

Graphical abstract
Keywords: Urease, Inhibitor design, Molecular modeling, Inhibitor-enzyme interactions
Abstract
Urease is a nickel-dependent metalloenzyme found in plants, some bacteria, and fungi. Bacterial enzyme is of special importance since it has been demonstrated as a potent virulence factor for some species. Especially it is central to Helicobacter pylori metabolism and virulence being necessary for its colonization of the gastric mucosa, and is a potent immunogen that elicits a vigorous immune response. Therefore, it is not surprising that efforts to design, synthesize and evaluate of new inhibitors of urease are and active field of medicinal chemistry. In this paper recent advances on this field are reviewed.
Introduction
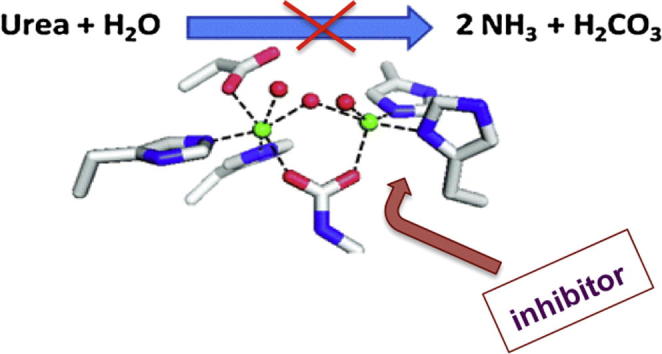
Being the first organic compound synthesized by Friedrich Wohler from inorganic components [1] urea has a unique role in history. Urea is an endogenous product of protein and amino acid catabolism. For example, approximately 20–35 g of urea is excreted in human urine per day. Urea is also used in huge quantities as fertilizer (being an exogenous source of ammonia for plants). This compound is hydrolytically stable and the half-life of non-enzymatic hydrolysis of urea is equal 3.6 years and the mechanism of this simple process is still disputable [2], [3]. In Nature it is hydrolyzed by an enzyme urease (urea aminohydrolase E.C.3.5.1.5), a multi-subunit nickel dependent metalloenzyme that catalyzes the hydrolysis of urea at a rate approximately 1014 times the rate of the un-catalyzed reaction [4], [5]. It is worth to express that the latter process is proceeding via different mechanism than this catalyzed by urease. This key enzyme of global nitrogen cycle converts urea to ammonia and carbamate, which in turn spontaneously generate carbon dioxide and next molecule of ammonia. Urease is the first enzyme, which was ever crystallized in 1926 by James B. Summer, who reported that a pure protein might function as an enzyme [6].
Bacteria, fungi, yeast, and plants produce urease where it catalyzes the urea degradation to supply these organisms with a source of nitrogen for growth. Urease is also a virulence factor found in various pathogenic bacteria. Therefore, it is not surprising that it is essential in colonization of a host organism and in maintenance of bacterial cells in tissues. Its activity leads to several implications such as appearance of urinary stones, catheters blocking, pyelonephritis, ammonia encephalopathy, hepatic coma as well as gastritis [7]. One of the most frequently studied bacterial urease is that from H. pylori, a causative agent of gastritis and peptic ulceration and stomach cancer [8], [9].
Ruminal microbial urease plays an important role in the nitrogen metabolism in ruminants such as cattle and sheep. The urea from diet or recycled from blood to rumen is hydrolyzed to ammonia by bacteria residing in this stomach. This causes poor nitrogen accumulation when diets contain a high urea content [10], [11].
Urea accounts significantly in total nitrogen fertilizers consumption worldwide. Its application is accompanied with large losses in ammonia, which is released upon action of bacterial ureases by its volatilization [12], [13].
Variable and important role of urease stimulate that this enzyme continued to be the focus of researchers around the world, in the fields of genetics, biochemistry and physiology [14], [15], [16]. Strategies based on urease inhibition are considered as a promising mean to treat the diseases caused by bacteria producing urease, as well as a mean to diminish nitrogen loss from urea used as fertilizer. Therefore, it is not surprising that inhibitors of urease have been recently reviewed [17], [18], [19], [20], [21]. In this paper the most recent discoveries leading to inhibitors of this enzyme will be reviewed in some detail.
Crystal and molecular structure of urease
Enzymes, especially those vital for pathogenesis, are considered to be the most effective and promising targets for small molecule interventions in human and animal therapy, as well for design of pesticides [22]. The process of development of new inhibitor of an enzyme is challenging, time consuming, expensive, and requires consideration of many aspects. To fulfill these challenges, several multidisciplinary approaches are required, which collectively would form the basis of rational design. Structure-guided methods are an integral part of such development with three-dimensional structure of a target enzyme, bound to its natural ligand or an effector of its activity (determined either by X-ray crystallography or by NMR), serving as a template to produce new inhibitors.
Plant and fungal ureases are homo-oligomeric proteins of 90-kDa identical subunits, while bacterial ureases are multimers of two (αβ) or three (αβγ) subunits of different molecular mass forming various complexes. Number of urease subunits is varied according to their sources. For example, Klebsiella aerogenes and Sporosarcina pasteurii enzymes are composed of an (αβγ)3 trimer with each α-subunit having an (αβ)8-barrel domain containing a bi-nickel active center [23]. Staphylococcus saprophyticus urease consists of these three subunits of (αβγ)4 stoichiometry [24], whereas urease from Helicobacter pylori consists of only two subunits (α and β) forming a spherical assembly of (αβ)12 stoichiometry [25]. There are an impressive number of papers dealing with determination of structures of ureases from various sources [26], [27], [28]. They revealed that, despite the difference in number of subunits, the structure of the active site in the vicinity of the nickel (II) ions is conserved and induces the same mechanism of catalytic activity [27], [29].
Also molecular modeling was used to understand better the mechanism of action of this enzyme [30]. The studies on two bacterial enzymes (Klebsiella aerogenes and Helicobacter pylori) have revealed experimentally unobserved wide-open flap state that, unlike the well-characterized closed and open states of the enzyme, allows ready access of inhibitors to the metal cluster in the active site [31], [32]. Molecular modeling was also used to predict the three-dimensional structure of Arabidopsis thaliana enzyme complexed with urea [33].
Crystal structures of ureases complexed with various ligands
Rational design of urease inhibitors is strongly enforced by the knowledge of crystal structures of this enzyme in its complexes with various inhibitors. Such structures have been determined and deposited in Protein Data Bank. The most of them consider Sporosarcina pasteurii urease complexes with the following ligands: β-mercaptoethanol (PDB 1UPB) [34], acetohydroxamate (PDB 4UPB) [35], phenylphosphorodiamidate (PDB 3UPB) [36], phosphate (PDB 1 IE7) [37] (N-(n-butyl)thiophosphoric triamide (PDB 4CU) [38], fluoride (PDB 4CEX) [39], sulfite (PDB 5A6T) [28], citrate (PDB 2UPB, Fig. 1) [27], boric acid (PDB 1S3T) [40], catechol (PDB 5G4H) [41] and 1,4-benzoquinone (PDB 5FSE) [42]. Other crystal structures are scarce and consider acetohydroxamate inhibited ureases from Helicobacter pylori urease complexed with acetohydroxamic acd (PDB 1E9Y) [25] and Klebsiella aerogenes (PDB 1FWE) [43] and jack bean urease complexed with phosphate (PDB 3LA4) [26].
Fig. 1.

Structural scheme (left panel) and model (right panel) of urease from S. pasteurii (pdb 4AC7) showing the requirements for the good inhibitor of the enzyme.
The crystal structures published recently indicate requirement for three indispensable elements for effective inhibitor: presence of nickel-complexing moiety alongside with properly placed network of hydrogen-bond donors and acceptors attached to flexible scaffold. Additionally, special attention should be paid to the proper protonation states of the designed ligands [27].
The process of design of urease inhibitors is also strongly dependent on their possible role – if considering potential drugs molecular scaffold of could be structurally complex since the drug might be expensive, whereas in the case of inhibition of decomposition of urea in soil inhibitor has to be of simple structure and thus substantially cheap.
Inhibitors bearing fragment of urea in their structures
Urea is a small molecule and natural substrate of urease. On the other hand, as indicated by crystallographic studies, the enzyme is quite flexible and is able to bind big scaffolds [27]. Therefore, compounds containing fragment of urea or thiourea are of natural choice for the construction of inhibitors of this enzyme. Such an example is 1-(4-chlorophenyl)-3-palmitoylthiourea (compound 1), the most potent amongst a series of effective inhibitors of jack bean urease obtained recently [44]. It appears to be uncompetitive inhibitor and its binding determined by molecular modeling is different than this expected since it is bound in a quite long distance from nickel ions (Fig. 2).
Fig. 2.

Structure of 1-(4-chlorophenyl)-3-palmitoylthiourea (1) and the mode of its binding by jack bean urease as remodeled by authors of this paper.
Barbiturates and thiobarbiturates could be also treated as compounds bearing urea fragment in their structures (see Fig. 3 for representative structures: compounds 2, 3, 4 and 5). They appeared to be moderate inhibitors, with inhibition constants in micromolar range. They are bound by ureases from jack bean and S. pasteurii in a manner analogous to the substrate with urea or thiourea fragment being complexed by two nickel (II) ions [45], [46], [47], [48].
Fig. 3.

Inhibitors of various ureases, which might be considered as expanded analogs of urea.
Representative structures of iminothiazolines (compound 6) [49], cyanoacetamides (compound 7) [50] and hydrazones (compound 8) [51], possessing structural fragments mimicking urea, are shown in Fig. 3. They appeared, however, to be weak to moderate uncompetitive or mixed inhibitors of jack bean and Helicobacter pylori enzymes, and have no practical value.
Quinolones
Quinolone antibiotics constitute an important class of large group of synthetic broad-spectrum antibacterial agents, which are nowadays the most successful clinically synthetic antibacterial drugs [52]. They inhibit DNA synthesis. Nearly all quinolone antibiotics in modern use are fluoroquinolones. Their two popular representatives – Levofloxacin and Ciprofloxacin (compounds 9 and 10, Fig. 4) [53], [54], as well as their analogs [55], appeared to be quite promising inhibitors of Helicobacter pylori and Proteus mirabilis enzymes. Molecular modeling suggests their binding with carboxylic group interacting with active site nickel ions. However, mechanism of additional covalent interaction with the enzymatic cysteine similar to this observed for simple quinones, cannot be ruled out [56]. Acetohydroxamic acid is a prescription medicine (Lithostat) that is used in patients with chronic urea-splitting urinary infection to prevent the excessive build-up of ammonia in the urine. It inhibits urease by complexing nickel ions and thus is also one of the compounds most intensively studied as the potential therapeutics for the treatment of ulcer caused by H. pylori [57]. Therefore, it is not surprising that modification of carboxylic group of fluoroquinolones by their conversion into hydroxyamic acid (compound 11, Fig. 4), hydrazide and amide yielded interesting classes of inhibitors of this enzyme [58].
Fig. 4.

Fluoroquinolones – inhibitors of urease.
Recently Moxifloxacin (compound 12) have been used for capping of silver and gold nanoparticles and appeared to be exceptional inhibitor of urease, more potent than antibiotic itself [59].
Flavonoids
It is well known that structural diversity and complexity within natural products stimulates research on their use as lead compounds for various diseases. Extracts of various plants, including green tea and cranberries often have been used to treat gastritis or urinary tract infections. This effect is believed to result from the action of (+)-catechin and (−)-epigallocatechin gallate as urease inhibitors [60]. Also flavonoids isolated from other plants: Daphne retusa (daphnretusic acid), Pistacia atlantica (transilitin and dihydro luteolin) and cotton (gossypol, gossypolone and apogossypol) appeared to be micromolar inhibitors of urease from jack bean [61], [62], [63]. These studies stimulated the efforts to analyze inhibitory potential of flavonoids in some detail. Thus, 11 natural and 19 synthetic compounds were screened against H. pylori urease [64]. They appear to be moderate competitive (micromolar range) to weak inhibitors of the enzyme with synthetic compounds 13 and 14, and quercetin (compound 15) (Fig. 5) [65] being the most active. Docking of the most active compound (13) into the crystal structure of H. pylori urease performed by the AutoDock program revealed the mode of binding of this inhibitor. In detail, the compound is oriented with its benzopyrone moiety in proximity to urea binding cavity, letting phenyl ring to locate at the mouth of the cavity. The channel to the active site for urea is therefore blocked off. Since catechol moiety of flavonoids does not bind nickel ion(s) there is a possibility of covalent interaction of this fragment of the molecule with one of cysteine residues present in the binding site. Such a mechanism has been determined and detail studied in the case of simple catechol [41].
Fig. 5.

Structures of flavonoid glycosides – inhibitors of H. pylori urease.
Radix Scutellariae, known as “Huang-Qin” in Chinese, is originated from the dried root of Scutellaria baicalensis. Its major bioactive compounds are flavone glycosides baicalin and scutellarin (Fig. 5, compounds 16 and 17). Baicalin was found to be a competitive, slow-binding and concentration-dependent inhibitor of jack bean and H. pylori ureases [66], [67], [68]. Kaempferol-3-O-β-D-glucopyranoside (compound 18) and kaempferol-3-O-α-L-rhamnopyranoside (Fig. 5, compound 19), isolated from the fruits of Syzygium alternifolium, appeared more potent inhibitors of H. pylori enzyme [69].
Molecular modeling revealed that these compounds are bound differently than flavonoids, with catechol being involved in complexation of nickel ion. However, the most important for inhibition seems to be interaction with cysteine located at the mobile flap covering the active site through its S—H…π interactions with aromatic fragment of these molecules (Fig. 6). The active site of ureases is of relatively small volume (related to the size of urea) and is covered by a movable flap. This flap contains a cysteine residue that could be targeted by inhibitors. This cysteine, besides being directly involved in the architecture of the active site, plays a vital role in positioning other key residues in the active site appropriately for the catalysis.
Fig. 6.

Mode of bonding of baicalin (16) to H. pylori urease as remodeled by authors of this paper.
Other natural products
Natural products (mostly secondary metabolites) have been the most successful source of potential drug leads so far. Even if these efforts somewhat decline in interest they continue to provide unique structural diversity of potential enzyme inhibitors. This is also the case if considering research on urease. In last several years there are several reviews on action of plant extracts [70], [71], [72] and isolated natural compounds [20], [73] towards this enzyme.
Representative examples of natural products of recently determined inhibitory action against urease are: boswellic acid (Fig. 7, compound 20) a component of African medicinal plant Boswellia carterii [74], palmatine (compound 21) and epiberberine (compound 22) from Coptis chinensis [75], [76], [77], a plant traditionally used in China for the treatment of gastrointestinal diseases, andrographolide (compound 23), the major diterpenoid lactone and the primary effective constituent of Chinese medicinal plant Andrographis aniculata [78] and a popular antibiotic from garlic – allicin (compound 24) [79], [80].
Fig. 7.

Representative examples of recently described natural products urease inhibitors.
Docking of palmitine to the ureases from jack bean and H. pylori revealed that this alkaloid well fills the active pockets of these ureases, tightly anchoring the helix-turn-helix motif over the active-site cavity (Fig. 8). This prevents the flap of the urease active-site cavity from backing to the close position, which results in the inhibition of its activity.
Fig. 8.

Docked conformation of palmitine in active site of H. pylori urease remodeled by authors of this paper.
It is worth to mention that there are quite intensive studies on influence of various honeys [81], [82], [83], honey fractions [84] and their combination with plant extracts [85] on the activity of urease from H. pyliori. These papers seem to indicate that regular daily consumption of these honeys can prevent gastric ulcers.
Heterocyclic compounds
The practice of random testing of a large number of newly synthesized molecules in hope to find a new drug candidate is still the most popular approach. This process of screening, though inefficient, has led to the identification of many new lead compounds. Aromatic heterocycles yielded the most interesting activity against ureases. All the compounds reported recently appear to be micromolar inhibitors of H. pylori or jack bean ureases. As suggested by molecular modeling, they are bound within the active site of the enzymes and their activity results from interaction of side chain of cysteine or methionine with π electrons of aromatic fragment of the molecule. In Fig. 9 the most representative examples of inhibitory benzimidazole (compound 25) [86], oxadiazole (compound 26) [87], ethyl tiazolidine-4-carboxylate (compound 27) [88] and dihydropyridone (compound 28) [89], [90]. Also thiadiazoles were considered as inhibitors of H. pylori urease, however enzymatic studies have not been carried out and this assumption was derived from their antibacterial activity supported by molecular modeling against this enzyme [91]. The combination of two inhibitory scaffolds, namely of benzimidazole with triazole (compound 29) or oxadiazole (compound 30) [92], as well as aminopyridine with carbazole (compound 31) [93] did not result in elevation of inhibitory activity.
Fig. 9.

Heterocyclic inhibitors of urease.
Inhibitors, which bind covalently to urease
These inhibitors are compounds designed to bind covalently to a specific molecular target and thereby suppress its biological function. They exhibit crucial advantage resulting from strong binding to the target and thus higher potency, extended duration of action and lower dose. However, they are also often considered as less attractive drug candidates because of drawbacks as general toxicity, immunogenicity and problems associated with degradation of inhibited proteins, issues that are of great concern. Therefore, it is not surprising that such inhibitors of urease have been scarcely studied.
Good candidates for such inhibitors are Michael acceptors. Thus, forty relatively simple molecules containing functional groups of various geometries (E and Z isomers) of substituted double bonds or containing linear triple bonds or allenes were screened for their inhibitory activities against S. pasteurii urease. This led to several compounds exhibiting potency in the nanomolar range [94]. All groups that are controlling the chemical reactivity of double/triple bonds contained carbonyl groups (carboxylic acids, their esters or ketones), with compounds 32 and 33 (Fig. 10) being the most potent. As shown by molecular modeling, compound 33 is the first example of an interesting mode of binding, which combines the formation of a covalent bond with the cysteine residue and interactions with two nickel ions (Fig. 10). Such a mode of binding seems to promote selectivity of the inhibitors toward this enzyme.
Fig. 10.

Two most potent Michael acceptor inhibitors of S. pasteurii urease and the mode of binding of compound 32.
Another example of covalent inhibitor of urease is Disulfiram (compound 34, Fig. 11), a drug used to support the treatment of chronic alcoholism by inhibiting acetaldehyde dehydrogenase. Kinetic experiments suggest that it carbamylates Citrullus vulgaris urease active site flap Cys695 in a manner similar to its action on dehydrogenase (Fig. 11) [95].
Fig. 11.

Structure of Disulfiram and its reaction with active site cysteine of urease.
Also novel selenoorganic bacterial urease inhibitors based on a 1,2-benzisoselenazol-3(2H)-one scaffold are acting by binding this sensitive cysteine in H. pylori and S. pasteurii enzymes [96]. The most active appeared to be ebselen (Fig. 12, compound 35), an agent of anti-inflammatory, anti-oxidant and cytoprotective activity studied as a potential drug against reperfusion injury, stroke, hearing loss, tinnitus and bipolar disorder. Molecular modeling had shown its preferable binding resulting from both complexation of nickel ion by carbonyl atom of the molecule and formation of sulfur-selenium bond with cysteine 322 (Fig. 12).
Fig. 12.

Structure of ebselen and the mode of its binding by S. pasteurii urease.
Organophosphorus compounds as transition state analogs
Competitive inhibition of urease by phosphate was first described as far as in 1934 [97] and intensively studied up to 2001 when its binding mode to urease from S. pasteurii was determined by crystallography [37]. It is a relatively weak inhibitor, whereas its amides (phosphoramidates) rank amongst the most active ones with their high efficiency being well justified by the crystal structures of complex of diamidophosphoric acid with S. pasteurii urease (compound 36, Fig. 13) [35]. This analysis had shown that high activity of this compound is apparently related to its close similarity to the transition state of the enzymatic reaction and tight binding to the active metallocenter.
Fig. 13.

Structures of phosphoramidates 36, 37, 38 and 39 and the mode of the binding of compound 36 by S. pasteurii urease.
Urea is a primary solid nitrogen fertilizer in the market because of the restriction against the use of ammonium nitrate, which may be employed as explosives, and the high price of ammonium sulfate. Its hydrolysis by bacterial ureases results in the loss of ammonia, which, besides the economic significance for the farmers, may have negative ecological impact on atmospheric quality. Since phosphoramidates are relatively cheap compounds they are considered as agents reducing the losses of ammonia from urease fertilizers. This is well exemplified by introduction of new formulation of an old inhibitor – N-(n-butyl)thiophosphoric triamide (NBPT, compound 37, ARM U™) to agriculture in 2017 [98], [99]. Recently evaluated binding of this inhibitor to S. pasteurii urease showed that NBPT, after binding to the enzyme, is hydrolyzed yielding monoamidothiophosphoric acid (MATP, compound 38), which is effectively bound to the two Ni(II) ions in the active site (Fig. 13) [38]. Thus, NBPT may be classified as suicide substrate of this enzyme.
Quite recently a big library of structurally variable phosphoramidates was prepared and studied against jack ban urease. Structure–activity relationship analyses suggest that the presence of cyclohexylamine group (see the structure of representative compound 39, Fig. 13) is an important feature associated with enhanced activities [100].
Unfortunately, the phosphoramidate P—N bond is not stable in aqueous solutions, which limits their further applications. Recently, compounds containing a carbon-to-phosphorus bond linkage (phosphonates and phosphinates) emerged as an alternative to overcome this hydrolytic liability. If considering that simple phosphoramidate (36) mimics the tetrahedral transition state of urea hydrolysis aminomethyl(P-methyl)phosphinic acid (Fig. 14, compound 40) might be treated as its extendent analog. Similarly to phosphoramidate 36 it appeared to be weak inhibitor of ureases from Proteus vulgaris and S. pasteurii. Further, enhanced by molecular modeling, modifications of its structure were done by derivatization of its amino moiety [101]. Indeed, Simple N-methylation of the parent structure to compound 41 gave a 20-fold increase in the inhibitory activity. Further modifications of the parent structure 40 resulted in several big libraries of phosphinate inhibitors with compounds 42, 43, 44 and 45 (Fig. 14) being the most potent, submicromolar inhibitors of the enzyme [102], [103], [104], [105].
Fig. 14.

Phosphinic acid inhibitors of urease.
The biological relevance of these inhibitors was verified in vitro against an ureolytically active Escherichia coli Rosetta host that expressed H. pylori urease and against a reference strain, H. pylori J99 [104]. The majority of the studied compounds exhibited urease-inhibiting activity in these whole-cell systems with bis(N-methylaminomethyl)phosphinic acid (Fig. 14, compound 46) being the most effective.
Basing on the results presented in a study describing the crystal structure of S. pasteurii urease complexed with citrate [27] a new scaffold of phosphonate (phosphinate)/carboxylate was proposed. It imitates the 1,2-dicarboxylate portion of citrate (Fig. 1). As a result, one of the most potent organophosphorus inhibitors of urease, α-phosphonomethyl-p-methylcinnamic acid (Fig. 15, compound 47), was identified [106].
Fig. 15.

Compound 47, an inhibitor of S. pasteurii urease and its binding to active site of the enzyme.
Molecular modeling has shown that it is so highly complementary to the enzyme active site that any modification of its structure resulted in diminished activity (Fig. 15).
Coordination complexes
Complexes of simple organic molecules with metal ions are applied as inhibitors of enzymes on the premise that they may either act through substitution of one of the ligands by specific amino acid side chains of the enzyme or by such preorganization of relatively simple molecules into complex scaffold that is complementary to the structure of binding sites of the enzyme. Most likely, in the case of urease, only this second mean has been used.
Complexation of copper (II) and zinc (II) ions by Schiff bases formed between simple analogs of salicylic aldehydes and phenylethylamines resulted in formation of either polymeric structures (these are not useful as inhbitiors) or dimeric ones, in which two molecules of ligand are bound to central copper ion (see the representative structure 48 in Fig. 16) [107]. The latter ones appeared far more effective inhibitors of jack bean urease than parent Schiff bases. Simple ternary cobalt (II) complexes with 1,2-bis(2-methoxy-6-formylphenoxy)ethane (obtained by reacting of vanillin with 1,2-dribromoethane) and phenylalanine, tryptophan (compound 49, Fig. 16) or methionine also appeared to be moderate inhibitors of jack bean urease [108]. Molecular modeling proved that they are well fitting to the binding cavity of this urease.
Fig. 16.

Metal ion complexes as inhibitors of urease.
Quite complex structure is a ternary chelate composed of two copper (II) ions with four molecules of ((E)-3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)acrylic acid (simple derivative of cinnamic acid) and two molecules of DMSO. It is potent, submicromolar inhibitor of jack bean urease [109].
For the construction of various supramolecular structures, silver as a d10 metal is quite frequently used because of its flexible coordination sphere and the fluid nature of interaction between silver and multifunctional ligands. Recently silver (I) carboxylate complexes based on the substituted trans-cinnamic acids, 1,4-benzodioxane-6-carboxylic acid and propyl-substituted imidazole-4,5-dicarboxylic acid (compound 50), which are the promising candidates for urease inhibitors [110], [111], [112]. In solution they form a polymeric structure and the mode of their binding do the enzyme was not evaluated.
Conclusions
Because of medicinal and agricultural importance of ureases the search for their inhibitors is quite extensive. In order to achieve this goal all he standard techniques of inhibitor design were applied. In many cases they were enforced by the application of computer-assisted inhibitor design. Despite of the detailed knowledge of the architecture of active and binding sites of ureases, the design, synthesis and evaluation of new inhibitors is still challenging and difficult. It is well illustrated by the fact that the most active ones exhibit submicromolar inhibitory constants. This results from that the binding sites are quite spacious and flexible and thus variable and difficult to predict mechanisms of inhibition might be utilized. The future perspective seems to relay on better understanding of binding preferences of the enzymes from different sources and on the application of computer-aided prediction of potentially active compounds.
Conflict of interest
The authors have declared no conflict of interest.
Compliance with Ethics Requirements
This article does not contain any studies with human or animal subjects.
Acknowledgements
This work was supported by statuary grants of Wrocław University of Science and Technology. The Biovia Discovery Studio package was used under a Polish country-wide license. The use of software resources (Biovia Discovery Studio program package) of the Wrocław Centre for Networking and Supercomputing is also kindly acknowledged.
Biographies

Paweł Kafarski was born in 1949. He studied chemistry at Wrocław University of Science and Technology where his scientific adventure started with M. Sc. Thesis, completed in 1971, followed by doctoral thesis (1977) both under the supervision of Prof. Przemysław Mastalerz. Prof. Mastalerz subsequently supervised his scientific career for many years. In his laboratory Paweł Kafarski worked on the synthesis of organophosphorus compounds and their potential biological activities. In 1976/1977 he interrupted his PhD studies and spent nine months at Marquette University at Milwaukee working in the laboratory of Prof. Sheldon E. Cremer on the synthesis of phosphetanes. In 1989 he spent six months in the laboratory of Prof. Henri-Jean Cristau at Ecole Nationale Superieure de Chimie at Montpellier elaborating the procedure for the synthesis of phosphono peptides containing P-N bond in their structures. Scientific activity of Paweł Kafarski was concentrated on elaboration of synthetic procedures suitable to produce phosphonate inhibitors (most likely in enantiomerically pure forms) of physiologically important enzymes, to mention only: aminopeptidases (targets for anti-cancer and anti-malarial drugs), cathepsin C (potential anti-tumor agents), glutamine synthetase (target for ant-tuberculosis agents), urease (antibacterials for treatment of stomach ulcer and stone formation in urinary tract) or L-phenylalanine ammonia lyase (potential herbicides). The design of ligands for these targets relied on knowledge of molecular mechanisms of the catalyzed reactions and on three-dimensional structures of the chosen proteins. He coauthored over 250 paper, which are well cited in the literature (over 5000 independent citations)

Michał Talma was born in 1991. He studied biotechnology at the Faculty of Chemistry, Wrocław University of Scince and Technology, Poland. He gained the M.Sc. degree in 2015 on immobilization of drugs in porous structures under supervision of Dr. Łukasz Radosiński. Currently, he is a Ph.D. student at the Department of Bioorganic Chemistry with Prof. Artur Mucha as supervisor. The topic of his thesis involves synthesis of bioactive phosphinic compounds starting from the Morita-Baylis-Hillman adducts.
Footnotes
Peer review under responsibility of Cairo University.
References
- 1.Wöhler F. Ueber künstliche Bildung des Harnstoffs. Ann Phys. 1828;88:253–256. [Google Scholar]
- 2.Yao M., Tung W., Chen X., Zhan C.G. Reaction pathways and free energy profiles for spontaneous hydrolysis of urea and tetramethylurea: unexpected substituent effects. Org Biomol Chem. 2013;11:7595–7605. doi: 10.1039/c3ob41055b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krajewska B., Ureases I. Functional, catalytic and kinetic properties: a review. J Mol Catal B. 2009;59:9–21. [Google Scholar]
- 4.Callahan B.P., Yuan Y., Wolfenden R.J. The burden borne by urease. Am Chem Soc. 2005;127:10828–10829. doi: 10.1021/ja0525399. [DOI] [PubMed] [Google Scholar]
- 5.Real-Guerra R, Stanisçuaski,F, Carlini CR. Chapter 15. Soybean urease: over a hundred years of knowledge. In: Board JE editor. A comprehensive survey of international soybean research – genetics, physiology, agronomy and nitrogen relationships. InTech; 2013, p. 318–39.
- 6.Sumner J.B. Isolation and crystallization of the enzyme urease. J Biol Chem. 1926;1926(69):435–441. [Google Scholar]
- 7.Konieczna I., Żarnowiec P., Kwinkowski M., Kolesińska B., Frączyk J., Kamiński Z. Bacterial urease and its role in long-lasting human diseases. Curr Pep Sep Sci. 2012;13:789–806. doi: 10.2174/138920312804871094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mobley HLT. Chapter 16. Urease. In: Mobley HLT, Mendz GL, Stuart L, editors. Helicobacter pylori: physiology and genetics. American Society of Microbiology Press; 2001.
- 9.Hassan S.T.S., Šudomová M. The development of urease inhibitors: what opportunities exist for better treatment of Helicobacter pylori infection in children? Children. 2017;4:art.2. doi: 10.3390/children4010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao S., Wang J., Zheng N., Bu D., Sun P., Yu Z. Reducing microbial ureolytic activity in the rumen by immunization against urease therein. Vet Res. 2015;11:art.94. doi: 10.1186/s12917-015-0409-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin D., Zhao S., Zheng N., Wang J. Urea metabolism and regulation by rumen bacterial urease in ruminants – a review. Ann Anim Sci. 2017 [Google Scholar]
- 12.Cameron K.C., Di H. Jj, Moir J.L. Nitrogen losses from the soil/plant system: a review. Ann Appl Biol. 2013;62:145–173. [Google Scholar]
- 13.Li Q., Cui, Liu X., Roelcke M., Pasda G., Zerulla W. A new urease-inhibiting formulation decreases ammonia volatilization and improves maize nitrogen utilization in North China Plain. Sci Rep. 2017;7:art.43853. doi: 10.1038/srep43853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krajewska B. A combined temperature-pH study of urease kinetics. Assigning pKa values to ionizable groups of the active site involved in the catalytic reaction. J Mol Catal. 2016;124:70–76. [Google Scholar]
- 15.Maroney M.J., Ciurli S. Nonredox nickel enzymes. Chem Rev. 2014;114:4206–4228. doi: 10.1021/cr4004488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hausinger RP, Karplus PA. Urease. In: Handbook on metalloproteins. Wiley Online Library; 2016.
- 17.Amtul Z., Rahman A., Siddiqui R., Choudhary M. Chemistry and mechanism of urease inhibition. Curr Med Chem. 2002;9:1323–1348. doi: 10.2174/0929867023369853. [DOI] [PubMed] [Google Scholar]
- 18.Upadhyay L.S.B. Urease inhibitors: a review. Ind J Biotechnol. 2012;11:381–388. [Google Scholar]
- 19.Macegoniuk K. Inhibitors of bacterial and plants urease. A review. Folia Bio Oecol. 2013;9:9–16. [Google Scholar]
- 20.Modolo L.V., de Souza A.X., Horta L.P., Araujo D.P., de Fátima A. An overview on the potential of natural products as ureases inhibitors: a review. J Adv Res. 2015;6:35–44. doi: 10.1016/j.jare.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kosikowska P., Berlicki Ł. Urease inhibitors as potential drugs for gastric and urinary tract infections: a patent review. Expert Opin Ther Pat. 2011;21:945–957. doi: 10.1517/13543776.2011.574615. [DOI] [PubMed] [Google Scholar]
- 22.Hughes J.P., Rees S., Kalindijan S.B., Philpott K.L. Principles in drug discovery. Brit J Pharmacol. 2011;162:1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jabri E., Carr M.B., Hausinger R.P., Karplus P.A. The crystal structure of urease from Klebsiella aerogenes. Science. 1995;268:998–1004. [PubMed] [Google Scholar]
- 24.Schäfer U.K., Kaltwasser H. Urease from Staphylococcus saprophyticus: purification, characterization and comparison to Staphylococcus xylosus urease. Arch Microbiol. 1994;161:393–399. doi: 10.1007/BF00288948. [DOI] [PubMed] [Google Scholar]
- 25.Ha N.C., Oh S.T., Sung J.Y., Cha K.A., Lee M.H., Oh B.H. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nature Struct Mol Biol. 2001;8:505–509. doi: 10.1038/88563. [DOI] [PubMed] [Google Scholar]
- 26.Balasubramanian A., Ponnuraj K. Crystal structure of the first plant urease from jackb ean: 83 years of journey from its first crystal tomolecular structure. J Mol Biol. 2010;400:274–283. doi: 10.1016/j.jmb.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 27.Benini S., Kosikowska P., Cianci M., Mazzei L., Gonzales Vara A., Berlicki Ł. The crystal structure of Sporosarcina pasteurii urease in a complex with citrate provides new hints for inhibitor design. J Biol Inorg Chem. 2013;18:391–399. doi: 10.1007/s00775-013-0983-7. [DOI] [PubMed] [Google Scholar]
- 28.Mazzei L., Cianci M., Benini S., Bertini L., Musiani F., Ciurli S. Kinetic and structural studies reveal a unique binding mode of sulfite to the nickel center in urease. J Inorg Biochem. 2016;154:42–49. doi: 10.1016/j.jinorgbio.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 29.Khan S., Karim A., Iqbal S. Helicobacter urease: Niche construction at the single molecule level. J Biosci. 2009;34:503–511. doi: 10.1007/s12038-009-0069-4. [DOI] [PubMed] [Google Scholar]
- 30.Carlsson H., Nordlander E. Computational modeling of the mechanism of urease. Bioorg Chem Appl. 2010:8. doi: 10.1155/2010/364891. art. 364891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roberts B.P., Miller B.R., III, Roitberg A.E., Mertz K.R., Jr. Wide-open flaps are key to urease activity. J Am Chem Soc. 2012;134:9934–9937. doi: 10.1021/ja3043239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minkara M.S., Ucisik M.L., Weaver M.N., Mertz K.R., Jr. Molecular dynamics study of Helicobacter pylori urease. J Chem Theory Comput. 2014;10:1852–1862. doi: 10.1021/ct5000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yata V.K., Thapa A., Mattaparthi V.S.K. Structural insight into the binding interactions of modeled structure of Arabidopsis thaliana urease with urea: an in silico study. J Biomol Struct Dyn. 2015;33:845–851. doi: 10.1080/07391102.2014.915765. [DOI] [PubMed] [Google Scholar]
- 34.Benini S., Rypniewski W.R., Wilson K.S., Ciurli S., Mangani S. The complex of Bacillus pasteurii urease with beta-mercaptoethanol from X-ray data at 1.65-A resolution.) J Biol Inorg Chem. 1998;3:268–273. doi: 10.1007/s007750050014. [DOI] [PubMed] [Google Scholar]
- 35.Benini S., Rypniewski W.R., Wilson K.S., Miletti S., Ciurli S., Mangani S. The complex of Bacillus pasteurii urease with acetohydroxamate anion from X-ray data at 1.55 Å resolution. J Biol Inorg Chem. 2000;5:110–118. doi: 10.1007/s007750050014. [DOI] [PubMed] [Google Scholar]
- 36.Benini S., Rypniewski W.R., Wilson K.S., Miletti S., Ciurli S., Mangani S. A new proposal for urease mechanism based on thecrystal structures of the native and inhibited enzyme from Bacillus pasteurii: Why urea hydrolysis costs two nickels. Struct Fold Des. 1999;7:205–216. doi: 10.1016/S0969-2126(99)80026-4. [DOI] [PubMed] [Google Scholar]
- 37.Benini S., Rypniewski W.R., Wilson K.S., Ciurli S., Mangani S. Structure-based rationalization of urease inhibition by phosphate: novel insights into the enzyme mechanism. J Biol Inorg Chem. 2001;6:778–780. doi: 10.1007/s007750100254. [DOI] [PubMed] [Google Scholar]
- 38.Mazzei L., Cianci M., Contaldo U., Musiani F., Ciurli S. Urease inhibition in the presence of N-(n-butyl)thiophosphoric triamide, a suicide substrate: structure and kinetics. Biochemistry. 2017;56:5391–5404. doi: 10.1021/acs.biochem.7b00750. [DOI] [PubMed] [Google Scholar]
- 39.Benini S., Cianci M., Mazzei L., Ciurli S. Fluoride inhibition of Sporosarcina pasteurii urease: structure and thermodynamics. J Biol Inorg Chem. 2014;19:1243–1261. doi: 10.1007/s00775-014-1182-x. [DOI] [PubMed] [Google Scholar]
- 40.Benini S., Rypniewski W.R., Wilson K.S., Mangani S., Ciurli S. Molecular details of urease inhibition by boric acid: insights into the catalytic mechanism. J Am Chem Soc. 2004;126:3714–3715. doi: 10.1021/ja049618p. [DOI] [PubMed] [Google Scholar]
- 41.Mazzei L., Cianci M., Musiani F., Lente G., Palombo M., Ciurli S. Inactivation of urease by catechol: kinetics and structure. J Inorg Chem. 2017;166:182–189. doi: 10.1016/j.jinorgbio.2016.11.016. [DOI] [PubMed] [Google Scholar]
- 42.Mazzei L., Cianci M., Musiani F., Ciurli S. Inactivation of urease by 1,4-benzoquinone: chemistry at the protein surface. Dalton Trans. 2016;45:5455–5459. doi: 10.1039/c6dt00652c. [DOI] [PubMed] [Google Scholar]
- 43.Pearson M.A., Michel L.O., Hausinger R.P., Karplus P.A. Structures of Cys319 variants and acetohydroxamate-inhibited Klebsiella aerogenes urease. Biochemistry. 1997;36:8164–8172. doi: 10.1021/bi970514j. [DOI] [PubMed] [Google Scholar]
- 44.Saeed A., ur-Rehman S., Channar P.A., Larik F.A., Abbas Q., Hasan M. Jack bean urease inhibitors, and antioxidant activity based on palmitic acid derived 1-acyl-3-arylthioureas: synthesis, kinetic mechanism and molecular docking studies. Drug Res (Stuttg) 2018;67:596–605. doi: 10.1055/s-0043-113832. [DOI] [PubMed] [Google Scholar]
- 45.Rauf A., Shahzad S., Bajda M., Yar M., Ahmed F., Hussain N. Design and synthesis of new barbituric- and thiobarbituric acid derivatives as potent urease inhibitors: structure activity relationship and molecular modeling studies. Bioorg Med Chem. 2015;23:6049–6058. doi: 10.1016/j.bmc.2015.05.038. [DOI] [PubMed] [Google Scholar]
- 46.Khan K.M., Ali M., Waldoof M., Zheer-ul-Haq, Khan M., Lodhi M.A. Molecular modeling-based antioxidant arylidene barbiturates as urease inhibitors. J Mol Graph Mod. 2011;30:153–156. doi: 10.1016/j.jmgm.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 47.Barakat A., SL-Majid A.M., Kofty G., Arshad F., Yousuf S., Iqbal Choudhary M. Synthesis and dynamics studies of barbituric acid derivatives as urease inhibitors. Chem Centr J. 2015;9:63–77. doi: 10.1186/s13065-015-0140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fazal R., Ali M., Ullah S., Rashid U., Ullah H., Taha M. Development of bis-thiobarbiturates as successful urease inhibitors and their molecular modeling studies. Chin Chem Lett. 2016;27:693–697. [Google Scholar]
- 49.Saeed A., Mahmood S.-U., RAfiq M., Asraf Z., Jebeen F., SEo S.-Y. Iminothiazoline-sulfonamide hybrids as jack beanurease inhibitors; Synthesis, kinetic mechanism and computational molecular modeling. Chem Biol Drug Des. 2016;87:434–443. doi: 10.1111/cbdd.12675. [DOI] [PubMed] [Google Scholar]
- 50.Rauf A., Nazish K.A., Nassim F.-U.H., Yaqoob A., Qureshi A.M. Synthesis of novel cyanoacetamides derivatives and their urease inhibition studies. Eur J Chem. 2015;6:163–168. [Google Scholar]
- 51.Sheng G.-H., Chen X.-F., Li J., Chen J., Xu Y., Han Y.-W. Synthesis, crystal structures and urease inhibition of N’-(2-Bromobenzylidene)-2-(4-nitrophenoxy)acetohydrazide and N’-(4-Nitrobenzy-lidene)-2-(4-nitrophenoxy)acetohydrazide. Acta Chim Slov. 2015;62:940–946. doi: 10.17344/acsi.2015.1770. [DOI] [PubMed] [Google Scholar]
- 52.Redgrave L.S., Sutton S.B., Webber M.A., Piddock L.V.J. Fluoroquinolone resistance: mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014;22:438–445. doi: 10.1016/j.tim.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 53.Abu-Sini M., Mayyas A., Al-Karablieh N., DArwish R., Al-Hiari Y., Aburjai T. Synthesis of 1,2,3-triazolo[4,5-h]quinolone derivatives with novel anti-microbial properties against Metronidazole resistant Helicobacter pylori. Molecules. 2017;22:841. doi: 10.3390/molecules22050841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abdullah M.A.A., El-Baky R.M.A., Hassan H.A., Abdelhafez E.-S.M.N., Abuo-Rahma G.E.-D.A. Fluoroquinolones as urease inhibitors: anti-Proteus mirabilis activity and molecular docking studies. Am J Microbiol Res. 2016;4:81–84. [Google Scholar]
- 55.Kathrotiya H.G., Patel M.P. Synthesis and identification of b-aryloxyquinoline based diversely fluorine substituted N-aryl quinolone derivatives as a new class of antimicrobial, antituberculosis and antioxidant agents. Eur J Med Chem. 2013;63:675–684. doi: 10.1016/j.ejmech.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 56.Zaborska W., Krajewska B., Kot M., Karcz W. Quinone-induced inhibition of urease: elucidation of its mechanisms by probing thiol groups of the enzyme. Bioorg Chem. 2007;35:233–242. doi: 10.1016/j.bioorg.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 57.Kosikowska P., Berlicki Ł. Urease inhibitors as potentialdrugs for gastric and urinary tract infections: a patent review. Exp Opin Therapeut Pat. 2011;21:945–957. doi: 10.1517/13543776.2011.574615. [DOI] [PubMed] [Google Scholar]
- 58.Abdullah M.A.A., Abuo-Rahma G.E.-D.A.A., Abdelhafez E.-S.M.N., Hassan H.A., El-Baky R.M.A. Design, synthesis, molecular docking, anti-Proteus mirabilis and urease inhibition of new fluoroquinolone carboxylic acid derivatives. Bioorg Chem. 2017;70:1–11. doi: 10.1016/j.bioorg.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 59.Nisar M., Ali Khan S., Raza Shah M., Khan A., FArooq U., Uddin G. Moxifloxacin-capped noble metal nanoparticles as potential urease inhibitors. New J Chem. 2015;39:8080–8086. [Google Scholar]
- 60.Loes A.N., Ruyle N., Arvizu M., Gresko A.L., Deutch C.E. Inhibition of urease activity in the urinary tract pathogen Staphylococcus saprophyticus. Lett Appl Microbiol. 2013;58:31–41. doi: 10.1111/lam.12153. [DOI] [PubMed] [Google Scholar]
- 61.Mansoor F., Anis I., Khan A., Marasini B.P., Iqbal Choudhary M., Raza Shah M. Urease inhibitory constituents from Daphne retusa. J Asian Nat Prod Res. 2014;16:210–215. doi: 10.1080/10286020.2013.837457. [DOI] [PubMed] [Google Scholar]
- 62.Uddin G., Ismail, Rauf A., Raza M., Khan H., Naruddin Urease inhibitory profile of extracts and chemical constituents of Pistacia atlantica ssp. cabulica Stocks. Nat Prod Res. 2016;12:1411–1416. doi: 10.1080/14786419.2015.1062378. [DOI] [PubMed] [Google Scholar]
- 63.Chen Y., Liao J., Chen M., Huang Q., Lu Q. Gossypol: new class of urease inhibitors, molecular docking and inhibition assay. J Chem Pharm Res. 2015;7:10–15. [Google Scholar]
- 64.Xiao Z.-P., Peng Z.-Y., Dong J.-J., He J., Ouyang H., Feng Y.-T. Synthesis, structureeactivity relationship analysis and kinetics study of reductive derivatives of flavonoids as Helicobacter pylori urease inhibitors. Eur J Med Chem. 2013;63:685–695. doi: 10.1016/j.ejmech.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 65.Xiao Z.-P., Wang X.-D., Peng Z.-Y., Huang S., Yang P., Li Q.-S. Molecular docking, kinetics study, and structure–activity a of quercetin and its analogous as Helicobacter pylori urease inhibitors. Agric Food Chem. 2012;60:10572–10577. doi: 10.1021/jf303393n. [DOI] [PubMed] [Google Scholar]
- 66.Tan L., Su J., Wu D., Yu X., Su Z., He J. Kinetics and mechanism study of competitive inhibition of jack-bean urease by baicalin. Sci World J. 2013:art.879501. doi: 10.1155/2013/879501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu X.-D., Zheng R.B., Xie J.H., Su J.-Y., Huang X.-Q., Wang Y.H. Biological evaluation and molecular docking of baicalin and scutellarin as Helicobacter pylori urease inhibitors. J Ethnopharmacol. 2015;162:69–78. doi: 10.1016/j.jep.2014.12.041. [DOI] [PubMed] [Google Scholar]
- 68.Lee B.W., Park I.H., Yim D., Coi S.S. Comprehensive evaluation of the anti-Helicobacter pylori activity of scutellariae radix. Nat Prod Sci. 2017;23:46–52. [Google Scholar]
- 69.Babu T.M.C., Rajesh S.S., Bhaskar B.V., Devi S., Rammohan A., Sivaraman T. Molecular docking, molecular dynamics simulation, biological evaluation and 2D QSAR analysis of flavonoids from Syzygium alternifolium as potent anti-Helicobacter pylori agents. RSC Adv. 2017;7:18277–18292. 46-53. [Google Scholar]
- 70.Amin M., Anwar F., Naz F., Mehmood T., Saari N. Anti-Helicobacter pylori and urease inhibition activities ofsome traditional medicinal plants. Molecules. 2013;18:2135–2149. doi: 10.3390/molecules18022135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mahernia S., Bagherzadeh K., Mojab F., Amanlou M. Urease inhibitory activities of some commonly consumed herbal medicines. Iran J Pharm Res. 2015;14:943–947. [PMC free article] [PubMed] [Google Scholar]
- 72.Bai S., Bharti P., Seasotiya L., Malik A., Dalal S. In vitro screening and evaluation of some Indian medicinal plants for their potential to inhibit Jack bean and bacterial ureases causing urinary infections. Pharm Biol. 2015;53:326–333. doi: 10.3109/13880209.2014.918158. [DOI] [PubMed] [Google Scholar]
- 73.Hassan S.T.S., Žemlička M. Plant-derived urease inhibitors as alternative chemotherapeutic agents. Arch Pharm. 2016;349:507–522. doi: 10.1002/ardp.201500019. [DOI] [PubMed] [Google Scholar]
- 74.Golbabei S., Bazl R., Golestanian S., Nabati F., Omrany Z.B., Yousefi B. Urease inhibitory activities of β- boswellic acid derivatives. J Pharm Sci. 2013;21:2. doi: 10.1186/2008-2231-21-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li C., Xie J., Chen X., Mo Z., Wu W., Liang Y. Comparison of Helicobacter pylori urease inhibition by rhizoma Coptidis, cortex Phellodendri and berberine: mechanisms of interaction with the sulfhydryl group. Planta Med. 2016;82:305–311. doi: 10.1055/s-0035-1558229. [DOI] [PubMed] [Google Scholar]
- 76.Zhou J.T., Li C.L., Tan L.H., Xu Y.F., Liu Y.H., Mo Z.Z. Inhibition of Helicobacter pylori and its associated urease by palmatine: pnvestigation on the potential mechanism. PLoS ONE. 2017;12:e0168944. doi: 10.1371/journal.pone.0168944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tan L., Li C., Chen H., Mo Z., Zhou J., Liu Y. Epiberberine, a natural protoberberine alkaloid, inhibits urease of Helicobacter pylori and jack bean: Susceptibility and mechanism. Eur J Pharm Sci. 2017;110:77–86. doi: 10.1016/j.ejps.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 78.Mo Z.-Z., Wang X.F., Zhang X., Su J.-Y., Chen H.-M., Liu Y.H. Andrographolide sodium bisulphite-induced inactivation of urease: inhibitory potency, kinetics and mechanism. BMC Compl Alternat Med. 2015;15:238. doi: 10.1186/s12906-015-0775-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ranjbar-Omid M., Arzanlou M., Amani M., Al-Hashem S.K.S., Mozafari N.A., Doghaheh H.P. Allicin from garlic inhibits the biofilm formation and urease activity of Proteus mirabilis in vitro. FEMS Microbiol Lett. 2015;362:fnv049. doi: 10.1093/femsle/fnv049. [DOI] [PubMed] [Google Scholar]
- 80.Mathialagan R., Mansor N., Al-Khateeb B., Mohamad M.H., Shamsuddin M.R. Evaluation of allicin as soil urease inhibitor. Procedia Eng. 2017;184:449–459. [Google Scholar]
- 81.Sahin H. Honey as an apitherapic product: its inhibitory effect on urease and xanthine oxidase. J Enz Inhib Med Chem. 2015;31:491–494. doi: 10.3109/14756366.2015.1039532. [DOI] [PubMed] [Google Scholar]
- 82.Rückriemen J., Klemm O., Henl T. Manuka honey (Leptospermum scoparium) inhibits jack bean urease activity due to methylglyoxal and dihydroxyacetone. Food Chem. 2017;230:540–546. doi: 10.1016/j.foodchem.2017.03.075. [DOI] [PubMed] [Google Scholar]
- 83.Kolyali S., Baltas N., Sahin H., Karaoglu S. Evaluation of anti-Helicobacter pylori activity and urease inhibition by some Turkish authentic honeys. J Sci Food Eng. 2017;7:67–73. [Google Scholar]
- 84.Matongo F., Nwodo U.U. In vitro asessment of Helicobacter pylori ureases inhibition by honey fractions. Arch Med Res. 2014;45:540–546. doi: 10.1016/j.arcmed.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 85.Hashem-Dabaghian F., Agah M., Taghavi-Shirazi M., Ghobadi A. Combination of Nigella sativa and honey in eradication of gastric Helicobacter pylori infection. Iran Red Crescent Med J. 2016;18:23771. doi: 10.5812/ircmj.23771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Arshad T., Khan K.M., Rasool N., Salar U., Hussain S., Asghar H. 5-Bromo-2-aryl benzimidazole derivatives as non-cytotoxic potential dual inhibitors of a-glucosidase and urease enzymes. Bioorg Chem. 2017;72:21–31. doi: 10.1016/j.bioorg.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 87.Hanif M., Shoaib K., Saleem M., Rama N.H., Zaib S., Iqbal J. Synthesis, urease inhibition, antioxidant, antibacterial, and molecular docking studies of 1,3,4-oxadiazole derivatives. ISRN Pharmacol. 2012:art.928901. doi: 10.5402/2012/928901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Laothi M.A., Shams S., Khan K.M. Thiazolidine esters: new potent urease inhibitors. J Chem Soc Pak. 2014;36:858–864. [Google Scholar]
- 89.Horta L.P., Mota Y.C.C., Barbosa G.M., Braga T.C., Marriel I.E., de Fátima A. J Braz Chem Soc. 2016;27:1512–1519. [Google Scholar]
- 90.Hakimi A.M., Lashgari N., Mahernia S., Ziarani G.M., Amanlou M. Facile one-pot four-component synthesis of 3,4-dihydro-2-pyridone derivatives: novel urease inhibitor scaffold. Res Pharm Sci. 2017;12:353–363. doi: 10.4103/1735-5362.213980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Alvandifar F., Tahghighi A., Sabourian R., Firoozpour L., Mahdavi M., Saniee P. J Chem Pharm Res. 2015;7:2512–2519. [Google Scholar]
- 92.Menteşe E., Bektaş H., Sokmen B.B., Emirik M., Çakir D., Kahvecii B. Synthesis and molecular docking study of some 5,6-dichloro-2- cyclopropyl-1H-benzimidazole derivatives bearing triazole, oxadiazole, and imine functionalities as potent inhibitors of urease. Bioorg Med Chem Lett. 2017;27:3014–3018. doi: 10.1016/j.bmcl.2017.05.019. [DOI] [PubMed] [Google Scholar]
- 93.Adsul L.K., Bandgar B.P., Chavan H.V., Jalde S.S., Dhakane V.D., Shirfule A.L. Synthesis and biological evaluation of novel series of aminopyrimidine derivatives as urease inhibitors and antimicrobial agents. J Enz Inhib Med Chem. 2013;28:1316–1323. doi: 10.3109/14756366.2012.740477. [DOI] [PubMed] [Google Scholar]
- 94.Macegoniuk K., Kowalczyk R., Rudzińska A., Psurski M., Wietrzyk J., Berlicki Ł. Potent covalent inhibitors of bacterial urease identified by activity-reactivity profiling. Bioorg Med Chem Lett. 2017;27:1346–1350. doi: 10.1016/j.bmcl.2017.02.022. [DOI] [PubMed] [Google Scholar]
- 95.Díaz-Sánchez Á.G., Alvarez-Parrilla E., Martínez-Martínez A., Aguirre-Reyes L., Orozpe-Olvera J.A., Ramos-Soto M.A. Inhibition of urease by Disulfiram, an FDA-approved thiol reagent used in humans. Molecules. 2016;21:1628. doi: 10.3390/molecules21121628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Macegoniuk K., Grela E., Palus J., Rudzińska-Szostak E., Grabowiecka A., Biernat M. 2-Benzisoselenazol-3(2H)-one derivatives as a new class of bacterial urease inhibitors. J Med Chem. 2016;59:8125–8133. doi: 10.1021/acs.jmedchem.6b00986. [DOI] [PubMed] [Google Scholar]
- 97.Howell S.P., Sumner J.B. The specific effects of buffers upon urease activity. J Biol Chem. 1934;104:619–626. [Google Scholar]
- 98.Grant C.A. Use of NBPT and ammonium thiosulphate as urease inhibitors with varying surface placement of urea and urea ammonium nitrate in production of hard red spring wheat under reduced tillage management. Can J Plant Sci. 2014;94:329–335. [Google Scholar]
- 99.Silva A.G.B., Sequeira C.H., Sermarini R.A., Otto R. Urease inhibitor NBPT on ammonia volatilization and crop productivity: a meta-analysis. Agron J. 2017;109:1–13. [Google Scholar]
- 100.Oliveira F.M., Barbosa L.C.A., Demuner A.J., Maltha C.R.A., Pereira S.R., Horta L.P. Synthesis, molecular properties and DFT studies of new phosphoramidates as potential urease inhibitors. Med Chem Res. 2014;23:5174–5187. [Google Scholar]
- 101.Vassiliou S., Grabowiecka A., Kosikowska P., Yiotakis A., Kafarski P., Berlicki Ł. Design, synthesis and evaluation of novel organophosphorus inhibitors of bacterial ureases. J Med Chem. 2008;51:5736–5744. doi: 10.1021/jm800570q. [DOI] [PubMed] [Google Scholar]
- 102.Vassiliou S., Kosikowska P., Grabowiecka A., Yiotakis A., Kafarski P., Berlicki Ł. Computer-aided optimization of phosphinic inhibitors of bacterial ureases. J Med Chem. 2010;53:5597–5606. doi: 10.1021/jm100340m. [DOI] [PubMed] [Google Scholar]
- 103.Vassiliou S., Grabowiecka A., Kosikowska P., Berlicki Ł. Three component Kabachnik-Fields condensation leading to substituted aminomethane-P-hydroxymethylphosphonic acids as a tool for screening of bacterial urease inhibitors. ARKIVOC. 2012:33–43. [Google Scholar]
- 104.Berlicki Ł., Bochno M., Grabowiecka A., Białas A., Kosikowska P., Kafarski P. N-Substituted aminomethanephosphonic and aminomethane-P-methylphosphinic acids as inhibitors of ureases. Amino Acids. 2012;42:1937–1945. doi: 10.1007/s00726-011-0920-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Macegoniuk K., Dziełak A., Mucha A., Berlicki Ł. Bis(aminomethyl)-phosphinic acid, a highly promising scaffold for the development of bacterial urease inhibitors. ACS Med Chem Lett. 2015;6:146–150. doi: 10.1021/ml500380f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ntatsopoulos V., Vassiliou S., Macegoniuk K., Berlicki Ł., Mucha A. Novel organophosphorus scaffolds of urease inhibitors obtained by substitution of Morita-Baylis-Hillman adducts with phosphorus nucleophiles. Eur J Med Chem. 2017;133:107–120. doi: 10.1016/j.ejmech.2017.03.070. [DOI] [PubMed] [Google Scholar]
- 107.Dong X., Li Y., Li Z., Cui Y., Zhu H. Synthesis, structures and urease inhibition studies of copper(II) and nickel(II) complexes with bidentate N, O-donor Schiff base ligands. J Inorg Biochem. 2012;108:22–29. doi: 10.1016/j.jinorgbio.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 108.Wang H., Zhang X., Zhao Y., Zhang D., Jin F., Fan Y. Three Co(II) complexes with a sexidentate N2O4-donor bis-Schiff base ligand: synthesis, crystal structures, DFT studies, urease inhibition and molecular docking studies. J Mol Struct. 2017;1148:496–504. [Google Scholar]
- 109.Chen X.-N., Wang C.F., Kong S., Zhou X., Zhang C.-Y., Sheng G.H. Structure and urease inhibitory activity of copper(II) complex with (E)-3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)acrylic acid. J Struct Chem. 2017;58:797–803. [Google Scholar]
- 110.Li X., Wang Y., Li Y., Gou Y., Wang Q. Synthesis, characterization and biological evaluation of two silver(I) trans-cinnamate complexes as urease inhibitors Z. Anorg Allg Chem. 2014;640:423–428. [Google Scholar]
- 111.Li Y., Jing H., Ma C., Wang Q. Synthesis, solid state structures and urease inhibitory activities of two silver(I) complexes with 1,4-benzodioxane-6 –carboxylate Transit. Met Chem. 2015;40:743–748. [Google Scholar]
- 112.Li Y., Lu X., Jing H., Wang Q., Cai Y. Synthesis, structures and antimicrobial activities of silver(I) complexes derived from 2-propyl-1H-imidazole-4,5-dicarboxylic acid. Inorg Chim Acta. 2017;467:117–122. [Google Scholar]