

Abstract
Cerebellar ataxias constitute a heterogeneous group of disorders that result in impaired speech, uncoordinated limb movements, and impaired balance, often ultimately resulting in wheelchair confinement. Motor dysfunction in ataxia can be attributed to dysfunction and degeneration of neurons in the cerebellum and its associated pathways. Recent work has suggested the importance of cerebellar neuronal dysfunction resulting from mutations in specific ion-channels that regulate membrane excitability in the pathogenesis of cerebellar ataxia in humans. Importantly, even in ataxias not directly due to ion-channel mutations, transcriptional changes resulting in ion-channel dysfunction are tied to motor dysfunction and degeneration in models of disease. In this review, we describe the role that ion-channel dysfunction plays in a variety of cerebellar ataxias, and postulate that a potential therapeutic strategy that targets specific ion-channels exists for cerebellar ataxia.
Keywords: Spinocerebellar ataxia, Ion channel, Purkinje neuron, Electrophysiology, Channel activator, Potassium channel
Introduction
Cerebellar ataxias are a large, heterogeneous group of movement disorders affecting neurons in the cerebellum and its associated pathways. Clinically, cerebellar dysfunction manifests as unsteady gait, abnormal eye movements, uncoordinated limb movements, and difficulties in speech. Although many cerebellar ataxias share clinical features, genetic causes are diverse and highlight the potential difficulty to diagnose and appropriately treat these disorders. For instance, there are over 30 known genetic mutations associated with autosomal-dominant spinocerebellar ataxia (SCA) that affect a wide variety of molecular pathways [27]. The recent discovery of several new disease-causing SCA mutations suggests that many undiscovered disease genes still remain [19, 32, 72, 95, 96].
Although disease-causing mutations in cerebellar ataxia are diverse, and expression of disease proteins are often widespread or ubiquitous throughout the central nervous system, cerebellar involvement is prominent. A subset of SCAs result from glutamine-encoding CAG repeats (the so-called polyglutamine SCAs: SCA1, 2, 3, 6, 7, 17). Although polyglutamine-expanded protein expression is widespread, and not necessarily restricted to just the nervous system, degeneration is restricted to specific neurons in the cerebellum and its associated pathways [87]. Purkinje neurons, brainstem neurons, and neurons of the cerebellar nuclei are particularly vulnerable to degeneration. Among these, Purkinje neurons are most prominently involved in SCAs [27]. The increased susceptibility of Purkinje neurons to degeneration in SCA suggests that these neurons may possess unique metabolic or physiologic properties that make them more vulnerable to a variety of insults. A unique feature of Purkinje neurons that may enhance their vulnerability is that they are autonomous pacemaker neurons which sustain firing 40 Hz even in the absence of synaptic input [36, 81]. Perturbations in ion-channel expression and function have the potential to greatly impact Purkinje neuron firing and drive motor impairment.
Growing evidence indicates that neuronal dysfunction is a central mechanism of disease across many etiologies of SCA. Conventional ion-channel mutations are known causes of several spinocerebellar ataxias and episodic ataxias [19, 25, 31, 32, 49, 57, 61, 72, 95, 96, 101, 106], while ion-channel dysfunction secondary to disease-causing mutations have been identified in several mouse models of polyglutamine SCA [21, 22, 54, 91]. This review will summarize current understanding of ion-channel dysfunction in cerebellar ataxia and explore ion-channel modulation as a potential strategy for the treatment of motor dysfunction.
Neuronal dysfunction in cerebellar ataxia
Purkinje neurons receive and integrate signals from several distinct neuronal pathways. Purkinje neuron intrinsic firing is modulated by synaptic activity to modify activity of downstream motor pathways. Under normal conditions, Purkinje neurons can sustain firing at a large dynamic range, up to several hundred spikes per second in vivo [100]. In order to properly transmit motor information, Purkinje neurons must be capable of fast modulation of this firing in order to encode information. There is debate as to whether Purkinje neurons use firing-rate coding, coding through synchronized Purkinje cell activity, or hybrid multiplexed coding to transmit output signals to motor nuclei [20, 37, 40]. Nevertheless, it is clear that rapid and precise modulation of Purkinje neuron membrane potential is necessary to encode coordinated motor output.
Purkinje neuron action potentials are dependent on precise, coordinated activity of a large complement of ion-channels in order to maintain autonomous repetitive spiking. Spontaneous action potentials are driven by resurgent sodium current carried by the voltage-gated sodium channel Nav1.6 [81]. Upon reaching threshold, Nav1.6 and Nav1.1 channels become maximally activated, generating the upstroke of the action potential. The falling phase of the action potential is driven by voltage-gated potassium channels, mostly Kv3 family members [67]. Upon membrane depolarization, voltage-gated calcium channels (mainly Cav2.1 and Cav3 family members) also become activated, allowing external calcium entry into Purkinje neurons [80, 97]. These voltage-gated calcium channels are tightly coupled to calcium-activated potassium channels (KCa channels), so that the net effect of calcium entry is an outward potassium current which hyperpolarizes the membrane potential [107]. The major KCa channels in Purkinje neurons are large-conductance calcium-activated potassium (BK, KCa1.1) channel and the small-conductance calcium-activated potassium (SK) channel (SK2, KCa2.2), which generate the after-hyperpolarization (AHP) [16, 28, 84]. The AHP is essential for de-activation of voltage-gated sodium and potassium channels, which allows for their activation during the subsequent action potential. The depolarization of the membrane potential during the interspike interval, which is necessary for autonomous spiking, is mediated by unique resurgent kinetics of voltage-gated sodium channels [81]. Finally, an assortment of subthreshold-activated potassium channels are active during the interspike interval [1, 10, 30, 53, 56, 79], while other channels such as TRPC3 and the inositol 1,4,5-trisphosphate receptor play important roles mediating calcium homeostasis and the modulation of cerebellar learning [8, 35, 55, 98]. Purkinje neurons spiking is therefore sensitive to perturbations in ion-channels, and mutations in any of these channels can cause motor dysfunction as outlined below.
Several studies have highlighted the importance of Purkinje neuron spike regularity and firing frequency for motor function. Early studies were performed in mutant mice with mutations in Cav2.1, the P/Q-type calcium channel encoded by the Cacna1a gene [24, 64, 103]. Normally, autonomous spiking in Purkinje neurons is very precise, with little variation in the duration of the interspike interval. Strikingly, Purkinje neurons in these mouse models show irregular spiking compared to wild-type controls, as evidenced by an increase in the coefficient of variation of the interspike interval between action potentials [39, 104]. Consistent with a role for calcium entry to regulate KCa channel activity, SK channel activators improve both Purkinje neuron spike regularity and motor performance [33, 104]. Additionally, the spiking of neurons of the deep cerebellar nuclei, which receive input from Purkinje neurons and act as the output of cerebellar motor processing, is also dependent upon KCa activity [90]. This suggests that there is a direct link between ion-channel function, Purkinje neuron spiking, and motor output from the cerebellum, and that pharmacologic agents which target ion-channel dysfunction may have therapeutic potential.
Conventional ion-channel mutations causing SCA
While disease-causing mutations in ataxia are diverse, many affected proteins are in related molecular pathways, suggesting that these pathways may be particularly important for neurons in the cerebellar motor circuit. There is some evidence that ataxia-related genes may cluster around pathways involving calcium homeostasis, synaptic integration, and membrane excitability [9]. Many of these potential ataxia-related genes show enriched expression in cerebellar Purkinje neurons, suggesting that not only are these pathways particularly important for cerebellar function, but also that Purkinje neuron dysfunction likely plays a central role in motor dysfunction in ataxia [9].
Indeed, conventional ion-channel mutations are known to result in ataxia. In general, these channelopathies tend to present as pure cerebellar ataxia as compared to more diverse symptoms in the polyglutamine SCAs [27]. Mouse models have provided valuable insight to the functional implications of disrupted ion-channel function in many of these ataxia-causing channelopathies. These models clearly demonstrate that electrophysiologic dysfunction contribute to motor impairment in ataxia.
In SCA13, point mutations in the KCNC3 gene result in the production of the voltage-gated potassium channel, Kv3.3, with either no functional current or altered kinetics [31, 106, 113]. Kv3.3 knockout mice serve as a mouse model for SCA13. In these mice, the lack of Kv3.3 current reduces the slope of action potential repolarization in cerebellar Purkinje neurons [3, 70]. Since full and efficient repolarization, which is mediated by Kv channels and calcium-activated potassium channels, allows for complete de-activation of voltage-gated sodium channels in preparation of the next action potential, the available resurgent sodium current through Nav1.6 is reduced in these neurons. Purkinje neurons from Kv3.3 knockout mice therefore display reduced firing frequency due to the altered interaction between Kv3.3 and other ion-channels that are active during the interspike interval [3]. Since Purkinje neuron-specific re-expression of Kv3.3 rescues spiking and motor function in global Kv3.3 null mice, Purkinje neuron electrophysiologic dysfunction is a primary source of behavioral impairment in these mice [44, 45]. These studies demonstrate the importance of considering interactions between ion-channels when exploring electrophysiologic phenotypes in mouse models of ataxia.
The ITPR1 locus, which encodes the inositol 1,4,5-trisphosphate (IP3) receptor gene, has emerged as a particularly important target gene site for SCA [98]. Currently, SCA15/16 is known to result from mutations in ITPR1 [49, 57, 101], and the recently identified nonprogressive congenital ataxia, SCA29, also maps to the ITPR1 locus [6, 43, 112]. Traditionally, ITPR1 mutations are associated with loss-of-function of the IP3 receptor. However, as has been demonstrated in at least one case, ITPR1 mutations can also cause cerebellar ataxia due to enhanced calcium release upon IP3 binding [12]. IP3 and diacylglycerol are produced upon postsynaptic metabotropic glutamate receptor (mGluR) activation, and IP3 subsequently binds to the IP3 receptor and promotes calcium release from internal stores [8]. In mice, full knockout of Itpr1 is fatal after postnatal day 23 [68] but before this time, cerebellar Purkinje neurons show a complete inability to produce synaptic long-term depression [47]. Additionally, Itpr1 heterozygous mice exhibit motor impairment on the rotarod [75], as do mGluR1 knockout mice [2], suggesting that synaptic dysregulation which occurs upon altered IP3 receptor function contributes to cerebellar ataxia.
In humans, mutations in the KCND3 gene, which encodes the A-type potassium channel Kv4.3, result in SCA19/22 [25, 61]. In heterologous cells, mutations in KCND3 tend to impair stability of Kv4.3-containing protein complexes, reduce Kv4.3 channel expression at the cell surface, and impair current density of these channels [25, 26, 61, 94]. Although Kv4.3-mutant mice have not been generated, it is probable that deletion of Kv4.3 would disrupt cerebellar processing. It is unclear how prominent a role Kv4.3 plays in Purkinje neuron function, and conflicting reports exist about the expression of Kv4.3 in adult Purkinje cells [42, 89]. However, Kv4.3 is observed in other adult cerebellar neuronal populations, including granule cells and molecular layer interneurons, suggesting that pre-synaptic alterations in cerebellar processing may disrupt Purkinje neuron integration and output from the cerebellar cortex [42]. Notably, Purkinje neuron degeneration in SCA19/22 patients suggests that these channels are indeed important for Purkinje neuron function [25].
Although ataxia is not the primary feature of disease, mutations in voltage-gated sodium (Nav) channels can result in ataxia in both humans and mouse models. In humans, mutations in SCN1A, the gene encoding Nav1.1, result in Dravet syndrome, a form of Severe Myoclonic Epilepsy in Infancy (SMEI) which can be accompanied by cerebellar ataxia [17]. In mice, Nav1.1 knockout causes ataxia associated with reduced Purkinje neuron firing frequency [52]. In addition to mutations in Nav channel pore-forming subunits, mutations in interactor proteins also contribute to Purkinje neuron dysfunction and ataxia. In humans, mutations in the FGF14 gene, encoding the fibroblast growth factor 14, result in SCA27 [102]. FGF14 has been shown to interact with voltage-gated sodium channels to modulate neuronal excitability [65], and disruptions in FGF14 expression cause ataxia in mice [105]. In FGF14−/− mice, Purkinje neuron spontaneous firing is greatly disrupted, with 80% of neurons appearing silent. Additionally, Nav 1.6 expression is reduced in Purkinje neurons of FGF14−/− mice, suggesting that this interaction is necessary for normal membrane expression [92]. The FGF14F145S mutation reduces Nav1.6 expression at the axon initial segment, and reduces sodium currents in hippocampal neurons, leading to early depolarization block upon current injections [58]. A recent study illustrates that FGF14 directly modulates resurgent sodium current mediated by Nav1.6 through interactions with the FGF14 N-terminus [109], suggesting that this function may be impaired and thereby drive a lack of spontaneous firing in FGF14−/− mice. Finally, there is evidence that FGF14 mutations may contribute to presynaptic changes that affect Purkinje neuron function. In granule cells, FGF14 mutations act in a dominant negative manner to suppress both sodium and calcium currents [58, 108]. AMPA receptor-mediated excitatory postsynaptic potentials (EPSCs) were reduced in Purkinje neurons following parallel fiber stimulation, suggesting that FGF14 mutations contribute to impaired neurotransmission from cerebellar granule cells [99]. Together, these data suggest that the FGF14-Nav interaction may be important in cerebellar neurons to modulate both intrinsic excitability and presynaptic activity through its interaction with voltage-gated sodium channels.
Mutations in the transient receptor potential channel type C3 (TRPC3) contribute to cerebellar ataxia in both mice and humans (SCA41). TRPC3 signaling is essential for mGluR1-mediated synaptic transmission and contributes to the induction of long-term depression in Purkinje neurons [35, 55]. A point mutation in the TRP domain of TRPC3, p.Arg762His, was identified in a patient with cerebellar ataxia and mild atrophy of the cerebellar vermis [32]. In moonwalker mice, a point mutation in Trpc3 results in motor impairment and progressive Purkinje neuron loss [7]. Notably, Purkinje neuron firing is markedly abnormal in moonwalker mice, with depolarization block of Purkinje neuron spiking [88]. Trpc3 activity may also play a normal role in regulating Purkinje neuron intrinsic firing frequency, particularly in the anterior cerebellum [114]. It is therefore possible that in addition to its roles in synaptic regulation, TRPC3 directly regulated Purkinje neuron firing frequency.
Recently, a point mutation in the CACNA1G gene was identified as the causative mutation in SCA42 [19, 72]. This mutation, p.Arg1715His, is located in the voltage-sensing S4 domain of the T-type calcium channel, Cav3.1 [19, 72]. When cloned into HEK293 cells, the p.Arg1715His mutation does not affect maximum channel conductance but does shift Cav3.1 activation to more positive membrane potentials [19, 72]. In mice, T-type calcium channel blockade reduces Purkinje neuron spike frequency in vitro, while mice lacking Cav3.1 in several brain regions, including the cerebellum, show increased Purkinje neuron spike frequency and irregularity in vivo [66, 77]. Synaptic dysfunction and the resulting impairment of motor learning is prominent in Cav3.1−/− mice, as these mice demonstrate an impaired ability to produce long-term potentiation at the parallel fiber-Purkinje neuron synapse, impaired performance on a rotarod, and impairments in the vestibulo-ocular reflex [66]. It is possible that reduced Cav3.1 activity may also reduce calcium availability for calcium-activated potassium channels, thereby impairing the generation of a normal spike after-hyperpolarization (AHP) and reducing spike regularity. However, the contribution of Cav3.1 as a calcium source for calcium-activated potassium channels remains controversial [107].
A recently discovered ion-channel mutation in the KCNMA1 gene, which encodes the BK channel, produces ataxia with cerebellar atrophy. In a heterologous cell expression system, G354S KCNMA1 greatly reduces macroscopic BK currents and acts in a dominant-negative fashion [95, 96]. Interestingly, both loss-of-function and gain-of-function BK channel mutations can cause epilepsy [73], indicating a narrow tolerable range of expression for BK channels. BK channel-null (BK−/−) mice display increased Purkinje neuron membrane excitability and exhibit motor impairment [84]. In global BK knockout mice, Purkinje neuron spontaneous firing is markedly impaired. A majority of Purkinje neurons from BK−/− mice are silent or fire in a burst pattern, while the remaining spontaneously firing cells do so at a greatly reduced frequency, in association with loss of the BK channel-dependent fast AHP [84]. Additionally, Purkinje neuron-specific deletion of BK channels also causes cerebellar ataxia, demonstrating the importance of BK channels for normal cerebellar physiology and motor function [13]. Together, this work highlights the importance of KCa channels in the maintenance of tonic firing in Purkinje neurons.
Ion-channel dysfunction in polyglutamine SCA
Although conventional channelopathies present a clear role for altered neuronal membrane excitability in ataxia, these forms of ataxia are less common and are estimated to be responsible for around ten percent of all cases of SCA [27]. Much more common are the polyglutamine SCAs, which result from expanded glutamine-encoding CAG repeat sequence in their respective causative genes. Apart from SCA6, which affects the α-subunit of the Cav2.1 voltage-gated calcium channel encoded by the CACNA1A gene, the disease-causing proteins in polyglutamine SCA are not directly associated with ion-channel function. ATXN1 (the disease protein in SCA1) is associated with transcriptional regulation and RNA splicing [48, 60, 111], ATXN2 (SCA2) plays a role in RNA metabolism [69, 74, 93], ATXN3 (SCA3) is de-ubiquitinating enzyme [18, 62], ATXN7 (SCA7) is a member of the SAGA transcriptional complex [38], and TBP (SCA17) is an essential component of tata box-based transcriptional initiation [110] (reviewed in [76]). These related functional roles suggest that transcriptional disruption may be an important initiating event in the polyglutamine SCAs.
Indeed, transcriptional disruption has been noted in mouse models of SCA. Gene expression analyses such as RNA sequencing and gene co-expression network analyses have been useful for the identification of molecular pathways which may be disrupted in SCA [9, 46, 78]. Interestingly, several genes show common downregulation of their mRNA transcripts in multiple SCA mouse models. These include several members of neuronal excitability pathways, including key ion-channels for Purkinje neuron function [9, 21, 22, 34, 46, 78]. Recent work has demonstrated that altered ion-channel expression in SCA can disrupt Purkinje neuron membrane excitability, and mouse models of polyglutamine SCA suggest that ion-channel modulators may represent a therapeutic strategy for both motor dysfunction and neurodegeneration.
In a mouse model of SCA1, disrupted Purkinje neuron membrane excitability is associated with reduced expression and function of two potassium channels, BK and the G-protein coupled inwardly-rectifying potassium (GIRK1) channel [22]. Functionally, Purkinje neurons from ATXN1[82Q] mice demonstrate a depolarized somatic membrane potential and a reduced fast afterhyperpolarization (AHP) amplitude early in disease, leading to a large proportion of non-firing cells. As disease progresses, dendritic degeneration reduces the size of ATXN1[82Q] Purkinje neurons, thereby increasing the current density of remaining BK and GIRK1 channels to restore spontaneous firing, although at a reduced frequency [22]. This suggests that neuronal remodeling during the degenerative process may actually modulate intrinsic excitability, and that dendritic degeneration may be a compensatory process to restore Purkinje neuron spiking. Interestingly, through a parallel process, loss of these channels results in a persistent increase in dendritic membrane excitability even in the presence of dendritic degeneration. Molecular pathways which influence dendritic excitability, such as protein kinase C activity, may act as targets for intervention in SCA [116]. Reducing dendritic hyperexcitability partially improves dendrite loss in ATXN1[82Q] mice [15]. A recent study has demonstrated that a combination of chlorzoxazone and baclofen, two potassium channel-activating drugs, improves both aberrant Purkinje neuron spiking and dendritic hyperexcitability in ATXN1[82Q] mice, thereby providing lasting improvements in motor dysfunction [117]. This combination of drugs is tolerated in human SCA patients and may improve symptoms [117]. A separate study has demonstrated that alterations in Purkinje neuron spiking can be corrected by aminopyridines [41], compounds which non-selectively block voltage-gated potassium channels and which have been previously shown to indirectly activate KCa channels [4]. Aminopyridines also improve motor performance in ATXN1[82Q] mice [41]. Together, these studies highlight a role for potassium channels in maintaining normal physiology in both the soma and dendrites of Purkinje neurons, and identify these channels as potential therapeutic targets.
Recent work in a mouse model of SCA2 has indicated that here too, altered potassium channel function underlies firing abnormalities. ATXN2[127Q] mice display motor impairment and dendritic degeneration, long preceding overt Purkinje neuron loss [34]. In addition, Purkinje neurons from these mice show progressive reductions in firing frequency with no change in spike regularity [21, 34]. These changes in firing are accompanied by a progressive reduction in the transcripts for Kcnma1 (encoding the BK channel) and Kcnc3 (encoding the voltage-gated potassium channel Kv3.3) which are important for Purkinje neuron repetitive spiking [21, 34]. Similar to ATXN1[82Q] mice (SCA1), early in disease, a significant fraction of ATXN2[127Q] Purkinje neurons display an absence of repetitive spiking in association with reduced BK and Kv3 channel function [21]. Later in disease, repetitive spiking is restored through the generation of a novel AHP likely mediated through subthreshold-activated potassium channels which can compensate for the loss of BK- and Kv3.3-mediated repolarizing currents during the interspike interval [21]. In the ATXN2[58Q] transgenic model of SCA2, where there is no prominent dendritic degeneration, aberrant Purkinje neuron bursting is seen both in vitro and in vivo [29, 54]. SK channel activators improve Purkinje neuron firing properties, and improve motor dysfunction in ATXN2[58Q] mice [54]. Additionally, a direct interaction between ATXN2 and the inositol 1,4,5-trisphosphate (IP3) receptor results in abnormal calcium signaling in ATXN2[58Q] mice which can be improved by treatment with dantrolene, a ryanodine receptor inhibitor [63]. Dantrolene also improves motor impairment in ATXN2[58Q] mice [63], suggesting that normalizing calcium signaling may either directly reduce calcium-mediated excitotoxicity or may improve the function of KCa channels to improve Purkinje neuron pacemaking. Overall, these studies indicate a clear role for potassium channel dysfunction which impairs Purkinje neuron spiking and thereby contributes to motor impairment in SCA2.
SCA3 is the most common dominantly inherited ataxia, and is caused by an expanded CAG repeat sequence in the ATXN3 gene [85]. Although SCA3 displays prominent involvement of neurons in the cerebellar nuclei in addition to extracerebellar involvement, Purkinje neuron pathology is sometimes a prominent feature of disease [87]. In the ATXN3[84Q] transgenic mouse model, changes in Purkinje neuron physiology accompany motor impairment [91]. Purkinje neurons from these mice display altered spiking in association with increased inactivation of Kv1 potassium channels [91]. The SK channel-activating compound SKA-31 improves spiking in ATXN3[84Q] Purkinje neurons and also improves motor performance, indicating that potassium channel dysfunction can be targeted pharmacologically in these mice [91]. Similar to ATXN2[58Q] mice, abnormal calcium signaling has been noted in ATXN3[84Q] mice. ATXN3 directly interacts with the IP3 receptor to increase calcium release events [14]. Inhibition of intracellular calcium release through dantrolene also improves motor performance and reduces Purkinje neuron degeneration [14], suggesting that a common disease mechanism may contribute to altered calcium homeostasis across mouse models of SCA2 and SCA3.
SCA6 results from an expanded CAG repeat in the CACNA1A gene which encodes the voltage-gated calcium channel Cav2.1 [115]. In homozygous SCA684Q/84Q knock-in mice, Purkinje neurons show increased spike irregularity and a reduction in firing frequency early in disease [50]. The compound 4-aminopyridine (4-AP), a potassium channel blocker which also indirectly activates KCa channels [5], restores spike regularity to SCA684Q/84Q Purkinje neurons both in vitro and in vivo [50]. Interestingly, chronic treatment with 4-AP improves motor function in SCA684Q/84Q mice [50]. These data suggest that Purkinje neuron spiking abnormalities are present in a mouse model of SCA6, and that these alterations in spiking may be targeted by potassium channel modulators.
These studies in SCA1, SCA2, SCA3, and SCA6 highlight a role for potassium channel dysfunction in altered Purkinje neuron physiology in ataxia. It is important to recognize, however, that alterations in different ion-channels can produce similar alterations in Purkinje neuron firing. It is therefore important to understand the specific ion-channel changes that underlie altered spiking in ataxia. Overall, activating calcium-activated potassium channels appears to correct altered spiking resulting from a variety of different etiologies, and represents a therapeutic target that is shared across multiple forms of ataxia.
Therapeutics based on ion-channel modulation
Designing effective therapies for SCA has proven difficult. Although most SCAs share clinical features, the underlying genetic mutations are diverse and in some cases remain unknown. Recent work has demonstrated the therapeutic potential of gene silencing therapies for ataxia. Among the most promising of these therapies are the antisense oligonucleotide (ASO)-based strategies in the polyglutamine SCAs. In mouse models of SCA2 and SCA3 [71, 86], ASOs have been shown to reduce expression of the respective disease-causing proteins, along with providing lasting improvements in motor performance in SCA2 mice [71, 86]. Additionally, ASO treatment improves firing abnormalities in two mouse models of SCA2, suggesting that transcriptional changes in ion-channels may be improved upon ASO treatment [86]. Although ASOs offer an exciting avenue of treatment for the polyglutamine SCAs, these therapies will likely offer limited therapeutic benefit SCAs in which disease-causing mutations are not autosomal dominant gain-of-function mutations, or are in cellular pathways where knocking down mutant protein is deleterious. In these cases, a more appropriate approach to therapy may be to identify shared features of disease which are observed across many etiologies of SCA. Emerging evidence presented in this review suggests that electrophysiologic dysfunction may be a shared feature of many SCAs.
Recent clinical trials with riluzole for the treatment of SCA suggest that shared features of neuronal dysfunction exist in human disease [82, 83]. While riluzole has several ion-channel targets, it is a known activator of KCa channels [11, 23]. KCa channel activators demonstrate therapeutic potential in the treatment of SCAs [33, 54, 59, 91]. A larger clinical trial with a pro-drug of riluzole is ongoing (ClinicalTrials.gov Identifier: NCT02960893). While yet preliminary, these trials suggest the promise of ion-channel modulators for the treatment of SCA. Future research should focus on the design of other ion-channel modulators with increased specificity and potency to correct symptoms that result from neuronal dysfunction.
Concluding remarks
The spinocerebellar ataxias are a large, diverse family of neurodegenerative disorders affecting the function of cerebellar pathways. Information to the cerebellum ultimately depends on proper coding by many different neuronal populations, all converging on cerebellar Purkinje neurons. Diverse ion-channel mutations result in cerebellar ataxia. In the more common polyglutamine ataxias, changes in ion-channel transcript levels result in altered ion-channel function. Recent work in rodent models of ataxia has highlighted the connection between Purkinje neuron dysfunction and motor impairment, suggesting that ion-channel modulation may be a promising therapeutic strategy for many forms of cerebellar ataxia.
Figure 1. Ion-channel dysfunction is associated with spinocerebellar ataxia in humans and rodent models.
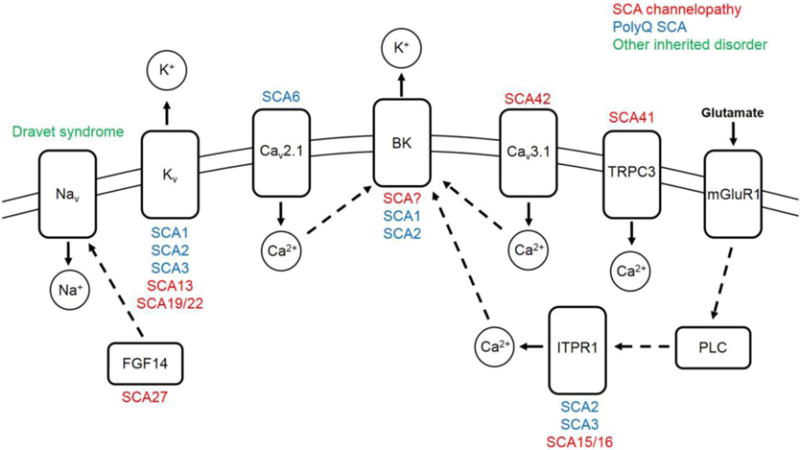
Ion-channels, which are displayed in the cell membrane, and other ion-channel associated proteins causing spinocerebellar ataxia in humans or rodent models of disease, are shown. SCAs associated with each protein are listed above or under each protein. Mutations which result in an SCA channelopathy are listed in red. Ion-channel dysfunction in mouse models of polyQ SCA are listed in blue. Dravet syndrome, a severe myoclonic epilepsy of infancy which can result in ataxia, is shown in green. Dashed arrows signify a protein-protein interaction. Solid arrows signify the direction of ion movement upon channel activation. Abbreviations: SCA, spinocerebellar ataxia; polyQ, polyglutamine; Nav, voltage-gated sodium channel; Kv, voltage-gated potassium channel; Cav, voltage-gated calcium channel; BK, large conductance calcium-activated potassium channel; TRPC3, transient receptor potential cation channel type 3; mGluR1, metabotropic glutamate receptor type 1; FGF14, fibroblast growth factor 14; ITPR1, inositol 1,4,5 trisphosphate receptor type 1; PLC, phospholipase C; Na+, sodium ion; K+, potassium ion; Ca2+, calcium ion.
Table 1. Ion-channel mutations resulting in spinocerebellar ataxia.
Known SCA channelopathies are listed. The associated gene is listed for each SCA, along with the known functional roles of each ion-channel or protein.
| Gene | Associated ataxia or inherited disorder | Encoded channel or protein | Normal function |
|---|---|---|---|
| CACNA1A | SCA6 [115], Episodic ataxia type 2 [51] | Cav2.1 Voltage-gated calcium channel, pore-forming subunit |
Inward calcium current (P/Q-type) upon depolarization Coupled to KCa channels to regulate spike frequency and regularity |
| KCNC3 | SCA13 [31, 106, 113] | Kv3.3 Voltage-gated potassium channel |
Potassium entry upon membrane depolarization, causing hyperpolarization |
| ITPR1 | SCA15 [57, 101], SCA16 [49], SCA29 [6, 43, 112] | Inositol 1,4,5-trisphosphate (IP3) receptor | Calcium release from internal stores upon IP3 binding |
| KCND3 | SCA19 [25], SCA22 [61] | Kv4.3 Voltage-gated potassium channel |
Potassium entry upon membrane depolarization, causing hyperpolarization |
| SCN8A | Dravet syndrome [17] | Nav1.6 Voltage-gated sodium channel, pore-forming subunit |
Sodium entry and membrane depolarization during the action potential |
| FGF14 | SCA27 [102] | Fibroblast growth factor 13 | Interacts with Nav to influence excitability |
| TRPC3 | SCA41 [32] | Transient receptor potential cation channel type 3 | Essential for mGlur1- mediated synaptic transmission, long-term depression |
| CACNA1G | SCA42 [19, 72] | Cav3.1 Voltage-gated calcium channel |
Inward calcium current (T-type) upon depolarization |
| KCNMA1 | Unnamed SCA [95, 96] | K 1.1 Ca Large conductance calcium-activated potassium (BK) channel |
Outward K current + upon activation Regulates spike frequency and regularity |
Highlights.
Motor impairment in cerebellar ataxia results in part from neuronal dysfunction
Mutations in specific ion-channels cause human cerebellar ataxia
Ion channel dysfunction is present in models of polyglutamine Spinocerebellar Ataxia
A therapeutic strategy targeting specific ion-channels exists for cerebellar ataxia
Acknowledgments
This work was supported by the NIH R01NS085054 (V.G.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Citations
- 1.Aguado C, Colon J, Ciruela F, Schlaudraff F, Cabanero MJ, Perry C, Watanabe M, Liss B, Wickman K, Lujan R. Cell type-specific subunit composition of G protein-gated potassium channels in the cerebellum. J Neurochem. 2008;105:497–511. doi: 10.1111/j.1471-4159.2007.05153.x. [DOI] [PubMed] [Google Scholar]
- 2.Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- 3.Akemann W, Knopfel T. Interaction of Kv3 potassium channels and resurgent sodium current influences the rate of spontaneous firing of Purkinje neurons. J Neurosci. 2006;26:4602–4612. doi: 10.1523/JNEUROSCI.5204-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alvina K, Khodakhah K. KCa channels as therapeutic targets in episodic ataxia type-2. J Neurosci. 2010;30:7249–7257. doi: 10.1523/JNEUROSCI.6341-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvina K, Khodakhah K. The therapeutic mode of action of 4-aminopyridine in cerebellar ataxia. J Neurosci. 2010;30:7258–7268. doi: 10.1523/JNEUROSCI.3582-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barresi S, Niceta M, Alfieri P, Brankovic V, Piccini G, Bruselles A, Barone MR, Cusmai R, Tartaglia M, Bertini E, Zanni G. Mutations in the IRBIT domain of ITPR1 are a frequent cause of autosomal dominant nonprogressive congenital ataxia. Clin Genet. 2017;91:86–91. doi: 10.1111/cge.12783. [DOI] [PubMed] [Google Scholar]
- 7.Becker EB, Oliver PL, Glitsch MD, Banks GT, Achilli F, Hardy A, Nolan PM, Fisher EM, Davies KE. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc Natl Acad Sci U S A. 2009;106:6706–6711. doi: 10.1073/pnas.0810599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 9.Bettencourt C, Ryten M, Forabosco P, Schorge S, Hersheson J, Hardy J, Houlden H, C. United Kingdom Brain Expression Insights from cerebellar transcriptomic analysis into the pathogenesis of ataxia. JAMA Neurol. 2014;71:831–839. doi: 10.1001/jamaneurol.2014.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bushell T, Clarke C, Mathie A, Robertson B. Pharmacological characterization of a non-inactivating outward current observed in mouse cerebellar Purkinje neurones. Br J Pharmacol. 2002;135:705–712. doi: 10.1038/sj.bjp.0704518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao YJ, Dreixler JC, Couey JJ, Houamed KM. Modulation of recombinant and native neuronal SK channels by the neuroprotective drug riluzole. Eur J Pharmacol. 2002;449:47–54. doi: 10.1016/s0014-2999(02)01987-8. [DOI] [PubMed] [Google Scholar]
- 12.Casey JP, Hirouchi T, Hisatsune C, Lynch B, Murphy R, Dunne AM, Miyamoto A, Ennis S, van der Spek N, O’Hici B, Mikoshiba K, Lynch SA. A novel gain-of-function mutation in the ITPR1 suppressor domain causes spinocerebellar ataxia with altered Ca2+ signal patterns. J Neurol. 2017;264:1444–1453. doi: 10.1007/s00415-017-8545-5. [DOI] [PubMed] [Google Scholar]
- 13.Chen X, Kovalchuk Y, Adelsberger H, Henning HA, Sausbier M, Wietzorrek G, Ruth P, Yarom Y, Konnerth A. Disruption of the olivo-cerebellar circuit by Purkinje neuron-specific ablation of BK channels. Proc Natl Acad Sci U S A. 2010;107:12323–12328. doi: 10.1073/pnas.1001745107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X, Tang TS, Tu H, Nelson O, Pook M, Hammer R, Nukina N, Bezprozvanny I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J Neurosci. 2008;28:12713–12724. doi: 10.1523/JNEUROSCI.3909-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chopra R, Wasserman AH, Bushart DD, Dell’Orco JM, Shakkottai VG. Increased dendritic excitability and calcium-dependent PKC activation: a novel mechanism underlying Purkinje neuron dendritic degeneration in cerebellar ataxias. Ann Neurol. 2016;80:S33–S34. [Google Scholar]
- 16.Cingolani LA, Gymnopoulos M, Boccaccio A, Stocker M, Pedarzani P. Developmental regulation of small-conductance Ca2+-activated K+ channel expression and function in rat Purkinje neurons. J Neurosci. 2002;22:4456–4467. doi: 10.1523/JNEUROSCI.22-11-04456.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327–1332. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Costa Mdo C, Paulson HL. Toward understanding Machado-Joseph disease. Prog Neurobiol. 2012;97:239–257. doi: 10.1016/j.pneurobio.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coutelier M, Blesneac I, Monteil A, Monin ML, Ando K, Mundwiller E, Brusco A, Le Ber I, Anheim M, Castrioto A, Duyckaerts C, Brice A, Durr A, Lory P, Stevanin G. A Recurrent Mutation in CACNA1G Alters Cav3.1 T-Type Calcium-Channel Conduction and Causes Autosomal-Dominant Cerebellar Ataxia. Am J Hum Genet. 2015;97:726–737. doi: 10.1016/j.ajhg.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Zeeuw CI, Hoebeek FE, Bosman LW, Schonewille M, Witter L, Koekkoek SK. Spatiotemporal firing patterns in the cerebellum. Nat Rev Neurosci. 2011;12:327–344. doi: 10.1038/nrn3011. [DOI] [PubMed] [Google Scholar]
- 21.Dell’Orco JM, Pulst SM, Shakkottai VG. Potassium channel dysfunction underlies Purkinje neuron spiking abnormalities in spinocerebellar ataxia type 2. Human Molecular Genetics. 2017 doi: 10.1093/hmg/ddx281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dell’Orco JM, Wasserman AH, Chopra R, Ingram MA, Hu YS, Singh V, Wulff H, Opal P, Orr HT, Shakkottai VG. Neuronal Atrophy Early in Degenerative Ataxia Is a Compensatory Mechanism to Regulate Membrane Excitability. J Neurosci. 2015;35:11292–11307. doi: 10.1523/JNEUROSCI.1357-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doble A. The pharmacology and mechanism of action of riluzole. Neurology. 1996;47:S233–241. doi: 10.1212/wnl.47.6_suppl_4.233s. [DOI] [PubMed] [Google Scholar]
- 24.Dove LS, Abbott LC, Griffith WH. Whole-cell and single-channel analysis of P-type calcium currents in cerebellar Purkinje cells of leaner mutant mice. J Neurosci. 1998;18:7687–7699. doi: 10.1523/JNEUROSCI.18-19-07687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duarri A, Jezierska J, Fokkens M, Meijer M, Schelhaas HJ, den Dunnen WF, van Dijk F, Verschuuren-Bemelmans C, Hageman G, van de Vlies P, Kusters B, van de Warrenburg BP, Kremer B, Wijmenga C, Sinke RJ, Swertz MA, Kampinga HH, Boddeke E, Verbeek DS. Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann Neurol. 2012;72:870–880. doi: 10.1002/ana.23700. [DOI] [PubMed] [Google Scholar]
- 26.Duarri A, Lin MC, Fokkens MR, Meijer M, Smeets CJ, Nibbeling EA, Boddeke E, Sinke RJ, Kampinga HH, Papazian DM, Verbeek DS. Spinocerebellar ataxia type 19/22 mutations alter heterocomplex Kv4.3 channel function and gating in a dominant manner. Cell Mol Life Sci. 2015;72:3387–3399. doi: 10.1007/s00018-015-1894-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–894. doi: 10.1016/S1474-4422(10)70183-6. [DOI] [PubMed] [Google Scholar]
- 28.Edgerton JR, Reinhart PH. Distinct contributions of small and large conductance Ca2+-activated K+ channels to rat Purkinje neuron function. J Physiol. 2003;548:53–69. doi: 10.1113/jphysiol.2002.027854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Egorova PA, Zakharova OA, Vlasova OL, Bezprozvanny IB. In vivo analysis of cerebellar Purkinje cell activity in SCA2 transgenic mouse model. J Neurophysiol. 2016;115:2840–2851. doi: 10.1152/jn.00913.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernandez-Alacid L, Aguado C, Ciruela F, Martin R, Colon J, Cabanero MJ, Gassmann M, Watanabe M, Shigemoto R, Wickman K, Bettler B, Sanchez-Prieto J, Lujan R. Subcellular compartment-specific molecular diversity of pre- and post-synaptic GABA-activated GIRK channels in Purkinje cells. J Neurochem. 2009;110:1363–1376. doi: 10.1111/j.1471-4159.2009.06229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Figueroa KP, Minassian NA, Stevanin G, Waters M, Garibyan V, Forlani S, Strzelczyk A, Burk K, Brice A, Durr A, Papazian DM, Pulst SM. KCNC3: phenotype, mutations, channel biophysics-a study of 260 familial ataxia patients. Hum Mutat. 2010;31:191–196. doi: 10.1002/humu.21165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fogel BL, Hanson SM, Becker EB. Do mutations in the murine ataxia gene TRPC3 cause cerebellar ataxia in humans? Mov Disord. 2015;30:284–286. doi: 10.1002/mds.26096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao Z, Todorov B, Barrett CF, van Dorp S, Ferrari MD, van den Maagdenberg AM, De Zeeuw CI, Hoebeek FE. Cerebellar ataxia by enhanced Ca(V)2.1 currents is alleviated by Ca2+-dependent K+-channel activators in Cacna1a(S218L) mutant mice. J Neurosci. 2012;32:15533–15546. doi: 10.1523/JNEUROSCI.2454-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansen ST, Meera P, Otis TS, Pulst SM. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet. 2013;22:271–283. doi: 10.1093/hmg/dds427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008;59:392–398. doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hausser M, Clark BA. Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron. 1997;19:665–678. doi: 10.1016/s0896-6273(00)80379-7. [DOI] [PubMed] [Google Scholar]
- 37.Heck DH, De Zeeuw CI, Jaeger D, Khodakhah K, Person AL. The neuronal code(s) of the cerebellum. J Neurosci. 2013;33:17603–17609. doi: 10.1523/JNEUROSCI.2759-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Helmlinger D, Hardy S, Sasorith S, Klein F, Robert F, Weber C, Miguet L, Potier N, Van-Dorsselaer A, Wurtz JM, Mandel JL, Tora L, Devys D. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum Mol Genet. 2004;13:1257–1265. doi: 10.1093/hmg/ddh139. [DOI] [PubMed] [Google Scholar]
- 39.Hoebeek FE, Stahl JS, van Alphen AM, Schonewille M, Luo C, Rutteman M, van den Maagdenberg AM, Molenaar PC, Goossens HH, Frens MA, De Zeeuw CI. Increased noise level of purkinje cell activities minimizes impact of their modulation during sensorimotor control. Neuron. 2005;45:953–965. doi: 10.1016/j.neuron.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 40.Hong S, Negrello M, Junker M, Smilgin A, Thier P, De Schutter E. Multiplexed coding by cerebellar Purkinje neurons. Elife. 2016;5 doi: 10.7554/eLife.13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hourez R, Servais L, Orduz D, Gall D, Millard I, de Kerchove d’Exaerde A, Cheron G, Orr HT, Pandolfo M, Schiffmann SN. Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci. 2011;31:11795–11807. doi: 10.1523/JNEUROSCI.0905-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsu YH, Huang HY, Tsaur ML. Contrasting expression of Kv4.3, an A-type K+ channel, in migrating Purkinje cells and other post-migratory cerebellar neurons. Eur J Neurosci. 2003;18:601–612. doi: 10.1046/j.1460-9568.2003.02786.x. [DOI] [PubMed] [Google Scholar]
- 43.Huang L, Chardon JW, Carter MT, Friend KL, Dudding TE, Schwartzentruber J, Zou R, Schofield PW, Douglas S, Bulman DE, Boycott KM. Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J Rare Dis. 2012;7:67. doi: 10.1186/1750-1172-7-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hurlock EC, Bose M, Pierce G, Joho RH. Rescue of motor coordination by Purkinje cell-targeted restoration of Kv3.3 channels in Kcnc3-null mice requires Kcnc1. J Neurosci. 2009;29:15735–15744. doi: 10.1523/JNEUROSCI.4048-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hurlock EC, McMahon A, Joho RH. Purkinje-cell-restricted restoration of Kv3.3 function restores complex spikes and rescues motor coordination in Kcnc3 mutants. J Neurosci. 2008;28:4640–4648. doi: 10.1523/JNEUROSCI.5486-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ingram M, Wozniak EAL, Duvick L, Yang R, Bergmann P, Carson R, O’Callaghan B, Zoghbi HY, Henzler C, Orr HT. Cerebellar Transcriptome Profiles of ATXN1 Transgenic Mice Reveal SCA1 Disease Progression and Protection Pathways. Neuron. 2016;89:1194–1207. doi: 10.1016/j.neuron.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Inoue T, Kato K, Kohda K, Mikoshiba K. Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J Neurosci. 1998;18:5366–5373. doi: 10.1523/JNEUROSCI.18-14-05366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Irwin S, Vandelft M, Pinchev D, Howell JL, Graczyk J, Orr HT, Truant R. RNA association and nucleocytoplasmic shuttling by ataxin-1. J Cell Sci. 2005;118:233–242. doi: 10.1242/jcs.01611. [DOI] [PubMed] [Google Scholar]
- 49.Iwaki A, Kawano Y, Miura S, Shibata H, Matsuse D, Li W, Furuya H, Ohyagi Y, Taniwaki T, Kira J, Fukumaki Y. Heterozygous deletion of ITPR1, but not SUMF1, in spinocerebellar ataxia type 16. J Med Genet. 2008;45:32–35. doi: 10.1136/jmg.2007.053942. [DOI] [PubMed] [Google Scholar]
- 50.Jayabal S, Chang HH, Cullen KE, Watt AJ. 4-aminopyridine reverses ataxia and cerebellar firing deficiency in a mouse model of spinocerebellar ataxia type 6. Sci Rep. 2016;6:29489. doi: 10.1038/srep29489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jodice C, Mantuano E, Veneziano L, Trettel F, Sabbadini G, Calandriello L, Francia A, Spadaro M, Pierelli F, Salvi F, Ophoff RA, Frants RR, Frontali M. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum Mol Genet. 1997;6:1973–1978. doi: 10.1093/hmg/6.11.1973. [DOI] [PubMed] [Google Scholar]
- 52.Kalume F, Yu FH, Westenbroek RE, Scheuer T, Catterall WA. Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci. 2007;27:11065–11074. doi: 10.1523/JNEUROSCI.2162-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanjhan R, Anselme AM, Noakes PG, Bellingham MC. Postnatal changes in TASK-1 and TREK-1 expression in rat brain stem and cerebellum. Neuroreport. 2004;15:1321–1324. doi: 10.1097/01.wnr.0000127462.15985.dc. [DOI] [PubMed] [Google Scholar]
- 54.Kasumu AW, Hougaard C, Rode F, Jacobsen TA, Sabatier JM, Eriksen BL, Strobaek D, Liang X, Egorova P, Vorontsova D, Christophersen P, Ronn LC, Bezprozvanny I. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem Biol. 2012;19:1340–1353. doi: 10.1016/j.chembiol.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim SJ. TRPC3 channel underlies cerebellar long-term depression. Cerebellum. 2013;12:334–337. doi: 10.1007/s12311-013-0455-1. [DOI] [PubMed] [Google Scholar]
- 56.Kindler CH, Pietruck C, Yost CS, Sampson ER, Gray AT. Localization of the tandem pore domain K+ channel TASK-1 in the rat central nervous system. Brain Res Mol Brain Res. 2000;80:99–108. doi: 10.1016/s0169-328x(00)00136-4. [DOI] [PubMed] [Google Scholar]
- 57.Knight MA, Kennerson ML, Anney RJ, Matsuura T, Nicholson GA, Salimi-Tari P, Gardner RJ, Storey E, Forrest SM. Spinocerebellar ataxia type 15 (sca15) maps to 3p24.2-3pter: exclusion of the ITPR1 gene, the human orthologue of an ataxic mouse mutant. Neurobiol Dis. 2003;13:147–157. doi: 10.1016/s0969-9961(03)00029-9. [DOI] [PubMed] [Google Scholar]
- 58.Laezza F, Gerber BR, Lou JY, Kozel MA, Hartman H, Craig AM, Ornitz DM, Nerbonne JM. The FGF14(F145S) mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J Neurosci. 2007;27:12033–12044. doi: 10.1523/JNEUROSCI.2282-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lam J, Coleman N, Garing AL, Wulff H. The therapeutic potential of small-conductance KCa2 channels in neurodegenerative and psychiatric diseases. Expert Opin Ther Targets. 2013;17:1203–1220. doi: 10.1517/14728222.2013.823161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD, Hyun ED, Duvick LA, Orr HT, Botas J, Zoghbi HY. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–1347. doi: 10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- 61.Lee YC, Durr A, Majczenko K, Huang YH, Liu YC, Lien CC, Tsai PC, Ichikawa Y, Goto J, Monin ML, Li JZ, Chung MY, Mundwiller E, Shakkottai V, Liu TT, Tesson C, Lu YC, Brice A, Tsuji S, Burmeister M, Stevanin G, Soong BW. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann Neurol. 2012;72:859–869. doi: 10.1002/ana.23701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li X, Liu H, Fischhaber PL, Tang TS. Toward therapeutic targets for SCA3: Insight into the role of Machado-Joseph disease protein ataxin-3 in misfolded proteins clearance. Prog Neurobiol. 2015;132:34–58. doi: 10.1016/j.pneurobio.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 63.Liu J, Tang TS, Tu H, Nelson O, Herndon E, Huynh DP, Pulst SM, Bezprozvanny I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci. 2009;29:9148–9162. doi: 10.1523/JNEUROSCI.0660-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lorenzon NM, Lutz CM, Frankel WN, Beam KG. Altered calcium channel currents in Purkinje cells of the neurological mutant mouse leaner. J Neurosci. 1998;18:4482–4489. doi: 10.1523/JNEUROSCI.18-12-04482.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lou JY, Laezza F, Gerber BR, Xiao M, Yamada KA, Hartmann H, Craig AM, Nerbonne JM, Ornitz DM. Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels. J Physiol. 2005;569:179–193. doi: 10.1113/jphysiol.2005.097220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ly R, Bouvier G, Schonewille M, Arabo A, Rondi-Reig L, Lena C, Casado M, De Zeeuw CI, Feltz A. T-type channel blockade impairs long-term potentiation at the parallel fiber-Purkinje cell synapse and cerebellar learning. Proc Natl Acad Sci U S A. 2013;110:20302–20307. doi: 10.1073/pnas.1311686110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martina M, Metz AE, Bean BP. Voltage-dependent potassium currents during fast spikes of rat cerebellar Purkinje neurons: inhibition by BDS-I toxin. J Neurophysiol. 2007;97:563–571. doi: 10.1152/jn.00269.2006. [DOI] [PubMed] [Google Scholar]
- 68.Matsumoto M, Nakagawa T, Inoue T, Nagata E, Tanaka K, Takano H, Minowa O, Kuno J, Sakakibara S, Yamada M, Yoneshima H, Miyawaki A, Fukuuchi Y, Furuichi T, Okano H, Mikoshiba K, Noda T. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature. 1996;379:168–171. doi: 10.1038/379168a0. [DOI] [PubMed] [Google Scholar]
- 69.McCann C, Holohan EE, Das S, Dervan A, Larkin A, Lee JA, Rodrigues V, Parker R, Ramaswami M. The Ataxin-2 protein is required for microRNA function and synapse-specific long-term olfactory habituation. Proc Natl Acad Sci U S A. 2011;108:E655–662. doi: 10.1073/pnas.1107198108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McMahon A, Fowler SC, Perney TM, Akemann W, Knopfel T, Joho RH. Allele-dependent changes of olivocerebellar circuit properties in the absence of the voltage-gated potassium channels Kv3.1 and Kv3.3. Eur J Neurosci. 2004;19:3317–3327. doi: 10.1111/j.0953-816X.2004.03385.x. [DOI] [PubMed] [Google Scholar]
- 71.Moore LR, Rajpal G, Dillingham IT, Qutob M, Blumenstein KG, Gattis D, Hung G, Kordasiewicz HB, Paulson HL, McLoughlin HS. Evaluation of Antisense Oligonucleotides Targeting ATXN3 in SCA3 Mouse Models. Mol Ther Nucleic Acids. 2017;7:200–210. doi: 10.1016/j.omtn.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morino H, Matsuda Y, Muguruma K, Miyamoto R, Ohsawa R, Ohtake T, Otobe R, Watanabe M, Maruyama H, Hashimoto K, Kawakami H. A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia. Mol Brain. 2015;8:89. doi: 10.1186/s13041-015-0180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.N’Gouemo P. Targeting BK (big potassium) channels in epilepsy. Expert Opin Ther Targets. 2011;15:1283–1295. doi: 10.1517/14728222.2011.620607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neuwald AF, Koonin EV. Ataxin-2, global regulators of bacterial gene expression, and spliceosomal snRNP proteins share a conserved domain. J Mol Med (Berl) 1998;76:3–5. doi: 10.1007/s001090050184. [DOI] [PubMed] [Google Scholar]
- 75.Ogura H, Matsumoto M, Mikoshiba K. Motor discoordination in mutant mice heterozygous for the type 1 inositol 1,4,5-trisphosphate receptor. Behav Brain Res. 2001;122:215–219. doi: 10.1016/s0166-4328(01)00187-5. [DOI] [PubMed] [Google Scholar]
- 76.Paulson HL, Shakkottai VG, Clark HB, Orr HT. Polyglutamine spinocerebellar ataxias - from genes to potential treatments. Nat Rev Neurosci. 2017;18:613–626. doi: 10.1038/nrn.2017.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petrenko AB, Tsujita M, Kohno T, Sakimura K, Baba H. Mutation of alpha1G T-type calcium channels in mice does not change anesthetic requirements for loss of the righting reflex and minimum alveolar concentration but delays the onset of anesthetic induction. Anesthesiology. 2007;106:1177–1185. doi: 10.1097/01.anes.0000267601.09764.e6. [DOI] [PubMed] [Google Scholar]
- 78.Pflieger LT, Dansithong W, Paul S, Scoles DR, Figueroa KP, Meera P, Otis TS, Facelli JC, Pulst SM. Gene co-expression network analysis for identifying modules and functionally enriched pathways in SCA2. Hum Mol Genet. 2017;26:3069–3080. doi: 10.1093/hmg/ddx191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pruss H, Derst C, Lommel R, Veh RW. Differential distribution of individual subunits of strongly inwardly rectifying potassium channels (Kir2 family) in rat brain. Brain Res Mol Brain Res. 2005;139:63–79. doi: 10.1016/j.molbrainres.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 80.Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19:1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ristori G, Romano S, Visconti A, Cannoni S, Spadaro M, Frontali M, Pontieri FE, Vanacore N, Salvetti M. Riluzole in cerebellar ataxia: a randomized, double-blind, placebo-controlled pilot trial. Neurology. 2010;74:839–845. doi: 10.1212/WNL.0b013e3181d31e23. [DOI] [PubMed] [Google Scholar]
- 83.Romano S, Coarelli G, Marcotulli C, Leonardi L, Piccolo F, Spadaro M, Frontali M, Ferraldeschi M, Vulpiani MC, Ponzelli F, Salvetti M, Orzi F, Petrucci A, Vanacore N, Casali C, Ristori G. Riluzole in patients with hereditary cerebellar ataxia: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14:985–991. doi: 10.1016/S1474-4422(15)00201-X. [DOI] [PubMed] [Google Scholar]
- 84.Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H, Sausbier U, Sailer CA, Feil R, Hofmann F, Korth M, Shipston MJ, Knaus HG, Wolfer DP, Pedroarena CM, Storm JF, Ruth P. Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci U S A. 2004;101:9474–9478. doi: 10.1073/pnas.0401702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schols L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291–304. doi: 10.1016/S1474-4422(04)00737-9. [DOI] [PubMed] [Google Scholar]
- 86.Scoles DR, Meera P, Schneider MD, Paul S, Dansithong W, Figueroa KP, Hung G, Rigo F, Bennett CF, Otis TS, Pulst SM. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature. 2017;544:362–366. doi: 10.1038/nature22044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Seidel K, Siswanto S, Brunt ER, den Dunnen W, Korf HW, Rub U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012;124:1–21. doi: 10.1007/s00401-012-1000-x. [DOI] [PubMed] [Google Scholar]
- 88.Sekerkova G, Kim JA, Nigro MJ, Becker EB, Hartmann J, Birnbaumer L, Mugnaini E, Martina M. Early onset of ataxia in moonwalker mice is accompanied by complete ablation of type II unipolar brush cells and Purkinje cell dysfunction. J Neurosci. 2013;33:19689–19694. doi: 10.1523/JNEUROSCI.2294-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Serodio P, Rudy B. Differential expression of Kv4 K+ channel subunits mediating subthreshold transient K+ (A-type) currents in rat brain. J Neurophysiol. 1998;79:1081–1091. doi: 10.1152/jn.1998.79.2.1081. [DOI] [PubMed] [Google Scholar]
- 90.Shakkottai VG, Chou CH, Oddo S, Sailer CA, Knaus HG, Gutman GA, Barish ME, LaFerla FM, Chandy KG. Enhanced neuronal excitability in the absence of neurodegeneration induces cerebellar ataxia. J Clin Invest. 2004;113:582–590. doi: 10.1172/JCI20216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shakkottai VG, do Carmo Costa M, Dell’Orco JM, Sankaranarayanan A, Wulff H, Paulson HL. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci. 2011;31:13002–13014. doi: 10.1523/JNEUROSCI.2789-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shakkottai VG, Xiao M, Xu L, Wong M, Nerbonne JM, Ornitz DM, Yamada KA. FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol Dis. 2009;33:81–88. doi: 10.1016/j.nbd.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shibata H, Huynh DP, Pulst SM. A novel protein with RNA-binding motifs interacts with ataxin-2. Hum Mol Genet. 2000;9:1303–1313. doi: 10.1093/hmg/9.9.1303. [DOI] [PubMed] [Google Scholar]
- 94.Smets K, Duarri A, Deconinck T, Ceulemans B, van de Warrenburg BP, Zuchner S, Gonzalez MA, Schule R, Synofzik M, Van der Aa N, De Jonghe P, Verbeek DS, Baets J. First de novo KCND3 mutation causes severe Kv4.3 channel dysfunction leading to early onset cerebellar ataxia, intellectual disability, oral apraxia and epilepsy. BMC Med Genet. 2015;16:51. doi: 10.1186/s12881-015-0200-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Staisch J, Du X, Carvalho-de-Souza J, Kubota T, Bezanilla F, Gomez C. A mutation causing reduced BK channel activity leads to cognitive impairment and progressive cerebellar ataxia. Neurology. 2016;86:P5.394. [Google Scholar]
- 96.Staisch J, Du X, Kubota T, de Souza J, Bezanilla F, Gomez C. A novel KNCMA1 mutation associated with progressive cerebellar ataxia. Neurology. 2015;84:P2.118. [Google Scholar]
- 97.Swensen AM, Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci. 2003;23:9650–9663. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tada M, Nishizawa M, Onodera O. Roles of inositol 1,4,5-trisphosphate receptors in spinocerebellar ataxias. Neurochem Int. 2016;94:1–8. doi: 10.1016/j.neuint.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 99.Tempia F, Hoxha E, Negro G, Alshammari MA, Alshammari TK, Panova-Elektronova N, Laezza F. Parallel fiber to Purkinje cell synaptic impairment in a mouse model of spinocerebellar ataxia type 27. Front Cell Neurosci. 2015;9:205. doi: 10.3389/fncel.2015.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Thach WT. Discharge of Purkinje and cerebellar nuclear neurons during rapidly alternating arm movements in the monkey. J Neurophysiol. 1968;31:785–797. doi: 10.1152/jn.1968.31.5.785. [DOI] [PubMed] [Google Scholar]
- 101.van de Leemput J, Chandran J, Knight MA, Holtzclaw LA, Scholz S, Cookson MR, Houlden H, Gwinn-Hardy K, Fung HC, Lin X, Hernandez D, Simon-Sanchez J, Wood NW, Giunti P, Rafferty I, Hardy J, Storey E, Gardner RJ, Forrest SM, Fisher EM, Russell JT, Cai H, Singleton AB. Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 2007;3:e108. doi: 10.1371/journal.pgen.0030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.van Swieten JC, Brusse E, de Graaf BM, Krieger E, van de Graaf R, de Koning I, Maat-Kievit A, Leegwater P, Dooijes D, Oostra BA, Heutink P. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected] Am J Hum Genet. 2003;72:191–199. doi: 10.1086/345488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wakamori M, Yamazaki K, Matsunodaira H, Teramoto T, Tanaka I, Niidome T, Sawada K, Nishizawa Y, Sekiguchi N, Mori E, Mori Y, Imoto K. Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J Biol Chem. 1998;273:34857–34867. doi: 10.1074/jbc.273.52.34857. [DOI] [PubMed] [Google Scholar]
- 104.Walter JT, Alvina K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- 105.Wang Q, Bardgett ME, Wong M, Wozniak DF, Lou J, McNeil BD, Chen C, Nardi A, Reid DC, Yamada K, Ornitz DM. Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron. 2002;35:25–38. doi: 10.1016/s0896-6273(02)00744-4. [DOI] [PubMed] [Google Scholar]
- 106.Waters MF, Minassian NA, Stevanin G, Figueroa KP, Bannister JP, Nolte D, Mock AF, Evidente VG, Fee DB, Muller U, Durr A, Brice A, Papazian DM, Pulst SM. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genet. 2006;38:447–451. doi: 10.1038/ng1758. [DOI] [PubMed] [Google Scholar]
- 107.Womack MD, Chevez C, Khodakhah K. Calcium-activated potassium channels are selectively coupled to P/Q-type calcium channels in cerebellar Purkinje neurons. J Neurosci. 2004;24:8818–8822. doi: 10.1523/JNEUROSCI.2915-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yan H, Pablo JL, Pitt GS. FGF14 regulates presynaptic Ca2+ channels and synaptic transmission. Cell Rep. 2013;4:66–75. doi: 10.1016/j.celrep.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yan H, Pablo JL, Wang C, Pitt GS. FGF14 modulates resurgent sodium current in mouse cerebellar Purkinje neurons. Elife. 2014;3:e04193. doi: 10.7554/eLife.04193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yang S, Li XJ, Li S. Molecular mechanisms underlying Spinocerebellar Ataxia 17 (SCA17) pathogenesis. Rare Dis. 2016;4:e1223580. doi: 10.1080/21675511.2016.1223580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yue MM, Lv K, Meredith SC, Martindale JL, Gorospe M, Schuger L. Novel RNA-binding protein P311 binds eukaryotic translation initiation factor 3 subunit b (eIF3b) to promote translation of transforming growth factor beta1-3 (TGF-beta1-3) J Biol Chem. 2014;289:33971–33983. doi: 10.1074/jbc.M114.609495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zambonin JL, Bellomo A, Ben-Pazi H, Everman DB, Frazer LM, Geraghty MT, Harper AD, Jones JR, Kamien B, Kernohan K, Koenig MK, Lines M, Palmer EE, Richardson R, Segel R, Tarnopolsky M, Vanstone JR, Gibbons M, Collins A, Fogel BL, Canada CCare4Rare, Dudding-Byth T, Boycott KM. Spinocerebellar ataxia type 29 due to mutations in ITPR1: a case series and review of this emerging congenital ataxia. Orphanet J Rare Dis. 2017;12:121. doi: 10.1186/s13023-017-0672-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang Y, Kaczmarek LK. Kv3.3 potassium channels and spinocerebellar ataxia. J Physiol. 2016;594:4677–4684. doi: 10.1113/JP271343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhou H, Lin Z, Voges K, Ju C, Gao Z, Bosman LW, Ruigrok TJ, Hoebeek FE, De Zeeuw CI, Schonewille M. Cerebellar modules operate at different frequencies. Elife. 2014;3:e02536. doi: 10.7554/eLife.02536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- 116.Chopra R, Wasserman AH, Pulst SM, De Zeeuw CI, Shakkottai VG. Protein kinase C activity is a protective modifier of Purkinje neuron degeneration in cerebellar ataxia. Hum Mol Genet. 2018 doi: 10.1093/hmg/ddy050. https://doi.org/10.1093/hmg/ddy050 In press. [DOI] [PMC free article] [PubMed]
- 117.Bushart DD, Chopra R, Singh V, Murphy GG, Wulff H, Shakkottai VG. Targeting potassium channels to treat cerebellar ataxia. Ann Clin Transl Neurol. 2018 doi: 10.1002/acn3.527. https://doi.org/10.1002/acn3.527 [DOI] [PMC free article] [PubMed]