

Abstract
Optical microscopy has generated great impact for modern research. While fluorescence microscopy provides the ultimate sensitivity, it generally lacks chemical information. Complementarily, vibrational imaging methods provide rich chemical-bond-specific contrasts. Nonetheless, they usually suffer from unsatisfying sensitivity or compromised biocompatibility. Recently, electronic preresonance stimulated Raman scattering (EPR-SRS) microscopy was reported, achieving simultaneous high detection sensitivity and superb vibrational specificity of chromophores. With newly synthesized Raman-active dyes, this method readily breaks the optical color barrier of fluorescence microscopy and is well-suited for supermultiplex imaging in biological samples. In this Perspective, we first review previous utilizations of electronic resonance in various Raman spectroscopy and microscopy. We then discuss the physical origin and uniqueness of the electronic preresonance region, followed by quantitative analysis of the enhancement factors involved in EPR-SRS microscopy. On this basis, we provide an outlook for future development as well as the broad applications in biophotonics.
Modern optical spectroscopy and microscopy methods have allowed researchers to study molecular processes in biological systems with unprecedented sensitivity and specificity. In particular, fluorescence microscopy is almost the method of choice for bioimaging applications. It offers robust single-molecule detectability, target specificity, and biocompatibility by probing the electronic resonance of versatile fluorescent probes and detecting the Stokes-shifted emission in a background-free manner.1−4 Complementary to fluorescence, Raman microscopy has become an increasingly valuable bioanalytical tool by providing rich chemical information derived from chemical-bond-specific vibrational transitions (Figure 1). However, the conventional spontaneous Raman scattering process is known to be about 10–14 orders of magnitude weaker than fluorescence (Figure 2a) and thus is highly restricted in its applications for live-cell imaging.5 In fact, the concern for sensitivity has almost always been associated with linear Raman detection when compared to fluorescence, with only one notable exception. That is the near-field-based techniques of surface-enhanced raman scattering (SERS) and tip-enhanced Raman scattering (TERS), which achieved single-molecule sensitivity about more than 20 and 10 years ago, respectively.6−10 It has long been thought (and debated) that the remarkable enhancement factor (EF) of 1010–1014 for SERS and TERS solely originated from plasmonic enhancement with the metallic nanostructures acting as optical antennas. It was only quantified much later that the resonance Raman effect, involving electronic resonance, played a significant role.11−13Considering that fluorescence is also a resonant process by definition, it seems that, to ensure high detection sensitivity (possibly down to single molecules), a shared feature here is by going electronic-resonant.
Figure 1.

Selective milestones in the development of Raman spectroscopy and bioimaging-oriented Raman microscopy.
Figure 2.
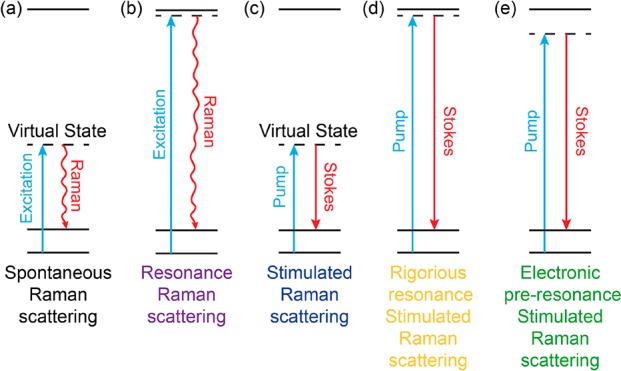
Energy diagrams of various Raman spectroscopy processes. (a) Spontaneous Raman scattering. (b) Resonance Raman scattering. (c) Nonresonant stimulated Raman scattering. (d) Rigorous resonance stimulated Raman scattering. (e) Electronic preresonance stimulated Raman scattering.
Indeed, the observation of the resonance Raman effect could be traced back to 1950s, even before the invention of lasers.14−16 Later, electronic resonance-enhanced Raman scattering proved to be particularly useful for spectroscopic studies. It could sensitively and selectively probe the chemical structures as well as the excited-state photophysics by bringing the laser excitation energy (ωexc) close to the molecular absorption peak (ω0) in both deep-UV and visible ranges (Figure 2b).17−19 This strategy exploits electronic and vibrational coupling in chromophores, in which the electronic resonance significantly promotes those nuclear vibrations that are coupled to the electronic transition. Although such a gain of Raman intensity offers good sensitivity for spectroscopic interrogation of light-absorbing chromophores, it often comes with several issues for imaging applications. First, the achievable Raman signals can be easily overwhelmed by the concomitant fluorescence backgrounds. This is because even with rigorous resonance, i.e., ωexc ≈ ω0, the corresponding resonance Raman cross section is still many orders (about 7–9 orders) of magnitude away from absorption cross section of a typical chromophore (σabs ≈ 10–16 cm2). Second, Raman detection under rigorous electronic resonance also suffers from fast photobleaching or degradation.
However, exceptions also exist for the electronic-resonant spontaneous Raman bioimaging if fluorescence emission is spectrally far away from the Raman scattering or the excited-state lifetime is extremely short (i.e., vanishing fluorescence quantum yield). For example, deep-ultraviolet resonant Raman imaging of a cell was demonstrated without any labeling at 257 nm excitation.20 For another example, resonance Raman imaging of cytochrome c in living cells has also been achieved by taking advantage of its short excited-state lifetime.21 Along a similar spectroscopic line but with probe engineering, recent utilization of a special class of probes, fluorescent quenchers with extremely low quantum yield, as resonance Raman reporters demonstrated specific organelle imaging capability by spontaneous Raman microscopy.22,23 Compared to nonresonant Raman (i.e., ωexc ≪ ω0), it showed 3 orders of magnitude signal enhancement.22,23 Such a Raman signal boost and fluorescent background reduction from resonance Raman reporters made spontaneous Raman imaging of certain targets possible in living cells. However, its sensitivity and imaging speed are still unsatisfying for visualizing a wide variety of biomolecules.
Toward higher sensitivity, SERS provides an ultrasensitive strategy. Its achievable up to 1014 giant EF made single-molecule vibrational spectroscopy possible, opening up many exciting applications in physical science.8,13 In addition, the required metallic surface in close proximity also effectively deprives the possible fluorescence background. However, as powerful and as sensitive as it is, there are a few near-field associated limitations. Biologically, the strict reliance on metallic nanostructures restricts the general applicability of SERS for biological targets. The close (angstrom-level) contact with the metal surface could often perturb the native properties of biomolecules, such as denaturing the proteins.8 Physically, SERS is usually difficult for precise quantitative analysis because the EFs offered by the surface plasmons vary substantially between different substrate–molecule configurations. With deeper understanding of the SERS mechanism, it was also gradually revealed that the resonance Raman effect played a significant role in the overall large EF. SERS of many chromophores could hence be more precisely regarded as surface-enhanced resonance Raman scattering (SERRS).8 Nonetheless, for a long time, the exact contribution of electronic enhancement was less clear due to a lack of proper tools to characterize the electronic resonance factors.
Evidenced by the evolutionary path of fluorescence microscopy, far-field spectroscopy provides more general biocompatibility compared to near-field methods. Nonlinear Raman scattering, the all-far-field advanced Raman spectroscopy, naturally takes the next lead in enhancing the Raman signals and exploiting electronic resonance in pursuit of higher sensitivity. Indeed, in 2003, femtosecond stimulated Raman scattering (FSRS) spectroscopy provided resonance-enhanced vibrational spectra for visible (e.g., Rhodamine 6G) and near-infrared (e.g., 3,3′-diethylthiatricarbocyanine iodide) dyes that are free from fluorescence background.24 In 2008, it was FSRS again that precisely quantified the vibronic features for the resonance Raman spectra of Rhodamine 6G. It unequivocally determined a 106 EF for the rigorous resonance (107 EF by integration over all Raman modes of Rhodamine 6G) compared to a standard reference, the spontaneous Raman cross section of C–O bond stretching of methanol (σspon, Raman ≈ 10–29 cm2).12 It was from then that the contributions between resonance Raman and surface plasmons could be explicitly separable for different SERRS substrate configurations. Later in 2008, triple-resonance coherent antistokes Raman scattering (CARS) microspectroscopy using femtosecond laser pulse shaping was also reported for detecting electronic resonance-enhanced nonlinear Raman signals from nonfluorescent molecules. This method offered a sensitivity approaching 100 molecules in solution with 3 s integration.25
However, the notion of resonance Raman has largely escaped from the radar of the modern nonlinear Raman imaging community, such as for CARS and the more recently developed stimulated Raman scattering (SRS) microscopy (Figure 2c). Both of them have been proven to be highly desirable for label-free imaging with even up to video-rate speed.26−32 A consensus has been reached in the field that SRS has superseded CARS microscopy in almost all technical aspects. In particular, SRS imaging offers linear dependence on concentration and shows identical spectra to spontaneous Raman with no interference complications from the nonresonance background as in CARS.27,28 By virtue of these advantages, SRS imaging, mostly in the label-free form, has made a major impact with exciting applications in biological and medical photonics.28,33−43 Such a label-free strategy is appealing and powerful as it introduces zero physical and chemical perturbation to biological systems. Going beyond the label-free concept, the coupling of SRS microscopy with small and bio-orthogonal vibrational tags, such as alkynes (i.e., carbon–carbon triple bond), has been recently demonstrated for the detection of small biomolecules in the cell-silent Raman spectral window.44,45 Such bio-orthogonal chemical imaging offers a powerful platform for functional metabolic imaging in live cells and animals.46−55 Its success underscores the importance of introducing vibrational probes to improve specificity and sensitivity of nonlinear Raman microscopy. However, even with extensive efforts of instrumentation improvement and small-tag optimization, the detection sensitivity of SRS is still in the range of 35 μM (i.e., diyne tags, double-conjugated alkynes)51 to 200 μM (small alkyne tags).44 Here, we note that nearly all of the previous CARS and SRS imagings were operated in the nonresonance region, in which the energy of the pump laser (ωpump) is well below that of the molecular absorption peak (i.e., ωpump ≪ ω0) (Figure 2c).
Realizing the possible large electronic resonance EF of 106 when compared to nonresonance Raman signals (e.g., C–O bond), Wei et al. introduced a new scheme of SRS microscopy by shifting from the commonly exploited nonresonance region (Figure 2c) to the electronic resonance (i.e., ωpump close to ω0) (Figure 2d,e).56 First, they directly explored the rigorous resonance SRS detection (Figure 2d). Because the Stokes laser wavelength (λStokes) is fixed at 1064 nm in the setup, the pump laser wavelength (λpump) for detecting typical electronically coupled vibrational modes (e.g., the total-symmetric vibration of conjugated double bonds, at ∼1600–1660 cm–1) is around 906 nm. Hence, a far-red absorbing molecule IR895 (λabs ≈ 900 nm) was chosen as a model compound for exploring the rigorous resonance SRS microspectroscopy. An intense but broad peak was observed without clearly identifiable vibrational signatures.56 The calculated cross section for this broad peak is about 108 of the standard Raman cross section of C–O bond in methanol acquired under the same SRS laser excitation conditions. Because the SRS signal is detected as pump laser intensity loss (i.e., the stimulated Raman loss) in the presence of Stokes photons, such a huge background does not result from fluorescence emission but a combination of the competing nonlinear optical processes.24,57 It might include other electronic resonance-enhanced four-wave mixing pathways such as resonant Rayleigh scattering and the absorption-based pump photon loss due to ground-state population restoration by stimulated emission from Stokes photons.
Comparing the measured EF of 108 from the broad peak of IR895 with the reported rigorous resonance Raman EF of 106, a signal-to-background (S/B) ratio of about 1% might be expected between its narrow-band vibrational signal and the broad-band electronic background. Such a small S/B ratio could be easily buried by background fluctuation. This was indeed the case where the narrow Raman features of IR895 were almost unobservable in the rigorous resonance SRS spectrum.56 As a reference, the solvent Raman peak of CH3 from pure methanol was still identifiable with a S/B of about 8%.56 It is worth mentioning that, in the case that Raman features are resolvable under rigorous electronic resonance SRS, normal Lorentzian-shaped Raman peaks should be inversed or partially dispersed depending on where exactly the laser is exciting on the absorption peak of the molecules.58 Similar broad electronic backgrounds also exist in FSRS spectra beneath the vibrational contrasts.12,24 Such a background is not a big issue for spectroscopic characterization with good signal-to-noise ratio but would largely complicate the interpretation for demanding imaging applications, in which unambiguous differentiation of on-resonance and off-resonance (ideally with vanishing contrasts) contributions is essential. Because the off-resonance background carries noise and is usually not spectrally flat beneath the on-resonance signal, a simple on–off subtraction may not work well for imaging analysis.
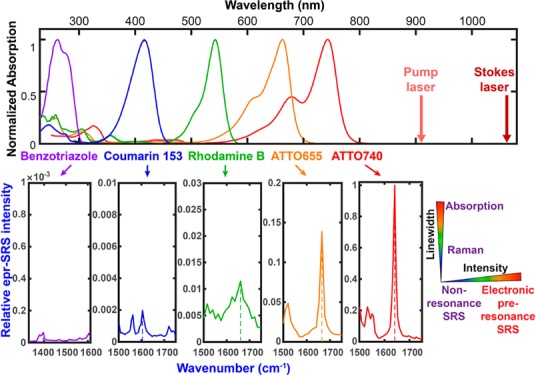
If rigorous resonance as in IR895 quickly brings up the background by evoking a combination of electronically enhanced multipathway backgrounds, would proper detuning away from the rigorous resonance help attenuate the electronic background and hence restore the chemical selectivity? Wei. et al. reported an electronic preresonance SRS (EPR-SRS) scheme by shifting the excitation to an electronic preresonance (EPR) window, in between the rigorous resonance and the nonresonance regions.56 In this scenario, the pump laser frequency (ωpump) is tuned away from the molecular absorption maximum (ω0) into a region of 2Γe< ω0 – ωpump < 6Γe (Γe is the homogeneous line width of the electronic transition, ∼700 cm–1) (Figure 2e). These two boundaries are set to ensure a fine balance between achievable EPR-Raman enhancement (with more then 104 EF) and fine chemical selectivity with a sufficiently attenuated electronic background (S/B > 5). A representative near-infrared absorbing dye in this EPR-SRS region is ATTO740 (ω0 – ωpump ≈ 3Γe), whose EPR-SRS spectrum is essentially free from electronic background. The high detection sensitivity, sharp Raman resonance, and vanishing background could be all evidenced from the EPR-SRS images of ATTO740 click-labeled EdU for newly synthesized DNA in mammalian cells (Figure 3a,b; the corresponding off-resonance image is nearly zero by tuning the λpump away for only 2 nm). Such exquisite chemical selectivity is well beyond what could be achieved by typical absorption or fluorescence detections in which a 2 nm shift in excitation wavelength would result in little difference in signal/image generation.
Figure 3.

EPR-SRS imaging of specific cellular targets. (a) On-resonance EPR-SRS imaging of ATTO740-labeled 5-ethynyl-2′-deoxyuridine (EdU) by click-chemistry for newly synthesized DNA in HeLa cells, targeting the double-bond vibrational peak of ATTO740 at 1640 cm–1. (b) Off-resonance (1616 cm–1) imaging of the same sample as that in (a) by tuning the pump wavelength away for 2 nm. (c–f) EPR-SRS imaging of ATTO740 immunolabeled α-tubulin (c), Tom20 (d, Mitochondria marker), Giantin (e, Golgi marker), and fibrillarin (f, Nucleoli marker) in HeLa cells. (g) EPR-SRS imaging of methylene blue, a known drug with low fluorescent quantum yield, in live HeLa cells. (h) EPR-SRS imaging of nonfluorescent oxidation product 4,4′-dichloro-5,5′-dibromoindigo from X-gal hydrolysis in live E. coli. Scale bar, 10 μm.
The achievable detection sensitivity of EPR-SRS could be down to 250 nM for ATTO740 (which is about 30–50 molecules in the laser focus) with a 1 ms time constant.56 As such, the sensitivity of EPR-SRS is approaching that of a commercial fluorescence microscope, opening up numerous possibilities for vibrational imaging of specific molecular targets in biological samples (Figure 3c–f). In addition, high photostability is another appealing factor for the EPR-SRS region because the pump laser is not directly exciting the absorption peak of the molecule. It was shown that 97% of ATTO740 was maintained even after 100 frames of continuous scanning,56 a feat difficult to obtain even with highly photostable dyes used for fluorescence imaging.59 In addition to fluorescent molecules, absorbing-only (nonfluorescent or with very low fluorescent quantum yield) chromophores could also be probed by EPR-SRS with good sensitivity and high specificity (Figure 3g,h).
It is natural to next ask what the physical origin is for such existence of a unique preresonance region. In particular, how is it possible to sufficiently amplify the electronically coupled vibrational signals without exciting much real electronic population? We could seek some insights from the excitation frequency dependence for both resonant Raman cross sections and electronic absorption cross sections, respectively. The theory of preresonance Raman was first worked out by Andreas C. Albrecht, who derived the Albrecht A-term preresonance approximation equation for totally symmetric transitions under a strong dominating electronic transition almost 60 years ago14,60
| 1 |
ωvib is the vibrational transition energy and K is a collection of frequency-independent factors of the Raman dyes. This equation could well describe the EPR-SRS measurements on dyes across a wide spectrum of absorptions by assuming dye-independent oscillator strength and ground-to-excited state Franck–Condon factors.56 We note that here the EPR-Raman cross sections are dependent on the frequency detuning (ω0 – ωpump) to its fourth power.
As a comparison, the molecular absorption spectrum in the condensed phase may be modeled by a pseudo-Voigt profile61,62
| 2 |
in which L(ν) is the Lorentzian distribution for homogeneous line broadening and G(ν) is the Gaussian distribution for inhomogeneous line broadening. Taking the measured absorption spectrum of ATTO740 in solution as a reference, Γe = 670 cm–1 and f = 0.98 best fit its absorption profile, largely following a Gaussian distribution, consistent with the known conclusion that absorption spectra in solution are largely inhomogeneously broadened. We then overplotted eqs 1 and 2 and normalized the values in reference to the numbers detuned away by 1.5Γe. It became obvious that the Raman signal from eq 1 decays much slower than the absorption signal (eq 2) in the preresonance region of the experiment (Figure 4, 820 nm (detuned away by 2Γe) to 920 nm (detuned away by 4Γe)). This comparison illustrates that virtual-state mediated preresonance Raman processes follow a slower decay as a function of frequency detuning in the defined preresonance regime compared to real-state mediated absorption transitions.
Figure 4.
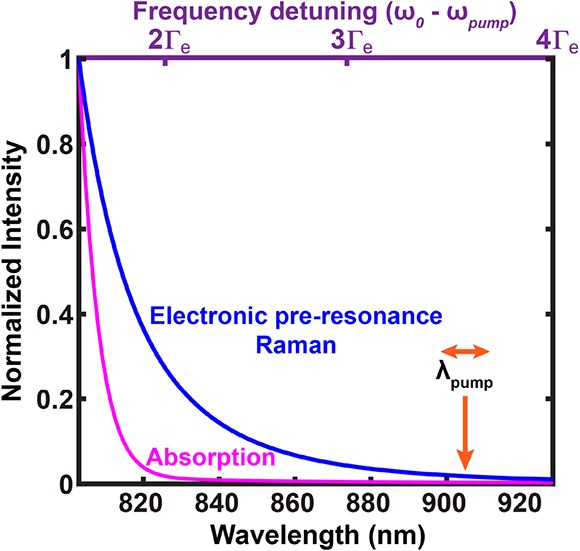
Signal dependence on the pump laser wavelength (also the frequency detuning (ω0 – ωpump)): Electronic preresonance Raman (blue) and absorption (red) spectra, normalized to 1.5Γe away from the absorption maximum.
Qualitatively, such slower signal decay behavior of EPR-SRS might also be understood by the frequency detuning dependence of absorption and Raman on χ(1) and χ(3), respectively. For the EPR-SRS process, under a strong electronic transition and the adiabatic approximation, χ(3) could be written as58
| 3 |
in which ωStokes is the Stokes photon frequency, Γvib is the line width of vibrational transition, and ωge is electronic transition frequency. g and e are ground and excited electronic states. |0), |1), and |υ) are ground, first, and intermediate vibrational states, respectively. M is the dipole moment operator.
Under (or close to) the Raman resonsance (i.e., ωpump – ωStokes ≈ ωvib), eq 3 could be rewritten as
| 4 |
Here, because the cross
section of SRS is
known to be proportional to the imaginary part of χ(3),58 it hence indicates that the EPR-SRS
signal is approximated to be proportional to the real part of the
curly bracketed terms of eq 4, i.e.,  for
summation over each υ and then
squared over.
for
summation over each υ and then
squared over.
As a comparison, linear absorption and dispersion each follow the imaginary- and real part changes of χ(1), respectively
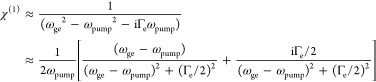 |
5 |
Except for the extra squared factor, which is related to χ(3) describing a higher-order optical process and also consistent with Albrecht A-term’s fourth-power dependence on the frequency detuning (eq 1), a qualitatively similar dependence of resonance Raman (the imaginary part of eq 4) versus linear dispersion (the real part of eq 5) on frequency detuning could be clearly spotted. Therefore, the differential frequency dependence between resonance Raman and linear absorption is analogous to the more familiar relationship between dispersion and absorption, in which the real part of χ(1) (dispersion) is well-known to decay much slower compared to the imaginary part of χ(1) (absorption) after a certain detuning range (e.g., in the EPR range) (Figure 5). Although being approximate and qualitative, this analogy could provide an intuitively physical picture for why such a peculiar EPR-Raman region exists with enhancement of electronically coupled vibrational signals over electronic backgrounds.
Figure 5.
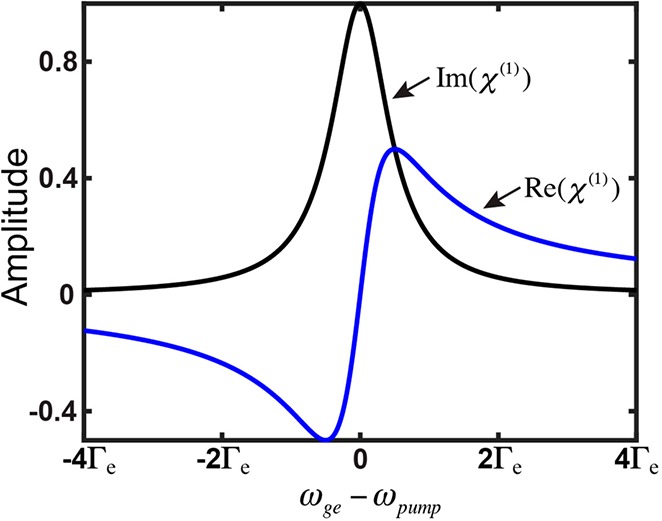
Dependence of the imaginary part (black line, i.e., absorption) and the real part (blue line, i.e., dispersion) of χ(1) on the laser frequency (ωpump) detuning from the electronic transition energy (ωge).
We next quantitatively analyze the EPR-SRS cross sections and the essential contributing factors for comprehensively understanding and better developing EPR-SRS microscopy. The spontaneous Raman cross section for the standard C–O vibration at 1030 cm–1 in methanol (σspon, Raman (C–O)) was reported to be 2.1 × 10–30 cm2 under 785 nm excitation.63 Extrapolating from 785 nm using the ω3pumpωStokes dependence to the SRS excitation wavelength of λpump ≈ 960 nm and λStokes ≈ 1064 nm, σspon, Raman(C–O) becomes 0.9 × 10–30 cm2. As shown in Figure 6a, the measured SRS signal of C–O in pure methanol (∼24.7 M in concentration) is (ΔIp/Ipump)SRS = 1.7 × 10–4 under a Stokes laser power (PStokes) of 120 mW. With a laser excitation volume of about 2 × 10–16 L, 24.7 M corresponds to 3 × 109 C–O bonds in the laser focus. With a laser waist area of about 2 × 10–9 cm2, the relative spontaneous Raman signal (ΔIp/I)spon, Raman for C–O in pure methanol would be (0.9 × 10–30 cm2) × (3 × 109)/(2 × 10–9 cm2) ≈ 1.35 × 10–12. We can then calculate the stimulated Raman EFSRS = (rSRS/rspon, Raman) to be (ΔIp/Ipump)SRS/(ΔIp/Ipump)spon, Raman ≈ (1.7 × 10–4)/(1.35 × 10–12) ≈ 1.3 × 108, comparable to that estimated before by both SRS microscopy and FSRS spectroscopy.24,27 This stimulated Raman amplification factor is physically meaningful as it describes the inherent rate acceleration of vibrational activation as a result of quantum stimulation from the Stokes beam and it is independent of the targeted vibrational modes. Under the microscopy configuration, the SRS cross section of C–O (σSRS(C–O)) thus becomes (0.9 × 10–30 cm2) × (1.3 × 108) = 1.2 × 10–22 cm2.
Figure 6.
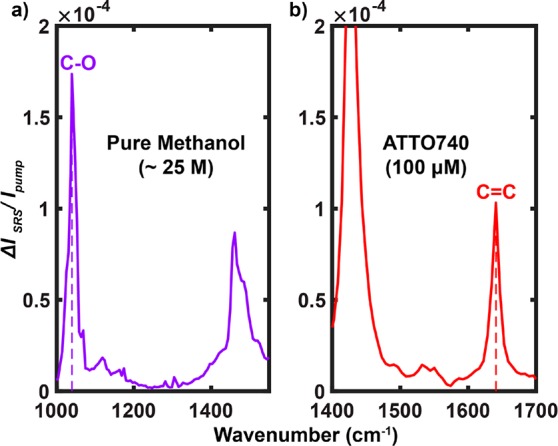
SRS spectrum of pure methanol (a) and EPR-SRS spectrum of 100 μM ATTO740 in DMSO (b) acquired with the same laser parameters. On-sample laser powers: Ppump = 24 mW and PStokes = 120 mW.
Under the same SRS laser excitation powers, the EPR-SRS signal of conjugated C=C vibration in ATTO740 (100 μM) is measured to be (ΔIp/I)SRS = 1.1 × 10–4 (Figure 6b). After scaling the concentrations, these two measurements (Figure 6a,b) yield EFEPR = [(ΔIp/I)SRS, ATTO740/(ΔIp/Ipump)SRS, C–O ] × [cC–O, methanol/cATTO740] of (1.1 × 10–4)/(1.7 × 10–4) × (24.7 M/100 μM) ≈ 1.6 × 105 when comparing ATTO740 to the C–O mode in methanol. This EFEPR value is close to that reported before of 106 when near resonance.12,63 It is worth mentioning again that, although this number is still 1 order of magnitude away from EFs under rigorous resonance conditions,12 such an EPR region is highly beneficial for attenuating the electronic backgrounds and achieving high vibrational selectivity, as evidenced by the EPR-SRS images (Figure 3). The overall EPR-SRS cross section of ATTO740 hence reaches (1.2 × 10–22 cm2) × (1.6 × 105) = 1.9 × 10–17 cm2, only an order of magnitude away from typical σabs, accounting for the superb EPR-SRS sensitivity down to 30–50 molecules. It is also worth mentioning that the overall EF for EPR-SRS (EFEPR-SRS = EFEPR × EFSRS) compared to the nonresonance spontaneous Raman thus reaches about (1.6 × 105) × (1.3 × 108) = 2.1 × 1013, a value very close to the highest achievable overall EF in SERRS. With further optimization of dyes and lasers (e.g., shorter pulse width or lower repetition rate) to gap the remaining 1 order of magnitude of sensitivity, EPR-SRS could serve as a complement or might even an alternative to fluorescence microscopy.
One of the distinctive applications for EPR-SRS microscopy is supermultiplex optical imaging to untangle the intrinsically complex biological systems, a highly sought-after technique in the incoming era of systems biology and big-data science and technology. Because the vibrational line width (∼10 cm–1) is about 100 times narrower compared to fluorescence microscopy (∼1500 cm–1 due to fast electronic dephasing), EPR-SRS in principle would offer 100 times more resolvable colors. Toward this goal, Wei et al. then devised a novel vibrational palette, in which each Raman-active dye bears a conjugated triple bond (i.e., nitrile or alkyne) and presents a single narrow EPR-SRS peak in the desired cell-silent Raman spectral window. This series of new dyes is termed MARS (MAnhanttan Raman Scattering) dyes.56 Using the newly developed MARS dyes in the near-infrared (∼650–800 nm) and merging with the orthogonal fluorophores in the visible range (∼400–650 nm), eight-color imaging on the same set of neuronal cell samples is demonstrated (Figure 7). This number could be further increased with future engineering of dye molecules to provide higher signals as well as better-resolved Raman peaks.64 Moreover, the supermultiplex capability of the EPR-SRS method could go beyond imaging to generating broad impacts in other fields of photonics, such as flow cytometry and data security.
Figure 7.
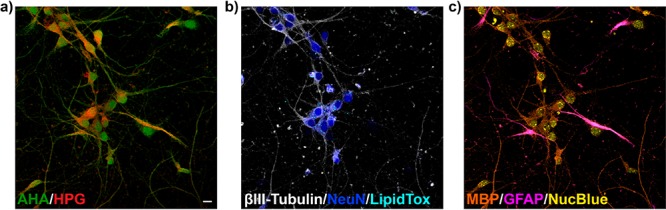
Representative eight-color tandem EPR-SRS and fluorescence imaging on the same set of neuronal cells. EPR-SRS targets: HPG (a, red, l-homopropargylglycine, for proteins synthesized in the pulse period for 12 h, labeled with MARS2228); AHA (a, green, l-azidohomoalanine, proteins synthesized in the chase period next for 10 h, labeled with Alexa 647); βIII-tubulin (b, gray, neurons, labeled with MARS2200); myelin basic protein (c, MBP, orange, oligodendrocytes, labeled with MARS2176); and glial fibrillary acidic protein (c, GFAP, magenta, astrocytes and neural stem cells, labeled with MARS2147). Fluorescence: NeuN (b, blue, neurons, labeled with Alexa568); LipidTox (b, cyan, lipid droplets); Nucblue (c, yellow, total DNA). Scale bars, 10 μm.
In retrospect, many biological discoveries were driven by technical innovations that explored less-charted spectroscopic principles, with assistance from novel matching reporters. The previously less-explored EPR-SRS imaging exhibits the desired combination of sub-μM high sensitivity and narrow chemical selectivity, merging the best of two worlds of electronic and vibrational microspectroscopy. We believe that EPR-SRS microscopy together with the newly developed MARS palette could become a valuable systems-method helping to elucidate complex biochemical and biophysical processes. Therefore, we hope this Perspective contributes to a deeper and more quantitative understanding of the fundamental physical principles underlying this new technique and help push its further development.
Acknowledgments
We are grateful for discussions with Lixue Shi, Zhixing Chen, Louis Brus, and Sunney Xie. W.M. acknowledges support from NIH Director’s New Innovator Award (1DP2EB016573) and R01 (EB020892) and the Camille and Henry Dreyfus Foundation.
Biographies
Lu Wei obtained her Ph.D. degree from Columbia University working with Prof. Wei Min, focusing on developing novel nonlinear optical spectroscopy and microscopy. Dr. Wei began her postdoctoral work at Columbia University in 2015 on developing supermultiplex microscopy for biomedical applications by electronic preresonance stimulated Raman scattering imaging of novel probes.
Wei Min graduated from Peking University with a bachelor’s degree in chemistry in 2003. He received his Ph.D. from Harvard University in 2008 studying single-molecule biophysics with Prof. Sunney Xie. After continuing his postdoctoral work in Xie’s group, Dr. Min joined the faculty of the Department of Chemistry at Columbia University in 2010 and was promoted to Full Professor there in 2017. He is also affiliated with the Kavli Institute for Brain Science and NeuroTechnology Center at Columbia University. Dr. Min’s current research interests focus on developing novel optical spectroscopy and microscopy technology to address biomedical problems. In particular, his group has contributed to the development of stimulated Raman scattering (SRS) microscopy and its broad application in biomedical imaging including bio-orthogonal chemical imaging and supermultiplex vibrational imaging.
The authors declare no competing financial interest.
References
- Lichtman J. W.; Conchello J.-A. Fluorescence Microscopy. Nat. Methods 2005, 2, 910–919. 10.1038/nmeth817. [DOI] [PubMed] [Google Scholar]
- Yuste R., Ed. Imaging: A Laboratory Manual; Cold Spring Harbor Press, New York, 2010. [Google Scholar]
- Zhang J.; Campbell R. E.; Ting A. Y.; Tsien R. Y. Creating New Fluorescent Probes for Cell Biology. Nat. Rev. Mol. Cell Biol. 2002, 3, 906–918. 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- Lavis L. D. Chemistry Is Dead. Long Live Chemistry!. Biochemistry 2017, 56, 5165–5170. 10.1021/acs.biochem.7b00529. [DOI] [PubMed] [Google Scholar]
- Suhalim J. L.; Boik J. C.; Tromberg B. J.; Potma E. O. The Need for Speed. J. Biophotonics 2012, 5, 387–395. 10.1002/jbio.201200002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneipp K.; Wang Y.; Kneipp H.; Perelman L. T.; Itzkan I.; Dasari R. R.; Feld M. S. Single Molecule Detection Using Surface-Enhanced Raman Scattering (SERS). Phys. Rev. Lett. 1997, 78, 1667–1670. 10.1103/PhysRevLett.78.1667. [DOI] [Google Scholar]
- Nie S.; Emory S. R. Probing Single Molecules and Single Nanoparticles by Surface-Enhanced Raman Scattering. Science 1997, 275, 1102–1106. 10.1126/science.275.5303.1102. [DOI] [PubMed] [Google Scholar]
- Zrimsek A. B.; Chiang N.; Mattei M.; Zaleski S.; McAnally M. O.; Chapman C. T.; Henry A.-I.; Schatz G. C.; Van Duyne R. P. Single-Molecule Chemistry with Surface- and Tip-Enhanced Raman Spectroscopy. Chem. Rev. 2017, 117, 7583–7613. 10.1021/acs.chemrev.6b00552. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Yeo B. S.; Schmid T.; Zenobi R. Single Molecule Tip-Enhanced Raman Spectroscopy with Silver Tips. J. Phys. Chem. C 2007, 111, 1733–1738. 10.1021/jp064740r. [DOI] [Google Scholar]
- Sonntag M. D.; Klingsporn J. M.; Garibay L. K.; Roberts J. M.; Dieringer J. A.; Seideman T.; Scheidt K. A.; Jensen L.; Schatz G. C.; Van Duyne R. P. Single-Molecule Tip-Enhanced Raman Spectroscopy. J. Phys. Chem. C 2012, 116, 478–483. 10.1021/jp209982h. [DOI] [Google Scholar]
- Dieringer J. A.; Wustholz K. L.; Masiello D. J.; Camden J. P.; Kleinman S. L.; Schatz G. C.; Van Duyne R. P. Surface-Enhanced Raman Excitation Spectroscopy of A Single Rhodamine 6G Molecule. J. Am. Chem. Soc. 2009, 131, 849–854. 10.1021/ja8080154. [DOI] [PubMed] [Google Scholar]
- Shim S.; Stuart C. M.; Mathies R. A. Resonance Raman Cross-Sections and Vibronic Analysis of Rhodamine 6G from Broadband Stimulated Raman Spectroscopy. ChemPhysChem 2008, 9, 697–699. 10.1002/cphc.200700856. [DOI] [PubMed] [Google Scholar]
- Le Ru E. C.; Etchegoin P. G. Single-Molecule Surface-Enhanced Raman Spectroscopy. Annu. Rev. Phys. Chem. 2012, 63, 65–87. 10.1146/annurev-physchem-032511-143757. [DOI] [PubMed] [Google Scholar]
- Asher S. A. UV Resonance Raman Studies of Molecular Structure and Dynamics: Applications in Physical and Biophysical Chemistry. Annu. Rev. Phys. Chem. 1988, 39, 537–588. 10.1146/annurev.pc.39.100188.002541. [DOI] [PubMed] [Google Scholar]
- Maier W.; Dorr F. Some Results of Resonance Raman Spectroscopy. Appl. Spectrosc. 1960, 14, 1–3. 10.1366/000370260774614670. [DOI] [Google Scholar]
- Johnson B. B.; Peticolas W. L. The Resonant Raman Effect. Annu. Rev. Phys. Chem. 1976, 27, 465–491. 10.1146/annurev.pc.27.100176.002341. [DOI] [Google Scholar]
- Schellenberg P.; Johnson E.; Esposito A. P.; Reid P. J.; Parson W. W. Resonance Raman Scattering by The Green Fluorescent Protein and An Analogue of Its Chromophore. J. Phys. Chem. B 2001, 105, 5316–5322. 10.1021/jp0046243. [DOI] [Google Scholar]
- Spiro T. G.; Strekas T. C. Resonance Raman Spectra of Hemoglobin and Cytochrome C: Inverse Polarization and Vibronic Scattering. Proc. Proc. Natl. Acad. Sci. U. S. A. 1972, 69, 2622–2626. 10.1073/pnas.69.9.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov E. V.; Ariese F.; Gooijer C. Achievements in Resonance Raman Spectroscopy Review of A Technique With a Distinct Analytical Chemistry Potential. Anal. Chim. Acta 2008, 606, 119–134. 10.1016/j.aca.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Kumamoto Y.; Taguchi A.; Smith N. I.; Kawata S. Deep Ultraviolet Resonant Raman Imaging of A Cell. J. Biomed. Opt. 2012, 17, 0760011. 10.1117/1.JBO.17.7.076001. [DOI] [PubMed] [Google Scholar]
- Okada M.; Smith N. I.; Palonpon A. F.; Endo H.; Kawata S.; Sodeoka M.; Fujita K. Label-free Raman Observation of Cytochrome C Dynamics During Apoptosis. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 28–32. 10.1073/pnas.1107524108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Heo J.; Lim C. K.; Pliss A.; Kachynski A. V.; Kuzmin A. N.; Kim S.; Prasad P. N. Organelle Specific Imaging in Live Cells and Immuno-Labeling Using Resonance Raman Probe. Biomaterials 2015, 53, 25–31. 10.1016/j.biomaterials.2015.02.056. [DOI] [PubMed] [Google Scholar]
- Kuzmin A. N.; Pliss A.; Lim C.-K.; Heo J.; Kim S.; Rzhevskii A.; Gu B.; Yong K. T.; Wen S.; Prasad P. N. Resonance Raman Probes for Organelle-Specific Labeling in Live Cells. Sci. Rep. 2016, 6, 28483. 10.1038/srep28483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCamant D. W.; Kukura P.; Mathies R. A. Femtosecond Broadband Stimulated Raman: A New Approach for High-Performance Vibrational Spectroscopy. Appl. Spectrosc. 2003, 57, 1317–1323. 10.1366/000370203322554455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min W.; Lu S.; Holtom G. R.; Xie X. S. Triple-Resonance Coherent Anti-Stokes Raman Scattering Microspectroscopy. ChemPhysChem 2009, 10, 344–347. 10.1002/cphc.200800502. [DOI] [PubMed] [Google Scholar]
- Saar B. G.; Freudiger C. W.; Reichman J.; Stanley C. M.; Holtom G. R.; Xie X. S. Video-Rate Molecular Imaging In Vivo with Stimulated Raman Scattering. Science 2010, 330, 1368–1370. 10.1126/science.1197236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min W.; Freudiger C. W.; Lu S.; Xie X. S. Coherent Nonlinear Optical Imaging: Beyond Fluorescence Microscopy. Annu. Rev. Phys. Chem. 2011, 62, 507–530. 10.1146/annurev.physchem.012809.103512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J.-X.; Xie X. S. Vibrational Spectroscopic Imaging of Living Systems: An Emerging Platform for Biology and Medicine. Science 2015, 350, aaa8870. 10.1126/science.aaa8870. [DOI] [PubMed] [Google Scholar]
- Ozeki Y.; Umemura W.; Otsuka Y.; Satoh S.; Hashimoto H.; Sumimura K.; Nishizawa N.; Fukui K.; Itoh K. High-Speed Molecular Spectral Imaging of Tissue with Stimulated Raman Scattering. Nat. Photonics 2012, 6, 845–851. 10.1038/nphoton.2012.263. [DOI] [Google Scholar]
- Liao C.-S.; Slipchenko M. N.; Wang P.; Li J.; Lee S.-Y.; Oglesbee R. A.; Cheng J.-X. Microsecond Scale Vibrational Spectroscopic Imaging by Multiplex Stimulated Raman Scattering Microscopy. Light: Sci. Appl. 2015, 4, e265. 10.1038/lsa.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp C. H. Jr.; Lee Y. J.; Heddleston J. M.; Hartshorn C. M.; Walker A. R. H.; Rich J. N.; Lathia J. D.; Cicerone M. T. High-Speed Coherent Raman Fingerprint Imaging of Biological tissues. Nat. Photonics 2014, 8, 627–634. 10.1038/nphoton.2014.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R.; Xu Y.; Zhang L.; Ma S.; Wang X.; Ye D.; Ji M. Dual-Phase Stimulated Raman Scattering Microscopy for Real-Time Two-Color Imaging. Optica 2017, 4, 44–47. 10.1364/OPTICA.4.000044. [DOI] [Google Scholar]
- Freudiger C. W.; Min W.; Saar B. G.; Lu S.; Holtom G. R.; He C.; Tsai J. C.; Kang J. X.; Xie X. S. Label-Free Biomedical Imaging with High Sensitivity by Stimulated Raman Scattering Microscopy. Science 2008, 322, 1857–1861. 10.1126/science.1165758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. C.; Min W.; Freudiger C. W.; Ruvkun G.; Xie X. S. RNAi Screening for Fat Regulatory Genes with SRS Microscopy. Nat. Methods 2011, 8, 135–138. 10.1038/nmeth.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min W. Label-free Optical Imaging of Nonfluorescent Molecules by Stimulated Radiation. Curr. Opin. Chem. Biol. 2011, 15, 831–837. 10.1016/j.cbpa.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Fu D.; Zhou J.; Zhu W. S.; Manley P. W.; Wang Y. K.; Hood T.; Wylie A.; Xie X. S. Imaging The Intracellular Distribution of Tyrosine Kinase Inhibitors in Living Cells with Quantitative Hyperspectral Stimulated Raman Scattering. Nat. Chem. 2014, 6, 614–622. 10.1038/nchem.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji M.; Lewis S.; Camelo-Piragua S.; Ramkissoon S. H.; Snuderl M.; Venneti S.; Fisher-Hubbard A.; Garrard M.; Fu D.; Wang A. C.; et al. Detection of Human Brain Tumor Infiltration with Quantitative Stimulated Raman Scattering Microscopy. Sci. Transl. Med. 2015, 7, 309ra163. 10.1126/scitranslmed.aab0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F.; Basu S.; Igras V.; Hoang M. P.; Ji M.; Fu D.; Holtom G. R.; Neel V. A.; Freudiger C. W.; Fisher D. E.; et al. Label-Free Imaging In Vivo with Stimulated Raman Scattering Microscopy. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 11624–11629. 10.1073/pnas.1515121112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F.; Yang W.; Mordes D. A.; Wang J. Y.; Salameh J. S.; Mok J.; Chew J.; Sharma A.; Leno-Duran E.; Suzuki-Uematsu S.; et al. Monitoring Peripheral Nerve Degeneration in ALS by Label-Free Stimulated Raman Scattering Imaging. Nat. Commun. 2016, 7, 13283. 10.1038/ncomms13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Condello S.; Thomes-Pepin J.; Ma X.; Xia Y.; Hurley T. D.; Matei D.; Cheng J.-X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314.e5. 10.1016/j.stem.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakisaka Y.; Suzuki Y.; Iwata O.; Nakashima A.; Ito T.; Hirose M.; Domon R.; Sugawara M.; Tsumura N.; Watarai H.; et al. Probing the Metabolic Heterogeneity of Live Euglena Gracilis with Stimulated Raman Scattering Microscopy. Nat. Microbiol. 2016, 1, 16124. 10.1038/nmicrobiol.2016.124. [DOI] [PubMed] [Google Scholar]
- Orringer D. A.; Pandian B.; Niknafs Y. S.; Hollon T. C.; Boyle J.; Lewis S.; Garrard M.; Hervey-Jumper S. L.; Garton H. J. L.; Maher C. O.; et al. Rapid Intraoperative Histology of Unprocessed Surgical Specimens via Fibre-Laser-Based Stimulated Raman Scattering Microscopy. Nat. Biomed. Eng. 2017, 1, 0027. 10.1038/s41551-016-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince R. C.; Frontiera R. R.; Potma E. O. Stimulated Raman Scattering: From Bulk to Nano. Chem. Rev. 2017, 117, 5070–5094. 10.1021/acs.chemrev.6b00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.; Hu F.; Shen Y.; Chen Z.; Yu Y.; Lin C.-C.; Wang M. C.; Min W. Live-Cell Imaging of Alkyne-Tagged Small Biomolecules by Stimulated Raman Scattering. Nat. Methods 2014, 11, 410–412. 10.1038/nmeth.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S.; Chen T.; Zhu Y.; Li A.; Huang Y.; Chen X. Live-cell Stimulated Raman Scattering Imaging of Alkyne-Tagged Biomolecules. Angew. Chem., Int. Ed. 2014, 53, 5827–5831. 10.1002/anie.201400328. [DOI] [PubMed] [Google Scholar]
- Wei L.; Hu F.; Chen Z.; Shen Y.; Zhang L.; Min W. Live-Cell Bioorthogonal Chemical Imaging: Stimulated Raman Scattering Microscopy of Vibrational Probes. Acc. Chem. Res. 2016, 49, 1494–1502. 10.1021/acs.accounts.6b00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z.; Shen Y.; Hu F.; Min W. Applications of Vibrational Tags in Biological Imaging by Raman Microscopy. Analyst 2017, 142, 4018–4029. 10.1039/C7AN01001J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F.; Chen Z.; Zhang L.; Shen Y.; Wei L.; Min W. Vibrational Imaging of Glucose Uptake Activity in Live Cells and Tissues by Stimulated Raman Scattering. Angew. Chem., Int. Ed. 2015, 54, 9821–9825. 10.1002/anie.201502543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F.; Lamprecht M. R.; Wei L.; Morrison B.; Min W. Bioorthogonal Chemical Imaging of Metabolic Activities in Live Mammalian Hippocampal Tissues with Stimulated Raman Scattering. Sci. Rep. 2016, 6, 39660. 10.1038/srep39660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.; Shen Y.; Xu F.; Hu F.; Harrington J. K.; Targoff K. L.; Min W. Imaging Complex Protein Metabolism in Live Organisms by Stimulated Raman Scattering Microscopy with Isotope Labeling. ACS Chem. Biol. 2015, 10, 901–908. 10.1021/cb500787b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. J.; Zhang W.; Zhang D.; Yang Y.; Liu B.; Barker E. L.; Buhman K. K.; Slipchenko L. V.; Dai M.; Cheng J.-X. Assessing Cholesterol Storage in Live Cells and C. Elegans by Stimulated Raman Scattering Imaging of Phenyl-Diyne Cholesterol. Sci. Rep. 2015, 5, 7930. 10.1038/srep07930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso-García A.; Pfisterer S. G.; Riezman H.; Ikonen E.; Potma E. O. D38-Cholesterol as a Raman Active Probe for Imaging Intracellular Cholesterol Storage. J. Biomed. Opt. 2016, 21, 061003. 10.1117/1.JBO.21.6.061003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Cheng J.-X. Direct Visualization of De Novo Lipogenesis in Single Living Cells. Sci. Rep. 2014, 4, 6807. 10.1038/srep06807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu D.; Yu Y.; Folick A.; Currie E.; Farese R. V. Jr.; Tsai T.-H.; Xie X. S.; Wang M. C. In Vivo Metabolic Fingerprinting of Neutral Lipids with Hyperspectral Stimulated Raman Scattering Microscopy. J. Am. Chem. Soc. 2014, 136, 8820–8828. 10.1021/ja504199s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando J.; Kinoshita M.; Cui J.; Yamakoshi H.; Dodo K.; Fujita K.; Murata M.; Sodeoka M. Sphingomyelin Distribution in Lipid Rafts of Artificial Monolayer Membranes Visualized by Raman Microscopy. Proc. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 4558–4563. 10.1073/pnas.1418088112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.; Chen Z.; Shi L.; Long R.; Anzalone A. V.; Zhang L.; Hu F.; Yuste R.; Cornish V. W.; Min W. Super-Multiplex Vibrational Imaging. Nature 2017, 544, 465–470. 10.1038/nature22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal A.; Erramilli S.; Ziegler L. D. Origin of Dispersive Line Shapes in Plasmonically Enhanced Femtosecond Stimulated Raman Spectra. J. Phys. Chem. C 2016, 120, 20998–21006. 10.1021/acs.jpcc.6b03303. [DOI] [Google Scholar]
- Takayanagi M.; Hamaguchi H.; Tasumi M. Probe-Frequency Dependence of The Resonant Inverse Raman Band Shape. J. Chem. Phys. 1988, 89, 3945–3950. 10.1063/1.454875. [DOI] [Google Scholar]
- Lukinavičius G.; Reymond L.; Umezawa K.; Sallin O.; D’Este E.; Göttfert F.; Ta H.; Hell S. W.; Urano Y.; Johnsson K. Fluorogenic Probes for Multicolor Imaging in Living Cells. J. Am. Chem. Soc. 2016, 138, 9365–9368. 10.1021/jacs.6b04782. [DOI] [PubMed] [Google Scholar]
- Albrecht A. C.; Hutley M. C. On The Dependence of Vibrational Raman Intensity on The Wavelength of Incident Light. J. Chem. Phys. 1971, 55, 4438–4443. 10.1063/1.1676771. [DOI] [Google Scholar]
- Stancik A. L.; Brauns E. B. A Simple Asymmetric Lineshape for Fitting Infrared Absorption Spectra. Vib. Spectrosc. 2008, 47, 66–69. 10.1016/j.vibspec.2008.02.009. [DOI] [Google Scholar]
- Wertheim G. K.; Butler M. A.; West K. W.; Buchanan D. N. E. Determination of The Gaussian and Lorentzian Content of Experimental Line Shapes. Rev. Sci. Instrum. 1974, 45, 1369. 10.1063/1.1686503. [DOI] [Google Scholar]
- Silva W. R.; Keller E. L.; Frontiera R. R. Determination of Resonance Raman Cross-Sections for Use in Biological SERS Sensing with Femtosecond Stimulated Raman Spectroscopy. Anal. Chem. 2014, 86, 7782–7787. 10.1021/ac501701h. [DOI] [PubMed] [Google Scholar]
- Hu F.; Zeng C.; Long R.; Miao Y.; Wei L.; Xu Q.; Min W. Supermultiplexed Optical Imaging and Barcoding with Engineered Polyynes. Nat. Methods 2018, 15, 194–200. 10.1038/nmeth.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]