

Abstract
Sepsis, a life-threatening organ dysfunction, results from a dysregulated host response to invading pathogens that may be characterized by overwhelming systemic inflammation or some sort of immune paralysis. Sepsis remains a major cause of morbidity and mortality. Treatment is nonspecific and relies on source control and organ support. Septic shock, the most severe form of sepsis is associated with the highest rate of mortality. Two large multicentre trials, undertaken 15 years apart, found that the combination of hydrocortisone and fludrocortisone significantly reduces mortality in septic shock. The corticosteroids family is composed of several molecules that are usually characterized according to their glucocorticoid and mineralocorticoid power, relative to hydrocortisone. While the immune effects of glucocorticoids whether mediated or not by the intracellular glucocorticoid receptor have been investigated for several decades, it is only very recently that potential immune effects of mineralocorticoids via non-renal mineralocorticoid receptors have gained popularity. We reviewed the respective role of glucocorticoids and mineralocorticoids in counteracting sepsis-associated dysregulated immune systems.
Keywords: glucocorticoids, mineralocorticoids, NF-κB, animal models, clinical trials, septic shock, sepsis, organ function
Introduction
Sepsis is defined by a life-threatening organ dysfunction resulting from deregulated host response to invading pathogens (1). The host–pathogen interaction in sepsis is associated with an excessive response of the innate immune system leading to systemic inflammation and organ failure (2). This excessive inflammatory response coexists with compensatory anti-inflammatory signaling (3). An initial immune response occurs after recognition of pathogen- or damage-associated molecular patterns by specific cellular receptors, leading to cellular activation and systemic inflammation (4, 5). In practice, patients with sepsis may present with a hyperimmune response, typically around the time of admission when the infectious process is not fully under control, or with an immune suppression state, which tends to occur at a later time (3). The resolution of inflammation is also an active process, partly mediated by lipid mediators such as eicosanoids, which exhibit pro-resolving proprieties, and lead to tissue reparation (6). Then, knowing the time course of the immune response to sepsis is likely a key factor for the success of immunomodulatory interventions (7). Sepsis is a leading cause of mortality and morbidity, with annual prevalence of sepsis estimated at 31.5 million and the annual number of deaths at 5.3 million, worldwide (8, 9). The incidence of sepsis is steadily rising (10). Approximately half of sepsis survivors suffer from physical and psychological sequel, directly impacting their quality of life (11, 12). Treatment of sepsis is based on source control and organs support (13). Corticosteroids are produced by the adrenal glands lying at the superior pole of the kidneys. Corticosteroids are synthesized by adrenal cortical cells from esterified cholesterol and possess four carbon rings. Each step of corticosteroid biosynthesis is controlled by a specific enzyme (Figure 1). Corticosteroids are divided into mineralocorticoids, which preferentially affect salt and water balance while glucocorticoids preferentially affect sugar metabolism and sex hormones. The adrenal cortex is divided into the zona glomerulosa, the outermost layer beneath the capsule, which secretes mineralocorticoids, the zona fasciculata, which secretes glucocorticoids, and the innermost layer, the zona reticularis, which secretes sex hormones. We will hereafter describe the biological effects of glucocorticoids and mineralocorticoids, without further mentioning sex hormones. Corticosteroids are commonly categorized according to their glucocorticoid and mineralocorticoid power, relative to hydrocortisone (Table 1). Glucocorticoids exhibit immune-modulating proprieties, in part through interaction with NF-κB (14). Thus, glucocorticoids have been used to treat patients with severe infections for more than 50 years. Much less information is available regarding immune effects of mineralocorticoids, yet the combination of fludrocortisone to hydrocortisone significantly reduced mortality from septic shock (15, 16). We herein reviewed the immune effects of glucocorticoids and mineralocorticoids that may be relevant to the management of sepsis.
Figure 1.
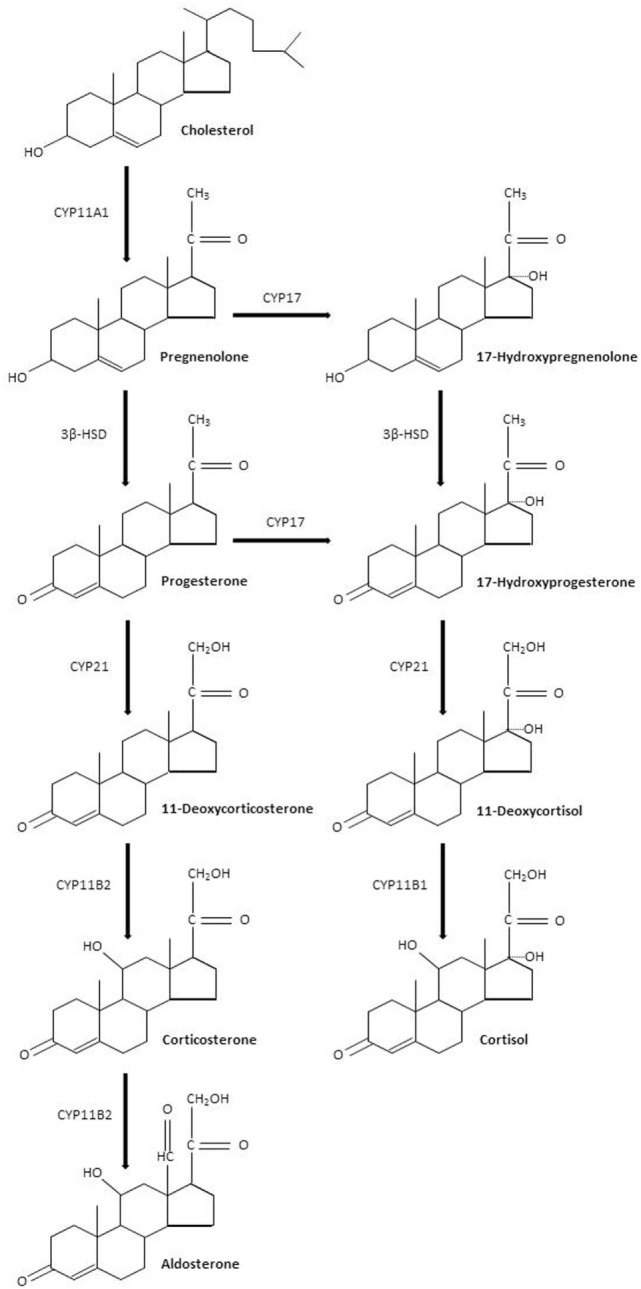
Corticosteroids biosynthesis. Abbreviations: CYP11A1, cholesterol desmolase; 3β-HSD, 3β hydroxysteroid dehydrogenase; CYP17, steroid 17α-hydroxylase; CYP21, steroid 21-hydroxylase; CYP11B2, aldosterone synthase; CYP11B1, steroid 11β-hydroxylase.
Table 1.
Relative potencies of natural and synthetic steroids.
| Compound | Glucocorticoid activity | Mineralocorticoid activity |
|---|---|---|
| Natural steroids | ||
| Cortisol | 1 | 1 |
| Corticosterone | 0.3 | 15 |
| Aldosterone | 0.3 | 3,000 |
| Deoxycorticosterone | 0.2 | 100 |
| Synthetic steroids | ||
| Cortisone | 0.8 | 0.8 |
| Fludrocortisone | 10 | 125 |
| Prednisone | 4 | 0.8 |
| Prednisolone | 4 | 0.8 |
| Methylprednisolone | 5 | 0.5 |
| Betamethasone | 25 | 0 |
| Dexamethasone | 25 | 0 |
Glucocorticoid and mineralocorticoid activity of natural and synthetic steroids, relative to cortisol.
Immune-Modulating Effects of Corticosteroids
Glucocorticoids
Molecular Mechanisms of Action
Glucocorticoids have anti-inflammatory effects through the production of anti-inflammatory proteins and inhibition of pro-inflammatory proteins. Glucocorticoids bind to a specific intracellular receptor, the glucocorticoid receptor (GR). The GR is a transcription factor belonging to the nuclear receptor superfamily encoded on chromosome 5q31-31 (17). Glucocorticoid-regulated transcription factors are 94-kDa proteins, composed of several specific domains. A ligand-binding domain made up of 12 α-helices is involved in the recognition and binding of corticosteroids, a DNA-binding domain composed of two zinc fingers for interaction of the hormone–receptor complex with specific DNA sequences, and a trans-activating domain for binding of transcriptional factors (18). Unbound GR located in the cytoplasm of almost all cells, are stabilized by chaperone proteins such as heat-shock proteins 70, heat-shock protein 90 (Hsp90), and immunophilin (19). Upon binding with glucocorticoids, the GR dissociates from chaperone proteins and translocates into the nucleus. Within the nucleus, homodimers of the glucocorticoid–GR complex interact with specific DNA sequences (glucocorticoid responsive elements) of the regulatory region of target genes (Figure 2). The expression of genes modulated by the hormone–GR complex occurs through chromatin remodeling (20, 21). Chromatin consists of nucleosomes; DNA associated with core histone proteins. Quiescent genes are composed of tightly wound DNA around histone proteins, hampering the ability of RNA polymerases to bind to DNA and to produce mRNA. Core histones may be acetylated, modifying the structure of nucleosomes, loosening the chromatin, and ultimately enhancing gene expression. Transcription factors such as NF-κB activate histone acetyltransferases (HATs), leading to acetylation of core histones. By contrast, histone deacetylases (HDACs) induce a tightening of the chromatin, repressing target genes expression. Activated GR inhibit HATs and activate HDACs, overall repressing the expression of pro-inflammatory genes. For instance, the expression of the IRF3 transcription factor, implicated in interferon production and viral protection, is downregulated by glucocorticoids (22, 23). The GR–glucocorticoid complex also inhibits the production of pro-inflammatory proteins by sequestration of NF-κB within the cytosol (24). NF-κB is implicated in the production of pro-inflammatory cytokines (25, 26). In a resting state, inactive NF-κB is bound to IκBα. Upon cellular activation, NF-κB and IκBα dissociate and NF-κB translocates into the cellular nucleus. Glucocorticoids increase the expression of the inhibitory protein IκBα, thereby sequestering NF-κB (27). Glucocorticoids also induce the expression of glucocorticoid-induced leucine zipper (GILZ) which inhibits NF-κB (28) as well as the anti-inflammatory protein MAP kinase phosphatase 1, which inhibits nuclear translocation of transcription factor GATA-3 implicated in Th2 type cytokine expression (29). In addition, glucocorticoids promote the production of annexin 1, which inhibits the expression of phospholipase A2. Phospholipase A2 catabolizes the production of arachidonic acid-derived elements, including prostaglandins and leukotrienes, which are implicated in pain and inflammatory responses (26). Annexin 1 is also implicated in the resolution of inflammation as well as in the phagocytosis by macrophages of apoptotic neutrophils (25).
Figure 2.

Mechanism of action of corticosteroids. Abbreviations: GR, glucocorticoid receptor; MR, mineralocorticoid receptor; HSP, heat-shock protein; HRE, hormonal response element.
The genomic effects of glucocorticoids take place only after several hours, following nuclear translocation of activated GR and gene regulation (30). This latency is explained by the time needed for mRNA production, protein synthesis, and transport (31).
Effect of Glucocorticoids in Health
Glucocorticoids suppress the production of acute phase reactants and of chemokines implicated in leukocyte chemo-attraction (32, 33), thereby reducing leukocytes migration into inflamed areas. Glucocorticoids suppress the expression of endothelial-leukocyte adhesion molecule 1, intracellular adhesion molecule 1 (ICAM-1), and vascular adhesion molecule 1 (VCAM-1) opposing to leukocytes trafficking through the endothelium (34, 35). Glucocorticoids affect both the innate and adaptive arms of the immune system. Target immune cells include (1) myeloid cells; macrophages, monocytes, dendritic cells (tissue-resident DCs, migratory DCs, and plasmacytoid DCs), as well as granulocytes and (2) lymphocytes, including CD8, T helper 1 (Th1), Th2, and Th17 as well as Treg and B cells (14). Broadly speaking, glucocorticoids repress the maturation, differentiation and proliferation of leukocytes of all subtypes.
Glucocorticoids attenuate fever by reducing monocytes and macrophages production of interleukin (IL)-1, TNF, IL-8, and MCP-1 (36). Glucocorticoids reduce the number of monocytes/macrophages, dendritic cells, and eosinophil and basophil granulocytes (37). Despite reducing the number of monocytes/macrophages, the capacity of glucocorticoid-treated macrophages for phagocytosis seems unaltered or even improved (38). Neutrophil granulocytes are not affected by the increased apoptosis induced by glucocorticoids, possibly because of the specific production of an inactive isoform of the GR (GR-beta) (39). Indeed, after treatment by glucocorticoids, the number of circulating neutrophil granulocytes increases, through an increased release by the bone marrow associated with increased demargination. However, these leukocytes may be functionally less efficient. Glucocorticoid-treated circulating polymorphonuclear leukocytes exhibit decreased levels of L-selectin receptors (40). Dexamethasone induces the expression by polymorphonuclear leukocytes of a decoy receptor for IL-1 (41). Glucocorticoids stabilize the lysosomal membranes, greatly reducing the amount of proteolytic enzymes released by lysosomes. Dendritic cells treated by glucocorticoids produce increased levels of the anti-inflammatory cytokines IL-10 and TGF-β (42). Glucocorticoids reduce the membrane expression of MHC class II and Fc receptors (43, 44) and suppress antigen presenting to T cells (45).
Activation, proliferation, and production of immunoglobulins by B cell lymphocytes are depressed by glucocorticoids (46, 47). They deplete thymic stroma cells and T cells by apoptosis (48–50). Circulating T-cell numbers are reduced with a shift from a pro-inflammatory Th1 phenotype to an anti-inflammatory Th2 phenotype (51–54). Glucocorticoids suppress the production by lymphocytes of the pro-inflammatory cytokines IL-2, IL-4, IL-5, IL-13, and INF (55, 56).
Mineralocorticoids
Molecular Mechanisms of Action
While the immune effects of glucocorticoids have been extensively investigated, those of mineralocorticoids have only recently gained attention. The biological activity of mineralocorticoids is mediated by interaction with a specific intracellular receptor, the mineralocorticoid receptor (MR). The MR is a transcription factors belonging to the nuclear receptor superfamily, encoded on chromosome 4, in the q31.1 region (57, 58). The structure of the mineralocorticoid-regulated transcription factor is highly similar to that of the GR and displays several specific domains, including a ligand-binding domain, a DNA-binding domain, and a trans-activating domain. The amino acid sequence of the DNA-binding domain of the GR and MR are approximately 94% similar, indicating that these two receptors may recognize and bind similar DNA sequences. The activated MR regulates the expression of a set of genes within target tissues, in a similar way to that of the activated GR. Unbound MR are stabilized in the cytoplasm by Hsp90. Activated MR will shed their chaperone proteins and translocate into the nucleus, form dimmers, and go on to recognize specific hormone recognizing elements of the DNA (Figure 2) (59). There is evidence that MR and GR may form functional heterodimers with specific properties (60, 61).
Surprisingly, cortisol and aldosterone bind the MR with equal affinity, indicating that the MR does not specifically recognize mineralocorticoids over glucocorticoids (57). However, the transcriptional response of the MR in response to aldosterone is approximately 100-fold higher than cortisol (62). Corticosteroid specificity is in part due to the activity of the type 2 isoenzyme of the 11β-hydroxysteroid dehydrogenase (11βHSD2), found in the kidney and in the colon and located near the MR. 11βHSD2 metabolizes glucocorticoids into an inactive derivative, cortisone. Since mineralocorticoids are unaffected by 11βHSD2 they are therefore able to interact with the MR (63). Therefore, the role of glucocorticoid binding of MR in non-epithelial tissues, which are devoid of 11βHSD2, is raised. The MR has several isoforms, some of which are able to bind both glucocorticoids and mineralocorticoids, while other isoforms bind exclusively mineralocorticoids (64). The density of GR and MR varies from one tissue to another. For instance, MR expression is higher than GR expression in the central nervous system; MR and GR are similarly expressed in the cardiovascular system while GR expression is higher than MR expression in the immune system (65). Finally, GR and MR also differ in their capacity to inhibit AP-1-mediated gene activation (66).
Effect of Mineralocorticoids on the Immune System in Health
Mineralocorticoid receptors play specific roles depending on their tissue expression. MRs located in the kidneys and the colon are implicated in NaCl reabsorption and K+ secretion (67, 68), where NaCl reabsorption is mediated by serum and glucocorticoid-induced kinase (SGK1), GILZ protein, and the epithelial sodium channel (69). Mineralocorticoid stimulation promotes the expression by endothelial cells of the VCAM-1, ICAM-1, and P-selectin membrane receptors, implicated in the adhesion of leukocytes to endothelial cells (70). In endothelial cells, mineralocorticoids also induce the production of reactive oxygen species via the activation of NADPH oxidase and Rac1 (71). In the brain, MRs are specifically located in the limbic system and are implicated in learning and memory (72). MRs are expressed in monocytes and macrophages (73), dendritic cells (74), and neutrophils (75). MR signaling in myeloid cells induces a pro-inflammatory response (76, 77). Indeed, macrophages exposed to mineralocorticoid agonists undergo a M1 type pro-inflammatory polarization associated with an increased production of TNF-α and of reactive oxygen species (78–80). In microglial cells, which are resident macrophages of the central nervous system, aldosterone activation induces an increased production of TNF-α and IL-6 in response to lipopolysaccharide stimulation (81). By contrast, MR knockout macrophages or macrophages treated by MR antagonists exhibit a M2 anti-inflammatory polarization (78). Mineralocorticoid agonists induce the activation of the mitogen-activated protein kinase pathway in dendritic cells, leading to the secretion of IL-6 and TGF-β1 (74). Mineralocorticoids indirectly lead to an increase in platelet cytosolic calcium concentrations, leading to platelet activation, thrombin formation, and platelet procoagulant activity (82, 83). Indeed, platelet cytosolic calcium entry is upregulated by the serum- and glucocorticoid-inducible kinase isoform SGK1, which is upregulated by mineralocorticoids (84). SGK1 upregulates IL-17-producing CD4+ helper T cells (Th17 cells). Th17 cells are dependent on IL-23 expression; SGK1 ensures the proper expression of the IL-23 receptor (85).
Mineralocorticoid receptor activation indirectly affects T lymphocyte phenotype. Indeed, dendritic cells activated by mineralocorticoid agonists impose a pro-inflammatory Th17 phenotype on CD4 T cells (74). Aldosterone stimulates IL-1β secretion by macrophages through NF-κB signaling and reactive oxygen species generation. Aldosterone also increases the expression of NLRP3, implicated in the formation of inflammasone and mature IL-1β in human peripheral blood mononuclear cells (86). Infusion of aldosterone in rodents results in elevated plasma IL-1β levels (86). Human blood mononuclear cells exposed to MR antagonists produce less cytokines, including TNF, IL-1α, IL-2, IL-6, INFγ, and GM-CSF (87). Aldosterone and MR agonists promote myocardial and kidney fibrosis (88, 89). Fludrocortisone, at high doses administered in vivo, paradoxically exhibits anti-inflammatory properties. Fludrocortisone inhibits histamine release by basophils (90) and IL-1 production by lung fragments (91).
Immunomodulation in Sepsis
Glucocorticoids
Animal Studies
The expression of the GR is upregulated in LPS-stimulated mouse macrophages and in mouse models of sepsis (92–94) (Table 2). However, others have shown that GR expression and protein levels decreased following a TNF challenge (95). The binding capacity of the GR decreases after an endotoxin challenge (94). The binding capacity of the GR may be altered through the action of nitric oxide (96, 97). LPS challenge in GR knockout mice induces higher mortality than in control animals (98). The endothelial GR regulates NF-κB in a model of endotoxin-induced sepsis (99). The deletion of the GR from endothelial cells, through the activation of NF-κB, is associated with higher mortality, higher nitric oxide levels, and higher levels of pro-inflammatory cytokines (TNF-α and IL-6) (99). In small animals with sepsis, high doses of dexamethasone and methylprednisolone significantly prolong survival (100–102). High doses of methylprednisolone continuously administered in a canine model of endotoxin shock also improve survival (103).
Table 2.
Main effects of glucocorticoid or mineralocorticoid administration during sepsis.
| Compound | Glucocorticoid | Mineralocorticoid |
|---|---|---|
| Small animal | ||
| Large animal |
|
|
| Man |
|
|
Studies in Humans
When LPS is administered to healthy subjects, the concomitant administration of hydrocortisone reduces plasma levels of TNF-α (112). The levels of E-selectin and of the soluble form of the receptor sE-selectin are also reduced following hydrocortisone therapy in sepsis (113, 114). During septic shock, hydrocortisone decreases the number of eosinophils (115), circulating levels of phospholipase A (116), serum levels of nitrite/nitrate, IL-6, IL-8, and markers of neutrophil activation (decreased expression of CD11b, CD64, and neutrophil elastase). Moreover, hydrocortisone lowers ex vivo whole blood production of IL-1 and IL-6 in response to LPS (113, 116–119). In hydrocortisone-treated patients with septic shock, monocyte mHLA-DR levels are depressed, while the capacity for phagocytosis of monocytes increases (113, 118). Glucocorticoids also attenuate LPS-stimulated monocyte production of migration inhibitory factor (120). In neutrophils of hydrocortisone-treated sepsis patients, the binding capacity of GR for glucocorticoid is reduced (121).
Clinical Trials
Short courses of high dose methylprednisolone or dexamethasone do not significantly reduce mortality and may even harm patients with sepsis (124–127). The introduction of the concept of sepsis-associated relative adrenal insufficiency in the 90s, led physicians and trialists to consider using prolonged courses of low doses of hydrocortisone (128). Several small size trials found that 200–400 mg of hydrocortisone per day for more than 3 days improved cardiovascular function in sepsis (117, 129, 130). GERINF05 was the first phase 3 trial that tested a prolonged course (7 days) of low to moderate doses (200 mg/day) of corticosteroids in septic shock with evidence of relative adrenal insufficiency (15). This trial found a significant improvement on survival and cardiovascular function in patients with septic shock and non-responders to a 250 µg ACTH test (Delta cortisol <9 μg/dl) (15). The CORTICUS trial could not reproduce the survival benefit of corticosteroids found in GERINF05 while confirming the benefit on cardiovascular homeostasis and organs function (131). A meta-analysis of the use of corticosteroids in sepsis published in 2015 concluded that corticosteroids reduced 28-day mortality, increased the rate of shock reversal, without increasing the risk of infection (122). More recently, the ADRENAL trial found no significant survival benefit from a continuous infusion of hydrocortisone in patients with septic shock (132). Nevertheless, the trial found that, when compared with placebo, hydrocortisone fasten the resolution of shock, shortened the duration of mechanical ventilation, and reduced the requirement for blood transfusion (132). In keeping with GERINF05 observations, the APROCCHSS trial found that the combination of hydrocortisone to fludrocortisone significantly reduced 90-day mortality (16). Likewise, corticosteroids hastened the resolution of shock and organs failure without causing major adverse events.
Mineralocorticoids
Animal Studies
In models of chronic cardiovascular diseases, aldosterone is associated with increased vascular and cardiac oxidative stress, inflammation, and fibrosis (133). In models of acute cardiovascular diseases, both exogenous aldosterone and overexpression of the MR increased blood pressure (134, 135). Aldosterone plays a role in salt appetite (136), and in coping behavior (137). MRs, contrary to GRs, when activated, play a neural antiapoptotic role by differentially influencing genes of the bcl-2 family (138). Similar to the renal epithelium, aldosterone may also favor the clearance of alveolar fluid by type II epithelial cells (139). By extrapolation, these responses may be of benefit during sepsis. However, very little is known about the MR or its regulation during sepsis. In small animals, sepsis is associated with downregulation of the MR in endothelial cells (92, 104), which is restored by supplementation by exogenous MR (104). In endotoxin shock, aldosterone reduces plasma levels of histamine, serotonin, bradykinin, and catecholamine (105). MR agonists reduce mortality in endotoxin-challenged small animals (107–109). In large animal sepsis models, aldosterone levels correlate with the severity of shock and with mortality (140), and mineralocorticoid supplementation improved survival and hastened shock reversal when administered prior to a bacterial challenge (110). The early administration of glucocorticoids plus mineralocorticoids improves the outcome in large animals with sepsis. In the sickest animals, glucocorticoids plus mineralocorticoids treatment hastened shock reversal and lowered plasma IL-6 levels (111).
Clinical Studies
The plasma concentrations of aldosterone were found to be unexpectedly low in meningococcal sepsis compared with ICU admissions for other reasons (141). Lower than expected aldosterone levels have also been reported in adults’ septic shock (142, 143). Inappropriately low aldosterone levels during septic shock were associated with increased ICU length of stay, an increased incidence of acute kidney failure (144), and increased mortality (145). Low aldosterone levels occurred despite high renin levels, suggesting impaired adrenal synthesis of aldosterone. In addition, a subset of patients with sepsis did not increase aldosterone levels in response to ACTH stimulation (146). Mineralocorticoid levels were found to correlate with IL-6 levels in meningococcal sepsis (147). The expression of the MR is downregulated in human endothelial cells exposed to TNF-α (104). The MR agonist fludrocortisone is administered orally, and there is no currently available intravenous formulation. The pharmacokinetics of fludrocortisone were assessed in healthy subjects (148), and in septic shock patients (149). However, there are still too few data on the direct effects of mineralocorticoids in patients with sepsis. A retrospective study in a pediatric population found that hydrocortisone combined with fludrocortisone was associated with shorter duration of vasopressor support in the most severe patients (123). Finally, it remains unclear whether or not part of the survival benefit from corticosteroids observed in the GERINF05 (15) and APROCCHSS (16) trials are directly related to fludrocortisone.
Future research should start with an individual patient data meta-analysis of all trials comparing hydrocortisone and/or fludrocortisone against placebo. Then, next trial should be designed as a two-by-two factorial placebo-controlled trial comparing hydrocortisone versus fludrocortisone versus hydrocortisone plus fludrocortisone (150).
Conclusion
In sepsis, there is sufficient evidence from animals and humans studies to support that glucocorticoids modulate innate immunity to promote the resolution of inflammation and organs failure. Much less is known about the immune effects of mineralocorticoids though increasing evidence from laboratory investigations suggested that they might favorably impact the outcome from sepsis. So far, survival benefits in adults with septic shock have been shown only for the combination of hydrocortisone plus fludrocortisone and not from the administration of hydrocortisone alone.
Author Contributions
DA has conceived the manuscript. All the authors have contributed to the literature search and writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA (2016) 315:801–10. 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The pathogenesis of sepsis. Annu Rev Pathol (2011) 6:19–48. 10.1146/annurev-pathol-011110-130327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent J-L. Sepsis and septic shock. Nat Rev Dis Primer (2016) 2:16045. 10.1038/nrdp.2016.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev (2012) 249:158–75. 10.1111/j.1600-065X.2012.01146.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of toll-like receptor signalling complexes. Nat Rev Immunol (2014) 14:546–58. 10.1038/nri3713 [DOI] [PubMed] [Google Scholar]
- 6.Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol (2009) 9:62–70. 10.1038/nri2470 [DOI] [PubMed] [Google Scholar]
- 7.Ahasic AM, Christiani DC. Personalized critical care medicine: how far away are we? Semin Respir Crit Care Med (2015) 36:809–22. 10.1055/s-0035-1564852 [DOI] [PubMed] [Google Scholar]
- 8.Fleischmann C, Scherag A, Adhikari NKJ, Hartog CS, Tsaganos T, Schlattmann P, et al. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med (2016) 193:259–72. 10.1164/rccm.201504-0781OC [DOI] [PubMed] [Google Scholar]
- 9.Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet (2015) 386:743–800. 10.1016/S0140-6736(15)60692-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med (2013) 41:1167–74. 10.1097/CCM.0b013e31827c09f8 [DOI] [PubMed] [Google Scholar]
- 11.Annane D, Sharshar T. Cognitive decline after sepsis. Lancet Respir Med (2015) 3:61–9. 10.1016/S2213-2600(14)70246-2 [DOI] [PubMed] [Google Scholar]
- 12.Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA (2010) 304:1787–94. 10.1001/jama.2010.1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med (2017) 43:304–77. 10.1007/s00134-017-4683-6 [DOI] [PubMed] [Google Scholar]
- 14.Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol (2017) 17:233–47. 10.1038/nri.2017.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Annane D, Sébille V, Charpentier C, Bollaert P-E, François B, Korach J-M, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA (2002) 288:862–71. 10.1001/jama.288.7.862 [DOI] [PubMed] [Google Scholar]
- 16.Annane D, Renault A, Brun-Buisson C, Megarbane B, Quenot J-P, Siami S, et al. Hydrocortisone plus fludrocortisone for adults with septic shock. N Engl J Med (2018) 378:809–18. 10.1056/NEJMoa1705716 [DOI] [PubMed] [Google Scholar]
- 17.Francke U, Foellmer BE. The glucocorticoid receptor gene is in 5q–q32. Genomics (1989) 4:610–2. 10.1016/0888-7543(89)90287-5 [DOI] [PubMed] [Google Scholar]
- 18.Weikum ER, Knuesel MT, Ortlund EA, Yamamoto KR. Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Rev Mol Cell Biol (2017) 18:159–74. 10.1038/nrm.2016.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Picard D, Khursheed B, Garabedian MJ, Fortin MG, Lindquist S, Yamamoto KR. Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature (1990) 348:166–8. 10.1038/348166a0 [DOI] [PubMed] [Google Scholar]
- 20.Nissen RM, Yamamoto KR. The glucocorticoid receptor inhibits NFkappaB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev (2000) 14:2314–29. 10.1101/gad.827900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luecke HF, Yamamoto KR. The glucocorticoid receptor blocks P-TEFb recruitment by NFkappaB to effect promoter-specific transcriptional repression. Genes Dev (2005) 19:1116–27. 10.1101/gad.1297105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reily MM, Pantoja C, Hu X, Chinenov Y, Rogatsky I. The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J (2006) 25:108–17. 10.1038/sj.emboj.7600919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell (2005) 122:707–21. 10.1016/j.cell.2005.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-κB and steroid receptor-signaling pathways. Endocr Rev (1999) 20:435–59. 10.1210/er.20.4.435 [DOI] [PubMed] [Google Scholar]
- 25.Vago JP, Nogueira CRC, Tavares LP, Soriani FM, Lopes F, Russo RC, et al. Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J Leukoc Biol (2012) 92:249–58. 10.1189/jlb.0112008 [DOI] [PubMed] [Google Scholar]
- 26.Hannon R, Croxtall JD, Getting SJ, Roviezzo F, Yona S, Paul-Clark MJ, et al. Aberrant inflammation and resistance to glucocorticoids in annexin 1-/- mouse. FASEB J (2003) 17:253–5. 10.1096/fj.02-0239fje [DOI] [PubMed] [Google Scholar]
- 27.Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science (1995) 270:286–90. 10.1126/science.270.5234.286 [DOI] [PubMed] [Google Scholar]
- 28.Berrebi D, Bruscoli S, Cohen N, Foussat A, Migliorati G, Bouchet-Delbos L, et al. Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: an anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood (2003) 101:729–38. 10.1182/blood-2002-02-0538 [DOI] [PubMed] [Google Scholar]
- 29.Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol (2002) 22:7802–11. 10.1128/MCB.22.22.7802-7811.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stahn C, Buttgereit F. Genomic and nongenomic effects of glucocorticoids. Nat Clin Pract Rheumatol (2008) 4:525–33. 10.1038/ncprheum0898 [DOI] [PubMed] [Google Scholar]
- 31.Wehling M. Specific, nongenomic actions of steroid hormones. Annu Rev Physiol (1997) 59:365–93. 10.1146/annurev.physiol.59.1.365 [DOI] [PubMed] [Google Scholar]
- 32.Miyamasu M, Misaki Y, Izumi S, Takaishi T, Morita Y, Nakamura H, et al. Glucocorticoids inhibit chemokine generation by human eosinophils. J Allergy Clin Immunol (1998) 101:75–83. 10.1016/S0091-6749(98)70196-4 [DOI] [PubMed] [Google Scholar]
- 33.Pype JL, Dupont LJ, Menten P, Van Coillie E, Opdenakker G, Van Damme J, et al. Expression of monocyte chemotactic protein (MCP)-1, MCP-2, and MCP-3 by human airway smooth-muscle cells. Modulation by corticosteroids and T-helper 2 cytokines. Am J Respir Cell Mol Biol (1999) 21:528–36. 10.1165/ajrcmb.21.4.3660 [DOI] [PubMed] [Google Scholar]
- 34.Cronstein BN, Kimmel SC, Levin RI, Martiniuk F, Weissmann G. A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci U S A (1992) 89:9991–5. 10.1073/pnas.89.21.9991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pitzalis C, Pipitone N, Bajocchi G, Hall M, Goulding N, Lee A, et al. Corticosteroids inhibit lymphocyte binding to endothelium and intercellular adhesion: an additional mechanism for their anti-inflammatory and immunosuppressive effect. J Immunol (1997) 158:5007–16. [PubMed] [Google Scholar]
- 36.Han J, Thompson P, Beutler B. Dexamethasone and pentoxifylline inhibit endotoxin-induced cachectin/tumor necrosis factor synthesis at separate points in the signaling pathway. J Exp Med (1990) 172:391–4. 10.1084/jem.172.1.391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boumpas DT, Chrousos GP, Wilder RL, Cupps TR, Balow JE. Glucocorticoid therapy for immune-mediated diseases: basic and clinical correlates. Ann Intern Med (1993) 119:1198–208. 10.7326/0003-4819-119-12-199312150-00007 [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Cousin JM, Hughes J, Van Damme J, Seckl JR, Haslett C, et al. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J Immunol (1999) 162:3639–46. [PubMed] [Google Scholar]
- 39.Strickland I, Kisich K, Hauk PJ, Vottero A, Chrousos GP, Klemm DJ, et al. High constitutive glucocorticoid receptor beta in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroids. J Exp Med (2001) 193:585–93. 10.1084/jem.193.5.585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakagawa M, Bondy GP, Waisman D, Minshall D, Hogg JC, van Eeden SF. The effect of glucocorticoids on the expression of L-selectin on polymor-phonuclear leukocyte. Blood (1999) 93:2730–7. [PubMed] [Google Scholar]
- 41.Re F, Muzio M, De Rossi M, Polentarutti N, Giri JG, Mantovani A, et al. The type II “receptor” as a decoy target for interleukin 1 in polymorphonuclear leukocytes: characterization of induction by dexamethasone and ligand binding properties of the released decoy receptor. J Exp Med (1994) 179:739–43. 10.1084/jem.179.2.739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stary G, Klein I, Bauer W, Koszik F, Reininger B, Kohlhofer S, et al. Glucocorticosteroids modify Langerhans cells to produce TGF-β and expand regulatory T cells. J Immunol (2011) 186:103–12. 10.4049/jimmunol.1002485 [DOI] [PubMed] [Google Scholar]
- 43.Bianchi M, Meng C, Ivashkiv LB. Inhibition of IL-2-induced Jak-STAT signaling by glucocorticoids. Proc Natl Acad Sci U S A (2000) 97:9573–8. 10.1073/pnas.160099797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piemonti L, Monti P, Allavena P, Sironi M, Soldini L, Leone BE, et al. Glucocorticoids affect human dendritic cell differentiation and maturation. J Immunol (1999) 162:6473–81. [PubMed] [Google Scholar]
- 45.DeKruyff RH, Fang Y, Umetsu DT. Corticosteroids enhance the capacity of macrophages to induce Th2 cytokine synthesis in CD4+ lymphocytes by inhibiting IL-12 production. J Immunol (1998) 160:2231–7. [PubMed] [Google Scholar]
- 46.Cupps TR, Edgar LC, Thomas CA, Fauci AS. Multiple mechanisms of B cell immunoregulation in man after administration of in vivo corticosteroids. J Immunol (1984) 132:170–5. [PubMed] [Google Scholar]
- 47.Cupps TR, Gerrard TL, Falkoff RJ, Whalen G, Fauci AS. Effects of in vitro corticosteroids on B cell activation, proliferation, and differentiation. J Clin Invest (1985) 75:754–61. 10.1172/JCI111757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Vliet E, Melis M, van Ewijk W. The influence of dexamethasone treatment on the lymphoid and stromal composition of the mouse thymus: a flowcytometric and immunohistological analysis. Cell Immunol (1986) 103:229–40. 10.1016/0008-8749(86)90086-9 [DOI] [PubMed] [Google Scholar]
- 49.Wang D, Müller N, McPherson KG, Reichardt HM. Glucocorticoids engage different signal transduction pathways to induce apoptosis in thymocytes and mature T cells. J Immunol (2006) 176:1695–702. 10.4049/jimmunol.176.3.1695 [DOI] [PubMed] [Google Scholar]
- 50.Woodward MJ, de Boer J, Heidorn S, Hubank M, Kioussis D, Williams O, et al. Tnfaip8 is an essential gene for the regulation of glucocorticoid-mediated apoptosis of thymocytes. Cell Death Differ (2010) 17:316–23. 10.1038/cdd.2009.125 [DOI] [PubMed] [Google Scholar]
- 51.Ramírez F, Fowell DJ, Puklavec M, Simmonds S, Mason D. Glucocorticoids promote a TH2 cytokine response by CD4+ T cells in vitro. J Immunol (1996) 156:2406–12. [PubMed] [Google Scholar]
- 52.Sun L, Yu G, Yang S, Zhang L. Effects of hydrocortisone on the differentiation of human T helper 2 cells. Scand J Immunol (2011) 73:208–14. 10.1111/j.1365-3083.2010.02500.x [DOI] [PubMed] [Google Scholar]
- 53.Franchimont D, Galon J, Gadina M, Visconti R, Zhou Y, Aringer M, et al. Inhibition of Th1 immune response by glucocorticoids: dexamethasone selectively inhibits IL-12-induced Stat4 phosphorylation in T lymphocytes. J Immunol (2000) 164:1768–74. 10.4049/jimmunol.164.4.1768 [DOI] [PubMed] [Google Scholar]
- 54.Liberman AC, Refojo D, Druker J, Toscano M, Rein T, Holsboer F, et al. The activated glucocorticoid receptor inhibits the transcription factor T-bet by direct protein-protein interaction. FASEB J (2007) 21:1177–88. 10.1096/fj.06-7452com [DOI] [PubMed] [Google Scholar]
- 55.Maneechotesuwan K, Yao X, Ito K, Jazrawi E, Usmani OS, Adcock IM, et al. Suppression of GATA-3 nuclear import and phosphorylation: a novel mechanism of corticosteroid action in allergic disease. PLoS Med (2009) 6:e1000076. 10.1371/journal.pmed.1000076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scheinman RI, Gualberto A, Jewell CM, Cidlowski JA, Baldwin AS. Characterization of mechanisms involved in transrepression of NF-kappa B by activated glucocorticoid receptors. Mol Cell Biol (1995) 15:943–53. 10.1128/MCB.15.2.943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science (1987) 237:268–75. 10.1126/science.3037703 [DOI] [PubMed] [Google Scholar]
- 58.Morrison N, Harrap SB, Arriza JL, Boyd E, Connor JM. Regional chromosomal assignment of the human mineralocorticoid receptor gene to 4q31.1. Hum Genet (1990) 85:130–2. 10.1007/BF00276340 [DOI] [PubMed] [Google Scholar]
- 59.Lombès M, Binart N, Oblin ME, Joulin V, Baulieu EE. Characterization of the interaction of the human mineralocorticosteroid receptor with hormone response elements. Biochem J (1993) 292(Pt 2):577–83. 10.1042/bj2920577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trapp T, Rupprecht R, Castrén M, Reul JM, Holsboer F. Heterodimerization between mineralocorticoid and glucocorticoid receptor: a new principle of glucocorticoid action in the CNS. Neuron (1994) 13:1457–62. 10.1016/0896-6273(94)90431-6 [DOI] [PubMed] [Google Scholar]
- 61.Mifsud KR, Reul JMHM. Acute stress enhances heterodimerization and binding of corticosteroid receptors at glucocorticoid target genes in the hippocampus. Proc Natl Acad Sci U S A (2016) 113:11336–41. 10.1073/pnas.1605246113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lombes M, Kenouch S, Souque A, Farman N, Rafestin-Oblin ME. The mineralocorticoid receptor discriminates aldosterone from glucocorticoids independently of the 11 beta-hydroxysteroid dehydrogenase. Endocrinology (1994) 135:834–40. 10.1210/endo.135.3.8070376 [DOI] [PubMed] [Google Scholar]
- 63.Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science (1988) 242:583–5. 10.1126/science.2845584 [DOI] [PubMed] [Google Scholar]
- 64.Zennaro MC, Le Menuet D, Lombès M. Characterization of the human mineralocorticoid receptor gene 5’-regulatory region: evidence for differential hormonal regulation of two alternative promoters via nonclassical mechanisms. Mol Endocrinol (1996) 10:1549–60. 10.1210/mend.10.12.8961265 [DOI] [PubMed] [Google Scholar]
- 65.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell (2006) 126:789–99. 10.1016/j.cell.2006.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pearce D, Yamamoto K. Mineralocorticoid and glucocorticoid receptor activities distinguished by nonreceptor factors at a composite response element. Science (1993) 259:1161–5. 10.1126/science.8382376 [DOI] [PubMed] [Google Scholar]
- 67.Ronzaud C, Loffing J, Bleich M, Gretz N, Grone H-J, Schutz G, et al. Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J Am Soc Nephrol (2007) 18:1679–87. 10.1681/ASN.2006090975 [DOI] [PubMed] [Google Scholar]
- 68.Terker AS, Yarbrough B, Ferdaus MZ, Lazelle RA, Erspamer KJ, Meermeier NP, et al. Direct and indirect mineralocorticoid effects determine distal salt transport. J Am Soc Nephrol (2016) 27:2436–45. 10.1681/ASN.2015070815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Soundararajan R, Zhang TT, Wang J, Vandewalle A, Pearce D. A novel role for glucocorticoid-induced leucine zipper protein in epithelial sodium channel-mediated sodium transport. J Biol Chem (2005) 280:39970–81. 10.1074/jbc.M508658200 [DOI] [PubMed] [Google Scholar]
- 70.Jeong Y, Chaupin DF, Matsushita K, Yamakuchi M, Cameron SJ, Morrell CN, et al. Aldosterone activates endothelial exocytosis. Proc Natl Acad Sci U S A (2009) 106:3782–7. 10.1073/pnas.0804037106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Iwashima F, Yoshimoto T, Minami I, Sakurada M, Hirono Y, Hirata Y. Aldosterone induces superoxide generation via Rac1 activation in endothelial cells. Endocrinology (2008) 149:1009–14. 10.1210/en.2007-0864 [DOI] [PubMed] [Google Scholar]
- 72.Berger S, Wolfer DP, Selbach O, Alter H, Erdmann G, Reichardt HM, et al. Loss of the limbic mineralocorticoid receptor impairs behavioral plasticity. Proc Natl Acad Sci U S A (2006) 103:195–200. 10.1073/pnas.0503878102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Armanini D, Strasser T, Weber PC. Characterization of aldosterone binding sites in circulating human mononuclear leukocytes. Am J Physiol (1985) 248:E388–90. [DOI] [PubMed] [Google Scholar]
- 74.Herrada AA, Contreras FJ, Marini NP, Amador CA, González PA, Cortés CM, et al. Aldosterone promotes autoimmune damage by enhancing Th17-mediated immunity. J Immunol (2010) 184:191–202. 10.4049/jimmunol.0802886 [DOI] [PubMed] [Google Scholar]
- 75.Bergmann A, Eulenberg C, Wellner M, Rolle S, Luft F, Kettritz R. Aldosterone abrogates nuclear factor kappaB-mediated tumor necrosis factor alpha production in human neutrophils via the mineralocorticoid receptor. Hypertension (2010) 55:370–9. 10.1161/HYPERTENSIONAHA.109.141309 [DOI] [PubMed] [Google Scholar]
- 76.Frieler RA, Ray JJ, Meng H, Ramnarayanan SP, Usher MG, Su EJ, et al. Myeloid mineralocorticoid receptor during experimental ischemic stroke: effects of model and sex. J Am Heart Assoc (2012) 1:e002584. 10.1161/JAHA.112.002584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rickard AJ, Morgan J, Bienvenu LA, Fletcher EK, Cranston GA, Shen JZ, et al. Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt-mediated inflammation and cardiac fibrosis. Hypertension (2012) 60:1443–50. 10.1161/HYPERTENSIONAHA.112.203158 [DOI] [PubMed] [Google Scholar]
- 78.Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schütz G, et al. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest (2010) 120:3350–64. 10.1172/JCI41080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Keidar S. Aldosterone administration to mice stimulates macrophage NADPH oxidase and increases atherosclerosis development: a possible role for angiotensin-converting enzyme and the receptors for angiotensin II and aldosterone. Circulation (2004) 109:2213–20. 10.1161/01.CIR.0000127949.05756.9D [DOI] [PubMed] [Google Scholar]
- 80.Łabuzek K, Liber S, Bułdak Ł, Machnik G, Liber J, Okopień B. Eplerenone promotes alternative activation in human monocyte-derived macrophages. Pharmacol Rep (2013) 65:226–34. 10.1016/S1734-1140(13)70983-6 [DOI] [PubMed] [Google Scholar]
- 81.Chantong B, Kratschmar DV, Nashev LG, Balazs Z, Odermatt A. Mineralocorticoid and glucocorticoid receptors differentially regulate NF-kappaB activity and pro-inflammatory cytokine production in murine BV-2 microglial cells. J Neuroinflammation (2012) 9:260. 10.1186/1742-2094-9-260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Braun A, Varga-Szabo D, Kleinschnitz C, Pleines I, Bender M, Austinat M, et al. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood (2009) 113:2056–63. 10.1182/blood-2008-07-171611 [DOI] [PubMed] [Google Scholar]
- 83.Mushtaq M, Nam T-S, Kim U-H. Critical role for CD38-mediated Ca2+ signaling in thrombin-induced procoagulant activity of mouse platelets and hemostasis. J Biol Chem (2011) 286:12952–8. 10.1074/jbc.M110.207100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Borst O, Schmidt E-M, Münzer P, Schönberger T, Towhid ST, Elvers M, et al. The serum- and glucocorticoid-inducible kinase 1 (SGK1) influences platelet calcium signaling and function by regulation of Orai1 expression in megakaryocytes. Blood (2012) 119:251–61. 10.1182/blood-2011-06-359976 [DOI] [PubMed] [Google Scholar]
- 85.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature (2013) 496:518–22. 10.1038/nature11868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bruder-Nascimento T, Ferreira NS, Zanotto CZ, Ramalho F, Pequeno IO, Olivon VC, et al. NLRP3 inflammasome mediates aldosterone-induced vascular damage. Circulation (2016) 134:1866–80. 10.1161/CIRCULATIONAHA.116.024369 [DOI] [PubMed] [Google Scholar]
- 87.Sønder SUS, Mikkelsen M, Rieneck K, Hedegaard CJ, Bendtzen K. Effects of spironolactone on human blood mononuclear cells: mineralocorticoid receptor independent effects on gene expression and late apoptosis induction. Br J Pharmacol (2006) 148:46–53. 10.1038/sj.bjp.0706700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nagai Y, Miyata K, Sun G-P, Rahman M, Kimura S, Miyatake A, et al. Aldosterone stimulates collagen gene expression and synthesis via activation of ERK1/2 in rat renal fibroblasts. Hypertension (2005) 46:1039–45. 10.1161/01.HYP.0000174593.88899.68 [DOI] [PubMed] [Google Scholar]
- 89.Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol (2013) 9:459–69. 10.1038/nrneph.2013.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schleimer RP, MacGlashan DW, Gillespie E, Lichtenstein LM. Inhibition of basophil histamine release by anti-inflammatory steroids. II. Studies on the mechanism of action. J Immunol (1982) 129:1632–6. [PubMed] [Google Scholar]
- 91.Bochner BS, Rutledge BK, Schleimer RP. Interleukin 1 production by human lung tissue. II. Inhibition by anti-inflammatory steroids. J Immunol (1987) 139:2303–7. [PubMed] [Google Scholar]
- 92.Barish GD, Downes M, Alaynick WA, Yu RT, Ocampo CB, Bookout AL, et al. A nuclear receptor atlas: macrophage activation. Mol Endocrinol (2005) 19:2466–77. 10.1210/me.2004-0529 [DOI] [PubMed] [Google Scholar]
- 93.Salkowski CA, Vogel SN. Lipopolysaccharide increases glucocorticoid receptor expression in murine macrophages. A possible mechanism for glucocorticoid-mediated suppression of endotoxicity. J Immunol (1992) 149:4041–7. [PubMed] [Google Scholar]
- 94.Bergquist M, Jirholt P, Nurkkala M, Rylander C, Hedenstierna G, Lindholm C. Glucocorticoid receptor function is decreased in neutrophils during endotoxic shock. J Infect (2014) 69:113–22. 10.1016/j.jinf.2014.03.011 [DOI] [PubMed] [Google Scholar]
- 95.Van Bogaert T, Vandevyver S, Dejager L, Van Hauwermeiren F, Pinheiro I, Petta I, et al. Tumor necrosis factor inhibits glucocorticoid receptor function in mice: a strong signal toward lethal shock. J Biol Chem (2011) 286:26555–67. 10.1074/jbc.M110.212365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Galigniana MD, Piwien-Pilipuk G, Assreuy J. Inhibition of glucocorticoid receptor binding by nitric oxide. Mol Pharmacol (1999) 55:317–23. 10.1124/mol.55.2.317 [DOI] [PubMed] [Google Scholar]
- 97.Duma D, Silva-Santos JE, Assreuy J. Inhibition of glucocorticoid receptor binding by nitric oxide in endotoxemic rats. Crit Care Med (2004) 32:2304–10. 10.1097/01.CCM.0000145996.57901.D7 [DOI] [PubMed] [Google Scholar]
- 98.Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ. Macrophage glucocorticoid receptors regulate toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood (2007) 109:4313–9. 10.1182/blood-2006-10-048215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Goodwin JE, Feng Y, Velazquez H, Sessa WC. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc Natl Acad Sci U S A (2013) 110:306–11. 10.1073/pnas.1210200110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pitcairn M, Schuler J, Erve PR, Holtzman S, Schumer W. Glucocorticoid and antibiotic effect on experimental gram-negative bacteremic shock. Arch Surg (1975) 110:1012–5. 10.1001/archsurg.1975.01360140156030 [DOI] [PubMed] [Google Scholar]
- 101.Ottosson J, Brandberg A, Erikson B, Hedman L, Dawidson I, Söderberg R. Experimental septic shock – effects of corticosteroids. Circ Shock (1982) 9:571–7. [PubMed] [Google Scholar]
- 102.Fabian TC, Patterson R. Steroid therapy in septic shock. Survival studies in a laboratory model. Am Surg (1982) 48:614–7. [PubMed] [Google Scholar]
- 103.White GL, Archer LT, Beller BK, Hinshaw LB. Increased survival with methylprednisolone treatment in canine endotoxin shock. J Surg Res (1978) 25:357–64. 10.1016/0022-4804(78)90131-2 [DOI] [PubMed] [Google Scholar]
- 104.Fadel F, André-Grégoire G, Gravez B, Bauvois B, Bouchet S, Sierra-Ramos C, et al. Aldosterone and vascular mineralocorticoid receptors in murine endotoxic and human septic shock. Crit Care Med (2017) 45:e954–62. 10.1097/CCM.0000000000002462 [DOI] [PubMed] [Google Scholar]
- 105.Ebata T, Hayasaka H. Effects of aldosterone and dexamethasone on blood chemical mediators in endotoxin shock. Jpn J Surg (1979) 9:79–85. 10.1007/BF02468720 [DOI] [PubMed] [Google Scholar]
- 106.Laviolle B, Nesseler N, Massart C, Bellissant E. Fludrocortisone and hydrocortisone, alone or in combination, on in vivo hemodynamics and in vitro vascular reactivity in normal and endotoxemic rats: a randomized factorial design study. J Cardiovasc Pharmacol (2014) 63:488–96. 10.1097/FJC.0000000000000072 [DOI] [PubMed] [Google Scholar]
- 107.Bein HJ, Jaques R. The antitoxic effect of aldosterone. Experientia (1960) 16:24–6. 10.1007/BF02201783 [DOI] [PubMed] [Google Scholar]
- 108.Bondoc CC, Beskid G, Wolferth CC, Howard JM, O’Malley JF. Cardiovascular and antitoxic effects of aldosterone on cats in endotoxin shock. Surg Gynecol Obstet (1962) 114:43–6. [PubMed] [Google Scholar]
- 109.Hayasaka H, O’malley JF, Howard JM. Antitoxic effect of aldosterone on cats in endotoxic shock. Further studies. Arch Surg (1963) 87:861–5. 10.1001/archsurg.1963.01310170147025 [DOI] [PubMed] [Google Scholar]
- 110.Hicks CW, Sweeney DA, Danner RL, Eichacker PQ, Suffredini AF, Feng J, et al. Efficacy of selective mineralocorticoid and glucocorticoid agonists in canine septic shock. Crit Care Med (2012) 40:199–207. 10.1097/CCM.0b013e31822efa14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hicks CW, Sweeney DA, Danner RL, Eichacker PQ, Suffredini AF, Feng J, et al. Beneficial effects of stress-dose corticosteroid therapy in canines depend on the severity of staphylococcal pneumonia. Intensive Care Med (2012) 38:2063–71. 10.1007/s00134-012-2735-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Barber AE, Coyle SM, Marano MA, Fischer E, Calvano SE, Fong Y, et al. Glucocorticoid therapy alters hormonal and cytokine responses to endotoxin in man. J Immunol (1993) 150:1999–2006. [PubMed] [Google Scholar]
- 113.Keh D, Boehnke T, Weber-Cartens S, Schulz C, Ahlers O, Bercker S, et al. Immunologic and hemodynamic effects of “low-dose” hydrocortisone in septic shock: a double-blind, randomized, placebo-controlled, crossover study. Am J Respir Crit Care Med (2003) 167:512–20. 10.1164/rccm.200205-446OC [DOI] [PubMed] [Google Scholar]
- 114.Leone M, Boutière-Albanèse B, Valette S, Camoin-Jau L, Barrau K, Albanèse J, et al. Cell adhesion molecules as a marker reflecting the reduction of endothelial activation induced by glucocorticoids. Shock (2004) 21:311–4. 10.1097/00024382-200404000-00004 [DOI] [PubMed] [Google Scholar]
- 115.Laviolle B, Annane D, Fougerou C, Bellissant E. Gluco- and mineralocorticoid biological effects of a 7-day treatment with low doses of hydrocortisone and fludrocortisone in septic shock. Intensive Care Med (2012) 38:1306–14. 10.1007/s00134-012-2585-1 [DOI] [PubMed] [Google Scholar]
- 116.Briegel J, Kellermann W, Forst H, Haller M, Bittl M, Hoffmann GE, et al. Low-dose hydrocortisone infusion attenuates the systemic inflammatory response syndrome. The Phospholipase A2 Study Group. Clin Investig (1994) 72:782–7. 10.1007/BF00180547 [DOI] [PubMed] [Google Scholar]
- 117.Oppert M, Schindler R, Husung C, Offermann K, Gräf K-J, Boenisch O, et al. Low-dose hydrocortisone improves shock reversal and reduces cytokine levels in early hyperdynamic septic shock. Crit Care Med (2005) 33:2457–64. 10.1097/01.CCM.0000186370.78639.23 [DOI] [PubMed] [Google Scholar]
- 118.Kaufmann I, Briegel J, Schliephake F, Hoelzl A, Chouker A, Hummel T, et al. Stress doses of hydrocortisone in septic shock: beneficial effects on opsonization-dependent neutrophil functions. Intensive Care Med (2008) 34:344–9. 10.1007/s00134-007-0868-8 [DOI] [PubMed] [Google Scholar]
- 119.Briegel J, Jochum M, Gippner-Steppert C, Thiel M. Immunomodulation in septic shock: hydrocortisone differentially regulates cytokine responses. J Am Soc Nephrol (2001) 12(Suppl 17):S70–4. [PubMed] [Google Scholar]
- 120.Maxime V, Fitting C, Annane D, Cavaillon J-M. Corticoids normalize leukocyte production of macrophage migration inhibitory factor in septic shock. J Infect Dis (2005) 191:138–44. 10.1086/426401 [DOI] [PubMed] [Google Scholar]
- 121.Bergquist M, Lindholm C, Strinnholm M, Hedenstierna G, Rylander C. Impairment of neutrophilic glucocorticoid receptor function in patients treated with steroids for septic shock. Intensive Care Med Exp (2015) 3:23. 10.1186/s40635-015-0059-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Annane D, Bellissant E, Bollaert PE, Briegel J, Keh D, Kupfer Y. Corticosteroids for treating sepsis. Cochrane Database Syst Rev (2015). 10.1002/14651858.CD002243.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hebbar KB, Stockwell JA, Fortenberry JD. Clinical effects of adding fludrocortisone to a hydrocortisone-based shock protocol in hypotensive critically ill children. Intensive Care Med (2011) 37:518–24. 10.1007/s00134-010-2090-3 [DOI] [PubMed] [Google Scholar]
- 124.Luce JM, Montgomery AB, Marks JD, Turner J, Metz CA, Murray JF. Ineffectiveness of high-dose methylprednisolone in preventing parenchymal lung injury and improving mortality in patients with septic shock. Am Rev Respir Dis (1988) 138:62–8. 10.1164/ajrccm/138.1.62 [DOI] [PubMed] [Google Scholar]
- 125.Veterans Administration Systemic Sepsis Cooperative Study Group. Effect of high-dose glucocorticoid therapy on mortality in patients with clinical signs of systemic sepsis. N Engl J Med (1987) 317:659–65. 10.1056/NEJM198709103171102 [DOI] [PubMed] [Google Scholar]
- 126.Sprung CL, Caralis PV, Marcial EH, Pierce M, Gelbard MA, Long WM, et al. The effects of high-dose corticosteroids in patients with septic shock. A prospective, controlled study. N Engl J Med (1984) 311:1137–43. 10.1056/NEJM198411013111801 [DOI] [PubMed] [Google Scholar]
- 127.Bone RC, Fisher CJ, Clemmer TP, Slotman GJ, Metz CA, Balk RA. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med (1987) 317:653–8. 10.1056/NEJM198709103171101 [DOI] [PubMed] [Google Scholar]
- 128.Rothwell PM, Udwadia ZF, Lawler PG. Cortisol response to corticotropin and survival in septic shock. Lancet (1991) 337:582–3. 10.1016/0140-6736(91)91641-7 [DOI] [PubMed] [Google Scholar]
- 129.Bollaert PE, Charpentier C, Levy B, Debouverie M, Audibert G, Larcan A. Reversal of late septic shock with supraphysiologic doses of hydrocortisone. Crit Care Med (1998) 26:645–50. 10.1097/00003246-199804000-00010 [DOI] [PubMed] [Google Scholar]
- 130.Briegel J, Forst H, Haller M, Schelling G, Kilger E, Kuprat G, et al. Stress doses of hydrocortisone reverse hyperdynamic septic shock: a prospective, randomized, double-blind, single-center study. Crit Care Med (1999) 27:723–32. 10.1097/00003246-199904000-00025 [DOI] [PubMed] [Google Scholar]
- 131.Sprung CL, Annane D, Keh D, Moreno R, Singer M, Freivogel K, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med (2008) 358:111–24. 10.1056/NEJMoa071366 [DOI] [PubMed] [Google Scholar]
- 132.Venkatesh B, Finfer S, Cohen J, Rajbhandari D, Arabi Y, Bellomo R, et al. Adjunctive glucocorticoid therapy in patients with septic shock. N Engl J Med (2018) 378:797–808. 10.1056/NEJMoa1705835 [DOI] [PubMed] [Google Scholar]
- 133.Rickard AJ, Morgan J, Chrissobolis S, Miller AA, Sobey CG, Young MJ. Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension (2014) 63:1033–40. 10.1161/HYPERTENSIONAHA.113.01803 [DOI] [PubMed] [Google Scholar]
- 134.Gomez-Sanchez EP. Intracerebroventricular infusion of aldosterone induces hypertension in rats. Endocrinology (1986) 118:819–23. 10.1210/endo-118-2-819 [DOI] [PubMed] [Google Scholar]
- 135.Cat AND, Griol-Charhbili V, Loufrani L, Labat C, Benjamin L, Farman N, et al. The endothelial mineralocorticoid receptor regulates vasoconstrictor tone and blood pressure. FASEB J (2010) 24:2454–63. 10.1096/fj.09-147926 [DOI] [PubMed] [Google Scholar]
- 136.Sakai RR, Nicolaïdis S, Epstein AN. Salt appetite is suppressed by interference with angiotensin II and aldosterone. Am J Physiol (1986) 251:R762–8. [DOI] [PubMed] [Google Scholar]
- 137.Oitzl MS, de Kloet ER. Selective corticosteroid antagonists modulate specific aspects of spatial orientation learning. Behav Neurosci (1992) 106:62–71. 10.1037/0735-7044.106.1.62 [DOI] [PubMed] [Google Scholar]
- 138.Almeida OF, Condé GL, Crochemore C, Demeneix BA, Fischer D, Hassan AH, et al. Subtle shifts in the ratio between pro- and antiapoptotic molecules after activation of corticosteroid receptors decide neuronal fate. FASEB J (2000) 14:779–90. 10.1096/fasebj.14.5.779 [DOI] [PubMed] [Google Scholar]
- 139.Suzuki S, Tsubochi H, Suzuki T, Darnel AD, Krozowski ZS, Sasano H, et al. Modulation of transalveolar fluid absorption by endogenous aldosterone in adult rats. Exp Lung Res (2001) 27:143–55. 10.1080/019021401750069384 [DOI] [PubMed] [Google Scholar]
- 140.Cortés-Puch I, Hicks CW, Sun J, Solomon SB, Eichacker PQ, Sweeney DA, et al. Hypothalamic-pituitary-adrenal axis in lethal canine Staphylococcus aureus pneumonia. Am J Physiol-Endocrinol Metab (2014) 307:E994–1008. 10.1152/ajpendo.00345.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lichtarowicz-Krynska EJ, Cole TJ, Camacho-Hubner C, Britto J, Levin M, Klein N, et al. Circulating aldosterone levels are unexpectedly low in children with acute meningococcal disease. J Clin Endocrinol Metab (2004) 89:1410–4. 10.1210/jc.2003-030505 [DOI] [PubMed] [Google Scholar]
- 142.Moraes RB, Friedman G, Viana MV, Tonietto T, Saltz H, Czepielewski MA. Aldosterone secretion in patients with septic shock: a prospective study. Arq Bras Endocrinol Metabol (2013) 57:636–41. 10.1590/S0004-27302013000800009 [DOI] [PubMed] [Google Scholar]
- 143.Tsai M-H, Chen Y-C, Lien J-M, Tian Y-C, Peng Y, Fang J-T, et al. Hemodynamics and metabolic studies on septic shock in patients with acute liver failure. J Crit Care (2008) 23:468–72. 10.1016/j.jcrc.2008.04.006 [DOI] [PubMed] [Google Scholar]
- 144.du Cheyron D, Lesage A, Daubin C, Ramakers M, Charbonneau P. Hyperreninemic hypoaldosteronism: a possible etiological factor of septic shock-induced acute renal failure. Intensive Care Med (2003) 29:1703–9. 10.1007/s00134-003-1986-6 [DOI] [PubMed] [Google Scholar]
- 145.Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab (1987) 64:592–5. 10.1210/jcem-64-3-592 [DOI] [PubMed] [Google Scholar]
- 146.Manglik S, Flores E, Lubarsky L, Fernandez F, Chhibber VL, Tayek JA. Glucocorticoid insufficiency in patients who present to the hospital with severe sepsis: a prospective clinical trial. Crit Care Med (2003) 31:1668–75. 10.1097/01.CCM.0000063447.37342.A9 [DOI] [PubMed] [Google Scholar]
- 147.den Brinker M, Joosten KFM, Liem O, de Jong FH, Hop WCJ, Hazelzet JA, et al. Adrenal insufficiency in meningococcal sepsis: bioavailable cortisol levels and impact of interleukin-6 levels and intubation with etomidate on adrenal function and mortality. J Clin Endocrinol Metab (2005) 90:5110–7. 10.1210/jc.2005-1107 [DOI] [PubMed] [Google Scholar]
- 148.Hamitouche N, Comets E, Ribot M, Alvarez J-C, Bellissant E, Laviolle B. Population pharmacokinetic-pharmacodynamic model of oral fludrocortisone and intravenous hydrocortisone in healthy volunteers. AAPS J (2017) 19:727–35. 10.1208/s12248-016-0041-9 [DOI] [PubMed] [Google Scholar]
- 149.Polito A, Hamitouche N, Ribot M, Polito A, Laviolle B, Bellissant E, et al. Pharmacokinetics of oral fludrocortisone in septic shock. Br J Clin Pharmacol (2016) 82:1509–16. 10.1111/bcp.13065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Druce LA, Thorpe CM, Wilton A. Mineralocorticoid effects due to cortisol inactivation overload explain the beneficial use of hydrocortisone in septic shock. Med Hypotheses (2008) 70:56–60. 10.1016/j.mehy.2007.04.031 [DOI] [PubMed] [Google Scholar]