

Abstract
Background/Aims
Recent research has highlighted the importance of interactions between commensal fungi and intestinal inflammation. However, there are few studies investigating whether commensal fungi contribute to inflammation in patients with Crohn's disease (CD). The aim of this study is to investigate reveal interactions between commensal fungi and host immune cells in CD.
Methods
CD14-positive monocytes were isolated from peripheral blood mononuclear cells from healthy human volunteers and then differentiated in the presence of macrophage colony-stimulating factor (M-CSF) (referred to as M-macrophages, M-Mϕs) or M-CSF and interferon-γ (IFN-γ) (referred to as M-gamma macrophages, Mγ-Mϕs). Cytokine production by these in vitro differentiated macrophages in response to β-(1,3)-glucan was analyzed by flow cytometry. Expression of Dectin-1 was examined using flow cytometry, western blotting, and quantitative reverse transcription-polymerase chain reaction. Cytokine production by in vitro differentiated macrophages in response to β-(1,3)-glucan was measured in the presence of an anti-Dectin-1 receptor antagonist, anti-Syr, or an anti-Fas-1 antibody. Cytokine production by lamina propria mononuclear cells (LPMCs) derived from CD patients in response to β-(1,3)-glucan was also analyzed.
Results
Mγ-Mϕs produced a large amount of tumor necrosis factor-α (TNF-α) and interleukin-6 in response to β-(1,3)-glucan. Dectin-1 expression was significantly higher in Mγ-Mϕs than in M-Mϕs. The increase in TNF-α production by Mγ-Mϕs stimulated with glucan was reversed by blocking Dectin-1, Syr or Fas-1. LPMCs derived from CD patients stimulated with β-(1,3)-glucan produced significantly higher amount of TNF-α than LPMCs derived from UC patients.
Conclusions
These results suggest that commensal fungal microbiota may contribute to the pathogenesis of CD by inducing macrophages-derived pro-inflammatory cytokines.
Keywords: Crohn disease, Candida albicans, Tumor necrosis factor-alpha, Dectin-1
INTRODUCTION
Crohn's disease (CD) is a chronic inflammatory disease that typically affects the terminal ileum and/or colon. Despite extensive study, the pathophysiology of CD remains unclear; nonetheless, host genetic factors, immune system dysregulation, and environmental factors are known to be involved.1,2 Importantly, intestinal bacterial communities play a critical role in the pathogenesis of the intestinal inflammation that occurs in CD,3,4,5 and dysbiosis, a major change in the composition of intestinal microbiota, can contribute to increased immune stimulation and mucosal barrier dysfunction.4
We have reported that macrophages differentiated in vitro by macrophage colony-stimulating factor (M-CSF) and interferon-γ (IFN-γ) (Mγ-Mϕs) from plasma blood mononuclear cells show a similar phenotype to that of intestinal Mϕs.6,7 Mγ-Mϕs and lamina propria CD14+ macrophages have the potential to produce inflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and IL-23, and this phenotype resembles that of M1 macrophages. In contrast to Mγ-Mϕs, the characteristics of macrophages differentiated by M-CSF (M-Mϕs) alone are similar to those of M2 macrophages, including the ability to produce a large amount of IL-10. Using these in vitro differentiated macrophages, we recently found that a receptor for bile acid signaling modulates immune response in CD patients.8
Anti-Saccharomyces cerevisiae antibodies (ASCAs) are known to be directed against the component of the yeast, and that has been proposed as serological markers for CD diagnosis.9 Importantly, Candida albicans has also been shown to generate ASCAs.10 Fungi, including C. albicans, represent another major community of the human intestine microbiota, and recent research has highlighted the importance of the fungi residing in the gut and the interaction between these resident fungi and intestinal inflammation.11,12,13,14,15,16,17 Candida species were found to be significantly more abundant in the feces of IBD patients compared to healthy controls,11,13,14,15,16,17 and another study indicated that bacterial diversity was increased and fungal diversity reduced in pouchitis patients who maintained clinical remission with probiotics.12 β-(1,3)-Glucans represent 40% of the cell wall of the yeast and C. albicans. C-type lectin receptors, including Dectin-1 and Dectin-2, recognize fungi and are involved in host defense mechanisms by initiating inflammatory and adaptive immune responses.18,19 Dectin-1 recognizes β-(1,3)-glucans and induces innate immune responses,20 whereas Dectin-2 mainly recognizes α-mannose.21 The effect of Candida on murine colitis has also been investigated. For example, infection significantly delayed colonic healing in mice,22 and it was also reported that colonization by C. albicans is promoted by DSS colitis in mice and enhances inflammatory responses through galectin-3.23
Because most investigations of the interaction between fungi and host cells have been performed using murine models, it is not clear whether C. albicans influences inflammation in human intestinal macrophages in a manner that impacts CD pathogenesis. Here, we investigate the response of macrophages to curdlan (β-(1,3)-glucan), one of the cell wall components of commensal fungi. The aim of this study is to reveal the interaction between commensal fungi and host immune cells in IBD.
METHODS
1. Collection of Peripheral Blood Cells and Intestinal Mucosa from Patients with CD
A total of 15 mL of peripheral blood was obtained from patients with CD and healthy controls. Colonic mucosa samples were obtained from surgically resected specimens from patients with CD and UC patients for this study. For all samples from CD patients, mucosa samples with macroscopically moderate to severe inflammation were obtained. All studies were approved by the Ethics Committee of Keio University Hospital (20090253) and written informed consent was obtained from all patients.
2. Isolation of Peripheral Blood Monocytes and In Vitro M and Mγ Differentiation
CD14-positive (CD14+) monocytes were isolated from peripheral blood mononuclear cells (PBMCs) using CD14+ MACS (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. The percentage of monocytes isolated using this method was evaluated by flow cytometry and was routinely >98%. For in vitro M-Mφ differentiation, CD14+ monocytes were cultured with 50 ng/mL recombinant human M-CSF (R&D Systems, Minneapolis, MN, USA) for 6 days. For in vitro Mγ-Mφ differentiation, CD14 monocytes were cultured with 50 ng/mL recombinant human M-CSF (R&D Systems) and 100 ng/mL recombinant human IFN-γ for 6 days.
3. Preparation of Lamina Propria Mononuclear Cells
Lamina propria mononuclear cells (LPMCs) were isolated from intestinal specimens using modifications of a previously described technique.6 Briefly, dissected mucosa samples were incubated in calcium and magnesium-free HBSS (Sigma-Aldrich, St. Louis, MO, USA) containing 2.5% heat-inactivated FBS (BioSource International, Camarillo, CA, USA) and 1 mM DTT (Sigma-Aldrich) to remove mucus. The mucosa was then incubated twice in HBSS containing 1 mM EDTA (Sigma-Aldrich) for 45 minutes at 37℃. Tissues were collected and incubated in HBSS containing 1 mg/mL collagenase type 3 and 0.1 mg/mL DNase I (Worthington Biochemical, Lakewood, NJ, USA) for 60 minutes at 37℃. The fraction was pelleted and resuspended in 40% Percoll solution (Amersham Biosciences) and layered onto 60% Percoll before centrifugation at 2000 rpm for 20 minutes at room temperature. Viable LPMCs were recovered from the 40% to 60% layer interface.
4. Isolation of Lamina Propria Mφ
Lamina propria (LP) CD14+ Mφs were isolated from LPMCs using EasySep human CD14 (Stem Cell Technologies, Vancouver, Canada). The percentage of each subset of cells isolated using this method was evaluated by flow cytometry and was routinely >95%.
5. Commensal Fungal Heat-Inactivated Antigens
Commensal fungal strains of C. albicans were cultured on YPD agar. Fungi were harvested and washed twice with ice-cold PBS. Next, fungal suspensions were heated at 5℃ for 30 minutes, washed, resuspended in PBS, and stored at −80℃. Complete killing was confirmed by 72 hours incubation on solid medium at 37℃.
6. Quantitative Real-Time RT-PCR Analysis
Total RNA was extracted using an RNeasy Micro kit (Qiagen, Hilden, Germany), and cDNA was synthesized using a QuantiTect RT kit (Qiagen) according to the manufacturer's instructions. Quantitative real-time RT-PCR was performed using the TaqMan Universal PCR Master mix (Applied Biosystems, Foster City, CA, USA) and on-demand gene-specific primers for human dectin-1. The results were analyzed using DNA Engine Opticon 2 System (MJ Research, Waltham, MA, USA). Relative quantification was achieved by normalizing to the values of the β-actin gene.
7. Cytokine Assay
TNF-α, IL-6, IL-1β, and IL-10 secretion was measured using a human inflammation cytometric beads array kit (BD Pharmingen, San Jose, CA, USA).
8. Statistical Analysis
Statistical analyses were performed using GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA).
RESULTS
1. Differentiated Mγ-Mφs Express CD206 and CD209
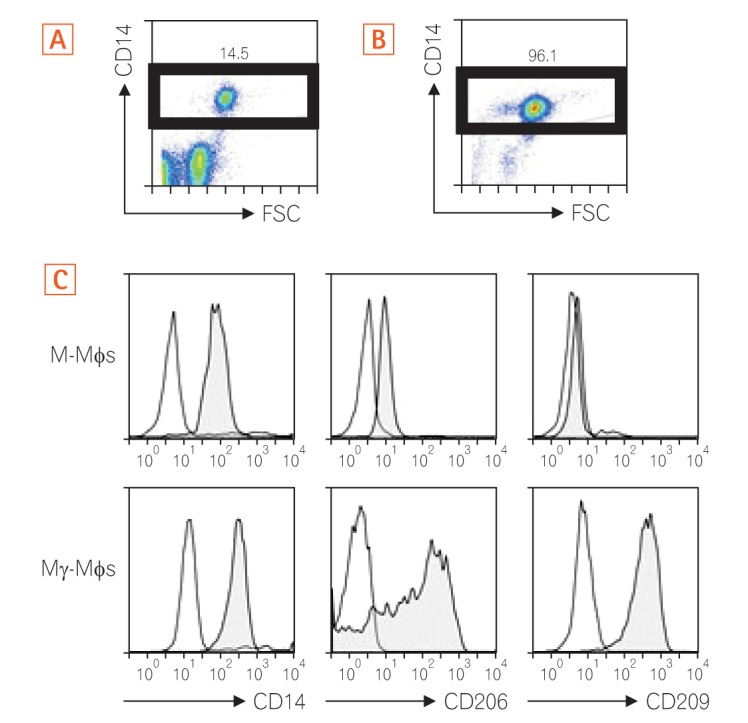
As shown in Fig. 1A and B, we enriched CD14+ monocytes using CD14+ MACS. The enriched CD14+ monocytes were then differentiated into M-Mφs or Mγ-Mφs according to our previous report.24 We investigated the surface markers of these cells to characterize their surface phenotypes. The Mγ-Mφs expressed higher levels of the mannose receptor (CD206) and C-type lectin CD209 than the M-Mφs (Fig. 1C).
Fig. 1. Differentiated Mγ-Mφs express CD206 and CD209. Cell surface staining. Peripheral blood mononuclear cells were gated on the monocyte gate forward scatter (FSC)/CD14+ before (A) and after (B) using CD14+ MACS. The numbers are the ratio of CD14+ cells in all monocytes. (C) Cell surface molecule of M-Mφs or Mγ-Mφs. The open histograms show the isotype control. The filled histograms show the cells. M-Mφs, macrophages differentiated in vitro by macrophage colony-stimulating factor alone; Mγ-Mφs, macrophages differentiated in vitro by macrophage colony-stimulating factor and interferon-γ.
2. Mγ-Mφs Produce a Large Amount of Inflammatory Cytokines in Response to β-(1,3)-Glucan
The cytokine profiles of macrophages derived from the peripheral blood cells of healthy humans in response to β-(1,3)-glucan (purchased from Wako Pure Chemical; Product code #034-09901) were analyzed. Secretion of TNF-α, IL-6, IL-1β from both M-Mφs and Mγ-Mφs in the presence of glucan were higher than secretion in the absence of glucan (Fig. 2). The pro-inflammatory cytokine levels from Mγ-Mφs were significantly higher compared to those from M-Mφs (P<0.001), whereas secretion of IL-10, an anti-inflammatory cytokine, from Mγ-Mφs was lower than from M-Mφs in the presence of glucan (Supplementary Fig. 1). Interestingly, increase of TNF-α was in a concentration dependent manner in response to glucan (data not shown). These results suggest that the cytokine-producing potential of Mγ-Mφs is consistent with that of M1 macrophages, producing high amount of pro-inflammatory cytokines in response to fungal components.
Fig. 2. Mγ-Mφs produce a large amount of inflammatory cytokines in response to β-(1,3)-glucan. Cytokine production (A, TNF-α; B, IL-6; C, IL-1β) by macrophages derived from peripheral blood cells from healthy controls (n=6) in response to β-(1,3)-glucan (A, 20 µg/mL; B and C, 100 µg/mL) was analyzed using a cytometric bead array kit. CD14+ monocytes were differentiated in the presence of M-CSF (M-macrophages, M-Mφs) or M-CSF and IFN-γ (M-gamma macrophages, Mγ-Mφs). M-Mφs and Mγ-Mφs (1×106 cells/mL) were stimulated with β-(1,3)-glucan for 24 hours. Statistical analysis was performed using the Mann-Whitney U-test. aP<0.001. TNF, tumor necrosis factor; IL, interleukin; M-CSF, macrophage colony-stimulating factor; IFN, interferon.

3. Glucan Induced Pro-inflammatory Cytokines in Mγ-Mφs via Dectin-1
Because the large amount of inflammatory cytokines produced by Mγ-Mφs was dependent on the concentration of β-(1,3)-glucan, we next assessed Dectin-1 expression. The mRNA levels of Dectin-1 in Mγ-Mφs were 6.7-fold higher than those in M-Mφs (P<0.01) (Fig. 3A). According to flow cytometry (Fig. 3B) and Western blotting (Fig. 3C), Dectin-1 expression was also markedly higher on Mγ-Mφs compared to M-Mφs. Previous reports have demonstrated that cytokines, including TNF-α, induction by β-glucans is dependent on Dectin-1 in bone marrow-derived dendritic cells25 and that Dectin-2 recognizes α-mannans and induces Th17 cell differentiation.26 TNF-α production by Mγ-Mφs was also examined and was markedly increased in the presence of heat-killed C. albicans. Furthermore, this higher production of TNF-α was largely reversed by an anti-Dectin-1 antibody but not an anti-Dectin-2 antibody (Fig. 3D). These results showed that Mγ-Mφs produced TNF-α through Dectin-1 not Dectin-2.
Fig. 3. Mγ-Mφs induction of proinflammatory cytokine via Dectin-1. (A) Expression of Dectin-1 mRNA in M-Mφs and Mγ-Mφs was examined using quantitative RT-PCR. Statistical analysis was performed using the Mann-Whitney U-test (aP<0.01). (B) Expression of Dectin-1, the receptor for β-(1,3)-glucan, was examined by flow cytometry. (C) Expression of Dectin-1 was also investigated by western blotting using CD14+ monocytes, M-Mφs, and Mγ-Mφs. Some isotypes of Dectin-1, Dectin-1A and 1B, comprise the 15 kDa band. (D) Mγ-Mφs from healthy controls (n=4) were cultured in the presence of isotype, 10 µg/mL anti-Dectin-1 antibody or 10 µg/mL anti-Dectin-2 antibody for 1 hour. Tumor necrosis factor-α (TNF-α) production by Mγ-Mφs in response to heat-killed Candida albicans was analyzed using a cytometric bead array kit. Statistical analysis was performed using the Mann-Whitney U-test (bP<0.05). M-Mφs, macrophages differentiated in vitro by macrophage colony-stimulating factor alone; Mγ-Mφs, macrophages differentiated in vitro by macrophage colony-stimulating factor and interferon-γ.

4. Dectin-1 Dependent Pathway Induces TNF-α Production in Mγ-Mφs and LP Macrophages from CD Patients
Next, TNF-α production by Mγ-Mφs, which were cultured in the presence of an antibody against Dectin-1, was analyzed to clarify whether the large amount of TNF-α produced by Mγ-Mφs occurs via interaction between glucan on the surface of various fungi and Dectin-1. The increased production of TNF-α by glucan-stimulated Mγ-Mφs was reversed by blocking Dectin-1, and this effect was not observed when Mγ-Mφs were co-cultured with lipopolysaccharide (LPS) (Fig. 4A). Although Gow et al.27 showed that spleen tyrosine kinase (Syk) inhibitors could not suppress TNF-α production in human PBMCs, TNF-α production by Mγ-Mφs was reversed by Syk or Raf-1 inhibitors (Fig. 4B). Taken together, TNF-α production by Mγ-Mφs occurs via Dectin-1-Syk/Raf-1 pathway, but not LPS dependent pathway.
Fig. 4. Dectin-1 induces tumor necrosis factor-α (TNF-α) production by Mγ-Mφs and lamina propria macrophages in a Syk and Raf-1-dependent manner. (A) Mγ-Mφs were cultured in the presence of isotype or 10 µg/mL anti-Dectin-1 antibody for 1 hour, and TNF-α production by Mγ-Mφs in response to β-(1,3)-glucan or lipopolysaccharide (LPS) was analyzed by flow cytometry (n=3). Statistical analysis was performed using the Mann-Whitney U-test (aP<0.05). (B) Mγ-Mφs were cultured in the presence of isotype, 10 µM Raf-1 inhibitor or 20 µM Syk inhibitor for 1 hour, and TNF-α production by Mγ-Mφs in response to β-(1,3)-glucan was analyzed by flow cytometry (n=4). Statistical analysis was performed using the Mann-Whitney U-test (aP<0.05). (C) CD3–CD14+ monocytes were isolated from lamina propria mononuclear cells from patients with CD (n=4) and cultured in the presence of isotype or 10 µg/mL anti-Dectin-1 antibody for 1 hour. TNF-α production in response to heat-killed Candida albicans or Enterococcus faecalis was analyzed using a cytometric bead array kit. Statistical analysis was performed using the Mann-Whitney U-test (bP<0.01). M-Mφs, macrophages differentiated in vitro by macrophage colony-stimulating factor alone; Mγ-Mφs, macrophages differentiated in vitro by macrophage colony-stimulating factor and interferon-γ; CA, Candida albicans; EF, Enterococcus faecalis.

Because we previously reported that Mγ-Mφs mimicked the phenotypes of intestinal macrophages in patients with CD,7 we next assessed cytokine production by LPMCs from patients with CD. TNF-α production by CD3–CD14+ LPMCs isolated from CD patients was high (2,076±323 pg/mL), even under un-stimulated conditions. TNF-α levels were also significantly higher under the condition of co-culture with C. albicans and Enterococcus faecalis compared to the unstimulated condition (P<0.01) (Fig. 4C). Although the anti-Dectin-1 antibody decreased TNF-α production by LPMCs when co-cultured with C. albicans, this effect was not observed after stimulation with E. faecalis. These results suggested that TNF-α production of LP macrophages induced by C. albicans in patients with CD was via Dectin-1 dependent pathway as Mγ-Mφs in vitro.
Because we previously reported that LP macrophages possess unique characteristics and influence pathogenesis of CD,7 we hypothesized that these TNF-α production pathways via Dectin-1 contribute to the pathogenesis of CD. We compared TNF-α and IL-6 production by LPMCs from patients with CD and UC using LPMCs isolated from surgical specimens. IL-6 production by LPMCs from patients with CD was significantly higher than in cells from patients with UC. In CD, IL-6 production by LPMCs stimulated with C. albicans was higher than by cells not stimulated with C. albicans. However, the difference was not significant (P=0.12). Although TNF-α production by LPMCs stimulated with C. albicans was higher for UC patients, this response was more than 10 times higher for CD patients (12,347±894 pg/mL) than UC patients (786±143 pg/mL) (Supplementary Fig. 2).
DISCUSSION
Although a number of studies investigating the role of microbiota,28,29,30,31 including commensal fungi,22,23 in intestinal inflammation have been conducted in models of experimental colitis, there are few studies on the contribution of commensal fungi to intestinal inflammation in patients with CD. Here, we demonstrate, for the first time, that C. albicans activated the production of TNF-α via Dectin-1, and not Dectin-2, in both in vitro differentiated macrophages and LP macrophages from patients with CD.
The role of glucan as a pro-inflammatory or anti-inflammatory stimulus is an ongoing debate. One study indicated that mice lacking Dectin-1 exhibited an increased susceptibility to chemically induced colitis, which was the result of altered responses to indigenous fungi. Furthermore, a polymorphism in the gene encoding Dectin-1 (CLEC7A) is strongly linked to a severe form of UC.32 These results suggest that the interaction between fungi and Dectin-1 expressed by immune cells influences health and protects against inflammatory diseases. The present studies indicate that glucan induced production of pro-inflammatory cytokines via dectin-1 on macrophages and our results are consisted with recent studies indicating that bacteria-derived glucan induced enteritis in murine models of IBD.22,23 These results may reflect that glucan of different origin-fungal versus bacterial- has different effects on enteritis.
Several studies also indicate that the composition of symbiotic fungi in the gut differs between CD patients and healthy controls. Importantly, the number of Candida genera in CD patients was increased compared to healthy controls.11,13,14,15,16,17 More recently, fungal dysbiosis was observed, with an increased Basidiomycota/Ascomycota ratio, a decreased proportion of S. cerevisiae and an increased proportion of C. albicans compared with healthy controls.17 Another recent study also indicated that Candida glabrata was overrepresented in CD and that S. cerevisiae and Filobasidium uniguttulatum were associated with non-inflamed mucosa in CD.15 Candida was also significantly more abundant in stool samples from pediatric patients with IBD, and it was also confirmed that pediatric IBD is associated with reduced diversity in both fungal and bacterial gut microbiota.11 These results suggest that fungi, including C. albicans, might play a role in pathogenesis of CD. However, these studies did not confirm whether commensal fungi interact with inflammatory cells within the intestinal lamina propria in humans.
In our study, we demonstrated that C. albicans affected the production of pro-inflammatory cytokines by CD3–CD14+ LPMCs from CD patients via Dectin-1. Saijo et al.25 demonstrated that Dectin-1 is not required for defense against C. albicans, suggesting that molecules other than β-1,3- and β-1,6-glucans are mainly involved in the induction of cytokines after infection of this fungus. Brown and Gordon33 discovered that Dectin-1 is expressed in each macrophage population. Dectin-1 induces cytokine production through the following 3 pathways: Toll-like receptor-Myd88-dependent production,20 Syk-dependent production,34,35,36 and Raf-1 dependent production.34 Furthermore, a recent study suggests that interactions between commensal fungi and Dectin-1 influence colitis.32 In our study, inhibition of Dectin-1, Syk, and Raf-1 failed to completely suppress TNF-α production, which suggested the existence of another pathway for TNF-α production.
In the murine model, colonization by C. albicans increases IL-17 and IL-23 production by gastric and oral tissues,37,38 indicating that Candida colonization might enhance inflammation by increasing the levels of these cytokines.13 Other recent studies indicate that this fungus exacerbates damage and delays healing of inflammatory lesions in animal models.22,23 C. albicans makes contact with intestinal epithelial cells and can also interact with immunologically competent cells in the gut. In the present study, we assessed pro-inflammatory cytokine production by LPMCs stimulated with heat-killed C. albicans in patients with UC and CD. Interestingly, TNF-α production was significantly higher in LP macrophages from CD patients compared to UC patients when the cells were stimulated with C. albicans. These results suggest that the responsiveness of LPMCs to the fungus differs between CD and UC, and thus, these antigens may differ in their contribution to disease.
This is the first study to show that LPMCs derived from CD patients stimulated with β-(1,3)-glucan produce a large amount of TNF-α compared with cells derived from UC patients. The findings indicate the possibility of utilizing C. albicans as an indicator of chronic inflammation in patients with CD. Although several therapies have been developed within the last decade, CD patients who are primary refractory or who have lost secondary responsiveness to medical treatment are still observed in the clinical setting. As recent report suggested,39 we also indicate that improvement of fungal dysbiosis and Dectin-1-Syk-Raf-1 signaling pathway may be a therapeutic target in CD.
ACKNOWLEDGEMENTS
The authors would like to express special thanks to Professor Peter Ernst (University of California, San Diego, CA, USA) for editing the manuscript and helpful discussions.
Footnotes
FINANCIAL SUPPORT: This study was supported in part by Health and Labour Sciences Research Grants for research on intractable diseases from the Japanese Ministry of Health.
CONFLICT OF INTEREST: No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION: Mori K, Naganuma M, Mizuno S, Suzuki H, Matsuoka K, Hisamatsu T and Kanai T participated in the design of the study; Kitazume M, Shimamura K and Sugita A collected samples of this study; FACS staining and PCR analysis was performed by Mori K, Suzuki H and Chiba S; Statistical analysis was done by Mori K and Hisamatsu T; Naganuma M, Mizuno S and Hisamatsu T drafted the first version of the manuscript; all authors were involved in the writing process, read, and approved the final manuscript.
SUPPLEMENTARY MATERIALS
M-Mφs produce a large amount of interleukin-10 (IL-10) in response to β-(1,3)-glucan. IL-10 production by macrophages derived from peripheral blood cells from healthy controls (n=6) in response to β-(1,3)-glucan (100 µg/mL) was analyzed using a cytometric bead array kit. CD14+ monocytes were differentiated in the presence of M-CSF (M-macrophages, M-Mφs) or M-CSF and IFN-γ (M-gamma macrophages, Mγ-Mφs). M-Mφs and Mγ-Mφs (1×106 cells/mL) were stimulated with β-(1,3)-glucan for 24 hours. Statistical analysis was performed using the Mann-Whitney U-test. aP<0.01. M-CSF, macrophage colony-stimulating factor; IFN, interferon.
Pro-inflammatory cytokine production after stimulation with Candida albicans is higher in lamina propria mononuclear cells (LPMCs) from CD patients than those from UC patients. Whole LPMCs from patients with UC or CD were collected and stimulated with β-(1,3)-glucan for 24 hours. IL-6 (A) and TNF-α (B) secretion by macrophages isolated from the intestine of subjects with either UC or CD was analyzed using a cytometric bead array kit. Statistical analysis was performed using the Mann-Whitney U-test. aP<0.05; bP<0.001. IL-6, interleukin-6; TNF, tumor necrosis factor.
References
- 1.Naganuma M, Sakuraba A, Hibi T. Ulcerative colitis: prevention of relapse. Expert Rev Gastroenterol Hepatol. 2013;7:341–351. doi: 10.1586/egh.13.18. [DOI] [PubMed] [Google Scholar]
- 2.Yun L, Hanauer S. Selecting appropriate anti-TNF agents in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol. 2009;3:235–248. doi: 10.1586/egh.09.20. [DOI] [PubMed] [Google Scholar]
- 3.Lewis JD, Chen EZ, Baldassano RN, et al. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn's disease. Cell Host Microbe. 2015;18:489–500. doi: 10.1016/j.chom.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sartor RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 5.Sartor RB. Therapeutic correction of bacterial dysbiosis discovered by molecular techniques. Proc Natl Acad Sci U S A. 2008;105:16413–16414. doi: 10.1073/pnas.0809363105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamada N, Hisamatsu T, Honda H, et al. Human CD14+ macrophages in intestinal lamina propria exhibit potent antigen-presenting ability. J Immunol. 2009;183:1724–1731. doi: 10.4049/jimmunol.0804369. [DOI] [PubMed] [Google Scholar]
- 7.Kamada N, Hisamatsu T, Okamoto S, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest. 2008;118:2269–2280. doi: 10.1172/JCI34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoneno K, Hisamatsu T, Shimamura K, et al. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn's disease. Immunology. 2013;139:19–29. doi: 10.1111/imm.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sendid B, Colombel JF, Jacquinot PM, et al. Specific antibody response to oligomannosidic epitopes in Crohn's disease. Clin Diagn Lab Immunol. 1996;3:219–226. doi: 10.1128/cdli.3.2.219-226.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Standaert-Vitse A, Jouault T, Vandewalle P, et al. Candida albicans is an immunogen for anti-Saccharomyces cerevisiae antibody markers of Crohn's disease. Gastroenterology. 2006;130:1764–1775. doi: 10.1053/j.gastro.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Chehoud C, Albenberg LG, Judge C, et al. Fungal signature in the gut microbiota of pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2015;21:1948–1956. doi: 10.1097/MIB.0000000000000454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kühbacher T, Ott SJ, Helwig U, et al. Bacterial and fungal microbiota in relation to probiotic therapy (VSL#3) in pouchitis. Gut. 2006;55:833–841. doi: 10.1136/gut.2005.078303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumamoto CA. Inflammation and gastrointestinal Candida colonization. Curr Opin Microbiol. 2011;14:386–391. doi: 10.1016/j.mib.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Q, Wang C, Tang C, He Q, Li N, Li J. Dysbiosis of gut fungal microbiota is associated with mucosal inflammation in Crohn's disease. J Clin Gastroenterol. 2014;48:513–523. doi: 10.1097/MCG.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liguori G, Lamas B, Richard ML, et al. Fungal dysbiosis in mucosa-associated microbiota of Crohn's disease patients. J Crohns Colitis. 2016;10:296–305. doi: 10.1093/ecco-jcc/jjv209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukhopadhya I, Hansen R, Meharg C, et al. The fungal microbiota of de-novo paediatric inflammatory bowel disease. Microbes Infect. 2015;17:304–310. doi: 10.1016/j.micinf.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sokol H, Leducq V, Aschard H, et al. Fungal microbiota dysbiosis in IBD. Gut. 2017;66:1039–1048. doi: 10.1136/gutjnl-2015-310746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dambuza IM, Brown GD. C-type lectins in immunity: recent developments. Curr Opin Immunol. 2015;32:21–27. doi: 10.1016/j.coi.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rizzetto L, De Filippo C, Rivero D, Riccadonna S, Beltrame L, Cavalieri D. Systems biology of host-mycobiota interactions: dissecting Dectin-1 and Dectin-2 signalling in immune cells with DC-ATLAS. Immunobiology. 2013;218:1428–1437. doi: 10.1016/j.imbio.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 20.Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. Dectin-1 mediates the biological effects of beta-glucans. J Exp Med. 2003;197:1119–1124. doi: 10.1084/jem.20021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009;9:465–479. doi: 10.1038/nri2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zwolinska-Wcislo M, Brzozowski T, Budak A, et al. Effect of Candida colonization on human ulcerative colitis and the healing of inflammatory changes of the colon in the experimental model of colitis ulcerosa. J Physiol Pharmacol. 2009;60:107–118. [PubMed] [Google Scholar]
- 23.Jawhara S, Thuru X, Standaert-Vitse A, et al. Colonization of mice by Candida albicans is promoted by chemically induced colitis and augments inflammatory responses through galectin-3. J Infect Dis. 2008;197:972–980. doi: 10.1086/528990. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki H, Hisamatsu T, Chiba S, et al. Glycolytic pathway affects differentiation of human monocytes to regulatory macrophages. Immunol Lett. 2016;176:18–27. doi: 10.1016/j.imlet.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Saijo S, Fujikado N, Furuta T, et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol. 2007;8:39–46. doi: 10.1038/ni1425. [DOI] [PubMed] [Google Scholar]
- 26.Saijo S, Ikeda S, Yamabe K, et al. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity. 2010;32:681–691. doi: 10.1016/j.immuni.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 27.Gow NA, Netea MG, Munro CA, et al. Immune recognition of Candida albicans beta-glucan by dectin-1. J Infect Dis. 2007;196:1565–1571. doi: 10.1086/523110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Atarashi K, Tanoue T, Oshima K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 29.Hayashi A, Sato T, Kamada N, et al. A single strain of Clostridium butyricum induces intestinal IL-10-producing macrophages to suppress acute experimental colitis in mice. Cell Host Microbe. 2013;13:711–722. doi: 10.1016/j.chom.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 30.Kashiwagi I, Morita R, Schichita T, et al. Smad2 and Smad3 inversely regulate TGF-beta autoinduction in Clostridium butyricum-activated dendritic cells. Immunity. 2015;43:65–79. doi: 10.1016/j.immuni.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 31.Sellon RK, Tonkonogy S, Schultz M, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iliev ID, Funari VA, Taylor KD, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science. 2012;336:1314–1317. doi: 10.1126/science.1221789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown GD, Gordon S. Immune recognition: a new receptor for beta-glucans. Nature. 2001;413:36–37. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- 34.Gringhuis SI, den Dunnen J, Litjens M, et al. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol. 2009;10:203–213. doi: 10.1038/ni.1692. [DOI] [PubMed] [Google Scholar]
- 35.LeibundGut-Landmann S, Gross O, Robinson MJ, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 36.Skrzypek F, Cenci E, Pietrella D, Rachini A, Bistoni F, Vecchiarelli A. Dectin-1 is required for human dendritic cells to initiate immune response to Candida albicans through Syk activation. Microbes Infect. 2009;11:661–670. doi: 10.1016/j.micinf.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 37.Conti HR, Shen F, Nayyar N, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saunus JM, Wagner SA, Matias MA, Hu Y, Zaini ZM, Farah CS. Early activation of the interleukin-23-17 axis in a murine model of oropharyngeal candidiasis. Mol Oral Microbiol. 2010;25:343–356. doi: 10.1111/j.2041-1014.2010.00570.x. [DOI] [PubMed] [Google Scholar]
- 39.Wheeler ML, Limon JJ, Bar AS, et al. Immunological consequences of intestinal fungal dysbiosis. Cell Host Microbe. 2016;19:865–873. doi: 10.1016/j.chom.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
M-Mφs produce a large amount of interleukin-10 (IL-10) in response to β-(1,3)-glucan. IL-10 production by macrophages derived from peripheral blood cells from healthy controls (n=6) in response to β-(1,3)-glucan (100 µg/mL) was analyzed using a cytometric bead array kit. CD14+ monocytes were differentiated in the presence of M-CSF (M-macrophages, M-Mφs) or M-CSF and IFN-γ (M-gamma macrophages, Mγ-Mφs). M-Mφs and Mγ-Mφs (1×106 cells/mL) were stimulated with β-(1,3)-glucan for 24 hours. Statistical analysis was performed using the Mann-Whitney U-test. aP<0.01. M-CSF, macrophage colony-stimulating factor; IFN, interferon.
Pro-inflammatory cytokine production after stimulation with Candida albicans is higher in lamina propria mononuclear cells (LPMCs) from CD patients than those from UC patients. Whole LPMCs from patients with UC or CD were collected and stimulated with β-(1,3)-glucan for 24 hours. IL-6 (A) and TNF-α (B) secretion by macrophages isolated from the intestine of subjects with either UC or CD was analyzed using a cytometric bead array kit. Statistical analysis was performed using the Mann-Whitney U-test. aP<0.05; bP<0.001. IL-6, interleukin-6; TNF, tumor necrosis factor.