

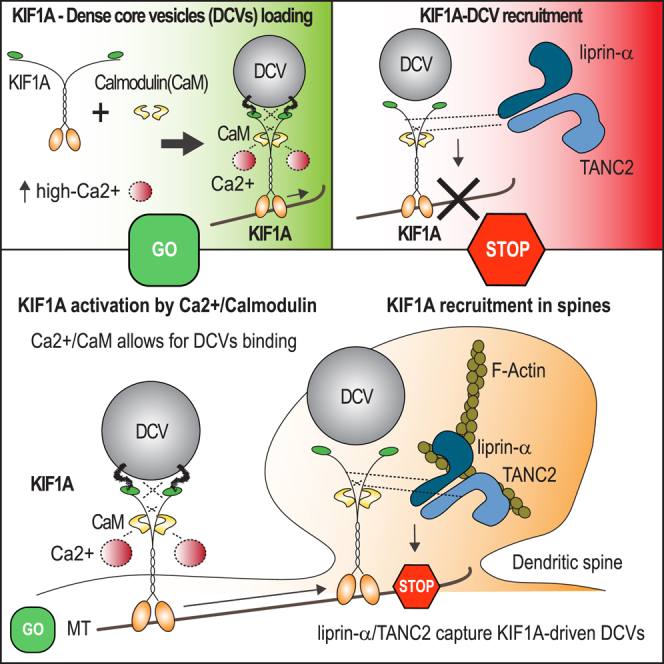
Summary
Tight regulation of neuronal transport allows for cargo binding and release at specific cellular locations. The mechanisms by which motor proteins are loaded on vesicles and how cargoes are captured at appropriate sites remain unclear. To better understand how KIF1A-driven dense core vesicle (DCV) transport is regulated, we identified the KIF1A interactome and focused on three binding partners, the calcium binding protein calmodulin (CaM) and two synaptic scaffolding proteins: liprin-α and TANC2. We showed that calcium, acting via CaM, enhances KIF1A binding to DCVs and increases vesicle motility. In contrast, liprin-α and TANC2 are not part of the KIF1A-cargo complex but capture DCVs at dendritic spines. Furthermore, we found that specific TANC2 mutations—reported in patients with different neuropsychiatric disorders—abolish the interaction with KIF1A. We propose a model in which Ca2+/CaM regulates cargo binding and liprin-α and TANC2 recruit KIF1A-transported vesicles.
Keywords: neuron, transport, dendritic spines, synapse, KIF1A, scaffold, calmodulin, calcium, TANC2, liprin-α
Graphical Abstract
Highlights
-
•
KIF1A directly interacts with CaM and with the scaffolds liprin-α and TANC2
-
•
KIF1A is regulated by a Ca2+/CaM-dependent mechanism, which allows for DCV loading
-
•
Liprin-α and TANC2 are static PSD proteins that are not part of the KIF1A-DCV complex
-
•
KIF1A-driven DCVs are recruited to dendritic spines by liprin-α and TANC2
Stucchi et al. show that KIF1A-dependent transport is regulated by CaM, liprin-α, and TANC2. KIF1A binding to DCVs is controlled by a Ca2+/CaM molecular mechanism, and KIF1A-driven DCVs are recruited in dendritic spines by the PSD scaffolds liprin-α and TANC2.
Introduction
Kinesins and dyneins are motor proteins responsible for cargo transport and delivery along microtubule tracks. Microtubule-based cargo transport is particularly important in cells with complex geometry, such as neurons (Hirokawa et al., 2010). The neuronal transport machinery delivers proteins, lipids, mRNA, and organelles to the distal sites, as well as controlling cargo degradation and recycling of building blocks. Various cargoes that are transported along microtubules move bidirectionally, exhibiting periods of rapid movements, pauses, and directional switches. The identification of mutations in genes encoding tubulin isoforms, motor proteins, or other components of the trafficking machinery in human patients strongly supports the view that defective cargo transport can directly trigger neurodegeneration (Millecamps and Julien, 2013). It is therefore important to understand how cargo trafficking is initiated and how cargoes are captured at their final destinations. In addition, transport deficits might arise through various indirect mechanisms. For example, the stalling of vesicles could trigger the formation of aggregates within axons that may result in neuronal dysfunction.
Several molecular mechanisms related to microtubules, motors, and cargo interactions were shown to regulate cargo pick-up and delivery (Maeder et al., 2014, Schlager and Hoogenraad, 2009). On one hand, microtubule orientation, length, and spacing in axons and dendrites can control cargo sorting and transport efficiency (Kapitein et al., 2010, Yogev et al., 2016). Additional motor-microtubule mechanisms include tubulin posttranslational modifications (PTMs), microtubule-associated proteins (MAPs), and tubulin isotypes (Gumy et al., 2017, Sirajuddin et al., 2014). On the other hand, transport is regulated on the site of motor-cargo interaction through motor interaction with phospholipids, receptors or integral membrane proteins, scaffolding proteins, and small Rab guanosine triphosphatase (GTPases) and their effector proteins (Maeder et al., 2014, Schlager and Hoogenraad, 2009). Moreover, local subcellular specializations and compartmentalization of specific structures can control local cargo trafficking. For instance, the axon initial segment (AIS) functions as a cargo filter that selectively prevents passage of dendritic vesicles while allowing the entry of axonal cargoes (Leterrier and Dargent, 2014). Furthermore, evidence suggests that during transient microtubule polymerization into dendritic spines, kinesin motors transport cargoes along microtubules (MTs) into spines (Esteves da Silva et al., 2015, McVicker et al., 2016). Despite growing evidence of transport regulation, little is known about how vesicles are loaded on motors and captured at the dendritic spines.
KIF1A, named UNC-104 in Caenorhabditis elegans (C. elegans) and Drosophila, was identified as the primary motor protein first for synaptic vesicles (SVs) (Niwa et al., 2008, Okada et al., 1995) and later for dense core vesicles (DCVs) (Barkus et al., 2008, Lo et al., 2011, Zahn et al., 2004). DCVs, also called secretory vesicles or post-Golgi vesicles, are transported from the Golgi apparatus to the plasma membrane in both axons and dendrites, unlike SVs (de Wit et al., 2006, Lochner et al., 2008). DCVs contain secretory proteins such as brain-derived neurotrophic factor (BDNF) and neuropeptide Y (NPY) and do not coincide with membrane protein transferrin receptor (TfR) (Lipka et al., 2016). The integral membrane protein synaptotagmin 4 (Syt4) is also present on DCVs (Dean et al., 2009). The scaffolding protein liprin-α interacts with the tail region of KIF1A (Shin et al., 2003), and liprin-α mutants in Drosophila impair cargo trafficking, suggesting that liprin-α is part of the KIF1A-cargo complex (Miller et al., 2005). However, in C. elegans, liprin-α is mainly localized to synaptic sites, and its distribution is not affected in KIF1A mutants (Sieburth et al., 2005). An additional model suggests that liprin-α can play an active role in clustering KIF1A-transported SVs (Wagner et al., 2009, Wu et al., 2013). Thus, whether liprin-α is a cargo adaptor linking KIF1A to vesicles or captures KIF1A-driven cargo to synaptic sites remains an open question.
To better understand how KIF1A-driven vesicle trafficking is controlled in neurons, we performed an interactome analysis of KIF1A. Based on those results, we focused on three KIF1A binding partners, the calcium binding protein calmodulin (CaM) and two synaptic scaffolding proteins: liprin-α and TANC2 (tetratricopeptide repeat, ankyrin repeat, and coiled coil containing 2). TANC2 was originally identified as a postsynaptic density (PSD)-95-interacting protein, regulating dendritic spine and synapse function (Han et al., 2010). TANC2 gene mutations are identified in patients with neurological disorders ranging from autism to schizophrenia (de Ligt et al., 2012, Fromer et al., 2014, Iossifov et al., 2012). To gain insights into the role of these three proteins in KIF1A-mediated transport, we used a combination of live-cell imaging and biochemical-proteomic methodology. This approach allowed us to demonstrate that calcium, acting via CaM, enhances KIF1A binding to DCVs and increases vesicle motility. In addition, we show that liprin-α and TANC2 are mainly localized in dendritic spines and recruit KIF1A-driven DCVs to synaptic sites. Altogether, our findings reveal a mechanism for pick-up and delivery of DCVs in dendrites.
Results
Kinesin-3 Family Member KIF1A Interacts with Liprin-α and TANC2
KIF1A contains a classical N-terminal motor domain, followed by three coiled coils, a stalk domain in the middle part, and a pleckstrin homology (PH) domain in the tail region, which in C. elegans is crucial for recognition of phosphatidylinositol 4,5-bisphosphate (PIP2) in cargo vesicle membranes (Figure 1A) (Klopfenstein et al., 2002). To identify KIF1A interactome, we set up a systematic affinity purification-mass spectrometry (AP-MS) screening using different KIF1A tail fragments (Figure 1A). BioGFP-KIF1A fragments were expressed in HEK293 cells, purified with streptavidin beads, and incubated with brain lysates. Co-isolated proteins were then analyzed by mass spectrometry (MS). Among the list of putative interacting proteins detected with this approach, we classified candidate KIF1A binding partners with a probability > 0.98, using SAINT (Significance Analysis of INTeractome) to score our AP-MS data (Figure S1A; Tables S1 and S2) (Choi et al., 2011). Most identified binding partners are associated with the stalk domain of KIF1A (amino acid 657-1105), and most of them belong to the postsynaptic density (PSD) (7.82 = −log10[corrected − p value] Gene Ontology [GO], cellular component) (Figures S1A and S1B). This stalk region is highly similar to MAGUK binding sites (MBSs) found in other kinesin-3 motors, such as KIF13B (Zhu et al., 2016). MBS regions mediate binding with the guanylate kinase (GK) domain, and in the case of KIF13B, MBS is required for the formation of a complex with the GK of the synaptic protein DLG1 (Hanada et al., 2000, Yamada et al., 2007). Although MBS regions are highly conserved, small amino acid sequence variations could drastically change their binding specificities for different scaffolding proteins. In line with this, our results indicate that the MBS domain of KIF1A interacts with a different group of postsynaptic scaffolds compared to KIF13B, confirming high selectivity of these domains (Figure S1A; Tables S1 and S2). As part of the interactome, we detected well-known interactors of KIF1A, such as liprin-α family proteins (Shin et al., 2003), as well as interactors such as TANC2. We observed a highly significant enrichment of TANC2 total spectral counts (Figure 1B) but not of counts for its homolog TANC1 (Figure 1C). Similarly, we found KIF1A associated only with TANC2 in our AP-MS experiment in which we immunoprecipitated TANC1 or TANC2 from rat brain extracts (Figure 1D). To validate our proteomic results, we performed affinity purification-Western blot (AP-WB) experiments in cells co-expressing bioGFP-KIF1A fragments (657-1105 and 657-1698), along with a hemagglutinin (HA)-tagged version of TANC1 and TANC2. As expected, both TANC2 and liprin-α bound the KIF1A middle fragment (657-1105), but not TANC1 (Figures 1E and 1F). Surprisingly, neither TANC2 nor liprin-α was able to bind a longer KIF1A fragment including the PH domain (Figures 1E and 1F), as shown by MS data (Figure 1B; Tables S1 and S2), indicating an auto-inhibitory role of the C-terminal portion of KIF1A. As final evidence, reverse AP-WB experiments confirmed that TANC2 and liprin-α are binding partners of KIF1A (Figures 1G and 1H), and in vitro competition assays showed that TANC2 and liprin-α2 partially compete for the same binding site on KIF1A (Figures S1C and S1D). In addition, we pinpointed their N-terminal domains as the regions mainly involved in the binding (Figures 1I and 1J; Figure S1F). We also found a potential association between TANC2 and liprin-α (Figures S1G and S1H); however, it is quite likely that the two proteins only indirectly interact by binding KIF1A within a macromolecular complex. Overall, these results identify the PSD proteins liprin-α and TANC2 as interactors of KIF1A (Figure 1K).
Figure 1.

KIF1A Binds to TANC2, Liprin-α, and CaM
(A) bioGFP-KIF1A(383-881, 657-1105, 657-1698, 950-1250, 1105-1698) used to perform AP-MS experiments.
(B) KIF1A interactors identified by MS. p values and spectral counts are graphically represented by colors and spheres, respectively. See also Figures S1A and S1B and Tables S1 and S2.
(C) TANC1 and TANC2 spectral counts detected by MS.
(D) TANC (TANC1 Ab, PanTANC Ab, and TANC2 Ab) immunoprecipitation-MS experiments. p values and spectral counts of KIF1A are graphically represented by colors and spheres, respectively.
(E) Western blots (WBs) of HA-TANC proteins and liprin-α1 in AP experiments of bioGFP-KIF1A(657-1105, 657-1698) from co-transfected HEK293 cells.
(F) WB of HA-liprin-α2 in AP of bioGFP-KIF1A(657-1105, 657-1698).
(G and H) WB of HA-KIF1A in AP of bioGFP-TANC2 (G) or bioGFP-liprin-α2 (H).
(I) WB of HA-KIF1A in AP of full-length GFP-TANC2 and GFP-TANC2 fragments (1-832, 833-1227, 1228-1500, and 1501-1900).
(J) WB of HA-KIF1A in AP of full-length GFP-liprin-α2 and GFP-liprin-α2 fragments (1-735 and 796-1257).
(K) Schematic representation of KIF1A in association with the PSD scaffolding proteins TANC2 and liprin-α2 and with the calcium sensor CaM.
(L) WB of HA-CaM in AP of bioGFP-KIF1A fragments (657-1105, 1105-1698, and 657-1698).
Kinesin-3 Family Member KIF1A Interacts with CaM
Among other identified candidate KIF1A interactors, we decided to further characterize the calcium regulator CaM. As suggested by the number of CaM spectra detected by MS (Figure 1B) and confirmed by subsequent AP-WB (Figure 1L), CaM is one of the few proteins capable of binding both the KIF1A middle tail region (657-1105) and the full tail (657-1698). Combined with CaM being calcium regulated, it represents an ideal modulator of KIF1A-based cargo trafficking (Figure 1K). Typically, calcium binding to the EF hands of CaM induces a conformational switch, which exposes the hydrophobic pockets present in the two lobes of CaM, allowing binding to target proteins (Zhang et al., 1995). To assess the ability of CaM to bind to KIF1A in response to calcium, we performed in vitro affinity purification (AP) assays using bioGFP-KIF1A fragments and HA-CaM in the presence of excessive calcium (2 mM) or EDTA (2 mM) (Wang and Schwarz, 2009). Western blot (WB) analysis revealed that the amount of CaM bound to KIF1A increased by ∼50% in calcium-supplemented samples (Figures 2A and 2B), indicating a stronger association between motor protein and CaM upon calcium addition.
Figure 2.

Ca2+ Promotes CaM-KIF1A Binding and Ca2+/CaM Modulates KIF1A Tail Conformation
(A) bioGFP-KIF1A(657-1105) was expressed in HEK293 cells, purified using streptavidin pull-downs, and incubated with protein extracts of cells expressing HA-CaM in the presence of 2 mM EDTA or 2 mM Ca2+. bioGFP-KIF1A(1105-1698) was used as negative control. Western blot detection was performed using HA and GFP antibodies.
(B) Quantification of CaM relative intensities shown in (A), calculated as the ratio of HA-CaM signals normalized on the affinity-purified bioGFP-KIF1A signals. n = 3 experiments per condition. The bars show mean ± SEM.
(C) bioGFP-KIF1A fragments (691-752, 800-896, and 986-1054) and mutant bioGFP-KIF1A(657-1105_5∗Ala) (W714A, W716A, Y717A, F719A, and L722A).
(D) WB of HA-CaM in AP experiments of bioGFP-KIF1A truncations (657-1105, 691-752, 800-896, and 986-1054) from co-transfected HEK293 cells.
(E) WB of HA-CaM in AP of bioGFP-KIF1A(657-1105_WT) or bioGFP-KIF1A(657-1105_5∗Ala).
(F) CaM spectral counts detected by MS in AP of bioGFP-KIF1A(657-1105_WT) or bioGFP-KIF1A(657-1105_5∗Ala).
(G) WB of HA-TANC2 and liprin-α1 in AP of bioGFP-KIF1A(657-1105, 691-752, 800-896, 986-1054).
(H) WB of HA-KIF1A(657-1105) in AP of bioGFP-KIF1A(1105-1698, 950-1250).
(I) bioGFP-KIF1A(1105-1698, 950-1250, 657-1698) were incubated with protein extracts of cells expressing HA-KIF1A(657-1105) in the presence of 2 mM EDTA or 2 mM Ca2+. WB detection was performed using HA and GFP antibodies. Ca2+ increases binding affinity between KIF1A fragments.
(J) bioGFP-KIF1A(1105-1698, 950-1250, 657-1698) were incubated with protein extracts of cells expressing HA-KIF1A(657-1105_WT) or HA-KIF1A(657-1105_5∗Ala). WB detection was performed using HA and GFP antibodies. Mutant KIF1A(657-1105_5∗Ala) shows reduced binding to KIF1A C-terminal fragments compared to WT.
(K and L) Crosslinking-mass spectrometry (XL-MS)-based analysis of purified bioGFP-KIF1A(657-1698_WT) (K) or bioGFP-KIF1A(657-1698_5∗Ala) (L). Schematic maps of intra-protein crosslinks are identified. Crosslinks detected in both WT and 5∗Ala are represented as black lines, WT-specific crosslinks are represented as red lines, and 5∗Ala-specific crosslinks are represented as blue lines.
The structure of Ca2+-bound CaM is flexible and dynamic, allowing it to recognize and modulate the activity of its substrates. As a consequence, the consensus sequence for CaM binding motifs is not well defined. Most Ca2+/CaM binding substrates can be broadly characterized by a short stretch of amino acids containing a high density of positively charged and hydrophobic residues (Yamniuk and Vogel, 2004). In search of potential CaM binding sites, we used two separate databases: the Calmodulin Target Database (http://calcium.uhnres.utoronto.ca) (Yap et al., 2000) and the Calmodulation Database and Meta-Analysis website (http://cam.umassmed.edu) (Mruk et al., 2014). This analysis revealed the existence of a conserved binding motif for CaM spanning residues 710–740 of KIF1A (Figure 2C). Therefore, we generated different KIF1A fragments of ∼60 amino acids each (Figure 2C) and found that KIF1A(691-752) interacted with CaM (Figure 2D). Consequently, we made a CaM binding-deficient bioGFP-KIF1A(657-1105) fragment by substituting five hydrophobic amino acids with alanine residues, as reported previously (Figure 2C) (Li and Sacks, 2003). The absence of HA-CaM signal and of CaM spectra in the bioGFP-KIF1A_5∗Ala pull-downs indicates that the hydrophobic residues are required for CaM recognition (Figures 2E and 2F). Altogether, these results indicate that CaM interacts with a short KIF1A fragment (691-752), in contrast to TANC2 and liprin-α, which require the entire region (657-1105) to associate with KIF1A (Figure 2G) and do not compete with CaM for KIF1A binding (Figures S1C–S1E).
CaM Binding to KIF1A Changes Intramolecular Interactions
Next, we wanted to address the question of the loss of KIF1A binding partners in the full-length fragment. As described earlier, TANC2 and liprin-α are able to bind to KIF1A(657-1105), but not to the longer fragment KIF1A(657-1698) (Figures 1B, 1E, and 1F; Figure S1A). One possible explanation is the presence of an inhibitory mechanism caused by the intramolecular interactions. To assess this, we performed AP-WB experiments using different bioGFP-KIF1A truncations, in combination with HA-KIF1A(657-1105). We only detected binding between HA-KIF1A(657-1105) and bioGFP-KIF1A(1105-1698), confirming an intramolecular interaction between the C terminus and the middle part of the tail (Figure 2H). This interaction is regulated by Ca2+/CaM binding (Figures 2I and 2J). To further characterize these intramolecular interactions, we applied crosslinking-mass spectrometry (XL-MS) to KIF1A(657-1698_WT) and mutant KIF1A(657-1698_5∗Ala). We detected nine intramolecular crosslinked peptides, of which 6 peptides were shared between the two proteins (in black) (Figures 2K and 2L). Three of those crosslinks were specific for either KIF1A-wild-type (WT) (highlighted in red) or KIF1A_5∗Ala (in blue). In the KIF1A_5∗Ala mutant, crucial intramolecular interactions were disturbed compared to the WT protein. In particular, crosslink 1044-1562 was not present in the absence of CaM and aberrant intramolecular crosslinks 720-1562 and 720-1684 were detected, confirming that the backfolding of the tail is governed by CaM binding.
Ca2+/CaM Regulates the Binding of KIF1A with DCVs
Altogether, our results highlight the importance of Ca2+/CaM in regulating KIF1A molecular dynamics. In the following steps, we decided to repeat our initial KIF1A AP-MS experiments in rat brain extracts, this time supplemented with either calcium or EDTA (Figures 3A–3C). The number of spectra detected for co-precipitated liprin-α and TANC2 markedly decreased in samples treated with calcium when compared to the EDTA condition (Figures 3A and 3C; Figure S2A), suggesting that calcium negatively affects the binding affinity of KIF1A for these scaffolds. Consistent with the in vitro AP data (Figures 2A and 2B), we detected more CaM spectra in the presence of calcium compared to the EDTA condition (Figures 3A–3C). Only in the presence of calcium KIF1A(657-1698) associated with specific DCV-related proteins such as Syt4 and synaptotagmin 11 (Syt11) (Figures 3B and 3C; Figure S2B). We obtained the same results for other proteins involved in PIP2 and phosphoinositide metabolism (Figure 3C; Figures S2C–S2E). Based on these results, we can conclude that calcium increases the binding between KIF1A and DCVs. These proteomic findings were confirmed by in vitro AP-WB experiments (Figures 3D–3I). Using a CaM binding-deficient KIF1A mutant (657-1698_5∗Ala), revealed a reduction in the amounts of co-precipitated Syt4 (Figure 3J) or Syt11 (Figure 3K), confirming a role for CaM in promoting the interaction between KIF1A tail and DCV proteins (Figure 3L). Our proteomic data did not allow us to derive any conclusion about SV-related proteins, because the number of spectra identified was not sufficient to show enrichment. Based on these considerations and taking into account that KIF1A(657-1105) was found associated with proteins enriched in the PSD (Figures S1A and S1B), we decided to further investigate the role of KIF1A in DCV trafficking in dendrites. First, we assess the role of CaM in the KIF1A-DCV interaction in hippocampal neurons. To this end, we co-expressed GFP-KIF1A_WT or GFP-KIF1A_5∗Ala with DCV proteins: NPY-RFP (Figures 4A and 4B) or mCherry-Syt4 (Figures 4C and 4D). KIF1A-WT co-localized with NPY and Syt4 on vesicles, whereas mutant KIF1A, which is unable to bind CaM, showed a diffuse cytoplasmic localization that did not coincide with DCVs (Figures 4A–4E). Because CaM-KIF1A binding affinity is already stimulated at low micromolar concentrations of calcium (Figures 4F and 4G) (comparable to physiological Ca2+ concentrations in vivo), we next wondered whether increasing calcium in neurons could activate KIF1A-mediated DCV transport. To test this, we increased intracellular concentration of calcium by treating neurons with bicuculline, which acutely enhances neuronal activity by inhibiting γ-aminobutyric acid (GABA) receptor activity. Bicuculline induced clustering of KIF1A and increased motility of KIF1A-NPY complexes within minutes (Figures 4H–4M). In addition, we examined the effect of calcium deprivation (BAPTA-AM treatment) on DCV motility, and BAPTA-AM-treated neurons showed lower motility of KIF1A, NPY, and Syt4 vesicles (Figures 4N–4P). These data strongly support a role for calcium in the regulation of KIF1A-DCV loading and motility in neurons. Altogether, these results indicate that binding of Ca2+/CaM to the KIF1A tail regulates DCV pick-up and trafficking (Figure 3L).
Figure 3.

KIF1A Binds DCVs in a Ca2+/CaM-Dependent Manner
(A) bioGFP-KIF1A(657-1105) was purified using streptavidin pull-downs and incubated with rat brain extracts in the presence of 2 mM EDTA or 2 mM Ca2+. KIF1A(657-1105) interactors were identified by MS. Mascot scores and spectral counts of selected proteins (TANC2, liprin-α, and CaM) are graphically represented by colors and spheres, respectively. See also (C) and Figure S2A.
(B) bioGFP-KIF1A(657-1698) was incubated with rat brain extracts in the presence of 2 mM EDTA or 2 mM Ca2+. KIF1A(657-1698) interactors were identified by MS. Mascot scores and spectral counts of selected proteins (Syt4, Syt11, and CaM) are graphically represented by colors and spheres, respectively. See also (C) and Figures S2B–S2E.
(C) Table represents the number of spectral counts detected in AP-MS experiments of bioGFP-KIF1A(657-1105) and bioGFP-KIF1A(657-1698) in the presence of 2 mM EDTA or 2 mM Ca2+ for selected co-purified proteins: TANC2, liprin-α, calmodulin (CaM), synaptotagmin (Syt), phosphatidylinositol 5-phosphate 4-kinase (Pip4k2), and inositol polyphosphate 5-phosphatase (Inpp5).
(D and E) bioGFP-KIF1A(657-1105) was incubated with protein extracts of cells expressing HA-TANC2 (D) or HA-liprin-α2 (E) in the presence of 2 mM EDTA or 2 mM Ca2+. WB detection was performed using HA and GFP antibodies.
(F and G) bioGFP-KIF1A(657-1698) was incubated with protein extracts of cells expressing mCherry-Syt4 (F) or mCherry-Syt11 (G) in the presence of 2 mM EDTA or 2 mM Ca2+. WB detection was performed using mCherry and GFP antibodies.
(H and I) Quantifications of Syt4 (H) and Syt11 (I) relative intensities shown in (F) and (G), calculated as the ratio of co-purified mCherry signals normalized on the bioGFP-KIF1A. n = 3 experiments per condition. The bars show mean ± SEM.
(J and K) bioGFP-KIF1A(657-1698_WT) or bioGFP-KIF1A(657-1698_5∗Ala) was incubated with protein extracts of cells expressing mCherry-Syt4 (J) or mCherry-Syt11 (K). WB detection was performed using mCherry and GFP antibodies.
(L) Schematic model illustrating the Ca2+-CaM mechanism acting on the KIF1A tail, leading to DCV loading and mobilization.
Figure 4.

KIF1A-Driven DCVs Transport Is Regulated by Ca2+/CaM
(A) Representative dendrites of rat hippocampal neurons (11–14 DIV) co-transfected with GFP-KIF1A_WT or GFP-KIF1A_5∗Ala (green) with NPY-RFP (red). Arrows point to co-localizing puncta. Scale bar, 5 μm.
(B) Line scans of fluorescence intensity of GFP-KIF1A and NPY-RFP channels shown in (A).
(C) Dendrites of neurons co-transfected with GFP-KIF1A_WT or GFP-KIF1A_5∗Ala (green) with mCherry-Syt4 (red). Arrows point to co-localizing puncta. Scale bar, 5 μm.
(D) Line scans of fluorescence intensity of GFP-KIF1A and mCherry-Syt4 channels shown in (C).
(E) Quantifications of GFP-KIF1A_WT and GFP-KIF1A_5∗Ala puncta in dendrites. The bars show mean ± SEM (n = 20–28 dendrites; ∗∗∗p < 0.001, t test).
(F) Ca2+ dependence of the in vitro binding of CaM to KIF1A. AP of CaM (anti-CaM beads) from lysates of cells transfected with HA-KIF1A(657-1105_WT) or HA-KIF1A(657-1105_5∗Ala). WB detection was performed using anti-HA antibody.
(G) Quantification of KIF1A binding to CaM shown in (F). The percentage of maximal binding to CaM was defined as the intensity of the co-precipitated band of KIF1A and set at 100% in 2 mM Ca2+. n = 3 experiments per condition. The bars show mean ± SEM.
(H and I) Representative kymographs showing trajectories of GFP-KIF1A and NPY-RFP vesicles in selected neurites of co-transfected neurons pre- and posttreatment with DMSO (H) or with 40 μm of bicuculline (I).
(J and K) Quantifications of trajectories of GFP-KIF1A co-localizing with NPY-RFP (see H and I) pre- and posttreatment with DMSO (J) or with bicuculline (K). The bars show mean ± SEM (n = 12 dendrites in J and n = 34 dendrites in K; ∗∗∗p < 0.001, paired t test).
(L) Intracellular calcium levels in a representative dendrite treated with bicuculline and visualized with the calcium indicator GCaMP6. Scale bar, 5 μm.
(M) GFP-KIF1A clustering in a representative dendrite treated with bicuculline. Scale bar, 5 μm.
(N–P) Quantifications of the percentage of mobile GFP-KIF1A (N), NPY-GFP vesicles (O), or mCherry-Syt4 vesicles (P) in neurons (11–14 DIV) treated with DMSO (CTR) or 10 μm of BAPTA-AM. Bars show the mean (n = 21–29 dendrites in N, n = 20–28 dendrites in O, and n = 41–50 dendrites in P; ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.001, t test).
KIF1A-Mediated DCV Trafficking Is Independent of TANC2 and Liprin-α
Next, we were interested whether KIF1A, TANC2, and/or liprin-α can also control DCV motility. We first systematically quantified the co-localization of KIF1A with DCV markers (NPY, Syt4, and Syt11) in hippocampal neurons (Figure 5A; Figures S3A and S3B). KIF1A showed ∼50% co-localization with NPY-, Syt4-, and Syt11-positive vesicles (Figures 5B and 5C). Next, we tested whether NPY and synaptotagmins label the same DCV population. Only 10% of the NPY-positive vesicles coincided with Syt4 or Syt11 (Figures 5D–5F; Figures S3C and S3D). In contrast, Syt4 and Syt11 showed ∼70% co-localization (Figures 5G–5I; Figures S3E and S3F), indicating that NPY and synaptotagmins label different subpopulations of DCVs.
Figure 5.

DCV Dendritic Trafficking Is KIF1A Dependent and TANC2/Liprin-α Independent
(A) Neurons (11–14 DIV) co-transfected with GFP-KIF1A (green), in combination with NPY-RFP, mCherry-Syt4, or mCherry-Syt11 (red). Arrows point to co-localizing puncta. Scale bar, 5 μm. See also Figures S3A and S3B.
(B and C) Percentage of co-localizing puncta between DCV proteins and KIF1A (B), and vice versa (C). The bars show mean ± SEM (n = 19–23 dendrites).
(D) NPY-GFP (green), in combination with mCherry-Syt4 or mCherry-Syt11 (red). Scale bar, 5 μm. See also Figures S3C and S3D.
(E and F) Co-localizing puncta between NPY and Syt4 or Syt11 (E), and vice versa (F). The bars show mean ± SEM (n = 21–31 dendrites).
(G) Phluorin-Syt4 (green) with mCherry-Syt11 (red) or Phluorin-Syt11 (green) with mCherry-Syt4 (red). Arrows point to co-localizing puncta. Scale bar, 5 μm. See also Figures S3E and S3F.
(H and I) Co-localizing puncta between Syt4 and Syt11 (H), and vice versa (I). The bars show mean ± SEM (n = 20–27 dendrites).
(J and K) Representative kymographs showing trajectories of NPY-RFP vesicles (J) or mCherry-Syt4 vesicles (K) (red) moving with GFP-KIF1A (green).
(L) Representative images acquired at 10 s intervals showing NPY-GFP motility in dendrites of neurons transfected with pSuper (right) or shRNA_KIF1A (left). In the merged images, red corresponds to 0 s, blue corresponds to 10 s, and green corresponds to 20 s. Scale bar, 5 μm.
(M) Quantification of the percentage of mobile NPY vesicles in neurons (11–14 DIV) co-transfected with NPY-GFP, in combination with pSuper, shRNA_KIF1A, shRNA_KIF1A + FL-HA-KIF1A, or shRNA_KIF1A + HA-KIF1A(1-1105). The bars show mean (n = 18–32 dendrites; ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.001, t test).
(N and O) Quantification of the percentage of mobile DCVs in neurons (11–14 DIV) co-transfected with NPY-GFP (N) or mCherry-Syt4 (O), in combination with pSuper, shRNA_KIF1A, shRNA_TANC2, shRNA_liprin-α2-3, or shRNA_liprin-α2-3 + shRNA_TANC2. The bars show mean (n = 17–58 dendrites in N and n = 17–39 dendrites in O; ∗∗∗p < 0.001, t test). See also Figures S3J–S3L.
Second, using live-cell imaging, we assessed whether NPY and DCVs are transported by KIF1A. Kymographs of those recordings showed robust co-movement of GFP-KIF1A and NPY-RFP, as well as mCherry-Syt4-labeled vesicles (Figures 5J and 5K). Next, we tested the effect of KIF1A knockdown on NPY or Syt4 vesicle motility. DCV motility was severely affected in neurons depleted of KIF1A, with ∼50% reduction of mobile NPY-GFP- or Syt4-Cherry-positive vesicles compared to pSuper control (Figures 5L–5O). We confirmed the specificity of this result with a rescue experiment in which expression of full-length KIF1A (but not of KIF1A lacking the C-terminal PH domain) restored NPY motility (Figure 5M). These data are in line with a previously described role for KIF1A in transporting DCVs (Arthur et al., 2010, Lipka et al., 2016, Lo et al., 2011, McVicker et al., 2016).
Finally, we assessed whether the depletion of TANC2 and/or liprin-α also affects DCV motility. We used previously described short hairpin RNAs (shRNAs) targeting liprin-α (Spangler et al., 2013) and generated TANC2-specific shRNAs, whose efficacy was tested by quantifying the intensity of the staining of endogenous TANC2 in neurons (Figures S3G–S3I). TANC2 or liprin-α knockdowns did not influence DCV motility, as shown by live-cell imaging of NPY or Syt4 (Figures 5N and 5O). In addition, other transport parameters, such as pausing frequency and duration, were not affected by this treatment (Figures S3J–S3L). These data suggest that TANC2 and liprin-α are not part of the KIF1A-DCV transport complex and do not work as classical cargo-adaptor proteins.
TANC2 and Liprin-α Are Scaffolding Proteins in Dendritic Spines
To better understand the roles of TANC2 and liprin-α in KIF1A-dependent cargo trafficking, we investigated their distribution in neurons. Both proteins localized in dendrites (Figure 6A), were enriched in dendritic protrusions, and co-localized with the postsynaptic markers PSD-95 and Homer (Figures 6B–6D). When co-expressed, TANC2 and liprin-α co-clustered in dendritic spines (Figure 6E). Analysis of the truncated isoforms revealed that the C-terminal part of TANC2 (1501-1900), which includes the PDZ binding domain, is responsible for the synaptic localization (Figure S4A). In contrast, for liprin-α, the N-terminal part (1-735) is required for the synaptic localization (Figure S4B). We followed those observations and characterized the interactomes of TANC2 and liprin-α (Tables S3 and S4). Based on this analysis, we identified postsynaptic density proteins (GO classification, cellular component), such as PSD-95(DLG4), SAP-97(DLG1), CASK, Scribble, Centaurin gamma2 and 3 (AGAP1 and AGAP3), and various subunits of the NMDA receptor (Grin1 and Grin2B), as the main interactors of TANC2 (Figure S4C; Table S3). These results are in line with previous findings that describe TANC2 as a synaptic scaffold, interacting and co-localizing with the synaptic anchoring proteins PSD-95 and Homer (Han et al., 2010) and with other PDZ domain proteins (Gasparini et al., 2017). Among the interacting partners of liprin-α2, we picked up both postsynaptic (Grip1, Grip2, Lin7, Trio, and CASK) (Wyszynski et al., 2002) and pre-synaptic proteins (Rims1/2, ERC1/2, and Munc13-1/2) involved in the maturation, docking and secretion of SVs and known to form macromolecular protein complexes with liprin-α in the axonal active zone (Figure S4D; Table S4) (Olsen et al., 2005, Spangler et al., 2013). Altogether, we can conclude that TANC2 and liprin-α are enriched in dendritic spines and interact with various synaptic proteins.
Figure 6.

TANC2 and Liprin-α2 Are PSD Scaffolds that Localize in Dendritic Spines
(A) Hippocampal neurons (11–14 DIV) transfected with GFP-TANC2 or GFP-liprin-α2 (green) and stained for MAP2 (red). Scale bar, 20 μm.
(B and C) GFP-TANC2 (B) or GFP-liprin-α2 (C) (green) co-localization with Homer and PSD-95 (red) in dendritic spines. Scale bar, 5 μm.
(D) Quantifications corresponding to the percentage of GFP-TANC2 (B) or GFP-liprin-α2 (C) puncta co-localizing with PSD-95 or Homer. The bars show mean ± SEM (n = 24–36 dendrites).
(E) Neurons co-expressing GFP-liprin-α2 and RFP-TANC2. Scale bar, 20 μm. Bottom panels correspond to higher magnification showing TANC2 and liprin-α2 co-localization in dendritic spines. Arrows point to co-localizing puncta. Scale bar, 5 μm.
(F) Dendritic protrusions of pSuper or TANC2-KD (knockdown) neurons (shRNA_TANC2#1, shRNA_TANC2#3, and shRNA_TANC2mix) co-transfected with MARCKS-GFP to visualize membrane morphology. Scale bar, 5 μm. Graphs show quantifications of protrusion density (number of protrusions per 10 μm) for total number of protrusions (left) or mushroom-shaped protrusions (right). The bars show mean ± SEM (n = 30–33 dendrites; ∗p < 0.05, ∗∗∗p < 0.001, t test).
(G) Dendritic protrusions in neurons co-transfected with pSuper, shRNA_TANC2#1, or shRNA_TANC2#1 + GFP-TANC2, in combination with MARCKS-GFP. Scale bar, 5 μm. The bars show mean ± SEM (n = 54–75 dendrites, ∗∗p < 0.01, ∗∗∗p < 0.001, t test).
(H) Dendritic protrusions in neurons transfected with pSuper, shRNA_KIF1A, shRNA_TANC2, shRNA_liprin-α2-3, or shRNA_liprin-α2-3 + shRNA_TANC2, together with MARCKS-GFP. Scale bar, 5 μm. The bars show mean ± SEM (n = 30–36 dendrites, ∗∗∗p < 0.001, t test).
TANC2, KIF1A, and Liprin-α Depletion Affects Spine Density and Morphology
Spine morphology is largely controlled by actin dynamics. Identifying TANC2 and liprin-α as enriched in dendritic spines led us to speculate that their distribution and accumulation may also be regulated by actin. Therefore, we transfected neurons with GFP-TANC2 or GFP-liprin-α2 and treated them with either latrunculin B to depolymerize the actin cytoskeleton or jasplakinolide to stabilize actin (Figures S4E and S4F). Latrunculin B treatment reduced the number of TANC2 and liprin-α2 clusters in spines by ∼30%, whereas jasplakinolide treatment resulted in an opposite phenotype, with ∼30% increase of clusters in spines (Figures S4G and S4H). These data suggest that the localization and clustering of TANC2 and liprin-α2 are affected by the actin cytoskeleton in dendritic spines. This hypothesis is strengthened by the ability of several TANC2- and liprin-α-interacting proteins to directly or indirectly associate with the actin cytoskeleton (Figure S4I). To get further insights into the roles of TANC2, KIF1A, and liprin-α in dendritic spine morphology, we performed knockdown experiments. TANC2 depletion caused a significant reduction in the total number of protrusions, particularly of mushroom spines (Figure 6F), and the effect was rescued by re-introducing full-length TANC2 (Figure 6F). KIF1A and liprin-α depletion showed a similar phenotype, severely affecting the total number of dendritic protrusions (Figure 6H). These data are consistent with previous findings (Han et al., 2010, McVicker et al., 2016) and show that TANC2, KIF1A, and liprin-α depletion affects spine number and morphology.
TANC2 and Liprin-α2 Act as Immobile Postsynaptic Posts Able to Recruit KIF1A in a Subset of Dendritic Spines
Based on the preceding results, we further examined the functional relationship between KIF1A and TANC2 or liprin-α in dendritic spines. Co-expression of HA-KIF1A with GFP-TANC2 or GFP-liprin-α2 showed that ∼10% KIF1A-FL co-clustered with TANC2 and liprin-α2 in dendritic spines (Figure 7A). In contrast, KIF1A(1-1105), lacking the PH domain but containing the TANC2 and liprin-α binding region, showed a rather diffuse cytoplasmic pattern (Figure 7B). Nevertheless, we noticed accumulation of KIF1A(1-1105) in dendritic spines that co-localized with TANC2 and liprin-α2 (Figure 7B), suggesting that KIF1A can co-localize with TANC2 and liprin-α2 in spines even without cargo binding. This effect was exacerbated when these proteins were expressed in COS7 cells (Figures S5A–S5F). Because KIF1A is a motor protein, we next examined the spatial and temporal dynamics of KIF1A, DCVs (by imaging NPY and Syt4), TANC2, and liprin-α. As we described earlier, KIF1A-, NPY-, and Syt4-positive vesicles showed high motility along the dendritic shaft. This was not the case for TANC2 and liprin-α, which were relatively static in dendritic spines (Figures 7C–7E). This finding supports our claim that TANC2 and liprin-α are not part of the KIF1A-DCV transport complex. Closer analysis of the vesicle dynamics revealed that DCVs frequently paused and stopped near TANC2- or liprin-α-positive clusters (Figures 7C–7E) and the number of DCV pauses at TANC2/liprin-α2 clusters was higher in mature neurons (17 days in vitro [DIV]) in which TANC2 and liprin-α2 were more accumulated in spines (Figures S5G–S5K). We also found DCV accumulation in dendritic spines, and in ∼10% of the spines, DCVs co-localized with TANC2 and liprin-α2 (Figures 7F and 7G). To gain additional functional insight, we analyzed the distribution of DCVs in TANC2- or liprin-α-depleted neurons. TANC2 and liprin-α knockdown, but not Cortactin knockdown, caused a reduction of KIF1A, NPY, and Syt4 clusters in spines by ∼40% compared to control (Figures 7H–7J). Based on these results, we propose that TANC2 and liprin-α recruit KIF1A-driven DCVs to dendritic spines (Figure 7K).
Figure 7.

TANC2 and Liprin-α Recruit KIF1A-Driven DCVs in Dendritic Spines
(A and B) Neurons co-expressing GFP-TANC2 or GFP-liprin-α2 (green), in combination with HA-KIF1A (A) or HA-KIF1A(1-1105) (B) (red) and stained for MAP2 (blue). Scale bar, 10 μm. Panels on the right correspond to higher magnifications showing dendritic TANC2 and liprin-α2 puncta co-localizing with KIF1A. Scale bar, 5 μm.
(C) Kymographs showing trajectories of GFP-KIF1A and RFP-TANC2 in a selected dendritic region of transfected hippocampal neurons (11–14 DIV).
(D) Trajectories of NPY-RFP (green) vesicles pausing at GFP-liprin-α2 (red) clusters (left panel); trajectories of NPY-GFP (green) vesicles pausing at RFP-TANC2 (red) clusters (right panel).
(E) mCherry-Syt4 (red) vesicles pausing at GFP-liprin-α2 (green) clusters (left panel) or at GFP-TANC2 (green) clusters (right panel).
(F and G) GFP-TANC2 (F) or GFP-liprin-α2 (G) co-localizing with NPY-RFP, mCherry-Syt4, or mCherry-Syt11 puncta in dendritic protrusions visualized with MARCKS-BFP. Scale bar, 2 μm.
(H–J) Quantifications showing the percentage of GFP-KIF1A (H), NPY-GFP (I), and mCherry-Syt4 (J) puncta in dendritic protrusions, measured in neurons transfected with pSuper, shRNA_Cortactin, shRNA_TANC2, and shRNA_liprin-α2-3 (11–14 DIV) and filled with MARCKS-BFP. The bars show mean ± SEM (n = 30–57 dendrites in H, n = 35–71 dendrites in I, and n = 43–90 dendrites in J; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, t test).
(K) Hypothetical model of KIF1A-dependent transport of DCVs in dendritic spines. KIF1A, in low Ca2+, is in an auto-inhibited conformation, unable to efficiently bind any cargo. In the presence of high Ca2+, KIF1A interacts with CaM, resulting in a conformational change of its C-terminal tail. Upon Ca2+/CaM binding, KIF1A is activated, allowing for DCV loading and motility. KIF1A-driven DCVs are recruited in dendritic spines by liprin-α and TANC2, which ensure a precise mechanism of synaptic tagging for the vesicles.
TANC2 Disease Mutations Abolish the Interaction with KIF1A and Affect KIF1A-DCV Recruitment
The preceding findings on the role of TANC2 in DCV transport turned our interest into potential consequences for neuronal dysfunction. TANC2 gene mutations R760C and R1066X (nonsense mutation) were found in patients with intellectual disorders and autism spectrum disorder, respectively (de Ligt et al., 2012, Iossifov et al., 2012). In the last part, we wanted to test whether those mutants may be affecting TANC2 localization to dendritic spines and/or its ability to bind KIF1A. While the localization of TANC2-R760C was similar to the WT TANC2, TANC2-R1066X failed to accumulate at the dendritic spines (Figures S6A and S6B). These results are in agreement with an important role of the C-terminal PDZ binding domain for TANC2 synaptic localization (Figure S4A). Furthermore, AP-WB experiments revealed strongly reduced binding between both TANC2 mutants and KIF1A (Figure S6C).
To directly assess the effect of TANC2 point mutation (R760C) on KIF1A and DCV transport, we next produced chimeric proteins, which are the result of the fusion between NF186 (Neurofascin) and TANC2(1-832_WT) or TANC2(1-832_R760C). As expected, both Neurofascin-fusion proteins mis-accumulate TANC2 at the AIS (Figures S6D and S6E). Neurons co-expressing a WT TANC2 chimera showed reduced run length (Figure S6F), speed (Figure S6G), and run duration (Figure S6H) of anterograde-transported DCVs at the AIS compared to a control NF186, whereas in the presence of a mutant R760C, DCV motility was not affected (Figures S6F–S6H). These data suggest that a single point mutation (within the N-terminal part of TANC2) found in patients with intellectual disorders, apart from being detrimental for binding with KIF1A (Figure S6C), drastically impairs the recruitment of KIF1A-transported vesicles in neurons. Altogether, these data suggest that the aberrant localization, the abolished interaction with KIF1A, and the deficient recruitment of KIF1A-driven vesicles of TANC2 mutants may contribute to the underlying molecular mechanisms that lead to neurological defects observed in patients.
Discussion
Ca2+/CaM Interacts with KIF1A and Allows DCV Binding and Motility
In neurons, calcium is a regulatory factor for multiple elements of the trafficking machinery. Calcium and calcium-activated proteins control motor protein processivity and motor-cargo binding (Hirokawa et al., 2010, Schlager and Hoogenraad, 2009). For example, Ca2+/CaM allows activation of actin-based motors, such as myosin Va (Krementsov et al., 2004), myosin VI (Batters et al., 2016), and myosin 1C (Lu et al., 2015). In those cases, calcium is required for an initial activation of the myosin motor protein, allowing the transition from a backfolded conformation to an active status. Another example is the activation of Ca2+/CaM-dependent protein kinase II (CaMKII) by calcium, which in turn phosphorylates KIF17, leading to the release of NMDA receptor-containing vesicles (Guillaud et al., 2008). Finally, the regulation of mitochondrial trafficking depends on calcium. Upon calcium binding, the adaptor protein Miro interacts with the motor domain of KIF5 motors, thereby preventing its microtubule interaction and inhibiting the transport of mitochondria (Macaskill et al., 2009, Wang and Schwarz, 2009). Similar to this mechanism, cytoplasmic KIF1A is in an auto-inhibited state and becomes active only upon cargo binding (Hammond et al., 2009, Niwa et al., 2016). In this study, we were able to decipher the role of calcium in KIF1A-dependent vesicle trafficking. We show that Ca2+/CaM-dependent modulation on KIF1A allows for binding to vesicular cargo. Our results indicate that at low calcium concentrations, the tail domain of KIF1A does not bind to vesicular cargo, whereas at high calcium concentrations, CaM binds KIF1A, allowing for subsequent DCV motility. Thus, we propose a mechanism in which Ca2+/CaM regulates the loading of DCVs. This model potentially represents a more general paradigm for other kinesins and cargoes in response to calcium. For example, KIF1Bβ contains a predicted CaM binding site in a highly conserved region and therefore could undergo similar Ca2+/CaM-dependent dynamics.
TANC2 and Liprin-α Capture KIF1A-Driven DCVs
Syt4 is present on DCVs and is transported by KIF1A along microtubules in the dendritic shaft (Arthur et al., 2010). Microtubule entry into dendritic spines was proposed as a mechanism of local delivery of KIF1A-mediated DCVs (McVicker et al., 2016). Our data point to two scaffolding proteins, TANC2 and liprin-α, present in dendritic spines as important players in the mechanism behind KIF1A-transported DCV capture. First, TANC2 and liprin-α bind the stalk domain of KIF1A. Second, they are stably present in dendritic spines while not being part of the KIF1A-cargo complex. Third, although TANC2 and liprin-α do not directly affect the motility of KIF1A-transported DCVs, they influence the cargo distribution in dendritic spines. Based on those points, we speculate that TANC2 and liprin-α, interacting with KIF1A, are able to stop and capture KIF1A-bound DCVs upon dendritic spine entry. In this model, TANC2 and liprin-α act as local signposts tethering KIF1A-transported DCVs. Consistent with our model, neurons lacking KIF1A show spine morphology defects, which can be directly caused by an altered transport of DCVs within the dendritic spines. In this study, we focused on KIF1A-dependent DCV transport in dendrites; however, the same general principles might be applied to the transport of SVs in the axonal compartment. Similar mechanisms have been described at the pre-synaptic axonal boutons, where static clusters of liprin-α can capture KIF1A-transported SVs (Olsen et al., 2005, Wu et al., 2013, Wu et al., 2016) or where actin pre-synaptic pools locally recruit DCVs (Bharat et al., 2017). Based on our biochemical results, DCV recruitment is favored by low calcium concentrations, thereby working as an opposing mechanism counteracting the Ca2+/CaM-induced DCV mobility. Myosin motors may play an additional role in trafficking DCVs into spines. Specifically, it was proposed that myosin V modulates DCV transport and delivery (Bittins et al., 2010) and myosin V activity is regulated by a similar Ca2+/CaM mechanism (Lu et al., 2006, Nguyen and Higuchi, 2005). Thus, local Ca2+ concentrations have an important role in the modulation of DCV trafficking and delivery in and out of dendritic spines.
In summary, we propose that Ca2+/CaM regulates cargo pick-up and scaffolding proteins liprin-α and TANC2 recruit KIF1A-driven DCVs into spines. Our findings reveal a potential general mechanism that depends on two basic elements: calcium to load motors to cargo and specific signposts to unload cargo. Given that alterations in cargo trafficking pathways were described in several neurological diseases, our findings that some TANC2 disease mutants do not interact with KIF1A are bringing to light additional molecular targets to investigate the trafficking machinery in neuropathological disease models.
Experimental Procedures
Animals
All experiments were approved by the DEC Dutch Animal Experiments Committee (Dier Experimenten Commissie), performed in line with institutional guidelines of Utrecht University, and conducted in agreement with Dutch law (1996 Wet op de Dierproeven) and European regulations (Directive 2010/63/EU). For details, see Supplemental Experimental Procedures. Female pregnant Wister rats were obtained from Janvier Laboratories. Hippocampal neurons were obtained from embryos of both genders at the embryonic day 18 (E18) stage of development.
Expression Vectors and shRNA Constructs
pebioGFP-KIF1A(657-1105, 657-1698) correspond to truncated versions of the KIF1A rat variant 2 (XM_003750741). HA-TANC2 and GFP-TANC2 were generated using FLAG-TANC2 as a template, as described in Han et al. (2010). The following shRNA sequences are used in this study: TANC2#1 (5′-CCTCAGTCAAGGGTCATAT-3′) targeting rat TANC2 mRNA (XM_008768351.1), shRNA_KIF1A (Kevenaar et al., 2016), and shRNA_liprin-α2/α3 (Spangler et al., 2013). All other constructs were created using PCR-based strategies. For details, see Supplemental Experimental Procedures.
Hippocampal Neuron Cultures, Transfections, and Treatments
Primary hippocampal cultures were prepared from E18 rat brains. Cells were plated on coverslips coated with poly-L-lysine (30 μg/mL) and laminin (2 μg/mL) at a density of 100,000/well. Hippocampal neurons were transfected using lipofectamine (Invitrogen). Neuron cultures were treated with 10 μM latrunculin B, 10 μM jasplakinolide, 10 μM BAPTA-AM, or 40 μM bicuculline and fixed or imaged from 0 to 1 hr after addition of the drugs. For details, see Supplemental Experimental Procedures.
AP-MS Using Biotin or GFP Pull-Down on Rat Brain Extracts
Brains were obtained from female adult rats and homogenized in tissue lysis buffer (50 mM Tris HCl, 150 mM NaCl, 0.1% SDS, 0.2% NP-40, and protease inhibitors). Brain lysates were centrifuged at 16,000 × g for 15 min at 4°C, and the supernatant was then incubated for 1 hr at 4°C with beads previously conjugated with the protein of interest. For details, see Supplemental Experimental Procedures.
Sample Preparation, Peptide Fractionation, MS, and Data Analysis
All samples were analyzed on an electron transfer dissociation (ETD)-enabled LTQ-Orbitrap Elite coupled to Proxeon EASY-nLC 1000 (Thermo Fisher Scientific) or on an Orbitrap Q-Exactive mass spectrometer (Thermo Fisher Scientific) coupled to an Agilent 1290 Infinity LC (Agilent Technologies). The full MS methods are available in the Supplemental Experimental Procedures.
Live-Cell Imaging Microscopy and Analysis
Live-cell imaging experiments were performed in an inverted spinning disk confocal microscope equipped with a Plan Apo VC 100×/60× NA 1.40 oil objective and an incubation chamber mounted on a motorized XYZ stage, which were all controlled using MetaMorph (Molecular Devices) software. For details, see Supplemental Experimental Procedures.
Statistical Methods
AP-MS data were analyzed using the SAINT (Significance Analysis of INTeractions, v.2.3.2) algorithm. Statistical significance was determined using Student’s t test assuming a two-tailed variation. The graphs represent mean ± SEM. For details, see Supplemental Experimental Procedures.
Acknowledgments
We thank Gary Banker for sharing bioGFP-KIF1A constructs, Camin Dean for sharing Syt4/Syt11 constructs, Mike Boxem for sharing human-CAM1 cDNA, Eunjoon Kim for sharing FLAG-TANC constructs and panTANC antibody, and Philipp Schätzle for cloning advice. This work was supported by the Netherlands Organisation for Scientific Research (865.10.010, NWO-ALW-VICI, CCH), the Netherlands Organisation for Health Research and Development (91215084, ZonMW-TOP, CCH), and the European Research Council (ERC) (617050, ERC-consolidator, CCH). G.B. was supported by the Deutsche Forschungsgemeinschaft Research Fellowship (807223).
Author Contributions
R.S. cloned the constructs; designed and performed proteomic, biochemical, and imaging experiments; and analyzed the data; G.P. and E.E.Z. conducted live imaging experiments and analyzed the data; J.H. and I.G.S.J. cloned DNA constructs and performed biochemical and imaging experiments; O.K. performed XL-MS experiments; R.A.S. supervised the XL-MS part; A.F.M.A. supervised the proteomic part; R.S. designed and assembled the figures; R.S. and C.C.H. wrote the manuscript with input from G.P.; and C.C.H. supervised and coordinated the study.
Declaration of Interests
The authors declare no competing interests.
Published: July 17, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and four tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.071.
Data and Software Availability
The accession number for the mass spectrometry proteomic data reported in this paper is PRIDE: PXD010080.
Supplemental Information
Contains data from affinity purification mass spectrometry analyses (AP-MS) (two biological replica) of bioGFP-KIF1A(657-1105) in rat brain extracts. PROTID = Uniprot accession code; gene name = corresponding gene name; Description = Protein name/description derived from ∗.fasta database; bioGFP_#PSM = number of PSM from bioGFP-only control AP; bioGFP_#peptides = number of peptides from bioGFP-only control AP; bioGFP(657-1105)_#PSM = number of PSM from bioGFP-KIF1A(657-1105); bioGFP(657-1105)_#peptides = number of peptides from bioGFP-KIF1A(657-1105); p value = probability score from SAINT (Significance Analysis of INTeractions, version 2.3.2) (Choi et al., 2011) analysis. 0 ≤ p ≤ 1, the higher the score the higher the probability for a given interaction; SAINT probability > 0.98 = proteins with a SAINT probability score > 0.98 are flagged as ‘TRUE’; AP-MS_HEK293_cells = value = #N/A, means that the homologous protein has not been identified in the control (CTR) AP-MS of bioGFP-KIF1A(657-1105) in HEK293 cells / value = gene name, means that the same homologous protein has been identified in the CTR AP-MS of bioGFP-KIF1A(657-1105) in HEK293cells.
Contains data from affinity purification mass spectrometry analyses (AP-MS) (two biological replica) of bioGFP-KIF1A(657-1698) in rat brain extracts. PROTID = Uniprot accession code; gene name = corresponding gene name; Description = Protein name/description derived from ∗.fasta database; bioGFP_#PSM = number of PSM from bioGFP-only control AP; bioGFP_#peptides = number of peptides from bioGFP-only control AP; bioGFP(657-1698)_#PSM = number of PSM from bioGFP-KIF1A(657-1698); bioGFP(657-1698)_#peptides = number of peptides from bioGFP-KIF1A(657-1698); p value = probability score from SAINT (Significance Analysis of INTeractions, version 2.3.2) (Choi et al., 2011) analysis. 0 ≤ p ≤ 1, the higher the score the higher the probability for a given interaction; SAINT probability > 0.98 = proteins with a SAINT probability score > 0.98 are flagged as ‘TRUE’; AP-MS_HEK293_cells = value = #N/A, means that the homologous protein has not been identified in the CTR AP-MS of bioGFP-KIF1A(657-1698) in HEK293 cells / value = gene name, means that the homologous protein has been identified in the CTR AP-MS of bioGFP-KIF1A(657-1698) in HEK293cells.
Contains data from affinity purification mass spectrometry analyses (AP-MS) (two biological replica) of bioGFP-TANC2 in rat brain extracts. PROTID = Uniprot accession code; gene name = corresponding gene name; Description = Protein name/description derived from ∗.fasta database; bioGFP_#PSM = number of PSM from bioGFP-only control AP; bioGFP_#peptides = number of peptides from bioGFP-only control AP; bioGFP-TANC2_#PSM = number of PSM from bioGFP-TANC2; bioGFP-TANC2#peptides = number of peptides from bioGFP-TANC2; p value = probability score from SAINT (Significance Analysis of INTeractions, version 2.3.2) (Choi et al., 2011) analysis. 0 ≤ p ≤ 1, the higher the score the higher the probability for a given interaction; SAINT probability > 0.90 = proteins with a SAINT probability score > 0.90 are flagged as ‘TRUE’; AP-MS_HEK293_cells = value = #N/A, means that the homologous protein has not been identified in the CTR AP-MS of bioGFP-TANC2 in HEK293 cells / value = gene name, means that the homologous protein has been identified in the CTR AP-MS of bioGFP-TANC2 in HEK293 cells.
Contains data from affinity purification mass spectrometry analysis (AP-MS) of bioGFP-liprin-α2 in rat brain extracts. PROTID = Uniprot accession code; gene name = corresponding gene name; Description = Protein name/description derived from ∗.fasta database; bioGFP_#PSM = number of PSM from bioGFP-only control AP; bioGFP_#peptides = number of peptides from bioGFP-only control AP; bioGFP-liprin-α2_#PSM = number of PSM from bioGFP-liprin-α2; bioGFP-liprin-α2#peptides = number of peptides from bioGFP-liprin-α2; p value = probability score from SAINT (Significance Analysis of INTeractions, version 2.3.2) (Choi et al., 2011) analysis. 0 ≤ p ≤ 1, the higher the score the higher the probability for a given interaction; SAINT probability > 0.99 = proteins with a SAINT probability score > 0.99 are flagged as ‘TRUE’; AP-MS_HEK293_cells = value = #N/A, means that the homologous protein has not been identified in the CTR AP-MS of bioGFP-liprin-α2 in HEK293 cells / value = gene name, means that the homologous protein has been identified in the CTR AP-MS of bioGFP-liprin-α2 in HEK293cells.
References
- Arthur C.P., Dean C., Pagratis M., Chapman E.R., Stowell M.H. Loss of synaptotagmin IV results in a reduction in synaptic vesicles and a distortion of the Golgi structure in cultured hippocampal neurons. Neuroscience. 2010;167:135–142. doi: 10.1016/j.neuroscience.2010.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkus R.V., Klyachko O., Horiuchi D., Dickson B.J., Saxton W.M. Identification of an axonal kinesin-3 motor for fast anterograde vesicle transport that facilitates retrograde transport of neuropeptides. Mol. Biol. Cell. 2008;19:274–283. doi: 10.1091/mbc.E07-03-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batters C., Brack D., Ellrich H., Averbeck B., Veigel C. Calcium can mobilize and activate myosin-VI. Proc. Natl. Acad. Sci. USA. 2016;113:E1162–E1169. doi: 10.1073/pnas.1519435113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharat V., Siebrecht M., Burk K., Ahmed S., Reissner C., Kohansal-Nodehi M., Steubler V., Zweckstetter M., Ting J.T., Dean C. Capture of dense core vesicles at synapses by JNK-dependent phosphorylation of synaptotagmin-4. Cell Rep. 2017;21:2118–2133. doi: 10.1016/j.celrep.2017.10.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittins C.M., Eichler T.W., Hammer J.A., 3rd, Gerdes H.H. Dominant-negative myosin Va impairs retrograde but not anterograde axonal transport of large dense core vesicles. Cell. Mol. Neurobiol. 2010;30:369–379. doi: 10.1007/s10571-009-9459-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H., Larsen B., Lin Z.Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z.S., Tyers M., Gingras A.C., Nesvizhskii A.I. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods. 2011;8:70–73. doi: 10.1038/nmeth.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ligt J., Willemsen M.H., van Bon B.W., Kleefstra T., Yntema H.G., Kroes T., Vulto-van Silfhout A.T., Koolen D.A., de Vries P., Gilissen C. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- de Wit J., Toonen R.F., Verhaagen J., Verhage M. Vesicular trafficking of semaphorin 3A is activity-dependent and differs between axons and dendrites. Traffic. 2006;7:1060–1077. doi: 10.1111/j.1600-0854.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- Dean C., Liu H., Dunning F.M., Chang P.Y., Jackson M.B., Chapman E.R. Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nat. Neurosci. 2009;12:767–776. doi: 10.1038/nn.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteves da Silva M., Adrian M., Schätzle P., Lipka J., Watanabe T., Cho S., Futai K., Wierenga C.J., Kapitein L.C., Hoogenraad C.C. Positioning of AMPA receptor-containing endosomes regulates synapse architecture. Cell Rep. 2015;13:933–943. doi: 10.1016/j.celrep.2015.09.062. [DOI] [PubMed] [Google Scholar]
- Fromer M., Pocklington A.J., Kavanagh D.H., Williams H.J., Dwyer S., Gormley P., Georgieva L., Rees E., Palta P., Ruderfer D.M. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini A., Tosatto S.C.E., Murgia A., Leonardi E. Dynamic scaffolds for neuronal signaling: in silico analysis of the TANC protein family. Sci. Rep. 2017;7:6829. doi: 10.1038/s41598-017-05748-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaud L., Wong R., Hirokawa N. Disruption of KIF17-Mint1 interaction by CaMKII-dependent phosphorylation: a molecular model of kinesin-cargo release. Nat. Cell Biol. 2008;10:19–29. doi: 10.1038/ncb1665. [DOI] [PubMed] [Google Scholar]
- Gumy L.F., Katrukha E.A., Grigoriev I., Jaarsma D., Kapitein L.C., Akhmanova A., Hoogenraad C.C. MAP2 defines a pre-axonal filtering zone to regulate KIF1- versus KIF5-dependent cargo transport in sensory neurons. Neuron. 2017;94:347–362. doi: 10.1016/j.neuron.2017.03.046. [DOI] [PubMed] [Google Scholar]
- Hammond J.W., Cai D., Blasius T.L., Li Z., Jiang Y., Jih G.T., Meyhofer E., Verhey K.J. Mammalian kinesin-3 motors are dimeric in vivo and move by processive motility upon release of autoinhibition. PLoS Biol. 2009;7:e72. doi: 10.1371/journal.pbio.1000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S., Nam J., Li Y., Kim S., Cho S.H., Cho Y.S., Choi S.Y., Choi J., Han K., Kim Y. Regulation of dendritic spines, spatial memory, and embryonic development by the TANC family of PSD-95-interacting proteins. J. Neurosci. 2010;30:15102–15112. doi: 10.1523/JNEUROSCI.3128-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T., Lin L., Tibaldi E.V., Reinherz E.L., Chishti A.H. GAKIN, a novel kinesin-like protein associates with the human homologue of the Drosophila discs large tumor suppressor in T lymphocytes. J. Biol. Chem. 2000;275:28774–28784. doi: 10.1074/jbc.M000715200. [DOI] [PubMed] [Google Scholar]
- Hirokawa N., Niwa S., Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein L.C., Schlager M.A., Kuijpers M., Wulf P.S., van Spronsen M., MacKintosh F.C., Hoogenraad C.C. Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr. Biol. 2010;20:290–299. doi: 10.1016/j.cub.2009.12.052. [DOI] [PubMed] [Google Scholar]
- Kevenaar J.T., Bianchi S., van Spronsen M., Olieric N., Lipka J., Frias C.P., Mikhaylova M., Harterink M., Keijzer N., Wulf P.S. Kinesin-binding protein controls microtubule dynamics and cargo trafficking by regulating kinesin motor activity. Curr. Biol. 2016;26:849–861. doi: 10.1016/j.cub.2016.01.048. [DOI] [PubMed] [Google Scholar]
- Klopfenstein D.R., Tomishige M., Stuurman N., Vale R.D. Role of phosphatidylinositol(4,5)bisphosphate organization in membrane transport by the Unc104 kinesin motor. Cell. 2002;109:347–358. doi: 10.1016/s0092-8674(02)00708-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krementsov D.N., Krementsova E.B., Trybus K.M. Myosin V: regulation by calcium, calmodulin, and the tail domain. J. Cell Biol. 2004;164:877–886. doi: 10.1083/jcb.200310065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leterrier C., Dargent B. No Pasaran! Role of the axon initial segment in the regulation of protein transport and the maintenance of axonal identity. Semin. Cell Dev. Biol. 2014;27:44–51. doi: 10.1016/j.semcdb.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Li Z., Sacks D.B. Elucidation of the interaction of calmodulin with the IQ motifs of IQGAP1. J. Biol. Chem. 2003;278:4347–4352. doi: 10.1074/jbc.M208579200. [DOI] [PubMed] [Google Scholar]
- Lipka J., Kapitein L.C., Jaworski J., Hoogenraad C.C. Microtubule-binding protein doublecortin-like kinase 1 (DCLK1) guides kinesin-3-mediated cargo transport to dendrites. EMBO J. 2016;35:302–318. doi: 10.15252/embj.201592929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo K.Y., Kuzmin A., Unger S.M., Petersen J.D., Silverman M.A. KIF1A is the primary anterograde motor protein required for the axonal transport of dense-core vesicles in cultured hippocampal neurons. Neurosci. Lett. 2011;491:168–173. doi: 10.1016/j.neulet.2011.01.018. [DOI] [PubMed] [Google Scholar]
- Lochner J.E., Spangler E., Chavarha M., Jacobs C., McAllister K., Schuttner L.C., Scalettar B.A. Efficient copackaging and cotransport yields postsynaptic colocalization of neuromodulators associated with synaptic plasticity. Dev. Neurobiol. 2008;68:1243–1256. doi: 10.1002/dneu.20650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Krementsova E.B., Trybus K.M. Regulation of myosin V processivity by calcium at the single molecule level. J. Biol. Chem. 2006;281:31987–31994. doi: 10.1074/jbc.M605181200. [DOI] [PubMed] [Google Scholar]
- Lu Q., Li J., Ye F., Zhang M. Structure of myosin-1c tail bound to calmodulin provides insights into calcium-mediated conformational coupling. Nat. Struct. Mol. Biol. 2015;22:81–88. doi: 10.1038/nsmb.2923. [DOI] [PubMed] [Google Scholar]
- Macaskill A.F., Rinholm J.E., Twelvetrees A.E., Arancibia-Carcamo I.L., Muir J., Fransson A., Aspenstrom P., Attwell D., Kittler J.T. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder C.I., Shen K., Hoogenraad C.C. Axon and dendritic trafficking. Curr. Opin. Neurobiol. 2014;27:165–170. doi: 10.1016/j.conb.2014.03.015. [DOI] [PubMed] [Google Scholar]
- McVicker D.P., Awe A.M., Richters K.E., Wilson R.L., Cowdrey D.A., Hu X., Chapman E.R., Dent E.W. Transport of a kinesin-cargo pair along microtubules into dendritic spines undergoing synaptic plasticity. Nat. Commun. 2016;7:12741. doi: 10.1038/ncomms12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S., Julien J.P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013;14:161–176. doi: 10.1038/nrn3380. [DOI] [PubMed] [Google Scholar]
- Miller K.E., DeProto J., Kaufmann N., Patel B.N., Duckworth A., Van Vactor D. Direct observation demonstrates that Liprin-alpha is required for trafficking of synaptic vesicles. Curr. Biol. 2005;15:684–689. doi: 10.1016/j.cub.2005.02.061. [DOI] [PubMed] [Google Scholar]
- Mruk K., Farley B.M., Ritacco A.W., Kobertz W.R. Calmodulation meta-analysis: predicting calmodulin binding via canonical motif clustering. J. Gen. Physiol. 2014;144:105–114. doi: 10.1085/jgp.201311140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H., Higuchi H. Motility of myosin V regulated by the dissociation of single calmodulin. Nat. Struct. Mol. Biol. 2005;12:127–132. doi: 10.1038/nsmb894. [DOI] [PubMed] [Google Scholar]
- Niwa S., Tanaka Y., Hirokawa N. KIF1Bbeta- and KIF1A-mediated axonal transport of presynaptic regulator Rab3 occurs in a GTP-dependent manner through DENN/MADD. Nat. Cell Biol. 2008;10:1269–1279. doi: 10.1038/ncb1785. [DOI] [PubMed] [Google Scholar]
- Niwa S., Lipton D.M., Morikawa M., Zhao C., Hirokawa N., Lu H., Shen K. Autoinhibition of a neuronal kinesin UNC-104/KIF1A regulates the size and density of synapses. Cell Rep. 2016;16:2129–2141. doi: 10.1016/j.celrep.2016.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y., Yamazaki H., Sekine-Aizawa Y., Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81:769–780. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- Olsen O., Moore K.A., Fukata M., Kazuta T., Trinidad J.C., Kauer F.W., Streuli M., Misawa H., Burlingame A.L., Nicoll R.A., Bredt D.S. Neurotransmitter release regulated by a MALS-liprin-alpha presynaptic complex. J. Cell Biol. 2005;170:1127–1134. doi: 10.1083/jcb.200503011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlager M.A., Hoogenraad C.C. Basic mechanisms for recognition and transport of synaptic cargos. Mol. Brain. 2009;2:25. doi: 10.1186/1756-6606-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H., Wyszynski M., Huh K.H., Valtschanoff J.G., Lee J.R., Ko J., Streuli M., Weinberg R.J., Sheng M., Kim E. Association of the kinesin motor KIF1A with the multimodular protein liprin-alpha. J. Biol. Chem. 2003;278:11393–11401. doi: 10.1074/jbc.M211874200. [DOI] [PubMed] [Google Scholar]
- Sieburth D., Ch’ng Q., Dybbs M., Tavazoie M., Kennedy S., Wang D., Dupuy D., Rual J.F., Hill D.E., Vidal M. Systematic analysis of genes required for synapse structure and function. Nature. 2005;436:510–517. doi: 10.1038/nature03809. [DOI] [PubMed] [Google Scholar]
- Sirajuddin M., Rice L.M., Vale R.D. Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat. Cell Biol. 2014;16:335–344. doi: 10.1038/ncb2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangler S.A., Schmitz S.K., Kevenaar J.T., de Graaff E., de Wit H., Demmers J., Toonen R.F., Hoogenraad C.C. Liprin-α2 promotes the presynaptic recruitment and turnover of RIM1/CASK to facilitate synaptic transmission. J. Cell Biol. 2013;201:915–928. doi: 10.1083/jcb.201301011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner O.I., Esposito A., Köhler B., Chen C.W., Shen C.P., Wu G.H., Butkevich E., Mandalapu S., Wenzel D., Wouters F.S., Klopfenstein D.R. Synaptic scaffolding protein SYD-2 clusters and activates kinesin-3 UNC-104 in C. elegans. Proc. Natl. Acad. Sci. USA. 2009;106:19605–19610. doi: 10.1073/pnas.0902949106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Schwarz T.L. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.E., Huo L., Maeder C.I., Feng W., Shen K. The balance between capture and dissociation of presynaptic proteins controls the spatial distribution of synapses. Neuron. 2013;78:994–1011. doi: 10.1016/j.neuron.2013.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G.H., Muthaiyan Shanmugam M., Bhan P., Huang Y.H., Wagner O.I. Identification and characterization of LIN-2(CASK) as a regulator of kinesin-3 UNC-104(KIF1A) motility and clustering in neurons. Traffic. 2016;17:891–907. doi: 10.1111/tra.12413. [DOI] [PubMed] [Google Scholar]
- Wyszynski M., Kim E., Dunah A.W., Passafaro M., Valtschanoff J.G., Serra-Pagès C., Streuli M., Weinberg R.J., Sheng M. Interaction between GRIP and liprin-alpha/SYD2 is required for AMPA receptor targeting. Neuron. 2002;34:39–52. doi: 10.1016/s0896-6273(02)00640-2. [DOI] [PubMed] [Google Scholar]
- Yamada K.H., Hanada T., Chishti A.H. The effector domain of human Dlg tumor suppressor acts as a switch that relieves autoinhibition of kinesin-3 motor GAKIN/KIF13B. Biochemistry. 2007;46:10039–10045. doi: 10.1021/bi701169w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamniuk A.P., Vogel H.J. Calmodulin’s flexibility allows for promiscuity in its interactions with target proteins and peptides. Mol. Biotechnol. 2004;27:33–57. doi: 10.1385/MB:27:1:33. [DOI] [PubMed] [Google Scholar]
- Yap K.L., Kim J., Truong K., Sherman M., Yuan T., Ikura M. Calmodulin target database. J. Struct. Funct. Genomics. 2000;1:8–14. doi: 10.1023/a:1011320027914. [DOI] [PubMed] [Google Scholar]
- Yogev S., Cooper R., Fetter R., Horowitz M., Shen K. Microtubule organization determines axonal transport dynamics. Neuron. 2016;92:449–460. doi: 10.1016/j.neuron.2016.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn T.R., Angleson J.K., MacMorris M.A., Domke E., Hutton J.F., Schwartz C., Hutton J.C. Dense core vesicle dynamics in Caenorhabditis elegans neurons and the role of kinesin UNC-104. Traffic. 2004;5:544–559. doi: 10.1111/j.1600-0854.2004.00195.x. [DOI] [PubMed] [Google Scholar]
- Zhang M., Tanaka T., Ikura M. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Biol. 1995;2:758–767. doi: 10.1038/nsb0995-758. [DOI] [PubMed] [Google Scholar]
- Zhu J., Shang Y., Xia Y., Zhang R., Zhang M. An atypical MAGUK GK target recognition mode revealed by the interaction between DLG and KIF13B. Structure. 2016;24:1876–1885. doi: 10.1016/j.str.2016.08.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Contains data from affinity purification mass spectrometry analyses (AP-MS) (two biological replica) of bioGFP-KIF1A(657-1105) in rat brain extracts. PROTID = Uniprot accession code; gene name = corresponding gene name; Description = Protein name/description derived from ∗.fasta database; bioGFP_#PSM = number of PSM from bioGFP-only control AP; bioGFP_#peptides = number of peptides from bioGFP-only control AP; bioGFP(657-1105)_#PSM = number of PSM from bioGFP-KIF1A(657-1105); bioGFP(657-1105)_#peptides = number of peptides from bioGFP-KIF1A(657-1105); p value = probability score from SAINT (Significance Analysis of INTeractions, version 2.3.2) (Choi et al., 2011) analysis. 0 ≤ p ≤ 1, the higher the score the higher the probability for a given interaction; SAINT probability > 0.98 = proteins with a SAINT probability score > 0.98 are flagged as ‘TRUE’; AP-MS_HEK293_cells = value = #N/A, means that the homologous protein has not been identified in the control (CTR) AP-MS of bioGFP-KIF1A(657-1105) in HEK293 cells / value = gene name, means that the same homologous protein has been identified in the CTR AP-MS of bioGFP-KIF1A(657-1105) in HEK293cells.
Contains data from affinity purification mass spectrometry analyses (AP-MS) (two biological replica) of bioGFP-KIF1A(657-1698) in rat brain extracts. PROTID = Uniprot accession code; gene name = corresponding gene name; Description = Protein name/description derived from ∗.fasta database; bioGFP_#PSM = number of PSM from bioGFP-only control AP; bioGFP_#peptides = number of peptides from bioGFP-only control AP; bioGFP(657-1698)_#PSM = number of PSM from bioGFP-KIF1A(657-1698); bioGFP(657-1698)_#peptides = number of peptides from bioGFP-KIF1A(657-1698); p value = probability score from SAINT (Significance Analysis of INTeractions, version 2.3.2) (Choi et al., 2011) analysis. 0 ≤ p ≤ 1, the higher the score the higher the probability for a given interaction; SAINT probability > 0.98 = proteins with a SAINT probability score > 0.98 are flagged as ‘TRUE’; AP-MS_HEK293_cells = value = #N/A, means that the homologous protein has not been identified in the CTR AP-MS of bioGFP-KIF1A(657-1698) in HEK293 cells / value = gene name, means that the homologous protein has been identified in the CTR AP-MS of bioGFP-KIF1A(657-1698) in HEK293cells.
Contains data from affinity purification mass spectrometry analyses (AP-MS) (two biological replica) of bioGFP-TANC2 in rat brain extracts. PROTID = Uniprot accession code; gene name = corresponding gene name; Description = Protein name/description derived from ∗.fasta database; bioGFP_#PSM = number of PSM from bioGFP-only control AP; bioGFP_#peptides = number of peptides from bioGFP-only control AP; bioGFP-TANC2_#PSM = number of PSM from bioGFP-TANC2; bioGFP-TANC2#peptides = number of peptides from bioGFP-TANC2; p value = probability score from SAINT (Significance Analysis of INTeractions, version 2.3.2) (Choi et al., 2011) analysis. 0 ≤ p ≤ 1, the higher the score the higher the probability for a given interaction; SAINT probability > 0.90 = proteins with a SAINT probability score > 0.90 are flagged as ‘TRUE’; AP-MS_HEK293_cells = value = #N/A, means that the homologous protein has not been identified in the CTR AP-MS of bioGFP-TANC2 in HEK293 cells / value = gene name, means that the homologous protein has been identified in the CTR AP-MS of bioGFP-TANC2 in HEK293 cells.
Contains data from affinity purification mass spectrometry analysis (AP-MS) of bioGFP-liprin-α2 in rat brain extracts. PROTID = Uniprot accession code; gene name = corresponding gene name; Description = Protein name/description derived from ∗.fasta database; bioGFP_#PSM = number of PSM from bioGFP-only control AP; bioGFP_#peptides = number of peptides from bioGFP-only control AP; bioGFP-liprin-α2_#PSM = number of PSM from bioGFP-liprin-α2; bioGFP-liprin-α2#peptides = number of peptides from bioGFP-liprin-α2; p value = probability score from SAINT (Significance Analysis of INTeractions, version 2.3.2) (Choi et al., 2011) analysis. 0 ≤ p ≤ 1, the higher the score the higher the probability for a given interaction; SAINT probability > 0.99 = proteins with a SAINT probability score > 0.99 are flagged as ‘TRUE’; AP-MS_HEK293_cells = value = #N/A, means that the homologous protein has not been identified in the CTR AP-MS of bioGFP-liprin-α2 in HEK293 cells / value = gene name, means that the homologous protein has been identified in the CTR AP-MS of bioGFP-liprin-α2 in HEK293cells.