

Abstract
Introduction:
Aldosterone decreases at high altitude (HA) but the effect of hypoxia on angiotensin-converting enzyme (ACE), a key step in the renin-angiotensin-aldosterone system, is unclear.
Methods:
We investigated the effects of exercise and acute normobaric hypoxia (NH, ~11.0% FiO2) on nine participants and six controls undertaking the same exercise at sea level (SL). NH exposure lasted 5 hours with 90 minutes of submaximal treadmill walking. Blood samples for aldosterone, ACE and cortisol were taken throughout exposure and at rest during a trek to HA (5140 m) in eight separate participants.
Results:
There was no difference in cortisol or aldosterone between groups pre-exercise. Aldosterone rose with exercise to a greater extent at SL than in NH (post-exercise: 700 ± 325 versus 335 ± 238 pmol/L, mean ± SD, p = 0.044). Conversely, cortisol rose to a greater extent in NH (post-exercise: 734 ± 165 versus 344 ± 159 nmol/L, mean ± SD, p = 0.001). There were no differences in ACE activity. During the trek to HA, resting aldosterone and cortisol reduced with no change in ACE.
Conclusions:
Acute NH subdues the exercise-associated rise in aldosteroe but stimulates cortisol, whereas prolonged exposure at HA reduces both resting aldosterone and cortisol. As ACE activity was unchanged in both environments, this is not the mechanism underlying the fall in aldosterone.
Keywords: Normobaric hypoxia, high altitude, ACE, cortisol, aldosterone, aldosterone synthase
Introduction
Angiotensin-converting enzyme (ACE) is a key component of the renin-angiotensin-aldosterone system (RAAS) that converts angiotensin I to angiotensin II, which has several actions, including the stimulation of aldosterone release from the adrenal gland via angiotensin type 1 receptors. Changes in the RAAS and fluid balance are central to early acclimatization at high altitude (HA).1,2
It is consistently reported in the literature that there is a reduction in resting aldosterone at HA.3–8 In addition, a relatively greater drop in aldosterone than renin occurs with hypoxia, reflecting a subdued aldosterone response to renin.9–11 This effect facilitates a beneficial natriuresis and diuresis, since a less subdued aldosterone at HA has been associated with acute mountain sickness (AMS) at HA.3
The literature regarding the effects of hypoxia and HA on ACE are less consistent however. Previous reports that acute hypoxia reduced ACE activity in dogs,12 the guinea pig foetal–placental unit13 and in human cultured endothelial cells14 were subsequently retracted in 198515–17 when the authors could not reproduce their findings.
In rats, altered pulmonary and systemic pressor responses to angiotensin I and II under hypoxic conditions have suggested an acute reduction in ACE activity.18 Similarly, chronic hypoxia in isolated rat lungs has shown reduced transpulmonary angiotensin I conversion19 that suggests either a reduction in ACE activity or alterations in contact time with ACE due to changes in pulmonary haemodynamics. An organ-specific effect was reported following the exposure of rats to normobaric hypoxia (NH) (FiO2 of 10%) over 14–28 days with a reduction in ACE activity in lung homogenates, an increase in renal homogenates, and no change in the homogenates from brain and testis.20 More recent work has shown that chronic hypoxia upregulates ACE mRNA expression in the rat carotid body21 and that, over 7–21 days, ACE activity significantly increases.22
In humans, it was initially reported that ACE activity was reduced while breathing a hypoxic gas mixture,23 but this article was retracted in 1987.24 Other workers have shown that reducing inspired oxygen in humans to produce saturations of 80–90% over 1–2 hours reduces aldosterone but not ACE,4 whereas in six subjects exercising for 1 hour at 65% V̇O2max, a rise in aldosterone occurred under both normoxic and hypoxic (equivalent to 3000 m) conditions with no change in ACE.25
More recently, it has been shown that ACE activity did not change in humans on exposure to 3800 m26 over 2 weeks. However, in 2016, it was reported that following rapid ascent (by car) to 3450 m no significant rise in ACE was seen, while ACE levels rose significantly in those given placebo or dexamethasone (regardless of the presence or absence of AMS) in those treated with budesonide.27
In summary, while the literature has consistently demonstrated a reduction in resting aldosterone at HA, the data regarding ACE activity are inconsistent and confusing. Therefore, we measured ACE activity, cortisol (as another marker of adrenal function) and aldosterone during acute NH, in a sea level (SL) control group and during a trek to HA. We hypothesized that ACE activity would not change but that aldosterone would be suppressed.
Materials and methods
This work was undertaken with ethical approval from the Ministry of Defence Research Ethics Committee (protocol 586). Written informed consent was provided by each participant prior to their inclusion in the study.
Experimental design
There were two parts to this study. The first was a laboratory study incorporating two groups (NH and SL) to evaluate the acute effects of a single 5-hour exposure to a simulated normobaric altitude of 4800 m against a seal level control condition. The second was an evaluation of a separate sample of volunteers undertaking a trek to HA. Subjects for the NH and SL groups were originally balanced for age, group size (n = 9), gender (three females and six males) and baseline V̇O2max at a simulated altitude of 4800 m. Due to technical difficulties with blood sampling, three subjects were lost from the SL group (two females and one male), leaving six in the SL group compared with nine in the NH group. Subjects in the NH and SL groups underwent all sessions in an environmental chamber and were blinded to conditions in the chamber throughout all sessions. Subjects studied on the trek to HA were a sample of volunteers already involved in the expedition. All subjects included in both aspects of the study were non-smoking lowlanders who had not been to altitudes > 1500 m in the 3 months prior to baseline testing.
Procedure
Laboratory study
Testing included four separate visits to the laboratory over a period of 2 weeks. Visit one included consent and screening. Visit two involved a preliminary NH testing session that was used to balance the subjects for the NH and SL groups, as previously outlined. Visit three was a repeat of visit two but under sea level conditions. The final visit was the 5-hour NH exposure or sea level control trial.
Preliminary NH session (visit 2)
All 15 subjects completed 1-hour NH exposure at a simulated altitude of 4800 m (~11.0% FiO2/ 77.3 mmHg PiO2, at 18°C and 45% relative humidity) in an environmental chamber (TISS, Alton, UK and Sporting Edge, Sherfield on Loddon, UK). The hour consisted of 15 minutes rest followed by an incremental 12-minute submaximal walk test on a treadmill (Woodway, Cranlea, Birmingham, UK) carrying a 10 kg rucksack walking at 1.5, 2.5, 3.5 and 4.5 km.h-1 for 3 minutes. Following 10 minutes rest, subjects completed a V̇O2max test at a self-selected running speed starting at 1% gradient, which increased by 1% each minute until volitional exhaustion. Throughout the submaximal walk and V̇O2max test, expired air was measured using an online gas analyser (MetaLyzer® 3B, Cortex, Leipzig, Germany), which had been calibrated prior to each trial in accordance with the manufacturer’s instructions. Visit three, which was a minimum of 48 hours after visit two, was a repeat of the preliminary hypoxic testing session but under sea level conditions, with a small modification to the speed and gradient used throughout the submaximal walk test (Table 1). Relative exercise intensities for the acute normobaric or sea level trials were interpolated using V̇O2 data from the submaximal walk and the V̇O2max (greatest 30s mean) score.
Table 1.
Submaximal walk protocol in preliminary testing for visits 2 and 3.
| Time (minutes) | Visit 2: 4800 m |
Visit 3: sea level |
||
|---|---|---|---|---|
| Speed (km.h-1) | Gradient (%) | Speed (km.h-1) | Gradient (%) | |
| 0–3 | 1.5 | 10 | 3 | 10 |
| 3–6 | 2.5 | 10 | 4 | 10 |
| 6–9 | 3.5 | 10 | 4 | 15 |
| 9–12 | 4.5 | 10 | 5 | 15 |
Acute NH
Nine subjects underwent an NH exposure at a simulated altitude of 4800 m (~11.0% FiO2/77.3 mmHg PiO2, at 18°C and 45% relative humidity) in the environmental chamber a minimum of 72 hours after visit three. The exposure lasted 5 hours and consisted of 150 minutes of rest followed by 90 minutes of graded submaximal exercise walking on a treadmill at 10–15% incline carrying a 10 kg rucksack (10 mins at 40%, 60 mins at 50%, 10 mins at 60% and 10 mins at 70% of altitude-specific V̇O2max), followed by a final 60-minute rest period. Venous blood samples (serum separator tube for ACE, potassium, osmolality, cortisol and ethylenediaminetetraacetic acid (EDTA) tube for plasma aldosterone) were taken from a cannulated forearm vein. Samples were taken at rest at baseline, prior to hypoxic exposure (T1), after 105 minutes hypoxic exposure (T2), immediately following exercise (T3) and at the end of the 5-hour exposure (T4) (Figure 1). Samples were centrifuged, separated and stored at −80°C until subsequent analysis for serum ACE activity, osmolality, potassium, cortisol and plasma aldosterone.
Figure 1.

Five-hour schematic of acute normobaric hypoxic exposure and sea level trial.
Six control subjects underwent the same protocol under sea level (normobaric normoxia, SL) conditions.
Terrestrial HA
A separate sample of eight subjects were subsequently studied at baseline and on an expedition to Nepal. These participants were part of a trekking team on the British Services Dhaulagiri Medical Research Expedition 2016. The trek started from Beni (899 m) and followed the Dhaulagiri circuit through to Marpha (2660 m). This took 14 days and reached a maximum altitude of 5360 m over the French Pass. Resting blood samples were taken by venepuncture in the morning the day after arrival at Dhaulagiri lower base camp (4600 m) on day 10 (average rate of ascent 376 m/day) and Hidden valley (5140 m) on day 12 (average rate of ascent 353 m/day). Samples were centrifuged, separated and stored in liquid nitrogen until return to the UK, where they were kept at −80°C until subsequent analysis.
Hormone assay
ACE (U/L) was assayed according to the manufacturer’s instructions using a photometric enzymatic assay (Bühlmann Laboratories, Schönenbuch, Switzerland). This assay gives a direct and quantitative determination of serum ACE activity with an intra-assay precision of 2.7% and an inter-assay precision of 8.1%. It uses the catalysis of hydrolysis of the synthetic substrate N-[3-(2-furyl)acryloyl]-L-phenylalanylglycine by ACE and the resulting decrease in absorbance at 340 nm to measure serum ACE activity.
Cortisol was assayed using the Roche Elecsys Cortisol I assay on serum samples. The test is a competitive chemiluminescence immunoassay (CLIA) and is fully automated, run on the Roche modular E unit (Roche Diagnostics, Burgess Hill, UK). The analytical range of the assay is 0.5–1750 nmol/L. Low levels are reported as ‘< 20 nmol/L’. The lower detection limit of 0.5 nmol/L represents the lowest measurable analyte level that can be distinguished from 0. There is an intra-assay coefficient of variation (CV) of 9.3–11.7%.
Aldosterone (pmol/L) was assayed using the IDS iSYS assay on plasma samples collected using EDTA vacutainers. The assay is a CLIA and is fully automated, run on the IDS iSYS immunoassay analyser (IDS PLC, Boldon, UK). It has an analytical range of 103–3656 pmol/L, with low levels being reported as ‘< 103 pmol/L’. The lower limit of quantification represents the lowest concentration of analyte that can be measured with acceptable precision. There is an intra-assay CV of 5.8–12.1%.
Aldosterone synthase was measured using the Cloud-Clone enzyme-linked immunosorbent assay (ELISA) for human aldosterone synthase (Cloud-Clone Corporation, Katy, TX, USA). Analysis was performed according to the manufacturer’s instructions. Briefly, the ELISA uses a microplate pre-coated with antibody against aldosterone synthase. Standards and samples are added to microplate wells along with a second, biotin-conjugated antibody against aldosterone synthase and avidin-conjugated horseradish peroxidase. After incubation and plate-washing, a tetramethylbenzidine (TMB) substrate is added and the enzyme/substrate reaction is measured spectrophotometrically. Aldosterone synthase concentrations were calculated in ng/mL. The manufacturer-quoted limit of detection is 0.054 ng/mL with an inter-assay CV of < 12% and intra-assay CV < 10%.
Osmolality was measured by micro-osmometer (Advanced Model 3320, Advanced Instruments, Norwood, MA, USA) using a suppression of freezing point method. Serum potassium was assayed using an automated Olympus AU 2700 system (Olympus Diagnostics, Hamburg, Germany). Assays were performed in the clinical biochemistry departments of the Royal Victoria Infirmary, Newcastle and Wansbeck General Hospital, Northumberland.
Statistical analysis
Statistical analysis was performed using SPSS 22. Data were tested for normality using a Shapiro–Wilk test and were shown to approximate to a normal distribution. A threshold of p < 0.05 was used as the level with which to decide statistical significance. Separate repeated measures analyses of variance were used for the SL versus NH groups (time (4) × group (2)) and the terrestrial HA (THA) group (time (3)). An independent Student’s t-test was used to compare the area under the curve for the four time points for aldosterone for the SL and NH groups. Paired and independent Student’s t-tests were used to make comparisons within and between groups. A Pearson product-moment correlation coefficient was used to assess if there was any association between ACE and oxygen saturation.
Results
Baseline characteristics
A total of 23 subjects took part in the study (nine in the NH chamber, six SL controls and eight on the trek to THA). There were no differences in baseline characteristics (p > 0.05) between groups (Table 2).
Table 2.
Baseline characteristics for the sea level group, normobaric hypoxic group and the terrestrial high altitude team.
| Height (cm) | Mass (kg) | Age (years) | HR (bpm) | Sp02 (%) | SBP (mmHg) | DBP (mmHg) | Male (n) | Female (n) |
V̇O2max at 4800 m (ml.kg.min-1) | |
|---|---|---|---|---|---|---|---|---|---|---|
| SL | 178 ± 10 | 79 ± 10 | 24 ± 2 | 64 ± 8 | 97 ± 1 | 128 ± 6 | 76 ± 6 | 5 | 1 | 35 |
| NH | 177 ± 10 | 76 ± 12 | 25 ± 4 | 66 ± 5 | 98 ± 1 | 130 ± 4 | 74 ± 10 | 6 | 3 | 36 |
| THA | 177 ± 4 | 76 ± 9 | 35 ± 9 | 59 ± 7 | 98 ± 1 | 130 ± 5 | 80 ± 5 | 7 | 1 | N/A |
All participants were non-smoking lowlanders and had not been to altitudes > 1500 m in the previous 3 months. No significant differences were detected between groups.
DBP: diastolic blood pressure; HR: heart rate; NH: normobaric hypoxia; SBP: systolic blood pressure; SL: sea level; THA: terrestrial high altitude.
Acute NH and SL controls
There was no significant difference in aldosterone at T1 or T2 between SL controls and NH (p = 0.600). Immediately post-exercise (T3), aldosterone levels were significantly greater in the SL than the NH group (700 ± 325 versus 335 ± 238 pmol/L, mean ± SD, p = 0.044). At the end of the 5-hour protocol, aldosterone remained greater in the SL controls than in the NH group (382 ± 196 versus 174 ± 105, pmol/L, mean ± SD, p = 0.049). Aldosterone increased following exercise in both the SL (p = 0.049) and NH (p = 0.010) groups (Figure 2), which was confirmed by the significant main effect of time on aldosterone (p = 0.000). Overall, using area under the curve across the 5-hour exposure, there was a significantly lower aldosterone response in the NH group compared to the SL group (p = 0.022, Figure 2).
Figure 2.
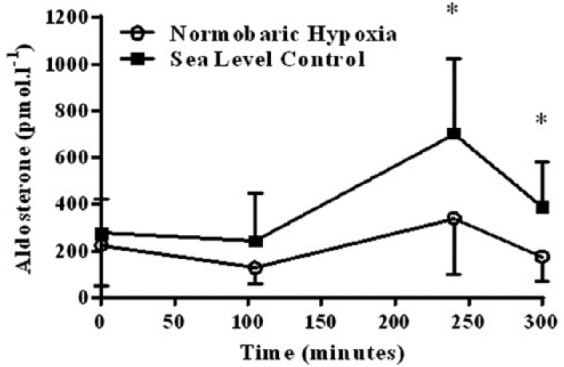
Serum aldosterone pre- and post-exercise in the normobaric hypoxia (n = 9) and sea leve (n = 6) groups (* = sea level significantly (p < 0.05) higher than the normobaric hypoxia group).
ACE activity did not change in response to exercise in the NH or at SL groups (Figure 3). There was no difference in ACE at any time point between NH and SL. There was no significant correlation between ACE and oxygen saturation across all three groups (SL, NH and THA) (r = −0.296, p = 0.171; r = −0.033, p = 0.85; and r = 0.171, p = 0.434, respectively). Data for aldosterone and ACE are shown in Table 3. Potassium, serum osmolality and aldosterone synthase values are also shown in Table 3. Results revealed no significant differences for the main effects of group or time for all three variables within laboratory-based testing (Table 3).
Figure 3.
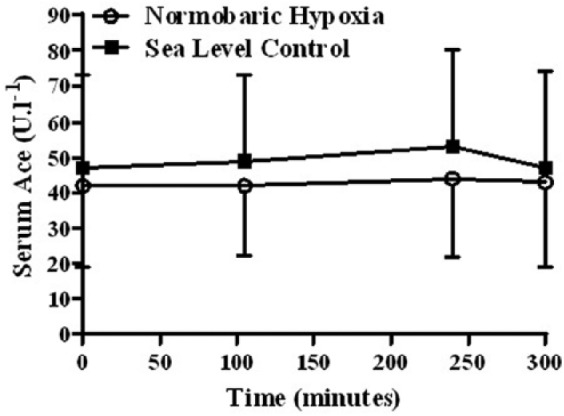
Angiotensin-converting enzyme pre- and post-exercise in the normobaric hypoxia (n = 9) and sea level (n = 6) groups.
Table 3.
Aldosterone, angiotensin-converting enzyme, aldosterone synthase, cortisol, potassium and serum osmolality (mean ± SD) pre- and post-exercise in the normobaric hypoxia and sea level groups.
| T1 | T2 | T3 | T4 | ||
|---|---|---|---|---|---|
| Aldosterone (pmol/L) | NH | 224 ± 176 | 127 ± 67 | 335 ± 238*,** | 174 ± 105* |
| SL | 271 ± 149 | 241 ± 204 | 700 ± 325** | 382 ± 196 | |
| ACE (U/L) | NH | 42 ± 23 | 42 ± 20 | 44 ± 22 | 43 ± 24 |
| SL | 47 ± 26 | 49 ± 24 | 53 ± 27 | 47 ± 27 | |
| Aldosterone synthase (ng/mL) | NH | 2.88 ± 0.59 | 3.04 ± 0.52 | 3.05 ± 0.63 | 2.99 ± 0.60 |
| SL | 2.39 ± 0.41 | 2.52 ± 0.43 | 2.63 ± 0.37 | 2.58 ± 0.41 | |
| Cortisol (nmol/L) | NH | 467 ± 117 | 477 ± 166 | 734 ± 165*,** | 632 ± 207* |
| SL | 562 ± 98 | 397 ± 80 | 344 ± 159 | 363 ± 117 | |
| K (mmol/L) | NH | 4.1 ± 0.2 | 4.3 ± 0.3 | 4.0 ± 0.4 | 3.5 ± 1.7 |
| SL | 4.1 ± 0.1 | 4.6 ± 0.4 | 4.5 ± 0.7 | 5.0 ± 0.5 | |
| Serum osmolality (mOsm/kg) | NH | 288 ± 4.6 | 289 ± 3.5 | 290 ± 3.5 | 289 ± 3.7 |
| SL | 288 ± 3.8 | 289 ± 4.0 | 290 ± 3.5 | 289 ± 3.7 |
= NH significantly different to SL, ** = significantly higher at T3 (post-exercise) compared with T2.
ACE: angiotensin-conerting enzyme; NH: normobaric hypoxia; SL: sea level; T: time point.
Assessing the cortisol results (Table 3) revealed a significant main effect of group (p = 0.012), but the main effect of time over the 5-hour exposure was not signifcant (p = 0.168). However, there was a signficant interaction between time and group (p = 0.000), indicating that both groups responded differently throughout the 5-hour exposure. There was no signifcant difference in cortisol concentration at T1 or T2 between SL controls and NH (p = 0.125 and p = 0.236) (Figure 4). As a result of the significant increase in cortisol in NH at T3 (p = 0.001), immediately post-exercise cortisol was significantly greater in NH than in SL (734 ± 165 versus 344 ± 159 nmol/L, mean ± SD, p = 0.001) (Figure 4). Although within the 1-hour recovery post-exercise to the end of exposure (T4) there was a significant decrease in cortisol in the NH group (p = 0.009), at the end of the 5-hour protocol cortisol remained greater in the NH group than in SL controls (632 ± 207 versus 363 ± 117, nmol/L, mean ± SD, p = 0.007).
Figure 4.
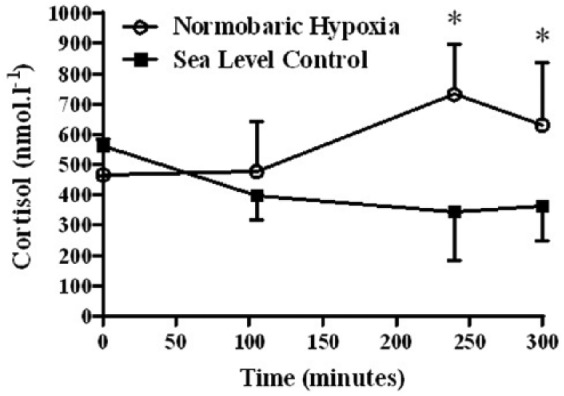
Cortisol pre- and post-exercise in the normobaric hypoxia (n = 9) and sea level (n = 6) groups (* = normobaric hypoxia significantly (p < 0.05) higher than sea level).
THA
On the expedition, baseline aldosterone at SL was 190 ± 66 pmol/L and dropped below the limit of detection at 4600 m and 5140 m. There was a significant main effect of time on aldosterone (p = 0.007). ACE activity (U/L, mean ± SD) was 43 ± 20 at SL and did not change at 4600 m (37 ± 19) or 5140 m (41 ± 17). The cortisol response throughout the expedition showed a significant main effect of time (p = 0.011). However, pairwise comparisons revealed that cortisol concentrations at 4600 m were significantly reduced in comparisons to baseline (p = 0.021), but that an increase in altitude to 5140 m resulted in no further significant change in cortisol concentration (p = 1.00) (Figure 5). Although the expedition aldosterone synthase response showed a significant main effect of time (p = 0.001), pairwise comparisons revealed that although there was a significant increase between baseline and 4600 m, aldosterone synthase had returned to baseline at 5140 m. There were no significant differences over time for either potassium or serum osmolality at THA (Table 4).
Figure 5.
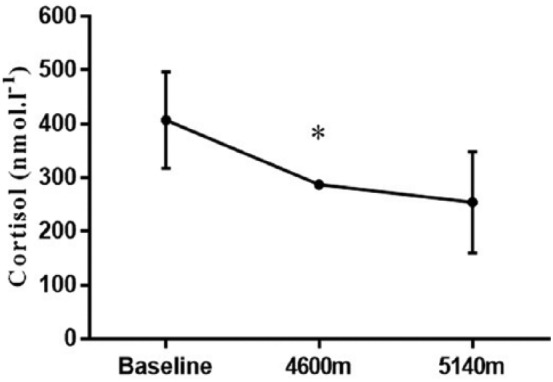
Resting cortisol (nmol/L) concentration at sea level baseline (n = 8) and on expedition, collected the morning of the day after arrival at Dhaulagiri lower base camp (4600 m) on day 10 (n = 8) and at Hidden valley (5140 m) on day 12 (n = 8) (* = significantly (p < 0.05) different to previous altitude).
Table 4.
Potassium, serum osmolality and aldosterone synthase (mean ± SD) for the terrestrial high altitude group.
| Baseline | 4600 m | 5140 m | |
|---|---|---|---|
| Potassium (mmol/L) | 4.16 ± 0.24 | 4.16 ± 0.28 | 3.96 ± 0.44 |
| Serum osmolality (mOsm/kg) | 289 ± 4 | 357 ± 153 | 477 ± 185 |
| Aldosterone synthase (ng/mL) | 3.21 ± 0.37 | 3.55 ± 0.30* | 3.20 ± 0.28* |
Collected at sea level baseline (n = 8) and on expedition, collected the morning of the day after arrival at Dhaulagiri lower base camp (4600 m) on day 10 (n = 8) and at Hidden valley (5140 m) on day 12 (n = 8). * = significantly different to previous altitude.
Discussion
Our data demonstrate that while exercise at SL and under NH conditions both induce a rise in aldosterone, this effect is suppressed under NH conditions compared to SL. This effect appears to occur independently of any differences in ACE activity. Trekking to HA is associated with a reduced resting aldosterone, also independent of any change in ACE activity. Taken together, our data suggest a dynamic response in aldosterone as a result of hypoxic exposure (normobaric, in the chamber, or hypobaric at THA) that occurs through a non-ACE-dependent mechanism. The subdued aldosterone under hypoxic conditions is probably a beneficial factor that prevents salt and water retention that may otherwise occur with exertion at HA, since a greater increase in exercise-associated aldosterone at HA has been associated with subsequent development of AMS.3
An increase in aldosterone with exercise is well established and is consistent with the fact that an exercise-associated rise in angiotensin II has been reported, at least in normoxic conditions.28 The subdued response of aldosterone to exercise under hypoxic conditions3 and the reduction in aldosterone at HA4,5,7,8 are also consistently reported. However, we have clearly demonstrated in both acute NH and at THA that this effect is not dependent on a reduction in ACE activity, and that it therefore must occur through a non-ACE-dependent mechanism.
The fact that we demonstrated no change in osmolality in any of the environments makes this an unlikely cause of the reduction in aldosterone. Although there was no statistically significant change in potassium at SL or under NH, there does appear to be a possible numerical change and so we are more circumspect about this, though we note that this occurs at the end of the exposure and would seem unlikely to account for the differences seen in aldosterone immediately post-exercise. Other mechanisms must therefore be considered. In rats, severe hypoxia specifically inhibits (in vitro) adrenal aldosterone synthesis,29,11 thought to be through a direct effect on aldosterone synthase mRNA (CYP11B2).11 In bovine adrenocortical cells, in vitro hypoxia directly inhibits angiotensin II and adrenocorticotropic hormone-stimulated aldosterone synthesis,10 and aldosterone has been found to reduce in proportion to hypoxia due to inhibition of the aldosterone synthase enzyme.30,31 However, we found no change in aldosterone synthase with exercise either at SL or in the hypoxic chamber. At THA, we found a statistically significant 11% rise in aldosterone synthase at 4600 m compared to SL, but this had returned to baseline by 5140 m. While we cannot explain the rise at 4600 m, there was certainly no evidence to suggest inhibition of aldosterone synthase at THA, and therefore this does not appear to be the mechanism behind a fall in aldosterone at THA in humans.
The findings in relation to cortisol may help suggest where in adrenal steroidogenesis the effect of hypoxia might lie. As cortisol rises acutely following exercise in acute NH but not at SL, whereas the aldosterone response to exercise appears subdued in acute NH versus normoxia, we suspect that any potential reduction in adrenal steroidogenesis must be downstream of CYP11A1 and CYP21A2, and therefore had suspected an effect on CYP11B2 (aldosterone synthase). With the more chronic exposure to hypoxia of THA, both resting morning aldosterone and cortisol are reduced. We have previously shown that with pharmacological stimulation of adrenal steroidogenesis using synacthen at HA, the cortisol response remains intact compared to SL but that the aldosterone response is subdued.32 Again, we had suspected that this reflects reduced activity of CYP11B2, but our subsequent analysis does not support this.
It is important to consider why some early work12–14 suggesting that hypoxia reduced ACE activity was subsequently retracted15–17. In one retraction letter, the authors stated that their original hypothesis that ACE activity was closely regulated by the partial pressure of oxygen was incorrect as they had failed to reproduce their findings in further experiments, but no other detail was given. In another retraction letter, they offer an honest apology that they had no scientific explanation as to why they could not reproduce their earlier findings.15 However, in acknowledgement of the advances in biochemistry, it must be noted that those authors had huge challenges in measuring ACE activity. In one experiment, they calculated ACE activity in the guinea pig foetal–placental unit.13 In order to do this, they infused their animal model with a solution containing bradykinin (as a substrate for ACE), with ACE activity calculated as the percent of substrate cleared by a single passage through the vascular bed while assuming equal perfusion inflow and effluent rates. We acknowledge the challenges they faced, applaud their honest retraction and are grateful for our current automated assay.
In humans, the initial report that ACE activity was reduced by hypoxia23 was subsequently retracted.24 In the original report, the authors had assayed ACE activity in serum using a fluorimetric method33 not that far removed from the modern assay. In their retraction, they state that they had repeated several studies of the effect of hypoxia on ACE, including one study in the field at 4100 m and five laboratory studies using hypoxic gas mixtures.23 They analysed these and other samples in several different laboratories using several different assays, and concluded that hypoxia had no effect on ACE activity. On balance therefore, and consistent with our findings, we perceive that the body of evidence favours no significant effect of hypoxia on ACE activity.
It seems reasonable to state that the reduction in aldosterone at THA is not due to a reduction in ACE activity, but also the hypothesis that this would be due to a reduction in aldosterone synthase activity is not the case. Alternative mechanisms must therefore be considered. Inhibition of adrenal aldosterone release due to elevated levels of adrenomedullin seen at HA34 or downregulation of adrenal angiotensin II receptors (observed in the lung after prolonged hypoxia35) are possible alternative explanations. Another possible factor could be an upregulation of ACE2 in hypoxia that would increase deactivation of angiotensin II (to angiotensin I–VII) and thus contribute to a reduction in aldosterone under hypoxic conditions. There is evidence for increased ACE2 expression in cultured human hepatocytes36 and human pulmonary arterial smooth muscle cells37 exposed to hypoxia. The finding that ACE2 expression is increased in response to myocardial ischaemia38 and hypoxic stress at the cellular level39 is supportive of the concept that ACE2 may be upregulated in response to tissue hypoxia, and could potentially explain our findings.
Limitations to our study include that we were not able to analyse angiotensin II or ACE2 due to the technical challenges involved in field research. A further limitation is that although the original groups for SL and NH were matched with nine participants each, the fact that we had blood sampling problems with three participants, all unfortunately from the SL group, meant we had fewer participants to compare than originally intended. This could have affected our statistical power, particularly in relation to any potential numerical changes in potassium during the SL and NH exposure.
Conclusion
In summary, we report that the aldosterone response to exercise is attenuated in NH compared to SL, and that resting aldosterone and cortisol are reduced during a trek to HA. Our data show no change in ACE activity with acute or more chronic hypoxic exposure, and suggest that the changes seen in aldosterone occur through a non-ACE-dependent mechanism. This mechanism does not appear to be through a reduction in aldosterone synthase activity and alternative explanations remain to be explored, such as a downregulation of adrenal angiotensin II receptors or an upregulation of ACE2.
Acknowledgments
We would like to thank all participants. We would also like to specifically thank the reviewers of the original manuscript; their diligent input allowed substantial improvement.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research reported in this study was supported by the Surgeon General and Joint Medical Command, UK, the Drummond Foundation, Leeds Beckett University, the Royal Navy Royal Marines Charity and the Mount Everest Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Defence Medical Services.
ORCID iD: Mark Cooke  https://orcid.org/0000-0003-0286-2731
https://orcid.org/0000-0003-0286-2731
References
- 1. Goldfarb-Rumyantzev AS, Alper SL. Short-term responses of the kidney to high altitude in mountain climbers. Nephrol Dial Transplant 2014; 29: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Woods DR, Montgomery HE. Angiotensin-converting enzyme and genetics at high altitude. High Alt Med Biol 2001; 2: 201–210. [DOI] [PubMed] [Google Scholar]
- 3. Bartsch P, Maggiorini M, Schobersberger W, et al. Enhanced exercise-induced rise of aldosterone and vasopressin preceding mountain sickness. J Appl Physiol 1991; 71: 136–143. [DOI] [PubMed] [Google Scholar]
- 4. Colice GL, Ramirez G. Effect of hypoxemia on the renin-angiotensin-aldosterone system in humans. J Appl Physiol 1985; 58: 724–730. [DOI] [PubMed] [Google Scholar]
- 5. De Angelis C, Ferri C, Urbani L, et al. Effect of acute exposure to hypoxia on electrolytes and water metabolism regulatory hormones. Aviat Space Environ Med 1996; 67: 746–750. [PubMed] [Google Scholar]
- 6. Milledge JS, Catley DM, Ward MP, et al. Renin-aldosterone and angiotensin-converting enzyme during prolonged altitude exposure. J Appl Physiol Respir Environ Exerc Physiol 1983; 55: 699–702. [DOI] [PubMed] [Google Scholar]
- 7. Ramirez G, Hammond M, Agosti SJ, et al. Effects of hypoxaemia at sea level and high altitude on sodium excretion and hormonal levels. Aviat Space Environ Med 1992; 63: 891–898. [PubMed] [Google Scholar]
- 8. Zaccaria M, Rocco S, Noventa D, et al. Sodium regulating hormones at high altitude: Basal and post-exercise levels. J Clin Endocrin Metab 1998; 83: 570–574. [DOI] [PubMed] [Google Scholar]
- 9. Milledge JS, Catley DM, Williams ES, et al. Effect of prolonged exercise at altitude on the renin-aldosterone system. J Appl Physiol Respir Environ Exerc Physiol 1983; 55: 413–418. [DOI] [PubMed] [Google Scholar]
- 10. Raff H, Ball DL, Goodfriend TL. Low oxygen selectively inhibits aldosterone secretion from bovine adrenocortical cells in vitro. Am J Phyisol 1989; 256: E640–E644. [DOI] [PubMed] [Google Scholar]
- 11. Raff H, Jankowski BM, Engeland WC, et al. Hypoxia in vivo inhibits aldosterone synthesis and aldosterone synthase mRNA in rats. J Appl Physiol 1996; 81: 604–610. [DOI] [PubMed] [Google Scholar]
- 12. Stalcup SA, Lipset JS, Legant PM, et al. Inhibition of converting enzyme activity by acute hypoxia in dogs. J Appl Physiol Respir Environ Exercise Physiol 1979; 46: 227–234. Retracted in J Appl Physiol 1985; 59: 1333. [DOI] [PubMed] [Google Scholar]
- 13. Davidson D, Stalcup SA, Mellins RB. Angiotensin-converting enzyme activity and its modulation by oxygen tension in the guinea pig fetal-placental unit. Circ Res 1981; 48: 286–291. [DOI] [PubMed] [Google Scholar]
- 14. Stalcup SA, Lipset JS, Woan JM, et al. Inhibition of angiotensin converting enzyme activity in cultured endothelial cells by hypoxia. J Clin Invest 1979; 63: 966–976. Retracted in J Clin Invest 1985; 75: 1745. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15. Stalcup SA, Lipset JS, Legant PM, et al. Retraction of two articles: Bradykinin. J Appl Physiol 1985; 59: 1333. [DOI] [PubMed] [Google Scholar]
- 16. Stalcup SA Lipset JS Woan JM et al.. Retraction. Inhibition of angiotensin converting enzyme activity in cultured endothelial cells by hypoxia. J Clin Invest 1985; 75: 1745. [PMC free article] [PubMed] [Google Scholar]
- 17. Stalcup SA, Lipset JS, Odya CE, et al. Retraction: Angiotensin-converting enzyme and oxygen tension. Circ Res 1985; 57: 646. [DOI] [PubMed] [Google Scholar]
- 18. Jin H, Oparil S, Ann HS, et al. Hypoxia-induced inhibition of converting enzyme activity: Role in vascular regulation. J Appl Physiol 1987; 63: 1012–1018. [DOI] [PubMed] [Google Scholar]
- 19. Jackson RM, Narkates AJ, Oparil S. Impaired pulmonary conversion of angiotensin I to angiotensin II in rats exposed to chronic hypoxia. J Appl Physiol 1986; 60: 1121–1127. [DOI] [PubMed] [Google Scholar]
- 20. Oparil S, Narkates AJ, Jackson RM, et al. Altered angiotensin-converting enzyme in lung and extrapulmonary tissues of hypoxia-adapted rats. J Appl Physiol 1988; 65: 218–227. [DOI] [PubMed] [Google Scholar]
- 21. Lam SY, Leung PS. Chronic hypoxia activates a local angiotensin-generating system in rat carotid body. Mol Cell Endocrinol 2003; 203: 147–153. [DOI] [PubMed] [Google Scholar]
- 22. Lam SY, Fung ML, Leung PS. Regulation of the angiotensin-converting enzyme activity by a time-course hypoxia in the carotid body. J Appl Physiol 2004; 96: 809–813. [DOI] [PubMed] [Google Scholar]
- 23. Milledge JS, Catley DM. Angiotensin converting enzyme response to hypoxia in man: Its role in altitude acclimatization. Clin Sci (Lond) 1984; 67: 453–456. Retracted in Clin Sci (Lond) 1987; 72: 149. [DOI] [PubMed] [Google Scholar]
- 24. Milledge JS, Catley DM. Angiotensin converting enzyme activity and hypoxia. Clin Sci (Lond) 1987; 72: 149. [DOI] [PubMed] [Google Scholar]
- 25. Bouissou P, Guezennec CY, Galen FX, et al. Dissociated response of aldosterone from plasma renin activity during prolonged exercise under hypoxia. Horm Metab Res 1988; 20: 517–521. [DOI] [PubMed] [Google Scholar]
- 26. Mason NP, Petersen M, Mélot C, et al. Changes in plasma bradykinin concentration and citric acid cough threshold at high altitude. Wilderness Environ Med 2009; 20: 353–358. [DOI] [PubMed] [Google Scholar]
- 27. Li HJ, Zheng CR, Chen GZ, et al. Budesonide, but not dexamethasone, blunted the response of aldosterone to renin elevation by suppressing angiotensin converting enzyme upon high-altitude exposure. J Renin Angiotensin Aldosterone Syst 2016; 17: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Woods D, Sanders J, Jones A, et al. The serum angiotensin-converting enzyme and angiotensin II response to altered posture and acute exercise, and the influence of ACE genotype. Eur J Appl Physiol 2004; 91: 342–348. [DOI] [PubMed] [Google Scholar]
- 29. Bruder ED, Nagler AK, Raff H. Oxygen-dependence of ACTH-stimulated aldosterone and corticosterone synthesis in the rat adrenal cortex: Developmental aspects. J Endocrinol 2002; 172: 595–604. [DOI] [PubMed] [Google Scholar]
- 30. Brickner RC, Jankowski BM, Raff H, et al. The conversion of corticosterone to aldosterone is the site of oxygen sensitivity of the bovine adrenal zona glomerulosa. Endocrinology 1992; 130: 88–92. [DOI] [PubMed] [Google Scholar]
- 31. Raff H, Kohandarvish S. The effect of oxygen on aldosterone release from bovine adrenocortical cells in vitro: PO2 vs. steroidogenesis. Endocrinology 1990; 127: 682–687. [DOI] [PubMed] [Google Scholar]
- 32. Mackey J, Mellor A, Watchorn J, et al. The adrenocortical response to synthetic ACTH following a trek to high altitude. Horm Metab Res 2016; 48: 658–663. [DOI] [PubMed] [Google Scholar]
- 33. Friedland J, Silverstein E. A sensitive fluorimetric assay for serum angiotensin-converting enzyme. Am J Clin Pathol 1976; 66: 416–424. [DOI] [PubMed] [Google Scholar]
- 34. Taylor MM, Samson WK. Adrenomedullin and the integrative physiology of fluid and electrolyte balance. Microsc Res Tech 2002; 57: 105–109. [DOI] [PubMed] [Google Scholar]
- 35. Chassagne C, Eddahibi S, Adamy C, et al. Modulation of angiotensin II receptor expression during development and regression of hypoxic pulmonary hypertension. Am J Respir Cell Mol Biol 2000; 22: 323–332. [DOI] [PubMed] [Google Scholar]
- 36. Paizis G, Tikellis C, Cooper ME, et al. Chronic liver injury in rats and humans up regulates the novel enzyme angiotensin converting enzyme 2. Gut 2005; 54: 1790–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang R, Wu Y, Zhao M, et al. Role of HIF-1alpha in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2009; 297: L631–L640. [DOI] [PubMed] [Google Scholar]
- 38. Burrell LM, Risvanis J, Kubota E, et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J 2005; 26: 369–375. [DOI] [PubMed] [Google Scholar]
- 39. Clarke NE, Belyaev ND, Lambert DW, et al. Epigenetic regulation of angiotensin-converting enzyme 2 (ACE2) by SIRT1 under conditions of cell energy stress. Clin Sci (Lond) 2014; 126: 507–516. [DOI] [PubMed] [Google Scholar]