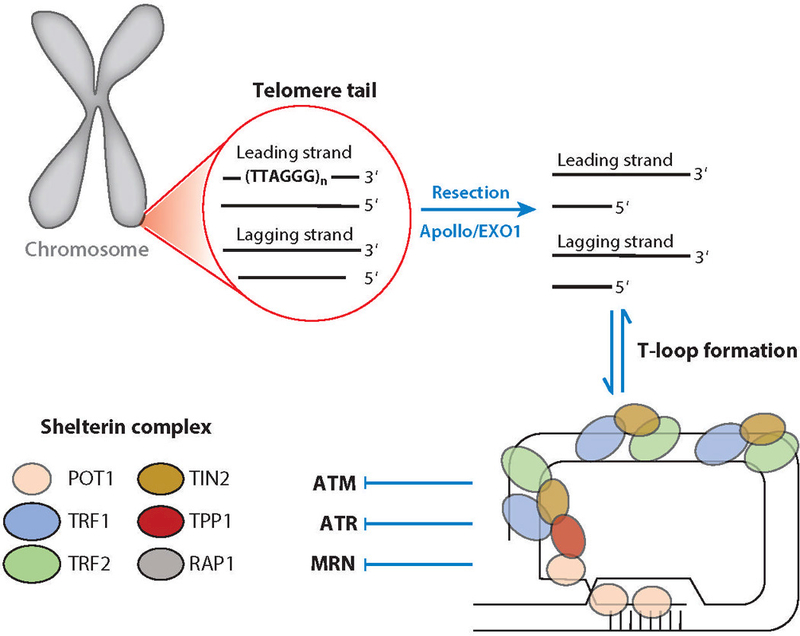
Telomere structure and dynamics regulate MRN interactions and distinguish chromosome ends from breaks. Human telomeres consist of a long stretch of repetitive TTAGGG sequences and interacting proteins. Telomeres forming from the replication of a leading strand result in tails with blunt ends, whereas the replication of a lagging strand generates telomeres with 3΄ overhangs. The 5΄ resection of a blunt-ended telomere inhibits telomere fusion through NHEJ. The telomeres with 3΄ overhangs form transient T-loop structures. The shelterin complex, which helps protect telomere ends, consists of telomeric repeat-binding factors 1 and 2 (TRF1 and TRF2), repressor and activator protein (RAP1), TRF1-interacting nuclear protein 2 (TIN2), protection of telomeres 1 (POT1), and TIN2- and POT1-interacting protein (TPP1). Abbreviations: ATM, ataxia-telangiectasia mutated; ATR, ataxia-telangiectasia and RAD3-related; CtIP, C-terminal binding protein–interacting protein; MRE11, meiotic recombination 11 homolog 1; MRN, MRE11–RAD50–NBS1 complex; NBS1, Nijmegen breakage syndrome protein 1; NHEJ, nonhomologous end joining; RAD50, DNA repair protein RAD50; T-loop, telomere loop.
In addition to the shelterin complex, the reverse transcriptase telomerase maintains telomere length in budding yeast, stem cells, and many cancer cells (164). Yet interestingly, in most somatic cells, telomerase is inactive, leading to a limited number of replicative cycles and thereby regulating mutational accumulation and tumorigenesis (165–167). In budding yeast, the MRX complex may act in recruiting telomerase to telomeres (168). Also, telomere elongation may depend on X-ray repair cross-complementing protein 3 (XRCC3) and NBS1 in stem cells (169). The MRX complex may also assist Rad6 and Bre1 ubiquitin-conjugating enzymes in successful telomere replication (170).
In humans, telomeres function in tumor suppression. Cells either enter a replicative senescence state or undergo apoptosis once telomere length becomes critical, to avoid losing essential genes from linear chromosome ends (166). This regulatory mechanism relies on MRN and other proteins that assist the shelterin complex to avoid DSB repair at functional telomeres (reviewed in 171, 172).
At functional telomeres, DNA ends are protected. Since telomeres are G-rich sequences, they can form non-B-DNA structures such as G-quadruplexes that protect against strand degradation (173). As a result, replication in telomeric regions may have more frequent fork stalling (174) that may require resolution by MRN, but this role is uncertain.
In addition, the 3΄ overhang can fold back on to the dsDNA to form a T-loop similar to strand invasion in HR (161, 175) (Figure 5). The six-protein shelterin complex contains telomeric repeat-binding factors 1 and 2 (TRF1 and TRF2), repressor and activator protein (RAP1), TRF1-interacting nuclear protein 2 (TIN2), protection of telomeres 1 (POT1), and TIN2- and POT1-interacting protein (TPP1) (176). TRF2 binds to dsDNA, protects the T-loop from helicases and resolvases (176), and inhibits ATM signaling, whereas POT1 preferentially binds ssDNA and inhibits ATR signaling (177) (Figure 5). Shelterin defects and telomerase abnormalities lead to dysfunctional telomeres, promoting MRN damage signaling and repair within a few cycles of replication (178).
The MRN complex may thus play dual roles at telomeres, where it may cause or inhibit genomic instability at dysfunctional telomeres by either promoting or inhibiting the NHEJ pathway, respectively (171). NBS1 co-crystallizes with TRF2 (179). NBS1 phosphorylation at S432 residue inhibits this interaction and promotes C-NHEJ at telomeres lacking TRF2 (179). Yet, NBS1 phosphorylation at the same site promotes A-NHEJ at telomeres lacking POT1–TPP1 (179). In addition to defective shelterin components, critically short or deleted ends lead to dysfunctional telomeres. Although most cancer cells use telomerase to replenish dysfunctional telomere lengths, some utilize MRE11-dependent and recombination-based alternative lengthening of telomere (ALT) for telomere maintenance (180, 181). Loss of both telomerase and the ALT pathway causes telomere dysfunction. In yeast cells with these characteristics, Mre11 opposes proliferation of cells lacking telomeres in the absence of Exo1 and checkpoint signaling (182).
In budding yeast lacking telomerase, the MRX complex promotes replicative senescence in association with Tel1 (ATM in humans) and RAP1-interacting factor 2 (Rif2) (183). Tel1 promotes MRX recruitment, whereas Rif2 acts as a negative regulator by enhancing ATP hydrolysis that opens Rad50, facilitating MRX release from DNA (184). In line with insights from yeast experiments, changes in expression and posttranslational modification of MRN and telomeric proteins are associated with various diseases in humans (185–187). In mouse embryonic fibro-blasts, the MRN complex is required for the response to telomere dysfunction, consistent with the idea that initiating this response shares common elements with the response to interstitial DNA damage (188). Interestingly, in patients with rheumatoid arthritis for which T cell aging occurs prematurely, low MRE11 levels lead to telomeric damage, juxtacentromeric heterochromatin unravelling, and upregulation of senescence marker. MRE11 inhibition in healthy T cells induced the aging phenotype, whereas MRE11 overexpression in rheumatoid arthritis T cells reverses it. The observation that the premature T cell aging is linked to MRE11 and telomere deprotection suggests MRE11 as a therapeutic target for immune aging and suppressing dysregulated inflammation (189). We expect that further knowledge of MRN biochemical mechanisms will be key to an improved understanding and intervention in multiple diseases including immune system aging.
MRN AND THE INNATE IMMUNE RESPONSE
MRN functions that sense, signal, and defend against viral infection (190) underscore the importance of deciphering the biochemical basis for its multifunctionality. MRN and other damage recognition receptors detect viral DNA and initiate a cascade of innate immune and DNA damage signaling (191) (Figure 6a). In the genome, activation of ATM by MRN is amplified over a mega-base region leading to formation of DNA damage foci, cell cycle arrest, and DNA repair (192). Upon viral infection, local but not global ATM activation and signaling are initiated by the MRN complex owing to the small size of viral DNA (193). This local ATM signaling avoids cell cycle arrest and global damage responses when a virus infects the cell (Figure 6a) (193). Thus, MRN biochemical responses allow cells to differentiate genome damage signaling (global ATM signaling) for DNA repair compared with foreign-genome signaling (local ATM signaling) to block viral replication in the nucleolus (193). As part of its repair-related activities, the MRN complex in association with NHEJ repair proteins can further limit viral DNA replication in the nucleus by forming long viral DNA concatemers (190, 194, 195), which are too large to be packaged. Concatemers also interfere with replication: Replication origins generally present at the viral DNA termini are buried inside heterogeneously fused viral DNA and mutated through error-prone NHEJ (190, 196, 197) (Figure 6a).
Figure 6.
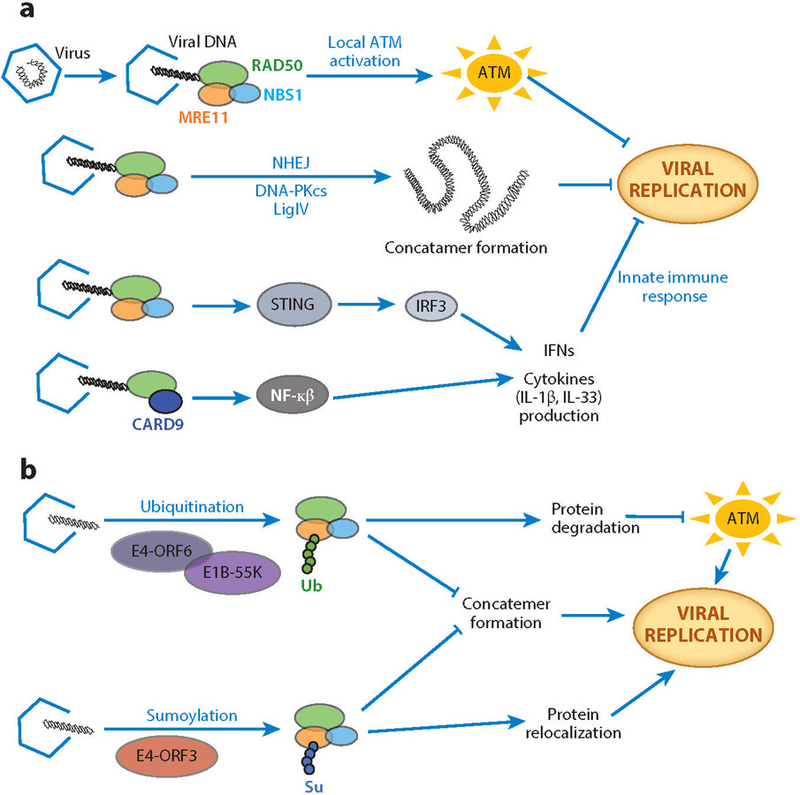
MRN in host and virus responses. (a) The MRN complex inhibits viral replication via pathways linking DNA repair proteins and the innate immune response. The interaction between the MRN complex and viral DNA leads to local ATM activation that avoids cell cycle arrest and apoptosis, which is characteristic of global ATM signaling in DNA damage. The MRN complex promotes the formation of viral DNA concatemers (randomly fused long chains of viral DNA) in association with NHEJ proteins to help inhibit viral replication. MRN components furthermore promote the innate immune response by inducing interferon and cytokine production through pathways involving STING, IRF3, and NF-κβ. (b) Viruses neutralize the MRN complex by relocalization and/or degradation. The viral protein complex of E4-ORF6 and E1B-55K directs the MRN complex for degradation through ubiquitination (Ub). The viral protein E4-ORF3 relocalizes the MRN complex to the nucleus through sumoylation (Su) to promote viral replication by limiting MRN-promoted viral DNA concatemer formation. Abbreviations: ATM, ataxia-telangiectasia mutated; CARD9, caspase recruitment domain-containing protein 9; DNA-PKcs, DNA-dependent protein kinase catalytic subunit; E1B-55K, a 55-kDa protein of adenovirus E1B gene; E4-ORF3, early region 4 of adenovirus open reading frame 3 protein; E4-ORF6, early region 4 of adenovirus open reading frame 6 protein; IFNs, type I interferons; IL-1β, interleukin 1 beta; IL-33, interleukin 33; IRF3, interferon regulatory factor 3; MRE11, meiotic recombination 11 homolog 1; MRN, MRE11–RAD50–NBS1 complex; NBS1, Nijmegen breakage syndrome protein 1; NF-κβ, nuclear factor κβ; NHEJ, nonhomologous end joining; RAD50, DNA repair protein RAD50; STING, stimulator of interferon genes.
DNA in the cytoplasm is recognized as a potential viral infection and induces an immune response. MRN binding to viral DNA in the cytoplasm leads to production of type I interferons (IFNs) through activation of nuclear factor κβ (NF-κβ), a stimulator of interferon genes (STING), and interferon regulatory factor 3 (IRF3) (Figure 6a) (198). Both MRE11 and RAD50, but not NBS1, are essential for producing IFNs. A cell line derived from an ATLD patient containing an MRE11 mutation that abrogates its DNA binding properties has defects in IFN production (198). Interestingly, RAD51 absence can also induce an innate response (199). RAD51 protects DNA inside the nucleus from MRE11-mediated nucleolytic degradation. Thus, in the absence of RAD51, MRE11 nuclease can degrade DNA, and the resulting DNA fragments are released into the cytoplasm where they activate the innate immune response. For interleukin-33 (IL-33), a member of the IL-1 superfamily of cytokines whose structure has been defined (200), stimulation upon adenoviral transduction depends on MRE11 status (201). Similarly, upon encountering cytosolic DNA, RAD50 forms a complex with innate immune system adaptor protein CARD9 in dendritic cells (202), which are the antigen-presenting cells that link the innate and adaptive immune systems. The cytosolic dsDNA-induced signaling complex (dsDNA–RAD50–CARD9) stimulates production of proinflammatory cytokine IL-1β through NF-κβ pathway activation (Figure 6a) (202). However, viruses encode specific proteins that bind MRN and other host DNA repair proteins rendering them inactive through protein relocalization and degradation (Figure 6b) by sumoylation and ubiquitination (190, 203). Yet, viruses can also rely on MRN and other host DNA repair proteins for successful lytic replication.
As a result, manipulation of MRN biochemistry by interactions with virus proteins is under active investigation, and several of these have been uncovered in human adenovirus (Ad) infection. Ad early region 4 (E4) encodes two proteins from open reading frame 3 (E4-ORF3) and open reading frame 6 (E4-ORF6). These proteins (with viral protein E1b-55K) inhibit concatemer formation by the MRN complex via relocalization and degradation (190, 204–207), avoiding MRN-mediated defects in late viral replication (205, 207). Upon recognition, E4-ORF3 protein localizes the MRN complex to specific nuclear locations in the host cells known as promyelocytic leukemia protein domains (or PML bodies), rendering the complex inactive (Figure 6b) (208). X-ray crystallography and mutational analyses show that E4-ORF3 assemble into both linear and branched polymeric structures by C-terminal tail swapping (209). This polymeric network spans the entire nuclear volume, forming multi-site interactions with PML bodies and MRN-binding interface in the C-terminal tail. Upon relocalization to PML, E4-ORF3 induces sumoylation of MRN complex components (210, 211). The E4-ORF6 in association with E1b-55K recruits other cellular proteins, such as cullin 5, Rbx1, and elongin B and C to form an active ubiquitin ligase complex (212, 213). This activated ligase complex ubiquitinates the MRN complex for proteasome-dependent degradation (190).
Building upon the Ad results, MRN roles are increasingly being elucidated in virus infection associated with human diseases—for example, high-risk human papillomavirus (HPV) that is implicated in cervical cancer and as well as in genital, head, and neck cancers (214). HPV-31 increases MRN complex half-life for productive viral replication (215). HPV-16 encodes transcription and replication factor E2 that interacts with RAD50-interacting protein 1 (Rint1) (216). Overexpression of exogenous Rint1 enhances HPV-16 replication, and a truncated mutant that lacks a RAD50-interacting domain reduces HPV-16 replication. Thus, HPV-16 may target MRN complex inactivation through Rint1 for productive replication. Similarly, the MRN complex inhibits replication of Kaposi sarcoma herpesvirus (KHSV) (217), which is implicated in Kaposi sarcoma, the most common cancer in HIV-infected individuals (218, 219). KHSV encodes a cytoplasmic isoform of latency-associated nuclear antigen (LANA). This isoform inhibits the host innate immune response by neutralizing MRN and cyclic GMP-AMP synthase DNA sensors (217).
The relationship linking viral infection, DNA repair, and innate immunity has biomedical importance, as oncolytic virus-based therapies are implemented in several cancers. For example, a modified oncolytic adenovirus mutant that lacks an antiapoptotic gene (E1B19K) sensitizes pancreatic cells in association with DNA-damage drugs (220), and the proposed mechanism is MRE11 inactivation. For oncolytic adenovirus type 5 being used in ovarian cancer cells (221), virus cytotoxicity is proposed to enable its ability to redistribute MRN (and other HR components). Overall, our evolving knowledge of interactions between viral infection, DNA repair, and the immune response requires defining multiple roles for MRN biochemistry.
SUMMARY AND PERSPECTIVES
A key aspect of the DNA double-helical structure, initially absent in the Watson and Crick model, is that it contains information not only for replication but also for repair of genomic damage (222) that is recognized by biologically conserved complexes such as MRN. Thus, MRN-DNA biochemistry is uncovering primary origins of normal and pathological cellular activities for DNA stress responses. Yet, this requires a path for a comprehensive characterization of proteins and nucleic acids, including their biologically relevant complexes and conformations (223). These observations have motivated our efforts to develop high-throughput and objective X-ray methods for comprehensive measurements of complexes and conformations and flexibility under near-physiological conditions (e.g., see 224–229). Thus, X-ray scattering and diffraction methods coupled to cryo-EM and single-molecule data are helping to define the MRN structural biochemistry described here.
At the cellular level, MRN defects can cause chromosome instability. At the systems level, we see the importance of maintaining the B-DNA double helix structure, as potential non-B-DNA structures (PONDS) occur at high frequency in cancer as translocations and deletion break points (230). Furthermore, at RFs where the double helix is opened for processing, precise control of DNA ends is needed to avoid not only mutations but also template switching and large repeat expansions (231, 232). These and other data provide compelling support for the notion that DNA structures at forks, ends, and breaks are intrinsic risk factors that complexes such as MRN are under strong evolutionary pressure to tightly control.
Combined genetic and biochemical data suggest that MRN has evolved to coordinate cellular network responses to DNA damage linked to mutation, cancer, and cell death. Its modular components are integrated into complexes and pathway networks in different ways, but MRN core structures and activities are maintained and conserved across all domains of life. Collective data show that multiple DNA stress responses are dynamically interrelated by MRN in ways that depend on its interactions and conformations. These results have broad implications for damage responses, as we are learning that even direct damage reversal enzymes, such as ALKBH3 (233), surprisingly function within large complexes for DNA and RNA integrity (234).
The evolutionarily conserved MRN complex is thus decoding a biological language of dynamic shapes controlling chemistry, connections, and complex functionalities from self-assembled, sequence-encoded building blocks whose fidelity is maintained by complexes such as MRN. For example, mutants modulating RAD50 dynamic conformations can switch biological outcomes from DNA excision to end joining (52). The data reviewed here demonstrate how MRN interactions and resulting activities feed into cellular decision making in DNA stress responses including viral defenses (typically considered to be only broadly related to the DDR). MRN subunit biochemical activities depend upon their three-dimensional folds, but MRN biological functions are governed by its complexes and conformationally regulated interaction networks. Thus, HR and MMEJ are activated in irradiated human cells owing to phosphorylation-dependent formation of either NBS1 or XRCC1 repair complexes with MRE11–RAD50 (93). In fact, the use of the same MRN subunits in multiple response networks provides a physical means to coordinate sensing, signaling, chromatin remodeling, and repair processing. This helps ensure appropriate responses to DSBs and stalled forks that avoid destructive interference and thus chromosome breakage and aberrations. As its cellular selectivity and function are determined partly by its assembly with partners and damaged DNA, we have focused on what we know and would like to know about dynamic MRN complexes and their control of biological outcomes. From the results to date, we can expect that defining the biochemical activities of MRN components and their assemblies will provide a critical foundation for understanding multiple DNA stress responses.
As a consequence of its dynamic conformations and assemblies, MRN responses are DNA structure specific rather than sequence specific. For example, MRN complexes confer specificity to distinct nuclease processing at DNA breaks and forks. These processes include an endonuclease nick and the initiation of exonuclease processing that is not adjacent to the DSB. Moreover, the resulting combined MRE11 endo- and exonuclease activities can act as go/no-go controls in the selection of HR versus NHEJ pathways (27). MRN can thus orchestrate DDRs as a sensor, regulator, and effector within distinct DSB repair, RF, and telomere rescue complexes as well as in viral response networks.
We propose that MRN conducts, coordinates, and connects responses to DNA stress largely by orchestrating three processes: assembly (localization and protection at damage), conformation (creation of a productive geometry for interactions and active site chemistry), and disassembly (controlling access and handoffs of DNA intermediates). In chemistry, transition state theory instructs us on the control of the reaction barrier as the expected rate-limiting step. In the cellular biochemistry of DNA replication and repair, however, the rate-limiting step is often not chemistry, which has been evolutionarily optimized for high efficiency. Instead, we see that protein and DNA binding energy is converted into conformational change to regulate network responses on the basis of the cell cycle state and the nature of the DNA damage. At the nanoscale of MRN, charge, chemical bonding, molecular mechanics, and thermal energy share similar scales: This allows their efficient interconservation to regulate specific activities within metastable MRN–DNA and MRN–protein intermediates. We propose that this ability to transfer binding energy into functionally important conformational states is an evolutionarily optimized and emergent MRN property. In practical terms, targeting conformational switching, as proposed here for RAD50 on the basis of the exceptional patient response in an otherwise failed clinical trial (60), suggests a novel biochemical strategy for precision medicine.
More broadly, current and emerging MRN results are aiding in addressing grand challenges of the postgenomic era for biochemistry and cancer biology: (a) an accurate prediction of pathology for damage response sequence variants, (b) an actionable mechanistic understanding of oncogenic replication and repair stress for biology and medicine, and (c) an advanced assessment of functional biochemical complexes and networks controlling outcomes from molecules to cells (235). Existing insights are already changing our concepts of damage detection, signaling, and repair with implications for strategies of controlling MRN activities for cancer, T cell aging, immune responses, and viral infections. Going forward, we will increasingly need to link structures of intact dynamic assemblies from cryo-EM and X-ray scattering studies to outcomes in cells and furthermore test this multiscale understanding with separation-of-function mutations and activity-specific chemical inhibitors, as was done for MRE11 (27). Emerging robust methods for the coexpression of large, multiprotein complexes provide enabling technologies for examining functional assemblies(75). The resulting foundational knowledge defining how these molecular machineries act may allow control of DNA stress responses for biology and medicine. We suggest that targeting MRN biochemical activities in DNA damage and the innate immune response together may overcome the mutational phenotype that characterizes cancer cells (236), analogously to how antibiotic knockdown of pathogens allows their clearance by our immune system.
ACKNOWLEDGMENTS
The authors thank Drs. Tanya T. Paull (University of Texas, Austin), Susan P. Lees-Miller (University of Calgary, Canada), Yunje Cho (Pohang University of Science and Technology, South Korea), R. Scott Williams (National Institute of Environmental Health Sciences, Durham), Gareth J. Williams (University of Calgary, Canada), Karl-Peter Hopfner (Ludwig Maximilian University, Germany), Titia de Lange (The Rockefeller University, New York), John H. Petrini (Memorial Sloan Kettering Cancer Center, New York), Ilya J. Finkelstein (University of Texas, Austin), Katharina Schlacher (MD Anderson Cancer Center, Houston), Walter J. Chazin (Vanderbilt University, Nashville), and David K. Cortez (Vanderbilt University, Nashville) for valuable suggestions. Our work on the MRN complex and DDR is supported by structural aid from Dr. Elizabeth D. Getzoff and Andrew S. Arvai plus the strong cell biology of our long-time collaborator Dr. Paul Russell (The Scripps Research Institute, La Jolla) along with other collaborators: Drs. Sankar Mitra (Houston Methodist, Houston), Alan Tomkinson (University of New Mexico, Albuquerque), Elena Petricci (University of Siena, Italy), Claire Wyman (Erasmus University Medical Center, The Netherlands), and Penny A. Jeggo (University of Sussex, United Kingdom). Our MRN research is supported in part by the National Institutes of Health grants R01CA117638, R01CA200231, and P01CA92584. Added support comes from the Robert A. Welch Distinguished Chair in Chemistry. J.A.T. acknowledges startup funds from the University of Texas STARs program. The work combining X-ray diffraction in crystals and solution at the Advanced Light Source SIBYLS beamline to define accurate structures, conformations, and assemblies of macromolecular machines is supported in part by the United States Department of Energy Integrated Diffraction Analysis Technologies (IDAT) program.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol. Cell 40:179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D’Amours D, Jackson SP. 2002. The MRE11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol 3:317–27 [DOI] [PubMed] [Google Scholar]
- 3.Connelly JC, Leach DRF. 2002. Tethering on the brink: the evolutionarily conserved Mre11–Rad50 complex. Trends Biochem. Sci 27:410–18 [DOI] [PubMed] [Google Scholar]
- 4.Stracker TH, Petrini JHJ. 2011. The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol 12:90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uchisaka N, Takahashi N, Sato M, Kikuchi A, Mochizuki S, et al. 2009. Two brothers with ataxia-telangiectasia-like disorder with lung adenocarcinoma. J. Pediatr 155:435–38 [DOI] [PubMed] [Google Scholar]
- 6.Matsumoto Y, Miyamoto T, Sakamoto H, Izumi H, Nakazawa Y, et al. 2011. Two unrelated patients with MRE11A mutations and Nijmegen breakage syndrome-like severe microcephaly. DNA Repair 10:314–21 [DOI] [PubMed] [Google Scholar]
- 7.Miyamoto R, Morino H, Yoshizawa A, Miyazaki Y, Maruyama H, et al. 2014. Exome sequencing reveals a novel MRE11 mutation in a patient with progressive myoclonic ataxia. J. Neurol. Sci 337:219–23 [DOI] [PubMed] [Google Scholar]
- 8.Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, et al. 1999. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99:577–87 [DOI] [PubMed] [Google Scholar]
- 9.Delia D 2004. MRE11 mutations and impaired ATM-dependent responses in an Italian family with ataxia-telangiectasia-like disorder. Hum. Mol. Genet 13:2155–63 [DOI] [PubMed] [Google Scholar]
- 10.Fernet M, Gribaa M, Salih MAM, Seidahmed MZ, Hall J, Koenig M. 2005. Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Hum. Mol. Genet 14:307–18 [DOI] [PubMed] [Google Scholar]
- 11.Brandt S, Samartzis EP, Zimmermann A-K, Fink D, Moch H, et al. 2017. Lack of MRE11-RAD50-NBS1 (MRN) complex detection occurs frequently in low-grade epithelial ovarian cancer. BMC Cancer 17:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gravells P, Grant E, Smith KM, James DI, Bryant HE. 2017. Specific killing of DNA damage-response deficient cells with inhibitors of poly(ADP-ribose) glycohydrolase. DNA Repair 52:81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim I-K, Kiefer JR, Ho CMW, Stegeman RA, Classen S, et al. 2012. Structure of mammalian poly(ADP-ribose) glycohydrolase reveals a flexible tyrosine clasp as a substrate-binding element. Nat. Struct. Mol. Biol 19:653–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Gudikote J, Giri U, Yan J, Deng W, et al. 2018. RAD50 expression is associated with poor clinical outcomes after radiotherapy for resected non–small cell lung cancer. Clin. Cancer Res 24:341–50 [DOI] [PubMed] [Google Scholar]
- 15.Lamarche BJ, Orazio NI, Weitzman MD. 2010. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett 584:3682–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams GJ, Lees-Miller SP, Tainer JA. 2010. Mre11–Rad50–Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair 9:1299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lavin M, Kozlov S, Gatei M, Kijas A. 2015. ATM-dependent phosphorylation of all three members of the MRN complex: from sensor to adaptor. Biomolecules 5:2877–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, et al. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316:1160–66 [DOI] [PubMed] [Google Scholar]
- 19.Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, et al. 2009. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 139:87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lloyd J, Chapman JR, Clapperton JA, Haire LF, Hartsuiker E, et al. 2009. A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell 139:100–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Sung S, Kim Y, Li F, Gwon G, et al. 2016. ATP-dependent DNA binding, unwinding, and resection by the Mre11/Rad50 complex. EMBO J 35:743–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sung S, Li F, Park YB, Kim JS, Kim AK, et al. 2014. DNA end recognition by the Mre11 nuclease dimer: insights into resection and repair of damaged DNA. EMBO J 33:2422–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schiller CB, Lammens K, Guerini I, Coordes B, Feldmann H, et al. 2012. Structure of Mre11–Nbs1 complex yields insights into ataxia-telangiectasia–like disease mutations and DNA damage signaling. Nat. Struct. Mol. Biol 19:693–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hammel M, Yu Y, Radhakrishnan SK, Chokshi C, Tsai M-S, et al. 2016. An intrinsically disordered APLF links Ku, DNA-PKcs, and XRCC4-DNA ligase IV in an extended flexible non-homologous end joining complex. J. Biol. Chem 291:26987–7006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavin MF. 2007. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 26:7749–58 [DOI] [PubMed] [Google Scholar]
- 26.Marechal A, Zou L. 2013. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol 5:a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, et al. 2014. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 53:7–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trenz K, Smith E, Smith S, Costanzo V. 2006. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J 25:1764–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lafrance-Vanasse J, Williams GJ, Tainer JA. 2015. Envisioning the dynamics and flexibility of Mre11-Rad50-Nbs1 complex to decipher its roles in DNA replication and repair. Prog. Biophys. Mol. Biol 117:182–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hopfner K-P, Karcher A, Shin DS, Craig L, Arthur LM, et al. 2000. Structural biology of Rad50 ATPase. Cell 101:789–800 [DOI] [PubMed] [Google Scholar]
- 31.Cahill D, Carney JP. 2007. Dimerization of the Rad50 protein is independent of the conserved hook domain. Mutagenesis 22:269–74 [DOI] [PubMed] [Google Scholar]
- 32.Hopfner K-P, Craig L, Moncalian G, Zinkel RA, Usui T, et al. 2002. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature 418:562–66 [DOI] [PubMed] [Google Scholar]
- 33.Park YB, Hohl M, Padjasek M, Jeong E, Jin KS, et al. 2017. Eukaryotic Rad50 functions as a rod-shaped dimer. Nat. Struct. Mol. Biol 24:248–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myler LR, Gallardo IF, Soniat MM, Deshpande RA, Gonzalez XB, et al. 2017. Single-molecule imaging reveals how Mre11-Rad50-Nbs1 initiates DNA break repair. Mol. Cell 67:891–98.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams GJ, Williams RS, Williams JS, Moncalian G, Arvai AS, et al. 2011. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat. Struct. Mol. Biol 18:423–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, et al. 2008. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135:97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das D, Moiani D, Axelrod HL, Miller MD, McMullan D, et al. 2010. Crystal structure of the first eubacterial Mre11 nuclease reveals novel features that may discriminate substrates during DNA repair. J. Mol. Biol 397:647–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seifert FU, Lammens K, Stoehr G, Kessler B, Hopfner KP. 2016. Structural mechanism of ATP-dependent DNA binding and DNA end bridging by eukaryotic Rad50. EMBO J 35:759–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seifert FU, Lammens K, Hopfner K-P. 2015. Structure of the catalytic domain of Mre11 from Chaetomium thermophilum. Acta Crystallogr. Sect. F 71:752–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA. 2001. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell 105:473–85 [DOI] [PubMed] [Google Scholar]
- 41.Lee JH. 2004. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304:93–96 [DOI] [PubMed] [Google Scholar]
- 42.Falck J, Coates J, Jackson SP. 2005. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434:605–11 [DOI] [PubMed] [Google Scholar]
- 43.You Z, Chahwan C, Bailis J, Hunter T, Russell P. 2005. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol 25:5363–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sibanda BL, Chirgadze DY, Ascher DB, Blundell TL. 2017. DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science 355:520–24 [DOI] [PubMed] [Google Scholar]
- 45.Stracker TH, Morales M, Couto SS, Hussein H, Petrini JHJ. 2007. The carboxy terminus of NBS1 is required for induction of apoptosis by the MRE11 complex. Nature 447:218–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Difilippantonio S, Celeste A, Kruhlak MJ, Lee Y, Difilippantonio MJ, et al. 2007. Distinct domains in Nbs1 regulate irradiation-induced checkpoints and apoptosis. J. Exp. Med 204:1003–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moiani D, Ronato DA, Brosey CA, Arvai AS, Syed A, et al. 2017. Targeting allostery with avatars to design inhibitors assessed by cell activity: dissecting MRE11 endo- and exonuclease activities. Methods Enzymol 601:205–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davies OR, Forment JV, Sun M, Belotserkovskaya R, Coates J, et al. 2015. CtIP tetramer assembly is required for DNA-end resection and repair. Nat. Struct. Mol. Biol 22:150–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andres SN, Appel CD, Westmoreland JW, Williams JS, Nguyen Y, et al. 2015. Tetrameric Ctp1 coordinates DNA binding and DNA bridging in DNA double-strand-break repair. Nat. Struct. Mol. Biol 22:158–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andres SN, Williams RS. 2017. CtIP/Ctp1/Sae2, molecular form fit for function. DNA Repair 56:109–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lammens K, Bemeleit DJ, Mockel C, Clausing E, Schele A, et al. 2011. The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell 145:54–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deshpande RA, Williams GJ, Limbo O, Williams RS, Kuhnlein J, et al. 2014. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J 33:482– 500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rojowska A, Lammens K, Seifert FU, Direnberger C, Feldmann H, Hopfner KP. 2014. Structure of the Rad50 DNA double-strand break repair protein in complex with DNA. EMBO J 33:2847–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He J, Shi LZ, Truong LN, Lu C-S, Razavian N, et al. 2012. Rad50 zinc hook is important for the Mre11 complex to bind chromosomal DNA double-stranded breaks and initiate various DNA damage responses. J. Biol. Chem 287:31747–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barfoot T, Herdendorf TJ, Behning BR, Stohr BA, Gao Y, et al. 2015. Functional analysis of the bacteriophage T4 Rad50 homolog (gp46) coiled-coil domain. J. Biol. Chem 290:23905–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roset R, Inagaki A, Hohl M, Brenet F, Lafrance-Vanasse J, et al. 2014. The Rad50 hook domain regulates DNA damage signaling and tumorigenesis. Genes Dev 28:451–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hohl M, Kwon Y, Galvan SM, Xue X, Tous C, et al. 2011. The Rad50 coiled-coil domain is indispensable for Mre11 complex functions. Nat. Struct. Mol. Biol 18:1124–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reindl S, Ghosh A, Williams GJ, Lassak K, Neiner T, et al. 2013. Insights into FlaI functions in archaeal motor assembly and motility from structures, conformations, and genetics. Mol. Cell 49:1069–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Acharya SN, Many AM, Schroeder AP, Kennedy FM, Savytskyy OP, et al. 2008. Coprinus cinereus Rad50 mutants reveal an essential structural role for Rad50 in axial element and synaptonemal complex formation, homolog pairing and meiotic recombination. Genetics 180:1889–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Al-Ahmadie H, Iyer G, Hohl M, Asthana S, Inagaki A, et al. 2014. Synthetic lethality in ATM-deficient RAD50-mutant tumors underlies outlier response to cancer therapy. Cancer Discov 4:1014–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao Y, Nelson SW. 2014. Autoinhibition of bacteriophage T4 Mre11 by its C-terminal domain. J. Biol. Chem 289:26505–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsutakawa SE, Lafrance-Vanasse J, Tainer JA. 2014. The cutting edges in DNA repair, licensing, and fidelity: DNA and RNA repair nucleases sculpt DNA to measure twice, cut once. DNA Repair 19:95–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim JH, Grosbart M, Anand R, Wyman C, Cejka P, Petrini JHJ. 2017. The Mre11-Nbs1 interface is essential for viability and tumor suppression. Cell Rep 18:496–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oh J, Al-Zain A, Cannavo E, Cejka P, Symington LS. 2016. Xrs2 dependent and independent functions of the Mre11-Rad50 complex. Mol. Cell 64:405–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crown KN, Savytskyy OP, Malik SB, Logsdon J, Williams RS, et al. 2013. A mutation in the FHA domain of Coprinus cinereus Nbs1 leads to Spo11-independent meiotic recombination and chromosome segregation. G3 3:1927–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Makharashvili N, Paull TT. 2015. CtIP: a DNA damage response protein at the intersection of DNA metabolism. DNA Repair 32:75–81 [DOI] [PubMed] [Google Scholar]
- 67.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, et al. 2007. Human CtIP promotes DNA end resection. Nature 450:509–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anand R, Ranjha L, Cannavo E, Cejka P. 2016. Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol. Cell 64:940–50 [DOI] [PubMed] [Google Scholar]
- 69.Liao S, Tammaro M, Yan H. 2016. The structure of ends determines the pathway choice and Mre11 nuclease dependency of DNA double-strand break repair. Nucleic Acids Res 44:5689–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jensen KL, Russell P. 2016. Ctp1-dependent clipping and resection of DNA double-strand breaks by Mre11 endonuclease complex are not genetically separable. Nucleic Acids Res 44:8241–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Q, Goldstein M, Alexander P, Wakeman TP, Sun T, et al. 2014. Rad17 recruits the MRE11-RAD50-NBS1 complex to regulate the cellular response to DNA double-strand breaks. EMBO J 33:862–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Deshpande RA, Lee J-H, Arora S, Paull TT. 2016. Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein-DNA adducts. Mol. Cell 64:593–606 [DOI] [PubMed] [Google Scholar]
- 73.Cannavo E, Cejka P. 2014. Sae2 promotes dsDNA endonuclease activity within Mre11–Rad50–Xrs2 to resect DNA breaks. Nature 514:122–25 [DOI] [PubMed] [Google Scholar]
- 74.Eisenstein M 2015. The field that came in from the cold. Nat. Methods 13:19–22 [DOI] [PubMed] [Google Scholar]
- 75.Gradia SD, Ishida JP, Tsai M-S, Jeans C, Tainer JA, Fuss JO. 2017. MacroBac: new technologies for robust and efficient large-scale production of recombinant multiprotein complexes. Methods Enzymol 592:1–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Williams GJ, Hammel M, Radhakrishnan SK, Ramsden D, Lees-Miller SP, Tainer JA. 2014. Structural insights into NHEJ: building up an integrated picture of the dynamic DSB repair super complex, one component and interaction at a time. DNA Repair 17:110–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jackson SP, Bartek J. 2009. The DNA-damage response in human biology and disease. Nature 461:1071–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lomax ME, Folkes LK, O’Neill P. 2013. Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin. Oncol 25:578–85 [DOI] [PubMed] [Google Scholar]
- 79.Paull TT. 2015. Mechanisms of ATM activation. Annu. Rev. Biochem 84:711–38 [DOI] [PubMed] [Google Scholar]
- 80.Wu L, Luo K, Lou Z, Chen J. 2008. MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. PNAS 105:11200–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pennisi R, Antoccia A, Leone S, Ascenzi P, di Masi A. 2017. Hsp90αregulates ATM and NBN functions in sensing and repair of DNA double-strand breaks. FEBS J 284:2378–95 [DOI] [PubMed] [Google Scholar]
- 82.von Morgen P, Burdova K, Flower TG, O’Reilly NJ, Boulton SJ, et al. 2017. MRE11 stability is regulated by CK2-dependent interaction with R2TP complex. Oncogene 36:4943–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lu C-S, Truong LN, Aslanian A, Shi LZ, Li Y, et al. 2012. The RING finger protein RNF8 ubiquiti-nates Nbs1 to promote DNA double-strand break repair by homologous recombination. J. Biol. Chem 287:43984–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li Z, Li J, Kong Y, Yan S, Ahmad N, Liu X. 2017. Plk1 phosphorylation of Mre11 antagonizes the DNA damage response. Cancer Res 77:3169–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang T, Penicud K, Bruhn C, Loizou JI, Kanu N, et al. 2012. Competition between NBS1 and ATMIN controls ATM signaling pathway choice. Cell Rep 2:1498–504 [DOI] [PubMed] [Google Scholar]
- 86.Zhang T, Cronshaw J, Kanu N, Snijders AP, Behrens A. 2014. UBR5-mediated ubiquitination of ATMIN is required for ionizing radiation-induced ATM signaling and function. PNAS 111:12091–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Staples CJ, Barone G, Myers KN, Ganesh A, Gibbs-Seymour I, et al. 2016. MRNIP/C5orf45 interacts with the MRN complex and contributes to the DNA damage response. Cell Rep 16:2565–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Abraham RT. 2001. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15:2177–96 [DOI] [PubMed] [Google Scholar]
- 89.Shin DS, Chahwan C, Huffman JL, Tainer JA. 2004. Structure and function of the double-strand break repair machinery. DNA Repair 3:863–73 [DOI] [PubMed] [Google Scholar]
- 90.Mahaney BL, Meek K, Lees-Miller SP. 2009. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J 417:639–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sfeir A, Symington LS. 2015. Microhomology-mediated end joining: A back-up survival mechanism or dedicated pathway? Trends Biochem. Sci 40:701–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Biehs R, Steinlage M, Barton O, Juhász S, Künzel J, et al. 2017. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol. Cell 65:671–84.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dutta A, Eckelmann B, Adhikari S, Ahmed KM, Sengupta S, et al. 2016. Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic Acids Res 45:2585–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC. 2015. Homology and enzymatic requirements of microhomology-dependent alternative end joining. Cell Death Dis 6:e1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shamanna RA, Lu H, de Freitas JK, Tian J, Croteau DL, Bohr VA. 2016. WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat. Commun 7:13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Grabarz A, Guirouilh-Barbat J, Barascu A, Pennarun G, Genet D, et al. 2013. A role for BLM in double-strand break repair pathway choice: prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep 5:21–28 [DOI] [PubMed] [Google Scholar]
- 97.Tomimatsu N, Mukherjee B, Deland K, Kurimasa A, Bolderson E, et al. 2012. Exo1 plays a major role in DNA end resection in humans and influences double-strand break repair and damage signaling decisions. DNA Repair 11:441–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tomimatsu N, Mukherjee B, Hardebeck MC, Ilcheva M, Camacho CV, et al. 2014. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat. Commun 5:3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Myler LR, Gallardo IF, Zhou Y, Gong F, Yang S-H, et al. 2016. Single-molecule imaging reveals the mechanism of Exo1 regulation by single-stranded DNA binding proteins. PNAS 113:E1170–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Genschel J, Modrich P. 2003. Mechanism of 5΄-directed excision in human mismatch repair. Mol. Cell 12:1077–86 [DOI] [PubMed] [Google Scholar]
- 101.Lu H, Shamanna RA, Keijzers G, Anand R, Rasmussen LJ, et al. 2016. RECQL4 promotes DNA end resection in repair of DNA double-strand breaks. Cell Rep 16:161–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Broderick R, Nieminuszczy J, Baddock HT, Deshpande RA, Gileadi O, et al. 2016. EXD2 promotes homologous recombination by facilitating DNA end resection. Nat. Cell Biol 18:271–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Silva J, Aivio S, Knobel PA, Bailey LJ, Casali A, et al. 2018. EXD2 governs germ stem cell homeostasis and lifespan by promoting mitoribosome integrity and translation. Nat. Cell Biol 20:162–74 [DOI] [PubMed] [Google Scholar]
- 104.Puddu F, Oelschlaegel T, Guerini I, Geisler NJ, Niu H, et al. 2015. Synthetic viability genomic screening defines Sae2 function in DNA repair. EMBO J 34:1509–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen H, Donnianni RA, Handa N, Deng SK, Oh J, et al. 2015. Sae2 promotes DNA damage resistance by removing the Mre11–Rad50–Xrs2 complex from DNA and attenuating Rad53 signaling. PNAS 112:E1880–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nitiss JL, Ferrari M, Dibitetto D, De Gregorio G, Eapen VV, et al. 2015. Functional interplay between the 53BP1-ortholog Rad9 and the Mre11 complex regulates resection, end-tethering and repair of a double-strand break. PLOS Genet 11:e1004928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bunting SF, Callen E, Wong N, Chen H-T, Polato F, et al. 2010. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141:243–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 2010. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med 207:855–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, et al. 2010. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol 17:688–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Daley JM, Sung P. 2014. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell. Biol 34:1380–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Thompson LH. 2012. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat. Res 751:158–246 [DOI] [PubMed] [Google Scholar]
- 112.Prakash R, Zhang Y, Feng W, Jasin M. 2015. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol 7:a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Symington LS. 2016. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol 51:195–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhao W, Steinfeld JB, Liang F, Chen X, Maranon DG, et al. 2017. BRCA1–BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 550:360–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Takeda S, Hoa NN, Sasanuma H. 2016. The role of the Mre11–Rad50–Nbs1 complex in double-strand break repair—facts and myths. J. Radiat. Res 57:i25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Branzei D, Foiani M. 2008. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol 9:297–308 [DOI] [PubMed] [Google Scholar]
- 117.Aceytuno RD, Piett CG, Havali-Shahriari Z, Edwards RA, Rey M, et al. 2017. Structural and functional characterization of the PNKP–XRCC4–LigIV DNA repair complex. Nucleic Acids Res 45:6238–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Aparicio T, Baer R, Gottesman M, Gautier J. 2016. MRN, CtIP, and BRCA1 mediate repair of topo-isomerase II–DNA adducts. J. Cell Biol 212:399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee KC, Padget K, Curtis H, Cowell IG, Moiani D, et al. 2012. MRE11 facilitates the removal of human topoisomerase II complexes from genomic DNA. Biol. Open 1:863–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang H, Li Y, Truong LN, Shi LZ, Hwang PY-H, et al. 2014. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol. Cell 54:1012–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zeman MK, Cimprich KA. 2014. Causes and consequences of replication stress. Nat. Cell Biol 16:2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Neelsen KJ, Lopes M. 2015. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol 16:207–20 [DOI] [PubMed] [Google Scholar]
- 123.Aze A, Zhou JC, Costa A, Costanzo V. 2013. DNA replication and homologous recombination factors: acting together to maintain genome stability. Chromosoma 122:401–13 [DOI] [PubMed] [Google Scholar]
- 124.Carr AM, Lambert S. 2013. Replication stress-induced genome instability: the dark side of replication maintenance by homologous recombination. J. Mol. Biol 425:4733–44 [DOI] [PubMed] [Google Scholar]
- 125.Kawabata T, Luebben SW, Yamaguchi S, Ilves I, Matise I, et al. 2011. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol. Cell 41:543–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Woodward AM, Göhler T, Luciani MG, Oehlmann M, Ge X, et al. 2006. Excess Mcm2–7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol 173:673–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ge XQ, Jackson DA, Blow JJ. 2007. Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Genes Dev 21:3331–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Blow JJ, Ge XQ, Jackson DA. 2011. How dormant origins promote complete genome replication. Trends Biochem. Sci 36:405–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xu X, Vaithiyalingam S, Glick GG, Mordes DA, Chazin WJ, Cortez D. 2008. The basic cleft of RPA70N binds multiple checkpoint proteins, including RAD9, to regulate ATR signaling. Mol. Cell. Biol 28:7345–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fanning E 2006. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res 34:4126–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bolderson E, Petermann E, Croft L, Suraweera A, Pandita RK, et al. 2014. Human single-stranded DNA binding protein 1 (hSSB1/NABP2) is required for the stability and repair of stalled replication forks. Nucleic Acids Res 42:6326–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Deng SK, Chen H, Symington LS. 2015. Replication protein A prevents promiscuous annealing between short sequence homologies: implications for genome integrity. BioEssays 37:305–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Oakley GG, Tillison K, Opiyo SA, Glanzer JG, Horn JM, Patrick SM. 2009. Physical interaction between replication protein A (RPA) and MRN: involvement of RPA2 phosphorylation and the N-terminus of RPA1. Biochemistry 48:7473–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cimprich KA, Cortez D. 2008. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol 9:616–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Duursma AM, Driscoll R, Elias JE, Cimprich KA. 2013. A role for the MRN complex in ATR activation via TOPBP1 recruitment. Mol. Cell 50:116–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee J, Dunphy WG. 2013. The Mre11-Rad50-Nbs1 (MRN) complex has a specific role in the activation of Chk1 in response to stalled replication forks. Mol. Biol. Cell 24:1343–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Myers JS, Cortez D. 2006. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J. Biol. Chem 281:9346–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GCM, et al. 2005. ATM-and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol 8:37–45 [DOI] [PubMed] [Google Scholar]
- 139.Kobayashi M, Hayashi N, Takata M, Yamamoto K-i. 2013. NBS1 directly activates ATR independently of MRE11 and TOPBP1. Genes Cells 18:238–46 [DOI] [PubMed] [Google Scholar]
- 140.Shiotani B, Nguyen Hai D, Håkansson P, Marechal A, Tse A, et al. 2013. Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep . 3:1651–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Seeber A, Hegnauer AM, Hustedt N, Deshpande I, Poli J, et al. 2016. RPA mediates recruitment of MRX to forks and double-strand breaks to hold sister chromatids together. Mol. Cell 64:951–66 [DOI] [PubMed] [Google Scholar]
- 142.Gatei M, Kijas AW, Biard D, Dörk T, Lavin MF. 2014. RAD50 phosphorylation promotes ATR down-stream signaling and DNA restart following replication stress. Hum. Mol. Genet 23:4232–48 [DOI] [PubMed] [Google Scholar]
- 143.Tittel-Elmer M, Lengronne A, Davidson MB, Bacal J, Francois P, et al. 2012. Cohesin association to replication sites depends on Rad50 and promotes fork restart. Mol. Cell 48:98–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Palermo V, Rinalducci S, Sanchez M, Grillini F, Sommers JA, et al. 2016. CDK1 phosphorylates WRN at collapsed replication forks. Nat. Commun 7:12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hashimoto Y, Chaudhuri AR, Lopes M, Costanzo V. 2010. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol 17:1305–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vallerga MB, Mansilla SF, Federico MB, Bertolin AP, Gottifredi V. 2015. Rad51 recombinase prevents Mre11 nuclease-dependent degradation and excessive PrimPol-mediated elongation of nascent DNA after UV irradiation. PNAS 112:E6624–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. 2011. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145:529–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schlacher K, Wu H, Jasin M. 2012. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22:106–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kim TM, Son MY, Dodds S, Hu L, Luo G, Hasty P. 2014. RECQL5 and BLM exhibit divergent functions in cells defective for the Fanconi anemia pathway. Nucleic Acids Res 43:893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Huh MS, Ivanochko D, Hashem LE, Curtin M, Delorme M, et al. 2016. Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis 7:e2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, et al. 2017. Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol. Cell 67:867–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pefani DE, O’Neill E. 2015. Safeguarding genome stability: RASSF1A tumor suppressor regulates BRCA2 at stalled forks. Cell Cycle 14:1624–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Leuzzi G, Marabitti V, Pichierri P, Franchitto A. 2016. WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. EMBO J 35:1437–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Trego KS, Chernikova SB, Davalos AR, Perry JJP, Finger LD, et al. 2014. The DNA repair endonuclease XPG interacts directly and functionally with the WRN helicase defective in Werner syndrome. Cell Cycle 10:1998–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Perry JJP, Asaithamby A, Barnebey A, Kiamanesch F, Chen DJ, et al. 2010. Identification of a coiled coil in Werner syndrome protein that facilitates multimerization and promotes exonuclease processivity. J. Biol. Chem 285:25699–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Perry JJP, Yannone SM, Holden LG, Hitomi C, Asaithamby A, et al. 2006. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat. Struct. Mol. Biol 13:414–22 [DOI] [PubMed] [Google Scholar]
- 157.Iannascoli C, Palermo V, Murfuni I, Franchitto A, Pichierri P. 2015. The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res 43:9788–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chaudhuri AR, Callen E, Ding X, Gogola E, Duarte AA, et al. 2016. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535:382–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Roy S, Tomaszowski K-H, Luzwick JW, Park S, Li J, et al. 2018. p53 orchestrates DNA replication restart homeostasis by suppressing mutagenic RAD52 and POLθ pathways. eLife 7:e31723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhu X-D, Küster B, Mann M, Petrini JHJ, de Lange T 2000. Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat. Genet 25:347–52 [DOI] [PubMed] [Google Scholar]
- 161.O’Sullivan RJ, Karlseder J. 2010. Telomeres: protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol 11:171–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Levy MZ, Allsopp RC, Futcher AB, Greider CW, Harley CB. 1992. Telomere end-replication problem and cell aging. J. Mol. Biol 225:951–60 [DOI] [PubMed] [Google Scholar]
- 163.Wu P, Takai H, de Lange T. 2012. Telomeric 3΄ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell 150:39–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shippen-Lentz D, Blackburn EH. 1990. Functional evidence for an RNA template in telomerase. Science 247:546–52 [DOI] [PubMed] [Google Scholar]
- 165.Hastie ND, Dempster M, Dunlop MG, Thompson AM, Green DK, Allshire RC. 1990. Telomere reduction in human colorectal carcinoma and with ageing. Nature 346:866–68 [DOI] [PubMed] [Google Scholar]
- 166.Harley CB, Futcher AB, Greider CW. 1990. Telomeres shorten during ageing of human fibroblasts. Nature 345:458–60 [DOI] [PubMed] [Google Scholar]
- 167.Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, et al. 1992. Telomere length predicts replicative capacity of human fibroblasts. PNAS 89:10114–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Takata H, Tanaka Y, Matsuura A. 2005. Late S phase-specific recruitment of Mre11 complex triggers hierarchical assembly of telomere replication proteins in Saccharomyces cerevisiae. Mol. Cell 17:573–83 [DOI] [PubMed] [Google Scholar]
- 169.Rivera T, Haggblom C, Cosconati S, Karlseder J. 2016. A balance between elongation and trimming regulates telomere stability in stem cells. Nat. Struct. Mol. Biol 24:30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wu Z, Liu J, Zhang Q-D, Lv D-K, Wu N-F, Zhou J-Q. 2017. Rad6–Bre1-mediated H2B ubiquitination regulates telomere replication by promoting telomere-end resection. Nucleic Acids Res 45:3308–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Marcand S 2014. How do telomeres and NHEJ coexist? Mol. Cell. Oncol 1:e963438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Marcomini I, Gasser SM. 2015. Nuclear organization in DNA end processing: telomeres versus double-strand breaks. DNA Repair 32:134–40 [DOI] [PubMed] [Google Scholar]
- 173.Williamson JR, Raghuraman MK, Cech TR. 1989. Monovalent cation-induced structure of telomeric DNA: the G-quartet model. Cell 59:871–80 [DOI] [PubMed] [Google Scholar]
- 174.Higa M, Fujita M, Yoshida K. 2017. DNA replication origins and fork progression at mammalian telomeres. Genes 8:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Denchi EL. 2009. Give me a break: How telomeres suppress the DNA damage response. DNA Repair 8:1118–26 [DOI] [PubMed] [Google Scholar]
- 176.Palm W, de Lange T. 2008. How shelterin protects mammalian telomeres. Annu. Rev. Genet 42:301–34 [DOI] [PubMed] [Google Scholar]
- 177.Denchi EL, de Lange T. 2007. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448:1068–71 [DOI] [PubMed] [Google Scholar]
- 178.Déjardin J, Kingston RE 2009. Purification of proteins associated with specific genomic loci. Cell 136:175–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rai R, Hu C, Broton C, Chen Y, Lei M, Chang S. 2017. NBS1 phosphorylation status dictates repair choice of dysfunctional telomeres. Mol. Cell 65:801–17.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cesare AJ, Reddel RR. 2010. Alternative lengthening of telomeres: models, mechanisms and implications. Nat. Rev. Genet 11:319–30 [DOI] [PubMed] [Google Scholar]
- 181.Shay JW, Reddel RR, Wright WE. 2012. Cancer and telomeres–an ALTernative to telomerase. Science 336:1388–90 [DOI] [PubMed] [Google Scholar]
- 182.Xue Y, Marvin ME, Ivanova IG, Lydall D, Louis EJ, Maringele L. 2016. Rif1 and Exo1 regulate the genomic instability following telomere losses. Aging Cell 15:553–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ballew BJ, Lundblad V. 2013. Multiple genetic pathways regulate replicative senescence in telomerase-deficient yeast. Aging Cell 12:719–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Durocher D, Cassani C, Gobbini E, Wang W, Niu H, et al. 2016. Tel1 and Rif2 regulate MRX functions in end-tethering and repair of DNA double-strand breaks. PLOS Biol 14:e1002387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Saretzki G, Panero J, Stella F, Schutz N, Fantl DB, Slavutsky I. 2015. Differential expression of non-shelterin genes associated with high telomerase levels and telomere shortening in plasma cell disorders. PLOS ONE 10:e0137972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hoxha M, Fabris S, Agnelli L, Bollati V, Cutrona G, et al. 2014. Relevance of telomere/telomerase system impairment in early stage chronic lymphocytic leukemia. Genes Chromosomes Cancer 53:612–21 [DOI] [PubMed] [Google Scholar]
- 187.Heng J, Zhang F, Guo X, Tang L, Peng L, et al. 2017. Integrated analysis of promoter methylation and expression of telomere related genes in breast cancer. Oncotarget 8:25442–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Attwooll CL, Akpinar M, Petrini JH. 2009. The Mre11 complex and the response to dysfunctional telomeres. Mol. Cell. Biol 29:5540–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Li Y, Shen Y, Hohensinner P, Ju J, Wen Z, et al. 2016. Deficient activity of the nuclease MRE11A induces T cell aging and promotes arthritogenic effector functions in patients with rheumatoid arthritis. Immunity 45:903–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Stracker TH, Carson CT, Weitzman MD. 2002. Adenovirus oncoproteins inactivate the Mre11–Rad50– NBS1 DNA repair complex. Nature 418:348–52 [DOI] [PubMed] [Google Scholar]
- 191.Lilley CE, Schwartz RA, Weitzman MD. 2007. Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol 15:119–26 [DOI] [PubMed] [Google Scholar]
- 192.Polo SE, Jackson SP. 2011. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 25:409–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shah GA, O’Shea CC. 2015. Viral and cellular genomes activate distinct DNA damage responses. Cell 162:987–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Weiden MD, Ginsberg HS. 1994. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. PNAS 91:153–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Boyer J, Rohleder K, Ketner G. 1999. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology 263:307–12 [DOI] [PubMed] [Google Scholar]
- 196.Rawlins DR, Rosenfeld PJ, Wides RJ, Challberg MD, Kelly TJ. 1984. Structure and function of the adenovirus origin of replication. Cell 37:309–19 [DOI] [PubMed] [Google Scholar]
- 197.Bernstein JA, Porter JM, Challberg MD. 1986. Template requirements for in vivo replication of adenovirus DNA. Mol. Cell. Biol 6:2115–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, et al. 2013. DNA damage sensor MRE11 rec-ognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. PNAS 110:2969–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Bhattacharya S, Srinivasan K, Abdisalaam S, Su F, Raj P, et al. 2017. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res 45:4590–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Liu X, Hammel M, He Y, Tainer JA, Jeng US, et al. 2013. Structural insights into the interaction of IL-33 with its receptors. PNAS 110:14918–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Stav-Noraas TE, Edelmann RJ, Poulsen LLC, Sundnes O, Phung D, et al. 2017. Endothelial IL-33 expression is augmented by adenoviral activation of the DNA damage machinery. J. Immunol 198:3318–25 [DOI] [PubMed] [Google Scholar]
- 202.Roth S, Rottach A, Lotz-Havla AS, Laux V, Muschaweckh A, et al. 2014. Rad50-CARD9 interactions link cytosolic DNA sensing to IL-1β production. Nat. Immunol 15:538–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Sohn S-Y, Hearing P. 2012. Adenovirus regulates Sumoylation of Mre11-Rad50-Nbs1 components through a paralog-specific mechanism. J. Virol 86:9656–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bridge E, Ketner G. 1989. Redundant control of adenovirus late gene expression by early region 4. J. Virol 63:631–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Halbert DN, Cutt JR, Shenk T. 1985. Adenovirus early region 4 encodes functions required for efficient DNA replication, late gene expression, and host cell shutoff. J. Virol 56:250–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Huang MM, Hearing P. 1989. Adenovirus early region 4 encodes two gene products with redundant effects in lytic infection. J. Virol 63:2605–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Weinberg DH, Ketner G. 1986. Adenoviral early region 4 is required for efficient viral DNA replication and for late gene expression. J. Virol 57:833–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Evans JD, Hearing P. 2005. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J. Virol 79:6207–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ou HD, Kwiatkowski W, Deerinck TJ, Noske A, Blain KY, et al. 2012. A structural basis for the assembly and functions of a viral polymer that inactivates multiple tumor suppressors. Cell 151:304–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sohn SY, Hearing P. 2012. Adenovirus regulates sumoylation of Mre11-Rad50-Nbs1 components through a paralog-specific mechanism. J. Virol 86:9656–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Bridges RG, Sohn SY, Wright J, Leppard KN, Hearing P. 2016. The adenovirus E4-ORF3 protein stimulates SUMOylation of general transcription factor TFII-I to direct proteasomal degradation. mBio 7:e02184–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Harada JN, Shevchenko A, Shevchenko A, Pallas DC, Berk AJ. 2002. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J. Virol 76:9194–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Querido E 2001. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev 15:3104–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.zur Hausen H 2009. Papillomaviruses in the causation of human cancers—a brief historical account. Virology 384:260–65 [DOI] [PubMed] [Google Scholar]
- 215.Johnson BA, Aloor HL, Moody CA. 2017. The Rb binding domain of HPV31 E7 is required to maintain high levels of DNA repair factors in infected cells. Virology 500:22–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Campos-León K, Wijendra K, Siddiqa A, Pentland I, Feeney KM, et al. 2017. Association of human papil-lomavirus 16 E2 with Rad50-interacting protein 1 enhances viral DNA replication. J. Virol 91:e02305–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Robertson ES, Mariggio` G, Koch S, Zhang G, Weidner-Glunde M, et al. 2017. Kaposi Sarcoma Her-pesvirus (KSHV) latency-associated nuclear antigen (LANA) recruits components of the MRN (Mre11-Rad50-NBS1) repair complex to modulate an innate immune signaling pathway and viral latency. PLOS Pathog 13:e1006335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, et al. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266:1865–69 [DOI] [PubMed] [Google Scholar]
- 219.Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, et al. 2009. A review of human carcinogens–Part B: biological agents. Lancet Oncol 10:321–22 [DOI] [PubMed] [Google Scholar]
- 220.Pantelidou C, Cherubini G, Lemoine NR, Halldén G 2016. The E1B19K-deleted oncolytic adenovirus mutant Ad∆19K sensitizes pancreatic cancer cells to drug-induced DNA-damage by down-regulating Claspin and Mre11. Oncotarget 7:15703–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Tookman LA, Browne AK, Connell CM, Bridge G, Ingemarsdotter CK, et al. 2015. RAD51 and BRCA2 enhance oncolytic adenovirus type 5 activity in ovarian cancer. Mol. Cancer Res 14:44–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Crick F 1974. The double helix: a personal view. Nature 248:766–69 [DOI] [PubMed] [Google Scholar]
- 223.Harrison SC. 2007. Comments on the NIGMS PSI. Structure 15:1344–46 [DOI] [PubMed] [Google Scholar]
- 224.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL II, et al. 2009. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat. Methods 6:606–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Rambo RP, Tainer JA. 2011. Characterizing flexible and intrinsically unstructured biological macro-molecules by SAS using the Porod-Debye law. Biopolymers 95:559–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hura GL, Budworth H, Dyer KN, Rambo RP, Hammel M, et al. 2013. Comprehensive macromolecular conformations mapped by quantitative SAXS analyses. Nat. Methods 10:453–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Rambo RP, Tainer JA. 2013. Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496:477–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Rambo RP, Tainer JA. 2013. Super-resolution in solution X-ray scattering and its applications to structural systems biology. Annu. Rev. Biophys 42:415–41 [DOI] [PubMed] [Google Scholar]
- 229.Brosey CA, Ho C, Long WZ, Singh S, Burnett K, et al. 2016. Defining NADH-driven allostery regulating apoptosis-inducing factor. Structure 24:2067–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Bacolla A, Tainer JA, Vasquez KM, Cooper DN. 2016. Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res 44:5673–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tsutakawa SE, Thompson MJ, Arvai AS, Neil AJ, Shaw SJ, et al. 2017. Phosphate steering by Flap Endonuclease 1 promotes 5΄-flap specificity and incision to prevent genome instability. Nat. Commun 8:15855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Rashid F, Harris PD, Zaher MS, Sobhy MA, Joudeh LI, et al. 2017. Single-molecule FRET unveils induced-fit mechanism for substrate selectivity in flap endonuclease 1. eLife 6:e21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Sundheim O, Vågbø CB, Bjørås M, Sousa MML, Talstad V, et al. 2006. Human ABH3 structure and key residues for oxidative demethylation to reverse DNA/RNA damage. EMBO J 25:3389–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Dango S, Mosammaparast N, Sowa ME, Xiong L-J, Wu F, et al. 2011. DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation. Mol. Cell 44:373–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Brosey CA, Ahmed Z, Lees-Miller SP, Tainer JA. 2017. What combined measurements from structures and imaging tell us about DNA damage responses. Methods Enzymol 592:417–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Loeb LA, Loeb KR, Anderson JP. 2003. Multiple mutations and cancer. PNAS 100:776–81 [DOI] [PMC free article] [PubMed] [Google Scholar]