

Abstract
Antarctic krill (Euphausia superba) is a crucial component of the Southern Ocean ecosystem, acting as the major link between primary production and higher trophic levels with an annual predator demand of up to 470 million tonnes. It also acts as an ecosystem engineer, affecting carbon sequestration and recycling iron and nitrogen, and has increasing importance as a commercial product in the aquaculture and health industries. Here we describe the creation of a de novo assembled head transcriptome for E. superba. As an example of its potential as a molecular resource, we relate its exploitation in identifying and characterizing numerous genes related to the circadian clock in E. superba, including the major components of the central feedback loop. We have made the transcriptome openly accessible for a wider audience of ecologists, molecular biologists, evolutionary geneticists, and others in a user‐friendly format at SuperbaSE, hosted at http://www.krill.le.ac.uk.
Keywords: Antarctic, circadian, crustacean, database, krill, transcriptome
1. INTRODUCTION
Antarctic krill Euphausia superba (Dana, 1852; hereafter “krill”) is a keystone species of the Southern Ocean ecosystem, a hugely abundant pelagic crustacean inhabiting a circumpolar belt between the Antarctic continent and the Polar Front (Nicol, Constable, & Pauly, 2000), with an estimated total biomass of 379 million tonnes (Mt) and postlarval production of 342–536 Mt/yr (Atkinson, Siegel, Pakhomov, Jessopp, & Loeb, 2009). It sits at the center of a “wasp‐waist” food web (Atkinson et al., 2014) providing the major link between primary production and higher trophic levels and so converting phytoplankton to animal protein. Krill make up to 70% of the food intake of Southern Ocean predators such as seals, seabirds, whales, squid, and fish (Murphy et al., 2007), with an estimated annual demand of 128–470 Mt in the Southern Ocean ecosystem as a whole (Mori & Butterworth, 2006). Krill also have an impact on the abiotic environment as a suggested “ecosystem engineer” (Murphy et al., 2013), playing an important role in the biological pump through sequestering carbon from the ocean surface to ocean interior (Perissinotto, Gurney, & Pakhomov, 2000; Tarling & Johnson, 2006), as well as facilitating the recycling of iron (Schmidt et al., 2011) and the production and uptake of ammonium (Whitehouse, Atkinson, & Rees, 2011). Recent studies have suggested that krill may be vulnerable to habitat loss as an effect of warming, with particular impact in areas accessible by predators and used by fisheries (Hill, Phillips, & Atkinson, 2013), and through ocean acidification (Kawaguchi et al., 2013). Recruitment, which is dependent on the survival of krill larvae overwintering under sea ice, is considered particularly vulnerable to climate change (Flores et al., 2012), and it will be vital to understand and predict the adaptability of krill to a changing environment and the potential consequences as it does so.
Commercially, krill provide the region's largest fishery by weight at 300,000 tonnes annually, mostly used in aquaculture as a bulk ingredient or nutritional additive in fish meal (Nicol & Foster, 2016). In recent years, there has been notable growth in the demand for products for human use in the nutraceutical industry, exploiting the high omega‐3 polyunsaturated fatty acid content in krill. Krill oil is now the second most popular source of omega‐3 after fish oil (Backes & Howard, 2014). The oil is taken as a treatment or preventative measure for conditions such as cardiovascular disease and arthritis; 29% of recent krill‐related patents are categorized as for medical usage (Nicol & Foster, 2016).
As a keystone species of clear ecological and economic significance, active research is thriving, encompassing a broad spectrum of approaches from the population level down to the molecular. To cite further examples from recent years, this interest ranges from modeling population dynamics (Groeneveld et al., 2015) and food webs (Atkinson et al., 2012), through swarming behavior (Tarling et al., 2009), diurnal migratory behavior (Cresswell et al., 2009; Gaten, Tarling, Dowse, Kyriacou, & Rosato, 2008), and physiological stressor responses (Auerswald, Freier, Lopata, & Meyer, 2008) to peptide evolution and phylogeny (Cascella et al., 2015) and changes in gene expression related to seasonal or molt status (Seear, Goodall‐Copestake, Fleming, Rosato, & Tarling, 2012; Seear et al., 2010).
The enthusiasm for krill research is not always matched by the tractability of the organism itself, however; in contrast to the fruit fly Drosophila melanogaster, a model organism of some repute and power (Jennings, 2011), krill are remote, difficult to transport and maintain, and each discovery is hard won. The krill genome presents further challenges. Estimated at 47 gigabases (Jeffery, 2012), the genome of E. superba is more than twice the size of the largest genome sequenced so far, that of the loblolly pine Pinus taeda (Neale et al., 2014), and a sequencing project is considered unfeasible until further reductions in sequencing costs and improvements in long‐read technologies are achieved (Jarman & Deagle, 2016). Alternative approaches to genetic questions have therefore been adopted for krill, such as expressed sequence tag (EST) libraries (De Pittà et al., 2008; Seear et al., 2009), microarrays (Seear et al., 2010) and, with increasing popularity as the costs of RNA‐seq continue to fall, transcriptomic data that can provide gene coding sequences from reconstruction of mRNA transcripts (Clark et al., 2011; De Pittà et al., 2013; Martins et al., 2015; Meyer et al., 2015; Sales et al., 2017).
In this study, we describe the creation and annotation of SuperbaSE, a new de novo assembled head transcriptome for E. superba that is openly available online to interested researchers. With the aim of reconstructing as comprehensive a set of accurate, full‐length transcripts as possible, we used multiple variations of the de Bruijn graph method of de novo assembly, in which reads are broken down into a library of k‐mers, “words” of length k, prior to reassembly (Compeau, Pevzner, & Tesler, 2011). The output generated by this method varies with k‐mer length, with low values favoring transcript diversity and reconstruction of lowly expressed genes at the cost of fragmentation and accuracy, while high values favor higher accuracy over a broader abundance range, at the expense of diversity (Robertson et al., 2010). De Bruijn assembly software packages can also vary in their output when using the same k‐mer length (Xie et al., 2014).
Our own research interests revolve around the circadian clock, a field which until recently was lacking genetic data for crustaceans (Strauss & Dircksen, 2010), although matters have improved in recent years (Christie, Fontanilla, Nesbit, & Lenz, 2013; Tilden, McCoole, Harmon, Baer, & Christie, 2011; Zhang et al., 2013) and the head transcriptome was created as a gene discovery resource to drive progress in this area. Prior to the decision to adopt an RNA‐seq approach, we cloned and Sanger sequenced a number of core circadian genes for the krill through degenerate PCR and RACE extension; for some, we obtained complete coding sequences while achieving only partial success with others due to difficulties with degenerate PCR and RACE extension. As an example of the utility and potential of SuperbaSE, we relate here its use in completing our partial sequences and identifying many others relevant to the molecular clock, including regulatory genes involved in the generation and maintenance of circadian periodicity (Özkaya & Rosato, 2012) and those downstream whose expression is regulated by the clock (clock‐controlled genes).
2. MATERIALS AND METHODS
2.1. Animal collection
The collection of E. superba samples took place during the Antarctic summer in February 2008, on the Discovery 2010 cruise JR177. Swarms were identified using an EK60 echosounder North‐West of South Georgia at 52°S and caught by target fishing using a pelagic RMT8 net. Catches were taken at 1 a.m., 6 a.m., 1 p.m., and 8 p.m. local time. Immediately after collection between 30 and 200 individuals from each net were flash frozen by placing them into tubes that were then plunged into methanol at −80°C, moved to a −80°C freezer until the end of the cruise and returned to the UK without thawing.
2.2. RNA extraction
For each wild catch, three frozen krill heads were combined and powdered with mortar and pestle. Samples were kept frozen by continuous addition of liquid N2 and by working on a bed of dry ice. Total RNA was extracted with TRIzol® reagent (Thermo Fisher Scientific, UK). RNA was resuspended in DEPC‐treated water and the concentration and purity [A(260 nm)/A(280 nm) >1.8] determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific). One microgram of total RNA was electrophoresed on a nondenaturing 1.5% (w/v) agarose gel to check for degradation. The Qiagen oligotex® mRNA extraction kit was used to isolate mRNA from one microgram of total RNA following the manufacturer's protocol. The mRNAs from each catch were combined into a single sample >10 μg and >250 ng/μl, which was delivered to BGI Tech Solutions (Hong Kong) Co Ltd (BGI) for generation of raw read data. The sample was subject to polyA enrichment using oligo‐dT beads, then fragmented and used to construct a cDNA library with an average fragment size of 200 base pairs (bp). This was primed with random hexamers and sequenced using the Illumina HiSeq 2000 platform to generate 100‐bp paired‐end reads.
2.3. De novo transcriptome assembly
The read data were subject to quality control by BGI, removing adapter sequences, contamination, and low‐quality reads, and once retrieved Fast QC v0.11.2 was used for further quality assessment and Trimmomatic 0.32 (Bolger, Lohse, & Usadel, 2014) to remove low quality leading and trailing bases.
Assembly was performed using Trinity r20140717 (Haas et al., 2013), Bridger r2014‐12‐01 (Chang et al., 2015), Trans‐ABySS 1.5.1 (Robertson et al., 2010), and SOAPdenovo‐Trans 1.03 (Xie et al., 2014). Trinity restricts the user to a k‐mer setting of 25. Bridger has a maximum permitted k‐mer of 31, and six assemblies were generated using k‐mers from 21 to 31 in two‐step increments. Trans‐ABySS and SOAPdenovo‐Trans can use higher k‐mer settings, and for each assembler, eight assemblies were created using k‐mers from 21 to 91 in 10‐step increments. A minimum contig length of 200 bp was specified for assembly.
For each assembler that permitted varying k‐mer settings, an assembler‐specific merged assembly was produced. For Trans‐ABySS, the built‐in function transabyss‐merge was used, while for the others the contig headers in each assembly were amended to avoid cross‐assembly duplicate IDs and the assemblies concatenated into a single FASTA file. These were then subject to the removal of redundancy using the dedupe.sh function of BBMap (http://sourceforge.net/projects/bbmap/), set to remove duplicates and containments at 100% identity including reverse complement comparisons.
Each merged assembly was assessed using the read metrics function of TransRate 1.01 (Smith‐Unna, Boursnell, Patro, Hibberd, & Kelly, 2016), which generates an output file of those contigs considered to be well assembled as supported by read‐mapping evidence. The “good contigs” files for each assembler were combined into a single file that was processed a second time using BBMap and TransRate to select the absolute best unique contigs across all assemblers. Going forward, this selection of contigs is referred to as the total assembly.
Transdecoder (Haas et al., 2013) was used to identify likely coding contigs with an open reading frame coding for peptides of 100 amino acids (aa) or longer—only the longest identified ORF per contig was retained. The output was processed using CD‐HIT (Fu, Niu, Zhu, Wu, & Li, 2012) at 100% identity to remove duplicates, resulting in the peptide assembly. The contigs from the total assembly that encode these peptides were extracted into a new database, hereafter referred to as the coding assembly.
2.4. Transcriptome annotation and analysis
The peptide assembly was queried against the SwissProt protein database from the UniProt Knowledgebase (UniProtKB) using the blastp function of BLAST+ 2.5.0 (Camacho et al., 2009). The output was set to return the single best result (‐max_target_seqs 1, ‐max_hsps 1) with an E‐value of 1.0e−6 or lower. All contigs from the peptide assembly that failed to return a result were subsequently queried against the arthropod protein database of the UniProtKB with the same criteria. Both protein databases were retrieved from ftp://ftp.ebi.ac.uk/pub/databases/fastafiles/uniprot/ on 26th November 2016. The coding assembly was also subject to sequence homology annotation using the blastx function of BLAST+ against the two protein databases using the same method. The accessions returned by these searches were used to retrieve associated Enzyme Codes, GO terms, KEGG, and InterProScan IDs from the UniProt website (http://www.uniprot.org/uploadlists/) using the Retrieve/ID mapping function. The peptide assembly was further annotated using functions built into Trinotate (Haas et al., 2013) to identify Pfam protein domains.
To provide an overview of the information present in our assembly and compare it to the current most extensive krill molecular resource, KrillDB (Sales et al., 2017), we used blastx annotation results from the high quality, manually reviewed SwissProt database as a metric. The SuperbaSE coding assembly, KrillDB (retrieved from http://krilldb.bio.unipd.it/ on 14 March 2017) and the mRNA transcripts derived from the genome of another crustacean, Daphnia pulex (Colbourne et al., 2011; retrieved from http://wfleabase.org/genome/Daphnia_pulex/current/mrna on 5th April 2017) were subject to this search using an E‐value cutoff of 1.0e−6 or lower, the output again set to return the single best result. Results were used to retrieve GO annotation from UniProtKB as above and summarized using WEGO (Ye et al., 2006). To identify the extent of the overlap between KrillDB and the coding assembly, the two resources were further subject to a reciprocal BLAST search using blastn, E‐value 1.0e−20 or lower, returning the single best result.
2.5. SuperbaSE: the online Euphausia superba transcriptome database
The transcript and annotation data of the peptide and coding assemblies were merged, processed and converted into an appropriate format for online viewing. Each entry was given sequential and consistently formatted gene, transcript and ORF IDs and assigned putative gene names based on BLAST annotation results. The site was created using a Catalyst framework to access annotated transcriptome data stored in a MySQL database, and the front‐end design makes use of template toolkit and Twitter Bootstrap.
2.6. Transcriptome mining
Coding sequences for E. superba bmal1 (Es‐bmal1, GenBank accession KX238952), clock (Es‐clk, KX238953), cryptochrome 1 (Es‐cry1, KX238951), and cryptochrome 2 (Es‐cry2, also reported as Escry by Mazzotta et al. (2010)) had already been successfully cloned and Sanger sequenced in full when the head transcriptome project was undertaken, while E. superba period (Es‐per, KX238955) and timeless (Es‐tim, KX238954) had both been partially cloned and sequenced (Table 1 and Supplementary Data). These sequences were used to query the total assembly to determine the extent and accuracy of their reconstruction in the transcriptome and, in the case of Es‐per and Es‐tim, to obtain the full coding sequences. The queries were performed by translation of the nucleotide sequences to putative peptides using ExPASy Translate (http://web.expasy.org/translate/), with these then used to mine the total assembly using the tblastn function of BLAST+ with an E‐value cutoff of 1.0e−3. For Es‐per and Es‐tim, the output was used to design primers for confirmation of the full coding sequences of both genes as outlined above (Supplementary Data, Table S2).
Table 1.
Core circadian genes cloned and Sanger sequenced for Euphausia superba. Es‐cryptochrome 2 has been previously reported as Escry (Mazzotta et al., 2010)
| Gene name | Accession | Length (nt/aa) | Top NCBI nr hit | ||||||
|---|---|---|---|---|---|---|---|---|---|
| mRNA | CDS | Peptide | Accession | Name | Species | E‐value | Identity (%) | ||
| Es‐bmal1 | KX238952 | 2,017 | 1,992 | 664 | AFV39705 | bmal1a | Pacifastacus leniusculus | 0 | 73 |
| Es‐clock | KX238953 | 4,044 | 4,032 | 1,344 | AAX44045 | clock | Macrobrachium rosenbergii | 0 | 79 |
| Es‐cryptochrome 1 | KX238951 | 1,644 | 1,599 | 533 | AJY53623 | cryptochrome 1 | Nilaparvata lugens | 0 | 53 |
| Es‐cryptochrome 2 | – | 1,720 | 1,635 | 545 | CAQ86665 | cryptochrome | Euphausia superba | 0 | 100 |
| Es‐period | KX238955 | 4,061 | 3,783 | 1,261 | ALC74274 | period‐like protein | Nephrops norvegicus | 0 | 67 |
| Es‐timeless | KX238954 | 4,222 | 3,933 | 1,311 | ALC74275 | timeless‐like protein | Nephrops norvegicus | 0 | 75 |
For regulatory and clock‐controlled genes, the peptide sequences of D. melanogaster/Mus musculus genes known to contribute to or interact with circadian timekeeping were used to search the total assembly for putative E. superba orthologs using the tblastn function of BLAST+ with an E‐value cutoff of 1.0e−3. To add additional support to such identification, candidate contigs were extracted from the assembly database and translated, and the peptide sequences blasted against D. melanogaster proteins curated in Flybase (Attrill et al., 2016) and against the NCBI nonredundant proteins database (excluding uncultured/environmental sample sequences and all entries with a title containing the words “putative,” “hypothetical,” or “predicted”). The same approach was taken in identifying a selection of neuropeptides (derived from Gard, Lenz, Shaw, & Christie, 2009; Ma et al., 2009; Christie, Durkin, Hartline, Ohno, & Lenz, 2010; Christie, Stemmler, & Dickinson, 2010; Ma et al., 2010; Christie, McCoole, Harmon, Baer, & Lenz, 2011; Christie, Nolan, Ohno, Hartline, & Lenz, 2011; Christie, Roncalli, et al., 2013) that might potentially be under the control of the biological clock in E. superba.
The derived peptide sequences obtained from this process were used to search KrillDB with the tblastn function with an E‐value cutoff of 1.0e−3 to compare the presence and extent of transcript reconstruction of circadian‐related genes in the two resources. In each case, KrillDB was also queried with the D.melanogaster/M.musculus ortholog used in the initial search process to search this resource for superior candidates.
3. RESULTS
3.1. Sequencing data and quality control
34,918,657 clean paired‐end 100‐bp reads (69,837,314 in total) were generated by the Illumina sequencing. After Trimmomatic processing for low‐quality bases at each end of all reads, the read lengths ranged between 81 and 100 bases.
3.2. Assembly and annotation metrics
The total assembly initially comprised 484,125 contigs. Submission to the NCBI Transcriptome Shotgun Assembly database required the assembly to be subject to a contamination screen, with 45 contigs removed as a result and 484,080 contigs submitted to the TSA under accession GFCS00000000. The total assembly achieved a TransRate score of 0.42, an improvement over all the individual multi‐ or single k‐mer assemblies produced by any one assembly package (Table 2). There were also improvements for the total assembly in the percentage of total read mappings, the percentage of good read mappings, the percentage of contigs with identified open reading frames (ORFs), and a reduction in the number of perceived bridges, which indicate transcript fragmentation. The contig mean length and assembly N50 of the total assembly were notably higher than the output of Trinity, SOAPdenovo‐Trans, and Trans‐ABySS, yet lower than the output of Bridger, signaling a proportionally large contribution of contigs from the latter; indeed, Bridger provided 50% of contigs in the total assembly, followed by Trans‐ABySS, SOAPdenovo‐Trans, and Trinity with 26%, 15%, and 9% respectively. From the total assembly, TransDecoder identified 147,450 contigs (the coding assembly) encoding putative peptides (the peptide assembly). Of these, 27,413 were deemed by TransDecoder to represent complete peptides; 60,826 were identified as internal fragments, 42,311 as missing the 5′ end and 16,900 as missing the 3′ end.
Table 2.
Comparison of de novo assembly quality using TransRate contig and read‐mapping metrics. Bridger, SOAPdenovo‐Trans, and Trans‐ABySS assemblies are multi‐k‐mer assemblies, see Methods for details. See Smith‐Unna et al. (2016) and http://hibberdlab.com/transrate/metrics.html for further details regarding TransRate metrics. Read‐mapping metrics were not considered appropriate for the coding assembly, which is a selective dataset
| Metric | Bridger | SOAPdenovo‐Trans | Trans‐ABySS | Trinity | Total assembly | Coding assembly |
|---|---|---|---|---|---|---|
| No. of contigs | 513,499 | 282,833 | 318,436 | 157,274 | 484,080 | 147,450 |
| Shortest | 189 | 200 | 200 | 201 | 200 | 297 |
| Longest | 27,543 | 17,265 | 37,689 | 11,148 | 34,468 | 34,468 |
| Mean length | 780 | 445 | 573 | 503 | 726 | 1,121 |
| No. with ORF | 138,937 | 51,336 | 72,597 | 31,566 | 135,969 | 100,823 |
| ORFs % | 27% | 18% | 23% | 20% | 28% | 81% |
| N50 | 1,218 | 480 | 738 | 602 | 1,071 | 1,518 |
| % of reads mapped | 97% | 93% | 92% | 82% | 98% | – |
| Good mappings | 30,880,238 | 28,292,720 | 28,688,483 | 22,012,681 | 31,833,986 | – |
| % good mappings | 88% | 81% | 82% | 63% | 91% | – |
| Potential bridges | 43,048 | 46,708 | 38,398 | 38,011 | 34,120 | – |
| % uncovered bases | 38% | 11% | 15% | 4% | 23% | – |
| % contigs with uncovered bases | 86% | 71% | 77% | 44% | 80% | – |
| % contigs uncovered | 45% | 16% | 18% | 4% | 29% | – |
| % contigs low‐covered | 93% | 75% | 80% | 76% | 89% | – |
| Transrate score | 0.13 | 0.26 | 0.25 | 0.21 | 0.42 | – |
BLAST annotation of the peptide assembly by sequence homology returned 67,762 peptide contigs with hits from the UniprotKB SwissProt dataset, with an additional 20,972 contigs annotated using the UniprotKB arthropod dataset. A further 3,467 contigs were annotated by querying SwissProt and the arthropod dataset with the coding assembly nucleotide sequences for an overall total of 92,201 contigs annotated in either peptide or nucleotide form. Of these contigs, 80,149 were subsequently annotated with at least one GO term, 57,094 with at least one KEGG identifier, and 86,405 with at least one InterProScan accession. Further to this, 67,782 contigs were annotated with Pfam domain identifiers (whole sequence E‐value of 1.0e−6 or lower), including 2,044 contigs that were not BLAST annotated.
The number of unique accessions returned by this BLAST annotation process was 23,931; of these, 10,762 were assigned to a single contig, while the rest were assigned to two contigs or more to reach the total number. The SwissProt entry Q9V7U0, for example, a pro‐resilin precursor from Drosophila melanogaster, annotated 1,148 contigs, and another 32 unique accessions were each assigned to 100 contigs or more. By way of comparison, a single assembly generated using Bridger at k‐mer = 25 (Bridger25) received 12,616 unique accessions from SwissProt (Supplementary Data, Table S3). Q9V7U0 was assigned to 274 contigs in this assembly, suggesting a duplication factor of 3.8 in the total assembly; of the SwissProt accessions assigned to 100 contigs or more in the peptide and coding assemblies, the duplication factor varied from 2.03 to 8.2.
Figure 1 shows the high‐level summary of the functional annotation of the coding assembly, KrillDB and the Daphnia pulex transcripts using SwissProt/GO mapping. The content of the two krill resources is highly similar at this level, and there appear to be no major functional categories missing or underpresented in comparison with the genome‐derived transcripts of D. pulex. Comparing the underlying SwissProt annotation, results showed that 67,206 coding assembly contigs were annotated by 15,441 unique accessions, 7,492 (48.5%) of which were not seen in the KrillDB results. KrillDB saw 65,248 annotated contigs in total with 10,887 unique accessions, 2,938 (27%) of which were not seen in the coding assembly results. In the results of the reciprocal BLAST, no match was found in KrillDB for 34,768 contigs from the coding assembly (23.6%), while 19,270 contigs from KrillDB had no match in the coding assembly (14.4%).
Figure 1.
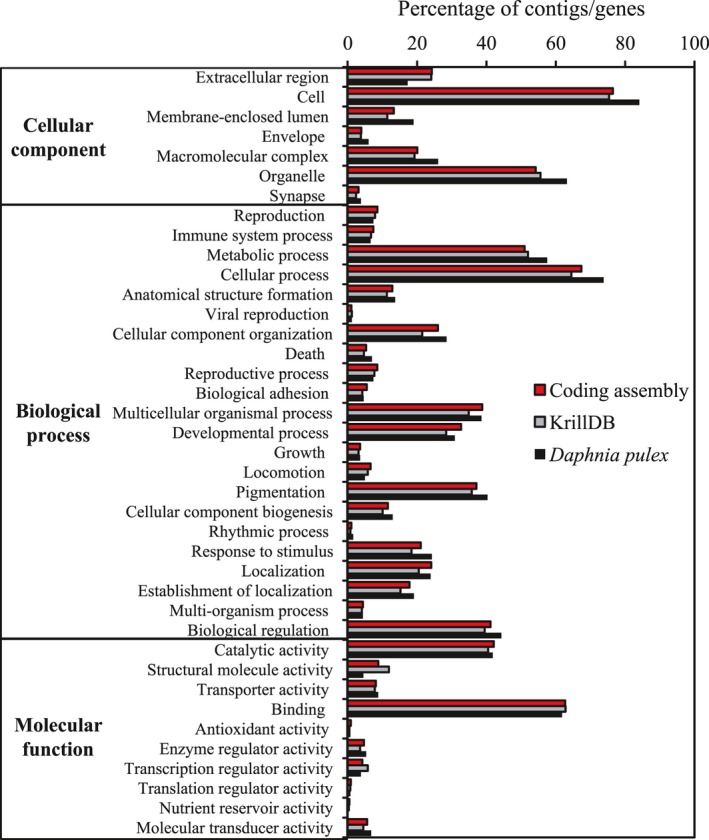
Functional characterization of SuperbaSE coding assembly, in comparison with KrillDB (also E. superba) and D. pulex mRNA transcripts. Chart depicts percentage of contigs/genes mapping to high‐level GO terms retrieved through BLAST annotation with SwissProt
3.3. Transcriptome mining
Searching the total assembly using the complete coding sequences of Es‐bmal1 (Figure 2), Es‐clk (Figure 3), Es‐cry1 (Figure 4), and Escry2 (Figure 5) returned transcripts representing between 88 and 100% of each sequence; in the case of Es‐cry2, this was a single, complete transcript, while the others returned 2–4 fragments. Searching the assembly using the fragments of Es‐per and Es‐tim returned a complete coding sequence for the former (Figure 6), and two large fragments, covering nearly the complete sequence, for the latter (Figure 7). In both cases, these full sequences were subsequently confirmed by PCR, cloned, and Sanger sequenced (Supplementary Data and Table 1). In some cases, minor variations were seen when comparing the peptide sequences derived from assembled transcripts to those confirmed by high fidelity PCR, and in the case of Es‐CLK, there is a notable stretch of disagreement with particular contigs from residues 561–641, and a smaller stretch later on (Figure 3).
Figure 2.

Es‐BMAL1 (accession KX238952) aligned with fragments mined from the total assembly. Yellow highlights represent sequence data not found in the total assembly
Figure 3.

Es‐CLOCK (accession KX238953) aligned with fragments mined from the total assembly. Yellow highlights represent sequence data not found in the total assembly. Red text highlights residues inconsistent between the confirmed PCR sequence and transcriptome sequences
Figure 4.

Es‐CRY1 (accession KX238951) aligned with fragments mined from the total assembly. Yellow highlights represent sequence data not found in the total assembly. Red text highlights residues inconsistent between the confirmed PCR sequence and transcriptome sequences
Figure 5.

Es‐CRY2 (accession CAQ86665, Mazzotta et al. (2010)) aligned with contig mined from the total assembly
Figure 6.

Es‐PERIOD (accession KX238955) aligned with contig mined from the total assembly. Red text highlights residues inconsistent between the confirmed PCR sequence and transcriptome sequence. Black arrows denote the point at which sequence data from 3′ RACE extension ends—beyond this the transcriptome was necessary to complete the sequence
Figure 7.

Es‐TIMELESS (accession KX238954) aligned with contig mined from the total assembly. Yellow highlights represent sequence data not found in the total assembly. Red text highlights residues inconsistent between the confirmed PCR sequence and transcriptome sequence. Black arrows denote the point at which sequence data from 3′ RACE extension ends—beyond this the transcriptome was necessary to complete the sequence
Table 3 shows the putative regulatory and clock‐controlled genes mined from the transcriptome, while Table 4 shows the results of BLAST searches against Flybase and NCBI nonredundant (NR) peptide databases using these output contigs to support their assigned identities. With the exception of jetlag, a putative ortholog was identified for all the 22 genes we attempted to identify, 18 of which were represented by full coding sequences. E. superba pigment‐dispersing hormone (PDH) has been previously reported (Toullec et al., 2013) as Eus‐PDH β, and the contig identified as Es‐pdh1 (ES.k51.S2746663) is in agreement with this. Table 5 shows the neuropeptides similarly identified and characterized using only the NCBI NR database; of the 31 preprohormones used as queries 21 returned results, all of which were accepted as putative E. superba orthologs. Candidate peptide sequences for E. superba allatostatin C, bursicon α, corazonin, and neuroparsin have previously been reported by Toullec et al. (2013), although none are in perfect agreement with our contigs.
Table 3.
Transcriptome mining: clock‐related query proteins and E. superba output contigs
| Query Protein | Accession | E. superba transcript/protein identifications | Length | Protein name | Length | |
|---|---|---|---|---|---|---|
| Protein name | Contig ID | E‐value | ||||
| CASEIN KINASE II α | AAN11415 | ES.21_comp3643_seq1 | 0 | 2556 | Es‐CKIIα | 350 |
| CASEIN KINASE II β | AAF48093 | ES.194593 | 4E‐119 | 1276 | Es‐CKIIβ | 219 |
| CLOCKWORK ORANGE | AAF54527 | ES.21_comp15386_seq1 | 2E‐26 | 2301 | Es‐CWO | 707 |
| CTRIP | AAF52092 | ES.31_comp9752_seq0 | 1E‐167 | 8148 | Es‐CTRIP | 2152 |
| DOUBLETIME | AAF57110 | ES.25_comp4349_seq1 | 0 | 2480 | Es‐DBT | 345 |
| JETLAG | AAF52178 | ES.21_comp16370_seq0 | 1E‐15 | 1901 | – | – |
| LARK | AAF50578 | ES.k31.S5232406 | 6E‐59 | 1199 | Es‐LARK | 326 |
| NEJIRE | AAF46516 | ES.31_comp5882_seq0 | 0 | 6924 | Es‐NEJ | 2072c |
| NEMO | AAF50497 | k21.S7956922 | 0 | 2038 | Es‐NEMO | 456c |
| PAR DOMAIN PROTEIN 1ε | AAF04509 | ES.k41.J3781389 | 8E‐05 | 1835 | Es‐PDP1 | 489 |
| PIGMENT DISPERSING FACTORa | AAF56593 | ES.k51.S2746663 | 8E‐06 | 387 | Es‐PDH1 | 74 |
| ES.c68106_g1_i1 | 3E‐05 | 531 | Es‐PDH2 | 79 | ||
| PIGMENT DISPERSING FACTOR RECEPTOR | AAF45788 | ES.21_comp12084_seq0 | 3E‐91 | 1588 | Es‐PDHR | 295 |
| PROTEIN PHOSPHATASE 1 | CAA39820 | ES.k51.J2636217 | 0 | 1870 | Es‐PP1 | 329 |
| PROTEIN PHOSPHATASE 2A ‐ MICROTUBULE STAR | AAF52567 | ES.23_comp1445_seq0 | 0 | 2263 | Es‐MTS | 309 |
| PROTEIN PHOSPHATASE 2A ‐ TWINS | AAF54498 | ES.21_comp997_seq2 | 0 | 3375 | Es‐TWS | 455 |
| PROTEIN PHOSPHATASE 2A ‐ WIDERBORST | AAF56720 | ES.104836 | 0 | 4190 | Es‐WBT | 461 |
| REV‐ERBαb | NP_663409 | ES.25_comp2884_seq0 | 1E‐45 | 3602 | Es‐E75 | 800 |
| SHAGGY | AAN09084 | ES.27_comp3209_seq0 | 0 | 2222 | Es‐SGG | 415 |
| SUPERNUMERARY LIMBS | AAF55853 | ES.25_comp34876_seq0 | 0 | 1843 | Es‐SLIMB | 612c |
| TAKEOUT | AAF56425 | ES.21_comp3588_seq0 | 1E‐11 | 1090 | Es‐TAKEOUT | 247 |
| TIMELESS (TIMEOUT)b | Q9R1X4 | ES.23_comp37221_seq0 | 2E‐82 | 934 | Es‐TIMEOUT | 1363d |
| ES.29_comp20079_seq0 | 4E‐94 | 3067 | ||||
| VRILLE | AAF52237 | ES.25_comp4277_seq0 | 3E‐43 | 2021 | Es‐VRI | 474 |
E. superba PDH also reported by Toullec et al. (2013).
Query protein used is a Mus musculus orthlog. All others from Drosophila melanogaster.
Es‐NEJIRE, Es‐NEMO and Es‐SLIMB are 3′ partial fragments.
Es‐TIMEOUT full protein sequence determined through PCR using fragments listed.
Table 4.
Results of blastp analyses of putative E. superba clock‐associated proteins against D. melanogaster protein database (Flybase) and NCBI nonredundant (nr) protein database
| Query | Top Flybase hit | Top NCBI nr hit | |||||
|---|---|---|---|---|---|---|---|
| Flybase No. | Associated gene name | E‐value | Accession | Name | Species | E‐value | |
| Es‐CKIIα | FBpp0070041 | casein kinase IIa | 1E‐162 | EFN84867 | Casein kinase II subunit alpha | Harpegnathos saltator | 0 |
| Es‐CKIIβ | FBpp0300427 | Casein kinase II ß | 6E‐114 | ELR50200 | Casein kinase II subunit beta | Bos mutus | 5E‐146 |
| Es‐CTRIP | FBpp0310477 | circadian trip | 1E‐173 | ACC99349 | ULF (TRIP12) | Homo sapiens | 0 |
| Es‐CWO | FBpp0081723 | clockwork orange | 8E‐27 | KDR16323 | Hairy/enhancer‐of‐split related with YRPW motif protein 2 | Zootermopsis nevadensis | 2E‐61 |
| Es‐DBT | FBpp0306615 | discs overgrown | 7E‐157 | AGV28719 | Casein kinase 1 epsilon | Eurydice pulchra | 0 |
| Es‐E75 | FBpp0074915 | Ecdysone‐induced protein 75B | 2E‐143 | AGS94407 | Ecdysteroid receptor E75 | Litopenaeus vannamei | 0 |
| Es‐LARK | FBpp0076555 | lark | 2E‐55 | NP_523957 | lark, isoform A | Drosophila melanogaster | 2E‐62 |
| Es‐NEJa | FBpp0305701 | nej | 0 | KDR19833 | CREB‐binding protein | Zootermopsis nevadensis | 0 |
| Es‐NEMOa | FBpp0076474 | nemo | 0 | NP_729316 | Nemo, isoform E | Drosophila melanogaster | 0 |
| Es‐PDP1 | FBpp0076495 | PAR‐domain protein 1 | 2E‐35 | EFN83234 | Hepatic leukemia factor | Harpegnathos saltator | 2E‐39 |
| Es‐PDH1 | FBpp0084396 | Pigment‐dispersing factor | 3E‐05 | JC4756 | PDH related peptide precursor 79 | Penaeus sp. | 9E‐27 |
| Es‐PDHR | FBpp0309084 | Pigment‐dispersing factor receptor | 9E‐97 | BAO01102 | Neuropeptide GPCR B2 | Nilaparvata lugens | 1E‐140 |
| Es‐PP1 | FBpp0306442 | Protein phosphatase 1a at 96A | 4E‐178 | EKC23784 | PP1‐alpha catalytic subunit | Crassostrea gigas | 0 |
| Es‐MTS | FBpp0310063 | microtubule star | 2E‐176 | KDR18186 | PP2A catalytic subunit alpha | Zootermopsis nevadensis | 0 |
| Es‐TWS | FBpp0081671 | twins | 0 | AFK24473 | PP2A regulatory subunit B | Scylla paramamosain | 0 |
| Es‐WBT | FBpp0084575 | widerborst | 0 | XP_002427654 | PP2A regulatory subunit epsilon | Pediculus humanus corporis | 0 |
| Es‐TAKEOUT | FBpp0297106 | CG2016 | 1E‐32 | ACO12182 | Circadian clock‐controlled protein precursor | Lepeophtheirus salmonis | 1.00E‐47 |
| Es‐TIMEOUT | FBpp0082180 | timeout | 0 | EEZ99220 | Timeout | Tribolium castaneum | 0 |
| Es‐SGG | FBpp0070450 | shaggy | 0 | AEO44887 | Shaggy | Tribolium castaneum | 0 |
| Es‐SLIMBa | FBpp0306059 | supernumerary limbs | 0 | KDR19729 | F‐box/WD repeat‐containing protein 1A | Zootermopsis nevadensis | 0 |
| Es‐VRI | FBpp0309715 | vrille | 4E‐42 | AAT86041 | Vrille | Danaus plexippus | 3E‐68 |
Fragment. Es‐PDH2 is not shown as it is a minor variant of Es‐PDH1.
Table 5.
Preprohormone query proteins with E. superba output contigs and subsequent blastp analysis against NCBI nonredundant protein database
| Peptide family (subfamily) | Output from total assembly using Accession as query | Output from NCBI NR using total assembly contig as query | |||||
|---|---|---|---|---|---|---|---|
| Accession | Contig | Size (nt) | E‐value | Accession | Description | E‐value | |
| Allatostatin A | ABS29318 | ES.27_comp22909_seq0 | 1239 | 4E‐19 | BAF64528 | allatostatin precursor protein Panulirus interruptus | 1E‐60 |
| Allatostatin B | AAF49354 | ES.27_comp14162_seq0 | 710 | 2E‐09 | AFV91538 | B‐type preproallatostatin I Pandalopsis japonica | 3E‐15 |
| Allatostatin Ca | AAF53063 | ES.23_comp57987_seq0 | 757 | 4E‐05 | BAO00935 | Allatostatin‐cc Nilaparvata lugens | 5E‐13 |
| Allatotropin | AAB08757 | – | – | – | – | – | – |
| Bursicon alphaa | EFX87546 | ES.21_comp33157_seq0 | 552 | 2E‐60 | AKJ74864 | Bursicon alpha subunit Penaeus monodon | 4E‐72 |
| Bursicon beta | EFX87749 | ES.21_comp28222_seq0 | 721 | 7E‐37 | AKJ74865 | Bursicon beta subunit Penaeus monodon | 9E‐63 |
| CHHamide | NP_001097784 | ES.25_comp21895_seq1 | 829 | 6E‐04 | EFX80320 | CCHamide‐like precursor Daphnia pulex | 3E‐08 |
| Corazonina | AAB32283 | ES.29_comp12038_seq1 | 365 | 2E‐04 | Q9GSA4 | Corazonin precursor‐related peptide Galleria mellonella | 2E‐06 |
| Crustacean cardioactive peptide | EFX70015 | ES.27_comp31900_seq0 | 685 | 2E‐02 | ABB46291 | Crustacean cardioactive peptide Carcinus maenas | 4E‐33 |
| Crustacean hyperglycemic hormone | ABQ41269 | ES.k41.R3771434 | 691 | 5E‐24 | ACN87216 | Crustacean hyperglycemic hormone precursor Charybdis japonica | 8E‐27 |
| Calcitonin‐like diuretic hormone | ACX46386 | ES.c66980_g1_i1 | 608 | 1E‐47 | ACX46386 | Prepro‐calcitonin‐like diuretic hormone Homarus americanus | 1E‐53 |
| Corticotropin‐releasing factor‐like diuretic hormone | AAF54421 | – | – | – | – | – | – |
| Ecdysis‐triggering hormone | AAF47275 | – | – | – | – | – | – |
| Eclosion hormone | AAA29310 | ES.21_comp1319_seq1 | 536 | 3E‐10 | BAO00951 | Eclosion hormone 2 Nilaparvata lugens | 2E‐16 |
| FMRFamide‐like peptide (myosuppressin) | ACX46385 | ES.265178 | 343 | 2E‐34 | BAG68789 | Myosuppressin‐like neuropeptide precursor Procambarus clarkii | 9E‐36 |
| FMRFamide‐like peptide (neuropeptide F) | AEC12204 | ES.23_comp4135_seq0 | 700 | 3E‐26 | AEC12204 | Preproneuropeptide F I Litopenaeus vannamei | 5E‐27 |
| FMRFamide‐like peptide (short neuropeptide F) | AAU87571 | ES.k61.S126078 | 378 | 4E‐08 | ETN63818 | Short neuropeptide F precursor Anopheles darlingi | 2E‐13 |
| FMRFamide‐like peptide (sulfakinin) | ABQ95346 | – | – | – | – | – | – |
| FMRFamide‐like peptide (other) | BAE06262 | ES.63534 | 521 | 6E‐06 | AAR19420 | FMRFamide‐like peptide precursor Periplaneta americana | 1E‐08 |
| Insulin‐like peptide | AAS65048 | – | – | – | – | – | – |
| Inotocin | ABX52000 | ES.c73008_g1_i1 | 538 | 3E‐17 | BAO00906 | Arginine vasotocin precursor Paralichthys olivaceus | 2E‐24 |
| Leucokinin | AAF49731 | – | – | – | – | – | – |
| Neuroparsina | ACO11224 | ES.31_comp27103_seq0 | 522 | 1E‐15 | ACO10490 | Neuroparsin‐A precursor Caligus rogercresseyi | 4E‐16 |
| Orcokinin | ACB41787 | ES.k51.J2644345 | 765 | 2E‐59 | Q9NL82 | Orcokinin‐like peptide 4 Precursor Procambarus clarkii | 1E‐77 |
| Periviscerokinin/pyrokinin | NP_001104182 | – | – | – | – | – | – |
| Proctolin | CAD30643 | – | – | – | – | – | – |
| Red pigment concentrating hormone | ABV46765 | ES.c39108_g1_i1 | 620 | 9E‐18 | ABV46765 | Red pigment concentrating hormone Macrobrachium rosenbergii | 4E‐17 |
| Adipokinetic hormone | EFX68649 | – | – | – | – | – | – |
| RYamide | EDP28140 | – | – | – | – | – | – |
| SIFamide | BAC55939 | ES.23_comp6805_seq0 | 935 | 7E‐25 | Q867W1 | SIFamide precursor Procambarus clarkii | 8E‐24 |
| Tachykinin‐related peptide | ACB41786 | ES.c73859_g1_i1 | 1289 | 6E‐37 | BAD06363 | Preprotachykinin B Panulirus interruptus | 1E‐37 |
Candidates for these genes also reported by Toullec et al. (2013).
Searching KrillDB for circadian‐related transcripts (core, regulatory, and clock‐controlled genes) generated largely similar results, albeit that the majority of transcripts returned from the total assembly were longer than their KrillDB counterparts (Table 6). In KrillDB, a 303 bp 3′ fragment was the only evidence found of Es‐bmal1, and Es‐per was represented by a 2,339 bp fragment, both less extensive than the results from the total assembly. Conversely, Es‐tim was represented by 4,047 bp contig in KrillDB and Es‐cry1 by a 1,819 bp contig, both covering the complete coding sequence in contrast to the shorter, fragmented transcripts in the total assembly. By visual inspection of the results, no equivalent was found in KrillDB for the total assembly contigs identified as representing the genes PP2A microtubule star, PP2A widerborst, and takeout—in the case of the first two genes the top result from KrillDB was a less compelling candidate compared to the output from the total assembly and each was also found to be present in the total assembly, but rejected in favor of the contigs listed in Table 3. In the case of takeout, no candidate contig was found in KrillDB.
Table 6.
Comparison of circadian‐related contigs from the total assembly of SuperbaSE and equivalents found in KrillDB. Longest accepted candidate contig shown for each assembly. “Agreement” shows percentage of identical residues in the largest HSP in each search result alignment
| Gene | Total assembly contig | Length | KrillDB contig | Length | Agreement (%) |
|---|---|---|---|---|---|
| Bmal1 | ES.21_comp39465_seq0 | 1,150 | ESS133965 | 303 | 100 |
| Clock | ES.21_comp24303_seq0 | 1,903 | ESS034516 | 1,795 | 100 |
| Period | ES.23_comp19428_seq0 | 4,476 | ESS133963 | 2,339 | 99 |
| Timeless | ES.23_comp16712_seq0 | 3,415 | ESS040526 | 4,047 | 99 |
| Cryptochrome 1 | ES.23_comp21133_seq0 | 1,100 | ESS023688 | 1,819 | 99 |
| Cryptochrome 2 | ES.154212 | 2,146 | ESS118473 | 1,855 | 100 |
| Casein kinase IIA | ES.21_comp3643_seq1 | 2,556 | ESS071624 | 1,825 | 91 |
| Casein kinase IIB | ES.194593 | 1,276 | ESS051147 | 805 | 97 |
| Clockwork orange | ES.21_comp15386_seq1 | 2,301 | ESS049809 | 2,111 | 95 |
| Ctrip | ES.31_comp9752_seq0 | 8,148 | ESS002511 | 2,454 | 100 |
| Doubletime | ES.25_comp4349_seq1 | 2,480 | ESS096454 | 1,925 | 91 |
| Lark | ES.k31.S5232406 | 1,199 | ESS098132 | 1,058 | 70 |
| Nejire | ES.31_comp5882_seq0 | 6,924 | ESS048098 | 2,180 | 98 |
| Nemo | k21.S7956922 | 2,038 | ESS021045 | 2,238 | 100 |
| Par Domain Protein 1 | ES.k41.J3781389 | 1,835 | ESS017186 | 465 | 75 |
| Pigment‐dispersing hormone | ES.k51.S2746663 | 387 | ESS069399 | 362 | 93 |
| PDH receptor | ES.21_comp12084_seq0 | 1,588 | ESS048516 | 1,557 | 100 |
| Protein phosphatase 1 | ES.k51.J2636217 | 1,870 | ESS073678 | 2,246 | 99 |
| PP2A ‐ Microtubule star | ES.23_comp1445_seq0 | 2,263 | ESS030735a | 1,334 | 58 |
| PP2A ‐ Twins | ES.21_comp997_seq2 | 3,375 | ESS126945 | 2,979 | 100 |
| PP2A ‐ Widerborst | ES.104836 | 4,190 | ESS023456a | 3,762 | 72 |
| Rev‐erb/E75 | ES.25_comp2884_seq0 | 3,602 | ESS094384 | 3,620 | 99 |
| Shaggy | ES.27_comp3209_seq0 | 2,222 | ESS074789 | 1,350 | 96 |
| Supernumerary limbs | ES.25_comp34876_seq0 | 1,843 | ESS133964 | 1,350 | 100 |
| Takeout | ES.21_comp3588_seq0 | 1,090 | – | – | – |
| Timeout | ES.23_comp37221_seq0 | 3,067 | ESS130300 | 2,078 | 99 |
| Vrille | ES.25_comp4277_seq0 | 2,021 | ESS123361 | 1,522 | 100 |
These contigs were rejected as equivalent to the query contigs on visual inspection.
3.4. SuperbaSE: The online Euphausia superba transcriptome database
SuperbaSE is hosted at http://www.krill.le.ac.uk. The site features a Quick Search which scans transcript/contig and GO fields for the input search text and an Advanced Search function with a wider set of options. Results can be exported as a raw FASTA file of nucleotide sequences, or individual results can be viewed on their own page further showing the predicted peptide sequence and hyperlinked annotation data that permits the retrieval of details from UniProt and other sources. Figure 8 shows the data for the circadian gene Es‐period as it is found on the website, including functional information. As an extra resource, the peptide assembly, coding assembly, and total assembly are available as searchable BLAST databases using the SequenceServer front end (Priyam et al., 2015), permitting fast, simple identification of genes of interest.
Figure 8.
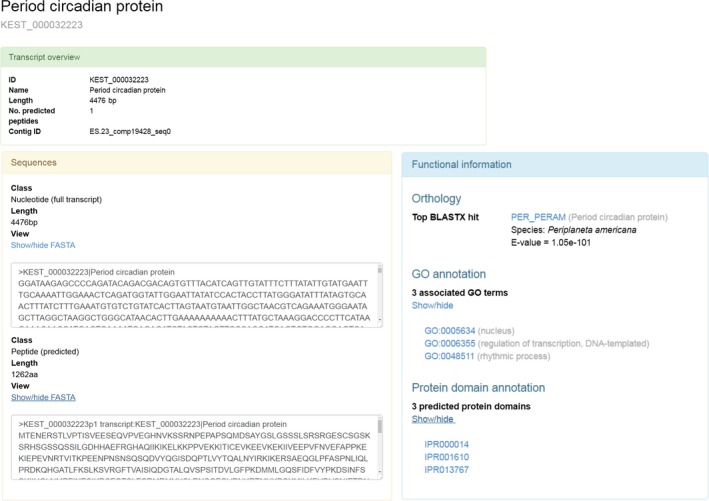
Sequence and annotation data found in SuperbaSE for circadian gene Es‐period
4. DISCUSSION
4.1. Assembly and assessment
SuperbaSE was created as a tool for gene discovery. As such, the pipeline adopted for assembly focused on generating a comprehensive set of correct and complete transcripts, and one of the major factors affecting the reconstruction of transcripts from short read data is the k‐mer length employed in assembly. Typically, as k‐mer length increases, so too does average contig length and the median coverage depth of contigs assembled, while contig number falls (Gibbons et al., 2009); low k‐mers therefore favor transcript diversity, while high k‐mers favor contiguity. Often an intermediate k‐mer is chosen as a compromise but from the perspective of gene discovery, in which diversity, contiguity, and accuracy are all prized, this is suboptimal. A number of studies have shown the value of a multiple k‐mer approach, showing that it increases the sequence information obtained for an individual transcript, increases the number of reads represented in the assembly, improves contiguity, and reconstructs transcripts not found in a single k‐mer assembly (Surget‐Groba & Montoya‐Burgos, 2010); that it increases the number of full‐length reconstructed transcripts (Zhao et al., 2011); and that for each k‐mer the resultant assembly contains unique contigs not present in others (Haznedaroglu, Reeves, Rismani‐Yazdi, & Peccia, 2012). Comparisons of different assembly packages have also shown variability in annotatable output when using the same dataset and k‐mer (Chang et al., 2015; Xie et al., 2014; Zhao et al., 2011). Our pipeline was inspired by such findings, employing a multi‐assembler, multi‐k‐mer approach to generate a large number of assemblies that were subsequently merged. We then used TransRate as a method of identifying the best‐assembled contigs among those in the merged assembly. As a test of the TransRate method, Smith‐Unna et al. (2016) performed a meta‐analysis of 155 published transcriptome assemblies in the NCBI TSA and determined that an assembly with a Transrate score of 0.22 would be superior to 50% of those assessed; while our initial assemblies each received scores of around that mark (Table 2), the total assembly consisting of the best contigs after merging and removal of duplicates was assigned a score of 0.42. Using this method increased the number of unique accessions retrieved by the coding assembly compared to various single assemblies (Supplementary Data, Table S3), despite the fact that, of these, only the coding assembly had been subject to processing to remove short, noncoding and low‐quality contigs which may otherwise have returned annotations, highlighting how information present in the read data can be missed if a comprehensive approach is not adopted.
Comparative methods of transcriptome assessment that use a related organism as a measure of completeness (O'Neil et al., 2013) focus on conserved sequence identification at the expense of novel and divergent transcripts and lose relevancy with increasing evolutionary distance between subject and reference. TransRate, in contrast, has the benefit of directly assessing contig quality based on read‐mapping metrics, identifying chimeras, gene family collapses, insertions, fragmentation, local misassemblies, and redundancy. This latter issue was further addressed through the use of BBMap's deduphe.sh at the nucleotide level and CD‐HIT at the peptide level to remove absolute duplicates. Despite these efforts, redundancy is still an issue. The total assembly contains nearly half a million contigs yet no individual assembly produced more than 160,000 contigs (data not shown). De novo assembly is a sensitive, error‐prone process and small differences in contig output across assemblers or k‐mers may produce many variants representing the same in vivo transcript. This is indicated in the number of accessions assigned to more than one contig, with those assigned to multiple contigs representing 55% of the total number. As illustrated by the example identified in the Results, that of the pro‐resilin precursor of Drosophila melanogaster, this can stretch to over a thousand contigs, although this particular annotation also appeared 274 times in a single assembly. This problem could be assuaged by relaxing the criteria for duplicates when running dedupe.sh and CD‐HIT ‐ by removing duplicates at 95% identity rather than 100%, for example—but again, given the focus on gene discovery, we preferred to tolerate redundancy over the possibility that correct transcripts may be lost, and we therefore offer this resource with the caveat that searches are likely to return multiple results worthy of investigation and that do not necessarily represent isoforms.
4.2. Functional and comparative analysis
The coding assembly of SuperbaSE compares favorably with the majority of existing molecular resources for E. superba in terms of the number of assembled contigs, annotated contigs, largest contig, average contig size, and N50 metric (Table 7). The recently published resource KrillDB (Sales et al., 2017) combines fragments from multiple adult libraries (Meyer et al., 2015) with Illumina sequencing of RNA libraries from krill larvae under varying CO2 levels and has similar metrics, including the number of predicted complete full‐length transcripts (27,928 for KrillDB, 27,413 for SuperbaSE). Both transcriptomes have a similar distribution of functional categories (Figure 1) that is not notably lacking in representation in comparison with the D. pulex genome.
Table 7.
Comparison of published E. superba de novo assemblies. “No. of reads” shows raw read count of new sequencing based on the samples described in this table, either as reported or obtained from NCBI SRA
| Clark et al. (2011) | De Pittà et al. (2013) | Martins et al. (2015) | Meyer et al. (2015) | Sales et al. (2017) | SuperbaSE (coding assembly) | |
|---|---|---|---|---|---|---|
| Sampling vicinity | South Orkney Islands | Ross Sea | Antarctic Peninsula | East Antarctica/Lazarev Sea | Indian Ocean | NW of South Georgia |
| Tissue type | Adult, whole | Adult, head, abdomen, thoracopods | Adult, head and eye stalks | Adult, whole/head, aquarium/wild | Larval, whole, aquarium‐bred | Adult, head |
| Experimental conditions/library type | Pooled time series | Pooled time series | Control/food deprivation/UV‐B stress | Pooled seasonal samples | Control/1000/2000 μatm pCO2 | Pooled time series |
| Sequencing technology | GS FLX Titanium (454) | GS FLX Titanium (454) | GS FLX Titanium (454) | GS FLX Titanium (454) | Illumina Genome Analyzer IIx | Illumina HiSeq 2000 |
| No. of reads | 943,817 | 96,803 | 377,442 | 1,771,572 | 368,287,158 | 69,837,314 |
| No. of reads used in assembly | 699,248 | 711,148 | 306,182 | 2,608,911 | 33,175,931 | 69,837,314 |
| No. of assembled contigs | 22,177 | 32,217 | 26,415 | 58,581 | 133,962 | 147,450 |
| Smallest contig | 137 | 300 | – | 300 | 200 | 200 |
| Largest contig | 8,515 | 8,558 | – | 11,127 | 14,396 | 34,468 |
| Average contig size | 492 | 890 | 593 | 691 | 964 | 1,121 |
| N50 | – | – | 666 | 716 | 1,294 | 1,518 |
| No. of reported BLAST annotations | 5,563 | 11,230 | 10,501 | 15,347 | 90,121 | 92,201 |
| BLAST annotation source | GenBank nonredundant | NCBI nonredundant, UniProtKB | NCBI nonredundant (arthropoda) | NCBI nucleotide, UniProtKB | NCBI nucleotide, UniProtKB/TREMBL | SwissProt, UniProt arthropoda |
| Notes | – | Final assembly includes publicly available ESTs and reads from Clark et al. (2011). | – | Final assembly includes reads from Clark et al. (2011) and De Pittà et al. (2013) | Reads were subject to digital normalization before assembly. Final assembly includes fragments from Meyer et al. (2015) | – |
Despite the broad overlap, SuperbaSE and KrillDB have some clear differences. The majority of KrillDB transcripts come from larval stage RNA extracted under experimental conditions–81% of assembled transcripts are from the larval sequencing project that this resource centers around. The other 19% come from previous adult transcriptomes assembled from a smaller number of reads generated through 454 pyrosequencing. SuperbaSE, however, derives entirely from wild‐caught adult head tissue, and it is clear that numerous transcripts are to be found in this resource and not in KrillDB and vice versa, with 23.6% of SuperbaSE contigs finding no match in KrillDB and 14.4% of KrillDB contigs having no match in SuperbaSE, based on a reciprocal blastn search using an E‐value threshold of 1.0e−20. Certain core circadian genes (Es‐bmal1, Es‐per) are less extensively reconstructed in KrillDB, while others (Es‐tim, Es‐cry1) were more fragmented in SuperbaSE. Both resources provide open access to their annotated datasets and it is apparent that they are complementary, serving to broaden the molecular information openly available for this species.
4.3. Circadian gene discovery
Using SuperbaSE, we have successfully identified the complete sequences for Es‐per and Es‐tim, two core circadian genes over 3.5 kb in length with low expression levels (Supplementary Data, Table S4). Previously cloned circadian genes of similarly low expression levels were well represented by extensive fragments, and deviations from the sequences obtained by high fidelity PCR were minimal with the exception of Es‐clk. We suggest that the causes of this particular exception are 1) a misassembly at the 3′ end of the contig ES.21_comp24303_seq0, as contig ES.31_comp16417_seq0 is in agreement with the PCR sequence, and 2) the inherent difficulty in reassembling a region showing a high level of repeats. Much of the disagreement is seen in or near an extraordinary long poly‐Q region, a common feature of Clock genes (Johnsen et al., 2007; O'Malley & Banks, 2008; Saleem, Anand, Jain, & Brahmachari, 2001); to our knowledge, the poly‐Q of Es‐CLK is the longest example yet found, surpassing that of the river prawn Macrobrachium rosenbergii (Yang, Dai, Yang, & Yang, 2006). Our findings show that E. superba possesses a full complement of core clock proteins in contrast to D. melanogaster, which lacks a CRY2, and M. musculus, which lacks TIMELESS and CRY1 (Reppert & Weaver, 2000). The krill clock shares this characteristic with a number of other arthropod species including the monarch butterfly Danaus plexippus (Zhu et al., 2008), and it has been suggested that this represents an ancestral form from which the fly and mouse clocks have been derived.
Further research is now needed to link the molecular clock to the rhythmic phenomena observed in E. superba. Diel vertical migration, in which krill rise to the surface at night to feed and return to deeper waters at dawn, is likely influenced by the biological clock (Gaten et al., 2008). Krill also exhibit annual rhythms with a molt‐shrink regression to a juvenile state over winter, returning to maturity in spring (Kawaguchi, Yoshida, Finley, Cramp, & Nicol, 2007), and seasonal cycling of metabolic rate (Meyer et al., 2010). At the genetic level, this may be governed by photoperiod (Seear et al., 2009), and circadian clock genes have been suggested to play a role in photoperiodic responses in D. melanogaster (Pegoraro, Gesto, Kyriacou, & Tauber, 2014). Es‐cry2 has shown rhythmic expression in both light–dark cycling conditions and in constant darkness with a short circadian period of 18 hr (Teschke, Wendt, Kawaguchi, Kramer, & Meyer, 2011), a finding interpreted as evidence of a clock with a wide range of entrainment and consistent with the extreme variation in photoperiod that E. superba is subject to, from constant light in summer to constant darkness during winter. With a full set of core circadian genes now cloned and sequenced, this intriguing system can be further characterized, and our preliminary results from assays on the interactions of the krill clock proteins indicate that the krill clock retains the functionality of both the fly and mouse mechanisms (data will be presented elsewhere) to an extent not seen in other species so far studied.
We have furthermore identified 18 putative full coding sequences for regulatory and clock‐controlled genes plus extensive fragments for three more such genes of interest and 21 preprohormone candidate contigs, the majority not previously reported. As an example of its usage outside the field of chronobiology, SuperbaSE has also been employed in the identification of an ancient conserved noncoding element in the 5′ region of the Not1 gene (Davies, Krusche, Tauber, & Ott, 2015).
5. CONCLUSION
We have described the assembly and annotation of SuperbaSE, a new transcriptome resource for Euphausia superba. Using this resource, we have successfully identified an extensive suite of circadian and clock‐related genes, including some that had proven difficult to clone using traditional methods. This has laid the groundwork for the molecular dissection of the circadian clock in this vital species in order to understand how these components interact to generate genetic, metabolic, physiological and behavioral rhythms, and perhaps even seasonal rhythmicity. From the total assembly, a subset of 147,450 coding contigs were identified, 92,201 of which were annotated by 23,931 unique UniProtKB accessions and then further annotated with GO terms, KEGG pathway IDs, Pfam domains, Enzyme Codes, and InterProScan identifiers. The annotated dataset and total assembly have been made freely available at SuperbaSE, a user‐friendly searchable database hosted at http://www.krill.le.ac.uk.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
BJH performed PCR, cloning and sequencing with contributions from PS, de novo transcriptome assembly and annotation, transcriptome analysis and wrote the manuscript with input from all authors. ÖÖ performed mRNA extraction, cDNA synthesis, degenerate PCR, cloning, and sequencing. NJD developed the website. EG and GT collected samples. ER, EG, CPK, and GT conceived the study.
6. DATA ACCESSIBILITY
Sequences of cloned circadian genes are available in GenBank under accessions KX238951, KX238952, KX238953, KX238954, and KX238955, as are the sequences of the total assembly under accession GFCS00000000. Read data were submitted to NCBI SRA under accession SRR4408478. Annotated coding and peptide assembly sequences are available at http://www.krill.le.ac.uk.
Supporting information
ACKNOWLEDGEMENTS
We thank the officers and the crew of the RRV “James Clark Ross” and the participants of cruise JR177, and two anonymous reviewers whose comments improved this manuscript. This work was funded by the Natural Environment Research Council (NERC) UK through an Antarctic Funding Initiative grant NE/D008719/1 (ÖÖ, PS, EG, CPK, GT, ER) and a PhD studentship (BJH). This research used the ALICE High Performance Computing Facility at the University of Leicester.
Hunt BJ, Özkaya Ö, Davies NJ, et al. The Euphausia superba transcriptome database, SuperbaSE: An online, open resource for researchers. Ecol Evol. 2017;7:6060–6077. 10.1002/ece3.3168
REFERENCES
- Atkinson, A. , Hill, S. L. , Barange, M. , Pakhomov, E. A. , Raubenheimer, D. , Schmidt, K. , … Reiss, C. (2014). Sardine cycles, krill declines, and locust plagues: Revisiting ‘wasp‐waist’ food webs. Trends in Ecology and Evolution, 29(6), 309–316. [DOI] [PubMed] [Google Scholar]
- Atkinson, A. , Nicol, S. , Kawaguchi, S. , Pakhomov, E. , Quetin, L. , Ross, R. , … Tarling, G. (2012). Fitting Euphausia Superba into Southern Ocean food‐web models: A review of data sources and their limitations. CCAMLR Science, 19, 219–245. [Google Scholar]
- Atkinson, A. , Siegel, V. , Pakhomov, E. A. , Jessopp, M. J. , & Loeb, V. (2009). A re‐appraisal of the total biomass and annual production of Antarctic krill. Deep‐Sea Research Part I: Oceanographic Research Papers, 56(5), 727–740. [Google Scholar]
- Attrill, H. , Falls, K. , Goodman, J. L. , Millburn, G. H. , Antonazzo, G. , Rey, A. J. , … the Flybase consortium . (2016). FlyBase: Establishing a Gene Group resource for Drosophila melanogaster . Nucleic Acids Research, 44(D1), D786–D792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerswald, L. , Freier, U. , Lopata, A. , & Meyer, B. (2008). Physiological and morphological colour change in Antarctic krill, Euphausia superba: A field study in the Lazarev Sea. Journal of Experimental Biology, 211(Pt 24), 3850–3858. [DOI] [PubMed] [Google Scholar]
- Backes, J. M. , & Howard, P. A. (2014). Krill oil for cardiovascular risk prevention: Is it for real? Hospital Pharmacy, 49(10), 907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. , & Madden, T. L. (2009). BLAST+: Architecture and applications. BMC Bioinformatics, 10(1), 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascella, K. , Jollivet, D. , Papot, C. , Léger, N. , Corre, E. , Ravaux, J. , … Toullec, J.‐Y. (2015). Diversification, evolution and sub‐functionalization of 70 kDa heat‐shock proteins in two sister species of Antarctic krill: Differences in thermal habitats, responses and implications under climate change. PLoS ONE, 10(4), e0121642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, Z. , Li, G. , Liu, J. , Zhang, Y. , Ashby, C. , Liu, D. , … Huang, X. (2015). Bridger: A new framework for de novo transcriptome assembly using RNA‐seq data. Genome Biology, 16(1), 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie, A. E. , Durkin, C. S. , Hartline, N. , Ohno, P. , & Lenz, P. H. (2010). Bioinformatic analyses of the publicly accessible crustacean expressed sequence tags (ESTs) reveal numerous novel neuropeptide‐encoding precursor proteins, including ones from members of several little studied taxa. General and Comparative Endocrinology, 167(1), 164–178. [DOI] [PubMed] [Google Scholar]
- Christie, A. E. , Fontanilla, T. M. , Nesbit, K. T. , & Lenz, P. H. (2013). Prediction of the protein components of a putative Calanus finmarchicus (Crustacea, Copepoda) circadian signaling system using a de novo assembled transcriptome. Comparative Biochemistry and Physiology. Part D, Genomics & Proteomics, 8(3), 165–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie, A. E. , McCoole, M. D. , Harmon, S. M. , Baer, K. N. , & Lenz, P. H. (2011). Genomic analyses of the Daphnia pulex peptidome. General and Comparative Endocrinology, 171(2), 131–150. [DOI] [PubMed] [Google Scholar]
- Christie, A. E. , Nolan, D. H. , Ohno, P. , Hartline, N. , & Lenz, P. H. (2011). Identification of chelicerate neuropeptides using bioinformatics of publicly accessible expressed sequence tags. General and Comparative Endocrinology, 170(1), 144–155. [DOI] [PubMed] [Google Scholar]
- Christie, A. E. , Roncalli, V. , Wu, L. S. , Ganote, C. L. , Doak, T. , & Lenz, P. H. (2013). Peptidergic signaling in Calanus finmarchicus (Crustacea, Copepoda): In silico identification of putative peptide hormones and their receptors using a de novo assembled transcriptome. General and Comparative Endocrinology, 187, 117–135. [DOI] [PubMed] [Google Scholar]
- Christie, A. E. , Stemmler, E. A. , & Dickinson, P. S. (2010). Crustacean neuropeptides. Cellular and Molecular Life Sciences, 67(24), 4135–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, M. S. , Thorne, M. A. S. , Toullec, J. Y. , Meng, Y. , Guan, L. L. , Peck, L. S. , & Moore, S. (2011). Antarctic krill 454 pyrosequencing reveals chaperone and stress transcriptome. PLoS ONE, 6(1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbourne, J. K. , Pfrender, M. E. , Gilbert, D. , Thomas, W. K. , Tucker, A. , Oakley, T. H. , … Boore, J. L. (2011). The ecoresponsive genome of Daphnia pulex . Science, 331(6017), 555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compeau, P. E. C. , Pevzner, P. A. , & Tesler, G. (2011). How to apply de Bruijn graphs to genome assembly. Nature Biotechnology, 29(11), 987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cresswell, K. A. , Tarling, G. A. , Thorpe, S. E. , Burrows, M. T. , Wiedenmann, J. , & Mangel, M. (2009). Diel vertical migration of Antarctic krill (Euphausia superba) is flexible during advection across the Scotia Sea. Journal of Plankton Research, 31(10), 1265–1281. [Google Scholar]
- Davies, N. J. , Krusche, P. , Tauber, E. , & Ott, S. (2015). Analysis of 5′ gene regions reveals extraordinary conservation of novel non‐coding sequences in a wide range of animals. BMC Evolutionary Biology, 15(1), 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pittà, C. , Bertolucci, C. , Mazzotta, G. M. , Bernante, F. , Rizzo, G. , De Nardi, B. , … Costa, R. (2008). Systematic sequencing of mRNA from the Antarctic krill (Euphausia superba) and first tissue specific transcriptional signature. BMC Genomics, 9, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pittà, C. , Biscontin, A. , Albiero, A. , Sales, G. , Millino, C. , Mazzotta, G. M. , … Costa, R. (2013). The Antarctic krill Euphausia superba shows diurnal cycles of transcription under natural conditions. PLoS ONE, 8(7), e68652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores, H. , Atkinson, A. , Kawaguchi, S. , Krafft, B. , Milinevsky, G. , Nicol, S. , … Werner, T. (2012). Impact of climate change on Antarctic krill. Marine Ecology Progress Series, 458, 1–19. [Google Scholar]
- Fu, L. , Niu, B. , Zhu, Z. , Wu, S. , & Li, W. (2012). CD‐HIT: Accelerated for clustering the next‐generation sequencing data. Bioinformatics, 28(23), 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gard, A. L. , Lenz, P. H. , Shaw, J. R. , & Christie, A. E. (2009). Identification of putative peptide paracrines/hormones in the water flea Daphnia pulex (Crustacea; Branchiopoda; Cladocera) using transcriptomics and immunohistochemistry. General and Comparative Endocrinology, 160(3), 271–287. [DOI] [PubMed] [Google Scholar]
- Gaten, E. , Tarling, G. , Dowse, H. , Kyriacou, C. , & Rosato, E. (2008). Is vertical migration in Antarctic krill (Euphausia superba) influenced by an underlying circadian rhythm? Journal of Genetics, 87(5), 473–483. [DOI] [PubMed] [Google Scholar]
- Gibbons, J. G. , Janson, E. M. , Hittinger, C. T. , Johnston, M. , Abbot, P. , & Rokas, A. (2009). Benchmarking next‐generation transcriptome sequencing for functional and evolutionary genomics. Molecular Biology and Evolution, 26(12), 2731–2744. [DOI] [PubMed] [Google Scholar]
- Groeneveld, J. , Johst, K. , Kawaguchi, S. , Meyer, B. , Teschke, M. , & Grimm, V. (2015). How biological clocks and changing environmental conditions determine local population growth and species distribution in Antarctic krill (Euphausia superba): A conceptual model. Ecological Modelling, 303, 78–86. [Google Scholar]
- Haas, B. J. , Papanicolaou, A. , Yassour, M. , Grabherr, M. , Blood, P. D. , Bowden, J. , … Regev, A. (2013). De novo transcript sequence reconstruction from RNA‐seq using the Trinity platform for reference generation and analysis. Nature Protocols, 8(8), 1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haznedaroglu, B. Z. , Reeves, D. , Rismani‐Yazdi, H. , & Peccia, J. (2012). Optimization of de novo transcriptome assembly from high‐throughput short read sequencing data improves functional annotation for non‐model organisms. BMC Bioinformatics, 13(1), 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, S. L. , Phillips, T. , & Atkinson, A. (2013). Potential climate change effects on the habitat of Antarctic krill in the weddell quadrant of the southern ocean. PLoS ONE, 8(8), e72246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarman, S. N. , & Deagle, B. E. (2016). Genetics of Antarctic krill In Volker Siegel (Ed.), Biology and ecology of Antarctic krill, advances in polar ecology (pp. 247–277). Switzerland: Springer International Publishing. [Google Scholar]
- Jeffery, N. W. (2012). The first genome size estimates for six species of krill (Malacostraca, Euphausiidae): Large genomes at the north and south poles. Polar Biology, 35(6), 959–962. [Google Scholar]
- Jennings, B. H. (2011). Drosophila – a versatile model in biology & medicine. Materials Today, 14(5), 190–195. [Google Scholar]
- Johnsen, A. , Fidler, A. E. , Kuhn, S. , Carter, K. L. , Hoffmann, A. , Barr, I. R. , … Kempenaers, B. (2007). Avian Clock gene polymorphism: Evidence for a latitudinal cline in allele frequencies. Molecular Ecology, 16(22), 4867–4880. [DOI] [PubMed] [Google Scholar]
- Kawaguchi, S. , Ishida, A. , King, R. , Raymond, B. , Waller, N. , Constable, A. , … Ishimatsu, A. (2013). Risk maps for Antarctic krill under projected Southern Ocean acidification. Nature Climate Change, 3(9), 843–847. [Google Scholar]
- Kawaguchi, S. , Yoshida, T. , Finley, L. , Cramp, P. , & Nicol, S. (2007). The krill maturity cycle: A conceptual model of the seasonal cycle in Antarctic krill. Polar Biology, 30, 689–698. [Google Scholar]
- Ma, M. , Bors, E. K. , Dickinson, E. S. , Kwiatkowski, M. A. , Sousa, G. L. , Henry, R. P. , … Li, L. (2009). Characterization of the Carcinus maenas neuropeptidome by mass spectrometry and functional genomics. General and Comparative Endocrinology, 161(3), 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, M. , Gard, A. L. , Xiang, F. , Wang, J. , Davoodian, N. , Lenz, P. H. , … Li, L. (2010). Combining in silico transcriptome mining and biological mass spectrometry for neuropeptide discovery in the Pacific white shrimp Litopenaeus vannamei . Peptides, 31(1), 27–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins, M. J. F. , Lago‐Leston, A. , Anjos, A. , Duarte, C. M. , Agusti, S. , Serrão, E. A. , & Pearson, G. A. (2015). A transcriptome resource for Antarctic krill (Euphausia superba Dana) exposed to short‐term stress. Marine Genomics, 23, 45–47. [DOI] [PubMed] [Google Scholar]
- Mazzotta, G. M. , De Pittà, C. , Benna, C. , Tosatto, S. C. E. , Lanfranchi, G. , Bertolucci, C. , & Costa, R. (2010). A cry from the krill. Chronobiology International, 27(3), 425–445. [DOI] [PubMed] [Google Scholar]
- Meyer, B. , Auerswald, L. , Siegel, V. , Spahic, S. , Pape, C. , Fach, B. , … Fuentes, V. (2010). Seasonal variation in body composition, metabolic activity, feeding, and growth of adult krill Euphausia superba in the Lazarev Sea. Marine Ecology Progress Series, 398, 1–18. [Google Scholar]
- Meyer, B. , Martini, P. , Biscontin, A. , De Pittà, C. , Romualdi, C. , Teschke, M. , … Kawaguchi, S. (2015). Pyrosequencing and de novo assembly of Antarctic krill (Euphausia superba) transcriptome to study the adaptability of krill to climate‐induced environmental changes. Molecular Ecology Resources, 15(6), 1460–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori, M. , & Butterworth, D. S. (2006). A first step towards modelling the krill‐predator dynamics of the antarctic ecosystem. CCAMLR Science, 13, 217–277. [Google Scholar]
- Murphy, E. J. , Hofmann, E. E. , Watkins, J. L. , Johnston, N. M. , Piñones, A. , Ballerini, T. , … Fretwell, P. (2013). Comparison of the structure and function of Southern Ocean regional ecosystems: The Antarctic Peninsula and South Georgia. Journal of Marine Systems, 109–110, 22–42. [Google Scholar]
- Murphy, E. , Watkins, J. , Trathan, P. , Reid, K. , Meredith, M. , Thorpe, S. , … Fleming, A. (2007). Spatial and temporal operation of the Scotia Sea ecosystem: A review of large‐scale links in a krill centred food web. Philosophical Transactions of the Royal Society B: Biological Sciences, 362(1477), 113–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale, D. B. , Wegrzyn, J. L. , Stevens, K. A. , Zimin, A. V. , Puiu, D. , Crepeau, M. W. , … Lewis, S. (2014). Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biology., 15(3), R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol, S. , Constable, A. J. , & Pauly, T. (2000). Estimates of circumpolar abundance of Antarctic krill based on recent acoustic density measurements. CCAMLR Science, 7, 87–99. [Google Scholar]
- Nicol, S. , & Foster, J. (2016).The fishery for Antarctic krill: Its current status and management regime In Volker Siegel (Ed.), Biology and ecology of Antarctic krill (pp. 387–421). Switzerland: Springer International Publishing. [Google Scholar]
- O'Malley, K. G. , & Banks, M. A. (2008). A latitudinal cline in the Chinook salmon (Oncorhynchus tshawytscha) Clock gene: Evidence for selection on PolyQ length variants. Proceedings of the Royal Society B: Biological Sciences, 275(1653), 2813–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neil, S. T. , Emrich, S. J. , Vera, C. , Wheat, C. , Fescemyer, H. , Frilander, M. , … Hendrickx, F. (2013). Assessing De Novo transcriptome assembly metrics for consistency and utility. BMC Genomics, 14(1), 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özkaya, Ö. , & Rosato, E. (2012). The Circadian clock of the fly: A neurogenetics journey through time. Advances in Genetics, 79–123. [DOI] [PubMed] [Google Scholar]
- Pegoraro, M. , Gesto, J. S. , Kyriacou, C. P. , & Tauber, E. (2014). Role for Circadian clock genes in seasonal timing: Testing the bünning hypothesis. PLoS Genetics, 10(9), e1004603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissinotto, R. , Gurney, L. , & Pakhomov, E. A. (2000). Contribution of heterotrophic material to diet and energy budget of Antarctic krill. Euphausia superba. Marine Biology, 136(1), 129–135. [Google Scholar]
- Priyam, A. , Woodcroft, B. J. , Rai, V. , Munagala, A. , Moghul, I. , Ter, F. , … Wurm, Y. (2015). Sequenceserver: A modern graphical user interface for custom BLAST databases. bioRxiv 033142. doi: 10.1101/033142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppert, S. M. , & Weaver, D. R. (2000). Comparing clockworks: Mouse versus fly. Journal of Biological Rhythms, 15(5), 357–364. [DOI] [PubMed] [Google Scholar]
- Robertson, G. , Schein, J. , Chiu, R. , Corbett, R. , Field, M. , Jackman, S. D. , … Birol, I. (2010). De novo assembly and analysis of RNA‐seq data. Nature Methods, 7(11), 909–912. [DOI] [PubMed] [Google Scholar]
- Saleem, Q. , Anand, A. , Jain, S. , & Brahmachari, S. K. (2001). The polyglutamine motif is highly conserved at the Clock locus in various organisms and is not polymorphic in humans. Human Genetics, 109(2), 136–142. [DOI] [PubMed] [Google Scholar]
- Sales, G. , Deagle, B. E. , Calura, E. , Martini, P. , Biscontin, A. , De Pittà, C. , … Jarman, S. (2017). KrillDB: A de novo transcriptome database for the Antarctic krill (Euphausia superba). PLoS ONE, 12(2), e0171908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, K. , Atkinson, A. , Steigenberger, S. , Fielding, S. , Lindsay, M. C. M. , Pond, D. W. , … Achterberg, E. P. (2011). Seabed foraging by Antarctic krill: Implications for stock assessment, bentho‐pelagic coupling, and the vertical transfer of iron. Limnology and Oceanography, 56(4), 1411–1428. [Google Scholar]
- Seear, P. J. , Goodall‐Copestake, W. , Fleming, A. , Rosato, E. , & Tarling, G. (2012). Seasonal and spatial influences on gene expression in Antarctic krill Euphausia superba . Marine Ecology Progress Series, 467, 61–75. [Google Scholar]
- Seear, P. J. , Tarling, G. A. , Burns, G. , Goodall‐Copestake, W. P. , Gaten, E. , Ozkaya, O. , & Rosato, E. (2010). Differential gene expression during the moult cycle of Antarctic krill (Euphausia superba). BMC Genomics, 11(1), 582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seear, P. J. , Tarling, G. A. , Teschke, M. , Meyer, B. , Thorne, M. A. S. , Clark, M. S. , … Rosato, E. (2009). Effects of simulated light regimes on gene expression in Antarctic krill (Euphausia superba Dana). Journal of Experimental Marine Biology and Ecology, 381(1), 57–64. [Google Scholar]
- Smith‐Unna, R. , Boursnell, C. , Patro, R. , Hibberd, J. M. , & Kelly, S. (2016). TransRate: Reference‐free quality assessment of de novo transcriptome assemblies. Genome Research, 26(8), 1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss, J. , & Dircksen, H. (2010). Circadian clocks in crustaceans: Identified neuronal and cellular systems. Frontiers in Bioscience, 15, 1040–1074. [DOI] [PubMed] [Google Scholar]
- Surget‐Groba, Y. , & Montoya‐Burgos, J. I. (2010). Optimization of de novo transcriptome assembly from next‐generation sequencing data. Genome Research, 20(10), 1432–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarling, G. A. , & Johnson, M. L. (2006). Satiation gives krill that sinking feeling. Current Biology, 16(3), R83–R84. [DOI] [PubMed] [Google Scholar]
- Tarling, G. A. , Klevjer, T. , Fielding, S. , Watkins, J. , Atkinson, A. , Murphy, E. , … Leaper, R. (2009). Variability and predictability of Antarctic krill swarm structure. Deep‐Sea Research Part I: Oceanographic Research Papers, 56(11), 1994–2012. [Google Scholar]
- Teschke, M. , Wendt, S. , Kawaguchi, S. , Kramer, A. , & Meyer, B. (2011). A Circadian clock in Antarctic Krill: An endogenous timing system governs metabolic output rhythms in the euphausid species Euphausia superba . PLoS ONE, 6(10), e26090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilden, A. R. , McCoole, M. D. , Harmon, S. M. , Baer, K. N. , & Christie, A. E. (2011). Genomic identification of a putative circadian system in the cladoceran crustacean Daphnia pulex . Comparative Biochemistry and Physiology Part D: Genomics and Proteomics, 6(3), 282–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toullec, J.‐Y. , Corre, E. , Bernay, B. , Thorne, M. A. S. , Cascella, K. , Ollivaux, C. , … Clark, M. S. (2013). Transcriptome and Peptidome Characterisation of the Main Neuropeptides and Peptidic Hormones of a Euphausiid: The Ice Krill, Euphausia crystallorophias . PLoS ONE, 8(8), e71609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse, M. J. , Atkinson, A. , & Rees, A. P. (2011). Close coupling between ammonium uptake by phytoplankton and excretion by Antarctic krill, Euphausia superba . Deep Sea Research Part I: Oceanographic Research Papers, 58(7), 725–732. [Google Scholar]
- Xie, Y. , Wu, G. , Tang, J. , Luo, R. , Patterson, J. , Liu, S. , … Wang, J. (2014). SOAPdenovo‐Trans: De novo transcriptome assembly with short RNA‐Seq reads. Bioinformatics, 30(12), 1660–1666. [DOI] [PubMed] [Google Scholar]
- Yang, J. S. , Dai, Z. M. , Yang, F. , & Yang, W. J. (2006). Molecular cloning of Clock cDNA from the prawn, Macrobrachium rosenbergii . Brain Research, 1067(1), 13–24. [DOI] [PubMed] [Google Scholar]
- Ye, J. , Fang, L. , Zheng, H. , Zhang, Y. , Chen, J. , Zhang, Z. , … Wang, J. (2006). WEGO: A web tool for plotting GO annotations. Nucleic Acids Research, 34(Web Server), W293–W297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Hastings, M. H. , Green, E. W. , Tauber, E. , Sladek, M. , Webster, S. G. , … Wilcockson, D. C. (2013). Dissociation of circadian and circatidal timekeeping in the marine crustacean Eurydice pulchra . Current Biology, 23(19), 1863–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Q.‐Y. , Wang, Y. , Kong, Y.‐M. , Luo, D. , Li, X. , & Hao, P. (2011). Optimizing de novo transcriptome assembly from short‐read RNA‐Seq data: A comparative study. BMC Bioinformatics, 12(Suppl 14), S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, H. , Sauman, I. , Yuan, Q. , Casselman, A. , Emery‐Le, M. , Emery, P. , & Reppert, S. M. (2008). Cryptochromes define a novel circadian clock mechanism in monarch butterflies that may underlie sun compass navigation. PLoS Biology, 6(1), 0138–0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequences of cloned circadian genes are available in GenBank under accessions KX238951, KX238952, KX238953, KX238954, and KX238955, as are the sequences of the total assembly under accession GFCS00000000. Read data were submitted to NCBI SRA under accession SRR4408478. Annotated coding and peptide assembly sequences are available at http://www.krill.le.ac.uk.