

Abstract
The glutathione redox system undergoes precise and dynamic changes during embryonic development, protecting against and mitigating oxidative insults. The antioxidant response is coordinately largely by the transcription factor Nuclear factor erythroid-2 (Nrf2), an endogenous sensor for cellular oxidative stress. We have previously demonstrated that impaired Nrf family signaling disrupts the glutathione redox system in the zebrafish embryo, and that impaired Nrf2 function increases embryonic sensitivity to environmental toxicants. Here, we investigated the persistent environmental toxicant and reported pro-oxidant perfluorooctanesulfonic acid (PFOS), and its impact on the embryonic glutathione-mediated redox environment. We further examined whether impaired Nrf2a function exacerbates PFOS-induced oxidative stress and embryotoxicity in the zebrafish, and the potential for Nrf2-PPAR crosstalk in the embryonic adaptive response. Wild-type and nrf2afh318−/− mutant embryos were exposed daily to 0 (0.01% v/v DMSO), 16, 32, or 64 µM PFOS beginning at 3 hours post fertilization (hpf). Embryonic glutathione and cysteine redox environments were examined at 72 hpf. Gross embryonic toxicity, antioxidant gene expression, and apoptosis were examined at 96 hpf. Mortality, pericardial edema, and yolk sac utilization were increased in wild-type embryos exposed to PFOS. Embryonic glutathione and cysteine redox couples and gene expression of Nrf2 pathway targets were modulated by both exposure and genotype. Apoptosis was increased in PFOS-exposed wild-type embryos, though not in nrf2a mutants. In silico examination of putative transcription factor binding site suggested potential crosstalk between Nrf2 and PPAR signaling, since expression of PPARs and gene targets was modulated by both PFOS exposure and Nrf2a genotype. Overall, this work demonstrates that nrf2a modulates the embryonic response to PFOS, and that PPAR signaling may play a role in the embryonic adaptive response to PFOS.
Keywords: glutathione, Nfe2l2, embryo, redox, perfluorinated, PFAS
1. Introduction
Early organogenesis is a tightly controlled process, taking cues from coordinated signaling pathways. If disrupted, this dysregulation can give rise to spontaneous abortion, birth defects, or predispose individuals to diseases that arise during childhood and adulthood. Oxidative stress, the change from a tightly controlled intracellular redox environment to predominantly oxidizing conditions and/or a loss of redox signaling and control, and classically concurrent with the generation of reactive oxygen species (ROS), is one of the most widely studied mechanisms of disrupted cellular signaling [1, 2]. Oxidative stress can range from mild to severe, manifesting as altered signal transduction to apoptosis and necrosis, respectively.
The embryo has innate antioxidant systems to prevent and protect against the damaging consequences of oxidation, as well as actively and dynamically coordinating embryonic cellular fate decisions which are vital for growth and tissue patterning. The endogenous reducing agent glutathione (GSH) is a tripeptide synthesized from glycine, cysteine, and glutamate by the enzyme glutathione synthetase, and under oxidizing cellular conditions, the active thiol group of the GSH molecule can preferentially reduce other important cellular proteins, protecting vital cellular functions. Following oxidation, glutathione dimerizes into glutathione disulfide (GSSG), which can later be recycled back into GSH via the enzyme glutathione reductase. The inducible and basal expression of genes encoding for these enzymes is coordinated largely by the Nuclear factor erythroid-2 (Nrf2) family of transcription factors [3, 4]. Nrf2 (Nfe2l2) is a constitutively expressed cytosolic protein, which is normally rendered inactive, ubiquitinated, and degraded by the ubiquitin-proteasome pathway [5]. During oxidative stress, Nrf2 translocates to the nucleus to serve as a transcription factor to increase expression of antioxidant enzymes [6].
Loss of Nrf2 function or mutation is not embryolethal and development proceeds normally, although these embryos may be less resilient and unable to cope with environmental stressors (as reviewed in [7]). This is due in part to the partially redundant function of other Nrf proteins (Nrf1, Nrf3) which can also activate the transcriptional antioxidant response. In the zebrafish, an evolutionary whole genome duplication adds further complexity to the pathway, as there are two Nrf2 paralogs—Nrf2a and Nrf2b. Though there is some redundancy in function, Nrf2a and Nrf2b have partitioned roles in the antioxidant response [8]. The zebrafish Nrf2a is functionally similar to the mammalian Nrf2, inducing the transcriptional activation of antioxidant enzymes such as those responsible for the synthesis of GSH.
Perfluorooctanesulfonic acid (PFOS) is a ubiquitous persistent organic pollutant, previously found in non-stick products until removal from production in the early 2000s in the United States. However, PFOS is still manufactured and used in consumer products in many other countries. It has been found in greater than 98% of human urinary samples, and has a half-life in the body of approximately 4.8 years [9-11]. PFOS has been repeatedly detected in fetal tissues and cord blood, demonstrating the ability to cross the placenta and expose the developing fetus [12, 13].
Exposures to PFOS during embryonic development have been associated with a number of congenital defects in mammalian and zebrafish models, including pericardial edema, craniofacial malformations, spinal malformations, enlargement of the liver, structural anomalies of the pancreas and gut, and decreased birth weight [14-18]. Other studies have found that PFOS exposures during embryogenesis are associated with decreased growth in the zebrafish [16, 19], and that PFOS increases embryonic ROS concentrations and induces oxidative stress in the developing embryo of several vertebrate species including zebrafish [16, 20]. While PFOS does not share the characteristics typical of a Nrf2 activator, it has been shown to induce Nrf2-ARE binding and subsequent upregulation of the antioxidant response in vitro [21] and to serve a protective function in PFOS-induced oxidative stress in early life stage zebrafish [22]. However, the relationship between Nrf2 and embryonic glutathione-mediated redox response following PFOS exposures requires characterization.
The goal of this study was to characterize the Nrf2-mediated antioxidant response to PFOS. While several other pathways, including the Peroxisome Proliferator-Activated Receptor (PPAR) signaling pathway, have been found to interact with Nrf2 signaling in response to toxicant exposures both in vitro and in vivo [23, 24], evidence suggests that Nrf2 functions as a first responder to toxicologically driven oxidative stress. We hypothesized that Nrf2a deficiency in zebrafish embryos would enhance PFOS-induced teratogenesis and redox dysregulation, and that these embryos would have weakened compensatory responses to these insults. The data indicate that that Nrf2a modulates the embryonic response to PFOS, and that PPAR signaling may play a role in the embryonic adaptive response to PFOS.
2. Materials & Methods
2.1 CHEMICALS
Heptadecafluorooctanesulfonic acid (PFOS) was obtained from Sigma-Aldrich (St. Louis, MO), and the solvent dimethyl sulfoxide (DMSO) was purchased from Fisher Scientific (Pittsburgh, PA). Stock solutions of 160, 320, and 640 mM for embryo exposures were prepared by dissolving PFOS into DMSO, and stored at room temperature in glass bottles inside of light-prohibitive, airtight containers until use. Acridine orange was purchased from Fisher Scientific, and stored at room temperature in a covered amber vial as a 10 mg/ml stock solution in 0.3X Danieau’s medium (17 mM NaCl, 2mM KCl, 0.12 mM MgSO4, 1.8 mM Ca(NO3)2, 1.5 mM HEPES; pH 7.6). All procedures were performed in strict accordance to prescribed safety precautions.
2.2 ANIMALS & HUSBANDRY
Homozygous Nrf2a wild-type and mutant (nrf2afh318−/−) fish crossed onto an AB strain background were obtained as homozygous populations from Dr. Mark Hahn at Woods Hole Oceanographic Institution, and genotypes were confirmed using PCR (as previously described in [25]). These fish were generated through the TILLING mutagenesis Project (R01HD076585) and originally obtained by Dr. Hahn as embryos from the Moens Laboratory (Fred Hutchinson Cancer Research Center, Seattle, WA, USA). The nrf2afh318−/− genotype is considered a loss-of-function mutation, since the point mutation produces a mutant amino acid sequence in the DNA binding domain of the Nrf2a protein, impairing its transcriptional activity. This mutation was originally characterized in [26], and further examined by our laboratory [25, 27].
Adult fish populations were maintained in an automated Aquaneering zebrafish system (San Diego, CA, USA). All animal procedures were performed in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and with approval from the University of Massachusetts Amherst Institutional Animal Care and Use Committee (Animal Welfare Assurance Number A3551-01). Fish were housed at 28.5°C and following at 14 h light:10 h dark cycle daily. Adult fish were fed the recommended amount of GEMMA Micro 300 (Skretting; Westbrook, ME) once daily in the morning. Breeding populations were housed at an appropriate density with a 2:1 female-to-male ratio.
Embryos for experiments were collected with 1 hour post-fertilization (hpf) from homozygous genotyped tanks, washed thoroughly, and confirmed for fertilization prior to experimental proceedings. Collected embryos were stored in polystyrene 100 mm petri dishes containing roughly 50 ml of 0.3x Danieau’s media.
2.3 EXPOSURES
Embryonic exposures were performed following the same procedures as previously published [18]. Briefly, pools of 10–15 mid-blastula stage embryos of each genotype were separately collected into fresh polystyrene petri dishes containing 20 ml of 0.3x Danieau’s media. Polystyrene plates were used because they have lower matrix retention/adherence rates for PFOS than other materials such as glass [28]. Stock solutions of PFOS or DMSO were added to the dishes at 0.01% v/v, resulting in exposures to 0 (DMSO control), 16, 32, or 64 µM PFOS. These concentrations were used in previously published zebrafish studies and are below the reported LC50 values; the concentrations in our study are also similar to those that have been shown to activate Nrf2 in cell culture [15, 17, 18, 21]. All exposure media was refreshed daily. At 24 hpf, all embryos were manually dechorionated using watchmakers’ forceps and chorion debris was removed from dishes. All experiments were repeated 3–4 times.
2.4 MICROSCOPY
Embryos were imaged at 96 hpf for embryonic morphology using a FBS10 Fluorescence Biological Microscope (Kramer Scientific; Amesbury, MA, USA). Embryos screened for morphological deformities were imaged at 5× magnification using transmitted light microscopy (2× eyepiece, 2.5× objective). Embryos were identically staged for microscopy in 3% methylcellulose, following extensive washing and anesthesia using 2% v/v MS-222 solution (prepared as 4 mg/ml tricaine powder in water, pH buffered, and stored at −20°C until use). Embryonic viability and swim bladder inflation percentages were recorded. For morphometry, all quantitative measures (fish length, pericardial area, yolk sac area) were performed on captured images using ZEN lite software (Carl Zeiss AG; Oberkochen, Germany).
2.5 QUANTIFICATION OF GLUTATHIONE AND CYSTEINE REDOX COUPLES
Embryonic redox state was characterized biochemically using reverse phase High Performance Liquid Chromatography (HPLC) with fluorescence detection, as previously described in [29]. Briefly, reduced (GSH) and oxidized (GSSG) glutathione, and reduced (Cys) and oxidized (CySS) cysteine concentrations were quantified. Each sample contained 25 pooled embryos. Samples were derivatized using methods previously described in [30, 31], and previously performed in [29, 32].
Soluble thiols were derivatized and precipitated, and injected into a Waters 2695 separations module equipped with a Supelcosil LC-NH2 column and fitted to a Waters 2475 fluorescence detector. Chromatographs were analyzed using Empower software (Waters; Milford, MA, USA). Excitation and emission wavelengths were 335 and 518 nm, respectively, and flow rate was 1.0 ml/min. A gradient method for two mobile phases was used: A) 80% methanol and 20% water, and B) 62.5% methanol, 12.5% glacial acetic acid, and 214 mg/ml sodium acetate trihydrate in water. Total glutathione (tGSH) and total cysteine (tCys) were calculated as [reduced + 2*oxidized]. The Nernst equation was used (pH 7.4) to calculate redox potential: Eh = E0 + (RT/nF) * log([GSSG]/[GSH]2), where E0 = −264 mV and (RT/nF) = 30. These calculations were normalized to cellular volume and sample protein concentration determined by BCA assay [33].
2.6 RNA ISOLATION & REVERSE TRANSCRIPTION
RNA was isolated from 96 hpf embryos in order to assess the effects of PFOS treatment and Nrf2 signaling on the expression of Nrf2 transcriptional target genes, other Nrf family genes, and expression of PPARs and their targets. At 96 hpf, embryos were collected into RNAlater (Fisher Scientific; Waltham, MA), snap frozen, and stored at −80°C until RNA isolation. To provide sufficient tissue, 5–7 embryos were pooled per sample, for a total of 4–5 samples per concentration/genotype combination. RNA was isolated using the GeneJET RNA Purification Kit (Fisher Scientific), following manufacturer instructions. RNA concentrations were quantified using a BioDrop µLITE spectrophotometer (BioDrop; Cambridge, UK), and 1 µg of RNA was reverse transcribed into cDNA using the iScript cDNA Synthesis Kit (Bio-Rad; Hercules, CA). cDNA was diluted to working stocks of 0.25 ng/µl, and was stored at −20°C until use.
2.7 QUANTITATIVE PCR
Quantitative PCR was performed using a CFX Connect Real-Time PCR Detection System (Bio-Rad). The total reaction mixture volume was 20 µl, containing 10 µl of 2X iQ SYBR Green Supermix (Bio-Rad), 5 pM of each primer, 5 µl of water, and 4 µl (1 ng) of cDNA. Sequences and optimization temperatures for the primers used in this study are provided in Supplemental Table 1. Data was analyzed using the CFX Manager software (Bio-Rad), and gene expression fold-changes were calculated using the ΔΔCT method [34]. Beta-actin (actb) and beta-2-macroglobulin (b2m) were used as housekeeping genes, and expression did not significantly change due to treatment or genotype. The arithmetic mean of the Cq value of both genes was used as the housekeeping index for use in the ΔΔCT model.
2.8 ACRIDINE ORANGE ASSAY
At 96 hpf, apoptosis was visualized using acridine orange. Embryos were thoroughly washed following PFOS exposures, and placed into 100 mm glass petri dishes containing 50 ml of 1 µg/ml acridine orange in 0.3x Danieau’s media. Several embryos remained unstained in a separate dish for use as controls for fluorescence. All dishes were placed in a light-prohibitive box for 30 minutes. After treatment, embryos were thoroughly washed 3 times. To qualitatively determine target regions or tissues of cell death, embryos were microscopically examined using a fluorescent GFP filter (excitation: 470 nm, emission: 525 nm). For quantitative measures, 3 embryos were pooled in a 1.5 ml polypropylene Eppendorf tube containing 160 µl of 50% ethanol in water and sonicated. Samples were transferred to black 96-well plates in triplicate wells, each containing 50 µl of homogenate. Unstained controls and blanks (50% ethanol) were also pipetted in triplicate for each plate. Samples were analyzed for GFP (excitation: 470 nm, emission: 525 nm) on a Cytation3 Cell Imaging Multi-Mode Reader (BioTek; Winooski, VT, USA).
2.9 IDENTIFICATION OF PUTATIVE TRANSCRIPTION FACTOR BINDING SITES
Promoters and gene regions of target genes were searched in silico for PPAR-responsive elements (PPREs). Zebrafish (GRCz10/danRer10 Assembly) target genes and promoters were examined using UCSC Genome Browser (http://genome.ucsc.edu/index.html), coupled with the JASPAR 2018 TFBS Track which utilizes the JASPAR 2018 CORE vertebrates collection database (http://jaspar.genereg.net/). Only putative binding sites with scores of 400 of higher (p-values<10−4) were considered.
2.10 STATISTICAL ANALYSES
Data is presented as the mean ± SEM. Chi-square tests of independence were used to examine differences in survival and swim bladder inflation. Two-way ANOVAs with Tukey post hoc tests were used to assess quantitative differences in trends between exposure, genotype, and their interaction using JMP Pro 13 (SAS Institute Inc., Cary, NC, USA). Nonparametric Kruskal-Wallis tests with Wilcoxon multiple comparisons were used to examine the effects of exposure for all endpoints. Nonparametric Mann-Whitney tests were used to compare wild-type and mutant embryos for each exposure concentration. A confidence level of 95% (α = 0.05) was used. For all experiments, 3–4 experimental replicates were performed.
3. Results
3.1 EMBRYONIC MORPHOMETRY
To assess embryotoxicity, we characterized embryonic mortality, fish length, pericardial area, yolk sac area, and swim bladder inflation in wild-type and Nrf2a mutant embryos exposed to PFOS. We have previously reported an absence of mortality and embryonic deformities in wild-type embryos resulting from these same exposures, though we had observed slightly lower rates of swim bladder inflation in PFOS-treated embryos [18]. Here, we confirmed that embryonic mortality was not significantly decreased by PFOS exposures in wild-type embryos (Fig. 1). Survival to 96 hpf was 95% for controls, and 91%, 91%, and 95% for embryos treated with 13, 32, and 64 µM PFOS, respectively. Control mutant embryos had reduced survival to 96 hpf (85%) compared to wild-type control embryos (p = 0.047). Though PFOS exposures reduced survival in Nrf2a mutant embryos by an additional 8–12%, these decreases were not statistically significant (p > 0.05). There was an inverted U-shape response in the wildtype fish with exposure to PFOS, but no dose-related changes in the mutants-rather the mutants were protected from the variations in length that were produced by PFOS in the wildtype embryos. Similarly, pericardial areas increased with increasing PFOS exposure in wild-type embryos (p<0.05 for 32 and 64 µM concentrations), though this effect was not observed in mutants. The only significant effect of PFOS on the mutant fish was in yolk sac area, where the mutants had a significantly smaller yolk sac than the mutant DMOS controls (p < 0.05), as well as the PFOS-exposed wild types. No significant changes were observed with swim bladder inflation (p = 0.07).
Figure 1. PFOS exposures disrupt embryogenesis, and Nrf2a modulates the response.
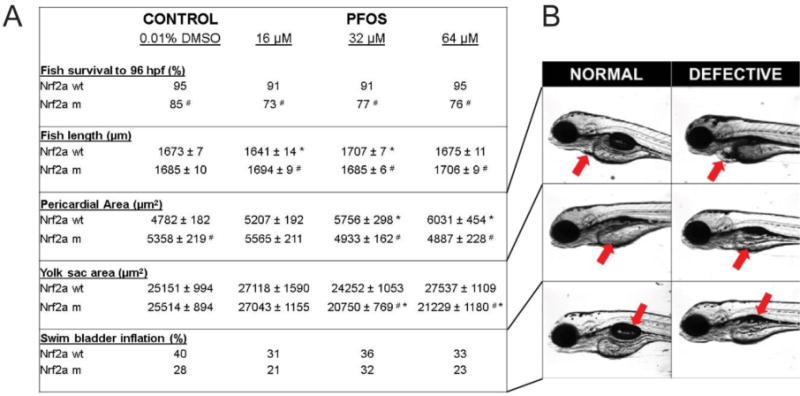
Survival was reduced in Nrf2a mutants compared to respective wild-type embryos, though PFOS exposures did not cause any significant reduction in survival for either genotype. Fish length was affected by PFOS exposures in wild-type fish, but not mutants. PFOS-exposed mutants had altered fish length compared to their wild-type, matched dose embryos. Pericardial area was increased in wild-type embryos exposed to PFOS, but decreased in mutants. Yolk sac area was increased at the lowest PFOS exposure concentration regardless of genotype, but mid and high concentrations were decreased in the mutants only. Swim bladder inflation was lower in all PFOS-exposed groups, except mutants at 32 µM. Asterisks (*) indicate a dose-related change compared to genotypic controls. Octothorpes (#) indicate a difference in response in mutants compared to wild-types. α = 0.05. n = 21-39 embryos from 3-4 experimental replicates
3.2 GENE EXPRESSION
Nrf2a is a master coordinator of the antioxidant response, and its induction results in the upregulation of a number of genes related to phase II detoxification and improving cellular stores of the antioxidant glutathione. To assess whether PFOS exposures impact regulation of these genes and to determine whether insufficient nrf2a exacerbates this response, expression of the genes gstp, gsta1, gclc, ggtb1, and the Nrf2b target p53 was examined in 96 hpf embryos (Fig. 2). Gene expression of the Nrf2a targets gstp, ggtb1, and gsta1 followed increasing trends for both wild-type and mutant embryos. Expression of gstp was significantly increased in wild-type embryos exposed to 32 µM PFOS (p=0.020), and mutants exposed to all PFOS concentrations (p=0.012 for all 3 concentrations). Expression of gsta1 was not significantly increased in wild-type embryos, though they were in mutants exposed to 32 and 64 µM PFOS (p=0.012 for both). Expression of ggt1b was only significantly increased by exposure to 64 µM PFOS in wild-type (p=0.037) and mutant (p=0.022) embryos, and this increase was more than doubled in mutants compared to wild-type embryos at the same exposure concentration (p=0.047). Exposures to 32 µM PFOS resulted in significantly greater gclc expression in wild-type embryos, and in all mutants exposed to PFOS (p=0.009). For gsta1 and ggt1b, genotype and exposure were both significant predictors of gene expression, (p<0.05 for both parameters), though both variables were independent (p>0.05 for interaction). Expression of gstp was significantly responsive to exposure alone. There was significant evidence of genotype-exposure interaction for both gclc and p53.
Figure 2. Gene expression of nrf target genes is modestly responsive to embryonic PFOS exposures.
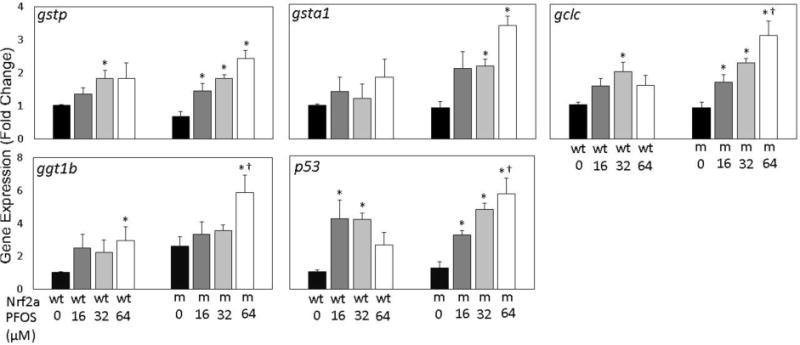
Expression of the Nrf2a target genes gstp, gsta1, gclc, ggt1b, and Nrf2b target p53 was examined using qPCR. For all genes, a dose-dependent increasing trend was observed in nrf2a mutant (m) embryos compared to wild-type (wt) embryos. Asterisks (*) indicate a dose-related change compared to genotypic controls. Daggers (†) indicate a change between wild-type and mutant embryos. α = 0.05. n=3-5 samples of 15 pooled embryos from 4 experimental replicates
To examine any compensation and redundancy occurring among nrf family members, gene expression of nrf1a paralogs (nrf1a, nrf1b), nrf2 paralogs (nrf2a, nrf2b), nrf3, and keap1 paralogs (keap1a, keap1b) was examined at 96 hpf in embryos across PFOS exposures and nrf2a genotypes (Fig. 3). Expression of nrf1a, nrf2a, and nrf2b was not significantly altered in wild-type embryos exposed to PFOS, nor was nrf1a expression in mutants. Expression of nrf1b was reduced by 16 µM PFOS exposures in both wild-type and mutant embryos (p=0.020 in wild-type; p=0.037 in mutants). All PFOS exposure concentrations in the mutants resulted in upregulation of nrf2a (p < 0.04) and nrf2b expression was significantly increased mutant embryos exposed to 32 and 64 µM PFOS (p=0.012 for both). Expression of nrf3 also followed this significantly increasing trend in mutants (p=0.012 and p=0.016 for 32 and 64 µM PFOS). However, in the wild type embryos, nrf3 followed an inverted U-shaped response, with significantly increased expression in 16 and 32 µM exposed embryos (p=0.020 for both). Similarly, expression of keap1a and keap1b both followed a U-shaped dose-response trend in wild-type embryos, significantly decreased for 16 and 32 µM for both genes (p=0.020 for all relationships). Only expression of keap1a was significantly decreased in mutants (p=0.022 and p=0.021 for 16 and 64 µM exposures, respectively). Expression of nrf1a, nrf2a, and nrf2b all had significant genotypic effects (p<0.05), while all genes had significant exposures effects. For both nrf2a and nrf3, there was evidence of genotype and exposure interaction effect on gene expression.
Figure 3. Gene expression of nrf and keap is modestly responsive to embryonic PFOS exposures.
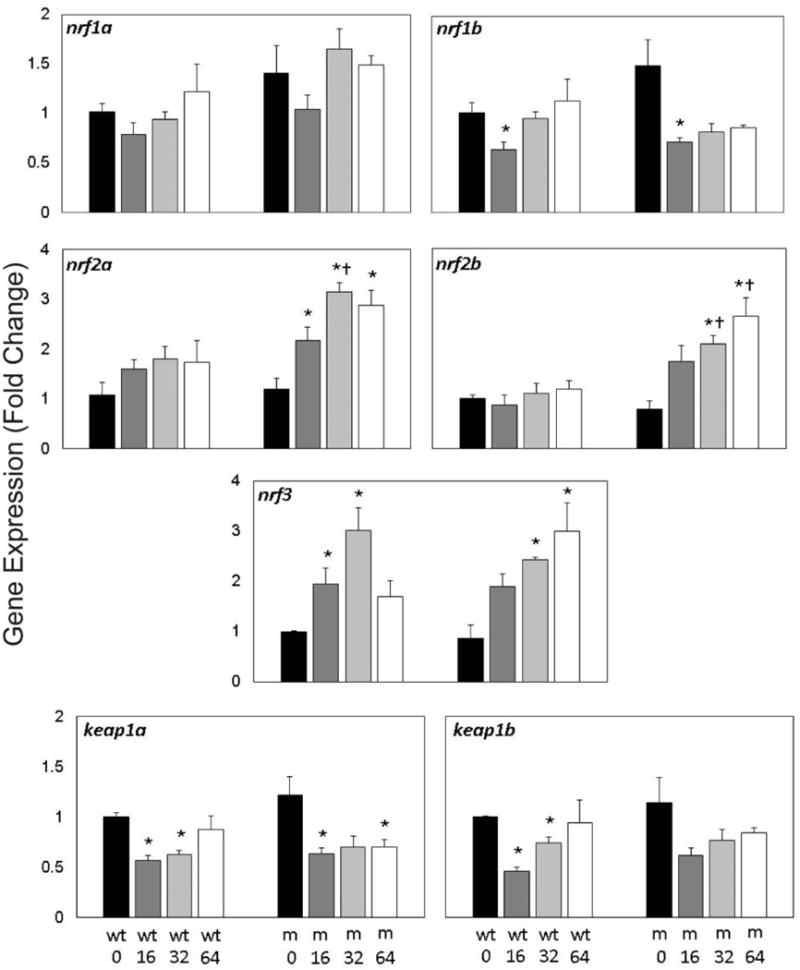
Expression of nrf (nrf1a, nrf2b, nrf2a, nrf2b, and nrf3) and keap (keap1a, keap1b) genes was measured in Nrf2a wild-type (wt) and mutant (m) using qPCR. Expression of gene paralogs was similar across exposures and genotypes. Expression of nrf1a, nrf1b, keap1a, and keap1b was significantly reduced by PFOS treatment, regardless of genotype. Expression of nrf2a, nrf2b, and nrf3 was increased in mutant embryos exposed to 64 µM PFOS. Asterisks (*) indicate a dose-related change compared to genotypic controls. Daggers (†) indicate a change between wild-type and mutant embryos. α=0.05. n=3-5 samples of 15 pooled embryos from 4 experimental replicates
3.3 EMBRYONIC GLUTATHIONE, CYSTEINE, AND REDOX POTENTIALS
To quantify redox changes, glutathione (GSH, GSSG; left) and cysteine (Cys, CySS; right) redox couples were quantitatively assessed using HPLC at 72 hpf (Fig. 4A). In wild-type embryos, embryonic exposures to 16, 32, and 64 µM PFOS increased oxidized GSSG concentrations (p=0.020, p=0.020, and p=0.027, respectively). Cys was decreased in wild-type embryos exposed to 32 µM PFOS only (p=0.002), though a modest decrease was observed due to all PFOS treatments. PFOS did not significantly change GSH or CySS concentrations in wild-type embryos (p>0.05). Embryonic exposure to PFOS did not significantly affect any of the GSH, GSSG, Cys, or CySS concentrations in Nrf2a mutant embryos (p>0.05).
Figure 4. Redox state is perturbed by PFOS treatment, and modulated by Nrf2 deficiency.
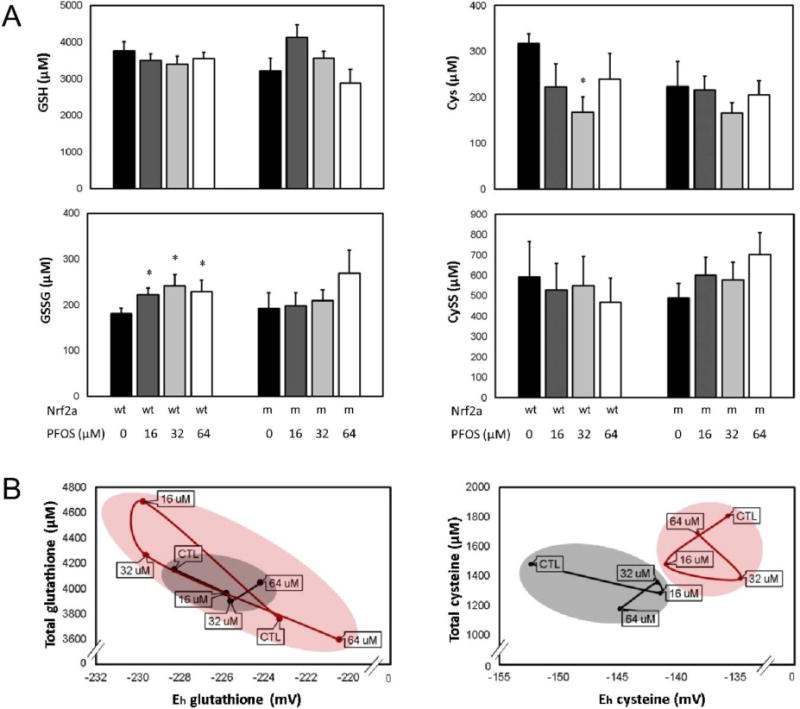
(A) Soluble thiol redox couples were quantified for the glutathione (GSH, GSSG; left) and cysteine (Cys, CySS; right) couples. PFOS increased oxidized GSSG and decreased Cys in wild-type (wt) embryos, though no effects were observed in mutants (m). (B) Total thiols and redox potential profiles were plotted to visualize the redox relationship between Nrf2a genotype and PFOS exposure for the glutathione (left) and cysteine (right) redox couples. In wild-type embryos (black), data were clustered more precisely and followed the expected trend—increasing PFOS exposures oxidized redox potentials. In Nrf2a mutants (red), two different profiles emerged for glutathione and cysteine. The total glutathione and glutathione redox potentials overlapped for wild-type and mutant embryos (left), though the range was much greater than in wild-type embryos. For cysteine (right), wild-type and mutant embryos had completely different profiles without overlap. Asterisks (*) indicate a dose-related change compared to genotypic controls. α = 0.05. n = 6-8 samples of 20 pooled embryos from 3-4 experimental replicates. Data presented are the mean and standard error of the mean (error bars in A, circle boundaries in B).
To examine the relationship between exposure, genotype, and redox environment, we plotted total glutathione or total cysteine (y-axis) measures against calculated redox potentials (x-axis) for all exposure and genotypic groups (Fig. 4B). Wild-type embryos (black line) responded to PFOS treatment in a manner supportive of our hypothesis, as PFOS treatment oxidized Eh for both the glutathione and cysteine redox couples. Nrf2a mutant embryos (red line) had differing responses for both the glutathione and cysteine redox couples. Glutathione Eh is reduced by 16 and 32 µM PFOS, likely due to an increase in total glutathione. As a result, there is a wider range of glutathione responses to PFOS in Nrf2a mutant embryos compared to wild-type embryos, and indicates a disruption of redox control. Total cysteine was modestly decreased by 16 and 32 µM PFOS in the wildtype embryos, but all mutant embryo samples had a more oxidized Cys:CySS redox potential, indicating genotype to be more influential for cysteine, with no overlap in plot area between wild-type and mutant embryos, regardless of exposures.
3.4 APOPTOSIS
Acridine orange staining was used to quantify and characterize apoptosis in PFOS-exposed wild-type and Nrf2a mutant embryos (Fig. 5). In wild type embryos, a general increase in apoptosis occurred with increasing PFOS concentration. Mean acridine orange staining concentrations were 270.0 ± 6.7 (arbitrary fluorescence units; AU) in control, 323.9 ± 9.8 in 16 µM (p<0.001), 586.8 ± 76.1 in 32 µM (p<0.001), and 344.3 ± 22.6 in 64 µM (p=0.005) exposed embryos. In mutant embryos, no change in acridine orange staining was measured due to any PFOS treatment (p>0.05) compared to controls. Mutant controls (321.0 ± 21.7) had elevated acridine orange staining compared to control wild-type embryos (p=0.046). However, staining was decreased in mutants exposed to 32 µM PFOS (306.7 ± 23.6) compared to the 32 µM wild-type embryos (p=0.010). In all cases, acridine orange staining was localized to several anatomical features, including the olfactory bulb, gill slits (which are also the means of acridine orange transport into the embryo), and near the heart or gut endoderm.
Figure 5. PFOS increases apoptosis in wild-type embryos.
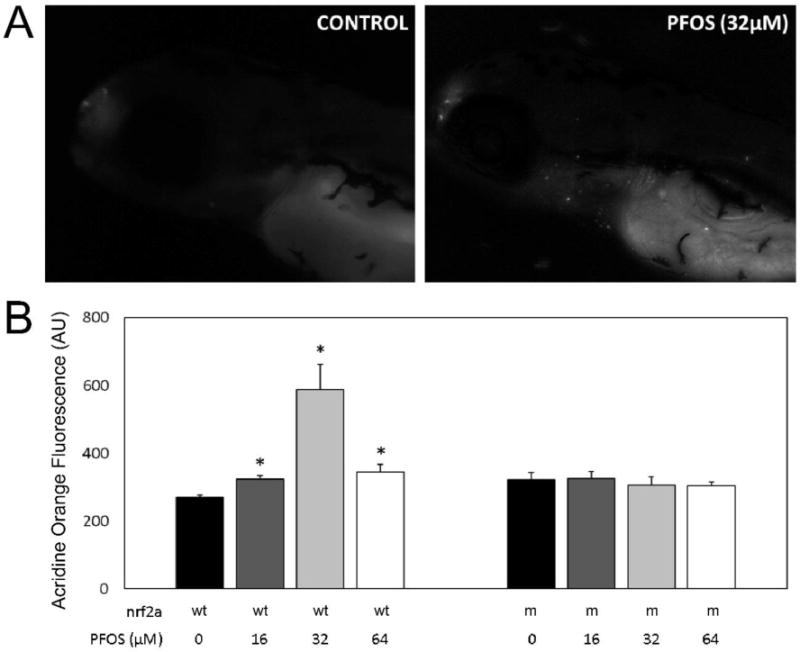
Acridine Orange staining was used to visualize apoptotic cells in vivo at 96 hpf in both wild-type (wt) and Nrf2a mutant (m) embryos. (A) Monochromatic epifluorescent images of control (left) and PFOS-treated (32 µM; right) embryos. An increased number of apoptotic cells can be seen in the rostrum, around the gills, and along the endodermal midline. (B) Homogenates of acridine orange stained embryos were used to quantitatively examine apoptosis. Nrf2a genotype for wild-type (+) and loss-of-function mutant (−) genotypes is provided below corresponding bars, as is PFOS exposure concentration. PFOS did not increase apoptosis in Nrf2a mutant embryos at any concentration. In wild-type embryos, PFOS increased apoptosis, with the greatest amount of apoptosis occurring in those exposed at 32 µM concentrations. Asterisks (*) indicate a dose-related change compared to genotypic controls. α = 0.05. n = 21-39 embryos from 3-4 experimental replicates
3.5 PUTATIVE TRANSCRIPTION FACTOR BINDING SITES (TFBS)
To investigate additional regulatory pathways that could comprise an additional adaptive response beyond ARE-regulation, we examined the promoters and intragenic regions of Nrf family and target genes for evidence of other regulatory elements. The scope of this search was narrowed to examine other transcription factors known to be responsive to PFOS exposures in the literature. Because this search was examining putative TFBS, rigor was increased by only including elements which correspond with complex consensus sequences when appropriate (i.e. an entire dimer consensus sequence rather than merely one of the two elements). A large number of peroxisome proliferator-activated receptor (PPAR) elements were found within the promoters and genes of all Nrf family members and targets, a pathway which has been widely demonstrated to respond to PFOS exposures [21, 35, 36]. Using JASPAR, these PPAR-responsive elements (PPREs) were divided into 3 categories: PPARA:RXRA, PPARG:RXRA, and sites for which both dimers may potentially bind (Figure 6A). All genes except for nrf3 contained multiple PPREs, and several had PPRE-rich domains in their promoter. The nrf2a promoter contains a PPARα:RXRα rich domain approximately 3,000 bp upstream of the transcription start site (TSS), with 8 alpha-specific PPREs. Several genes, including nrf1a and nrf2b also have PPREs flanking the TSS, including a PPARα-specific PPRE region flanking exon 1 within the nrf1a gene. For gstp, there was a cluster of PPARγ-specific PPREs immediately following exon 1.
Figure 6. Putative TFBS found within Nrf family genes, PPARs, and their targets suggest potential for crosstalk in the adaptive response to PFOS.

(A) Promoters (−5,000bp) and genes for Nrf family genes and their examined targets were searched for PPARA:RXRA and PPARG:RXRA specific putative binding sites using JASPAR. Sites were found all genes, except for nrf3. (B) Expression of pparaa and pparg is more sensitive to PFOS-induced modulation in mutants than in wild-type embryos. PPARα target fabp1a is decreased by PFOS exposures, while PPARγ target fabp1b1 is increased. (C) Promoters (−5,000bp) and genes of pparaa and pparg were examined for putative MAF:Nfe2 (ARE) TFBS using JASPAR. Asterisks (*) indicate a dose-related change compared to genotypic controls. Daggers (†) indicate a change between wild-type and mutant embryos. α=0.05. n=3-5 samples of 15 pooled embryos from 4 experimental replicates
Due to the potential crosstalk between Nrf2 and PPARs, pparaa and pparg were also examined for putative Maf:Nfe2 TFBS (Figure 6C). Though the promoter of pparaa did not contain any putative Maf:Nfe2 TFBS, several were present flanking exons within the gene— including two within or immediately following exon 1. The promoter region of pparg contains two putative Maf:Nfe2 TFBS, and the gene contains an additional 12 sites including 7 between exons 1 and 2.
3.6 GENE EXPRESSION OF PPARs AND TARGET GENES
We measured the gene expression of PPARα (pparaa) and PPARγ (pparg), as well as several of their target genes including lipid transporter apolipoprotein A1 (apoa1a), and fatty acid binding proteins 1a (fabp1a) and 1b1 (fabp1b1) in response to PFOS exposures in Nrf2a wild-type and mutant embryos (Figure 6B). Expression of fabp1a has been previously shown to be indicative of PPARα activation in the zebrafish, while fabp1b1 was shown to be indicative of PPARγ activation [37]. Expression of pparaa and pparg was not significantly changed in wild-type embryos in response to PFOS exposures. However, pparg expression was increased with PFOS exposures by more than 2–3 fold in mutant embryos (p=0.060, 0.012, and 0.012 from 16, 32, and 64 µM exposures, respectively). Though there was an increasing trend for pparaa in mutant embryos, this increase was only statistically significant due to the 32 µM exposure (p=0.037). Expression of the target apoa1a was elevated in wild-type embryos exposed to 32 µM PFOS (p=0.020), and in mutants exposed to 32 or 64 µM PFOS (p=0.012). Expression of PPARα-specific target fabp1a was reduced in wild-type embryos exposed to 16 and 32 µM PFOS (p=0.037) and by all concentrations in mutants (p=0.012). Control mutants has nearly double the expression of fabp1a compared to wild-type controls (p=0.028). The trend of PPARγ-specific fabp1b1 gene expression followed an opposite trend. In both wild-type and mutant embryos, fabp1b1 expression increased with PFOS concentration, though these changes were only statistically significant for 32 µM PFOS exposure in wild-type (p=0.020) and for 32 and 64 µM PFOS in mutants (p=0.012).
4. Discussion
Understanding the consequences of developmental pro-oxidant exposures is an important challenge, due to the high prevalence and persistence of compounds such as PFOS in humans, wildlife, and the environment. We have previously demonstrated that Nrf2a could modify embryotoxicity resulting from developmental exposures to polychlorinated biphenyls [25], and that impaired nrf function is sufficient to perturb embryonic redox state [3]. In this study, we hypothesized that impairment of Nrf2a function in zebrafish embryos would enhance PFOS-induced teratogenesis and redox dysregulation, and that these embryos would have weakened compensatory responses. The results of these experiments confirm that PFOS disrupts embryogenesis, and that Nrf2a modulates these effects—though not necessarily as predicted. The formation of PFOS-induced pericardial edema, reduced growth, and apoptosis were ameliorated by the deficient Nrf2a genotype, while accelerated yolk utilization was enhanced in mutant embryos with deficient Nrf2a. One interpretation of these findings is that Nrf2a does not provide the protective effect predicted with respect to teratogenesis and apoptosis, and yet, Nrf2a did provide some protection from the PFOS-induced accelerated yolk usage. However, if Nrf2a is a primary defense, then the secondary defenses are likely to be enhanced in the Nrf2a deficient embryos. These data provide evidence of additional signaling pathways which might be more effective at mitigating PFOS embryotoxicity, including other members of the Nrf-family and PPARs, which are indeed enhanced in the Nrf2a mutant fish.
Several morphometric endpoints were altered by PFOS treatment (Figure 1). With respect to fish length, there was an inverted U-shape response in the wildtype fish, but no dose-related changes in the mutants-rather the mutants were protected from the variations in length that were produced by PFOS in the wildtype embryos. A similar finding was noted with respect to pericardial area The only significant morphometric effect of PFOS on the mutant fish was in yolk sac area, where the mutants had a significantly smaller yolk sac than the mutant DMOS controls, as well as the PFOS-exposed wild types. Decreased yolk sac area indicates increased nutrient utilization, which has been identified in numerous studies as a sensitive indicator of developmental toxicity [38].
We assessed apoptosis in response to PFOS exposures with acridine orange staining, and examined modification of this relationship by Nrf2a (Fig. 5). Apoptosis was increased by PFOS exposures in wild-type embryos only. We also observed upregulation of the tumor suppressor and cell cycle regulatory gene p53 by PFOS in both genotypes, confirming results from a study conducted by Shi et al. that observed increased p53 expression in wild-type embryos exposed to PFOS [16]. Interestingly, no change in apoptosis was observed for Nrf2a mutant embryos in this study, despite the concordant p53 upregulation. However, it is important to note that decreased survival occurred in Nrf2a mutant embryos overall. Therefore, it is possible that the lack of apoptosis in mutants may be at least in part attributed to increased embryonic lethality. The timing of PFOS exposures may also contribute to some of the genotypic effects observed in this endpoint. Because PFOS exposures were last renewed 24 h preceding the assay or microscopy, it is possible that mutant embryos experienced increased apoptosis more swiftly after renewed exposures than wild-type embryos, and thus increased apoptosis was only detected in wild-type embryos 24 h later. However, the acridine orange apoptosis assay showed an inverse U-shaped dose response, which suggests a compensatory response that can ameliorate this increase in apoptosis at the highest exposure concentration. The lack of apoptosis in the mutants exposed to PFOS suggests that the apoptosis pathway initiated by the exposure is Nrf2a-dependent, and that perhaps the compensatory pathway initiated in the highest exposure group is already being activated in the Nrf2a mutants. Additional studies are necessary to better understand these findings.
In wild-type embryos, reduced Cysteine and GSH concentrations followed U-shaped trends and PFOS-exposed embryos were substantially more oxidized than controls (Figure 4). Our data complement findings previously reported by Shi et al., which demonstrated that PFOS exposures increased oxidative stress and induced the antioxidant response in zebrafish embryos. In this study, reduced Cysteine concentrations decreased by nearly 50% in embryos exposed to 32 µM PFOS. This depleted cysteine concentration occurs despite the increased gene expression of ggt1b, a plasma membrane transporter directly responsible for cellular uptake of Cys (Figure 2). It is possible that the reduced cysteine concentrations in wild-type embryos could be due to preferentially increased GSH biosynthesis and recycling, as Cys is the rate-limiting substrate for GSH synthesis. Here, total glutathione was increased and Eh was reduced by PFOS treatment in wild-type embryos, also suggesting that increased GSH biosynthesis has occurred. In future studies, an embryonic ontogeny of the redox response following PFOS treatment could illuminate the sensitivity and responsiveness of the embryo to PFOS as a pro-oxidant, as glutathione concentrations and synthesis greatly change throughout embryogenesis [40].
We previously demonstrated that morpholino-induced loss of nrf function was sufficient to disrupt the embryonic glutathione redox environment [3]. In this study, we confirmed this finding using nrf2a loss-of-function mutant embryos, finding oxidized redox potentials in mutants (Fig. 4). In a study reported by Shi et al., partial morpholino knockdown of Nrf2a exacerbated PFOS-induced oxidative stress [22]. In the current study, we differentially explored the glutathione and cysteine redox couples to better understand these relationships. Interestingly, the glutathione and cysteine redox couples responded differently to impaired Nrf2a signaling. The glutathione redox profiles of wild-type and mutant embryos are overlapping, though redox potentials and total glutathione concentrations in mutant embryos were much more variable and dysregulated in nrf2a mutants. Interestingly, PFOS-exposed mutant embryos had increased total glutathione concentrations, typically indicative of an antioxidant response and often inducible Nrf family function. Therefore, it is possible that the modest increase of nrf2b and nrf3 gene expression, coupled with decreased keap1a/b expression may be sufficient to maintain this compensatory response. One the other hand, cysteine redox profiles of wild-type and nrf2a mutant embryos are very distinct, and control mutant redox potentials are more oxidized than even exposed wild-type redox potentials. Together, these data suggest that the cysteine redox couple of nrf2a mutants is more sensitive to PFOS perturbation than wild-type embryos.
To evaluate the induction of Nrf family targets involved in the antioxidant response, gene expression of ARE-inducible genes was examined (Fig. 2). Though there were more modest changes of target gene expression in wild-type embryos, there was a larger dose-dependent increase of gene expression observed in nrf2a mutant embryos. This confirms our previous findings examining Nrf signaling in nrf1a, nrf1b, nrf2a, and nrf2b morphants [3]. Several Nrf family members have somewhat redundant or overlapping function, and this could potentially be due to functional compensation by other Nrf proteins. To assess whether this may be the case, we also examined nrf1a, nrf1b, nrf2a, nrf2b, nrf3, keap1a, and keap1b gene expression in these embryos (Fig. 3). PFOS exposures drastically reduced gene expression of both nrf1 paralogs and both keap1 paralogs. However, nrf2b and nrf3 expression was increased by PFOS treatment in mutants. It is possible that the upregulation of nrf2b and nrf3, as well as increased expression of the still minimally functional nrf2a, is capable of maintaining induction of some of these targets to compensate for deficient Nrf2a function [41].
Collectively, these data suggest that the inducible antioxidant response championed by other Nrf family members may be able to mitigate oxidative stress in the embryo. However, this response alone does not explain the totality of the embryonic response to PFOS in wild-type and nrf2a mutant embryos. For this reason, we examined the promoters and genes of ARE-targets for other transcriptional regulatory elements known to be labile due to PFOS exposures. After examination, the density of PPREs throughout the majority of these genes emerged as a potential modulator of the PFOS response in the embryo (Figure 6). Several studies have demonstrated that PFOS alters PPAR signaling, and that Nrf2a function may enhance or regulate PPAR expression during the response to toxicants or pharmacological agents via crosstalk mechanisms [23, 24, 42, 43]. In silico analysis of Nrf family and target genes supported this crosstalk, with PPREs in all Nrf-family sequences analyzed except in nrf3. Expression of pparg and its targets apoa1a and fabp1b1 was significantly increased in a dose-dependent manner in nrf2a mutant embryos. This data, complemented by observed putative AREs in the pparaa and pparg promoters suggests that PPARγ may be another mediator of the adaptive response to PFOS during deficient or inadequate Nrf2 signaling.
In conclusion, PFOS disrupts the glutathione and cysteine redox environments and increases apoptosis during embryonic development. Impaired nrf2a signaling further perturbs embryonic redox state, but decreases PFOS-induced apoptosis. The antioxidant response is enhanced by PFOS exposure in Nrf2a-deficient embryos, likely due to the induction of other redundant Nrf function or via indirect mechanisms such as crosstalk with PPAR signaling. This work demonstrates that Nrf2 is a first responder to PFOS-induced injury, and alternate signaling pathways, such as PPARs, provide a second level of defense that are more easily activated in the case where Nrf2a function is impaired.
Supplementary Material
Highlights.
Nrf2a protects against accelerated yolk usage but not pericardial edema or apoptosis induced by PFOS
PFOS oxidizes the glutathione redox potential in zebrafish embryos
Compensatory mechanisms upregulate antioxidant responses in Nrf2a mutant embryos exposed to PFOS
PPAR and other Nrf-family members are likely contributors to the compensatory antioxidant responses that are enhanced in Nrf2a mutant embryos.
Acknowledgments
Funding
This work was funded by the National Institute of Environmental Health Sciences (R01 ES025748 and R01 ES028201 to ART-L; F32 ES028085 to KES). We thank Craig Harris (University of Michigan) for use of his HPLC and facilities for redox profiling experiments, and Diana Franks (Woods Hole Oceanographic Institution) for her design of several of the primers utilized in this study. This work was possible due to the exceptional animal care and laboratory assistance of Aviraj Basnet, Katrina Borofski, Christopher Clark, Shana Fleischman, Sadia Islam, Yankel Karasik, David Nava, Archit Rastogi, Monika Roy, Christopher Sparages, Olivia Venezia, and Felicia Wang.
Abbreviations
- ROS
reactive oxygen species
- GSH
glutathione
- GSSG
glutathione disulfide
- PFOS
Perfluorooctanesulfonic acid
- DMSO
dimethyl sulfoxide
- ARE
antioxidant response element
- PPAR
peroxisome proliferator activated receptor
- PPRE
PPAR response element
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hansen JM, Harris C. Redox control of teratogenesis. Reproductive Toxicology. 2013;35:165–179. doi: 10.1016/j.reprotox.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Jones DP. Redefining oxidative stress. Antioxidants & redox signaling. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 3.Sant KE, Hansen JM, Williams LM, Tran NL, Goldstone JV, Stegeman JJ, Hahn ME, Timme-Laragy A. The role of Nrf1 and Nrf2 in the regulation of glutathione and redox dynamics in the developing zebrafish embryo. Redox biology. 2017;13:207–218. doi: 10.1016/j.redox.2017.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi M, Li L, Iwamoto N, Nakajima-Takagi Y, Kaneko H, Nakayama Y, Eguchi M, Wada Y, Kumagai Y, Yamamoto M. The Antioxidant Defense System Keap1-Nrf2 Comprises a Multiple Sensing Mechanism for Responding to a Wide Range of Chemical Compounds. Molecular and cellular biology. 2009;29:493–502. doi: 10.1128/MCB.01080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Molecular and cellular biology. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nguyen T, Nioi P, Pickett CB. The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. Journal of biological chemistry. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hahn ME, Timme-Laragy AR, Karchner SI, Stegeman JJ. Nrf2 and Nrf2-related proteins in development and developmental toxicity: Insights from studies in zebrafish (Danio rerio) Free Radical Biology and Medicine. 2015;88(Part B):275–289. doi: 10.1016/j.freeradbiomed.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Timme-Laragy AR, Karchner SI, Franks DG, Jenny MJ, Harbeitner RC, Goldstone JV, McArthur AG, Hahn ME. Nrf2b, novel zebrafish paralog of oxidant-responsive transcription factor NF-E2-related factor 2 (NRF2) J Biol Chem. 2012;287:4609–4627. doi: 10.1074/jbc.M111.260125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calafat AM, Wong LY, Kuklenyik Z, Reidy JA, Needham LL. Polyfluoroalkyl chemicals in the U.S. population: data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000. Environ Health Perspect. 2007;115:1596–1602. doi: 10.1289/ehp.10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olsen GW, Burris JM, Ehresman DJ, Froehlich JW, Seacat AM, Butenhoff JL, Zobel LR. Half-life of serum elimination of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ Health Perspect. 2007;115:1298–1305. doi: 10.1289/ehp.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Tully JS, Needham LL. Serum concentrations of 11 polyfluoroalkyl compounds in the u.s. population: data from the national health and nutrition examination survey (NHANES) Environ Sci Technol. 2007;41:2237–2242. doi: 10.1021/es062686m. [DOI] [PubMed] [Google Scholar]
- 12.Inoue K, Okada F, Ito R, Kato S, Sasaki S, Nakajima S, Uno A, Saijo Y, Sata F, Yoshimura Y, Kishi R, Nakazawa H. Perfluorooctane Sulfonate (PFOS) and Related Perfluorinated Compounds in Human Maternal and Cord Blood Samples: Assessment of PFOS Exposure in a Susceptible Population during Pregnancy. Environ Health Perspect. 2004;112:1204–1207. doi: 10.1289/ehp.6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen MH, Ha EH, Wen TW, Su YN, Lien GW, Chen CY, Chen PC, Hsieh WS. Perfluorinated compounds in umbilical cord blood and adverse birth outcomes. PLoS ONE. 2012;7:e42474. doi: 10.1371/journal.pone.0042474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thibodeaux JR, Hanson RG, Rogers JM, Grey BE, Barbee BD, Richards JH, Butenhoff JL, Stevenson LA, Lau C. Exposure to Perfluorooctane Sulfonate during Pregnancy in Rat and Mouse. I: Maternal and Prenatal Evaluations. Toxicological Sciences. 2003;74:369–381. doi: 10.1093/toxsci/kfg121. [DOI] [PubMed] [Google Scholar]
- 15.Zheng XM, Liu HL, Shi W, Wei S, Giesy JP, Yu HX. Effects of perfluorinated compounds on development of zebrafish embryos. Environ Sci Pollut Res Int. 2011;19:2498–2505. doi: 10.1007/s11356-012-0977-y. [DOI] [PubMed] [Google Scholar]
- 16.Shi X, Du Y, Lam PK, Wu RS, Zhou B. Developmental toxicity and alteration of gene expression in zebrafish embryos exposed to PFOS. Toxicol Appl Pharmacol. 2008;230:23–32. doi: 10.1016/j.taap.2008.01.043. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Tanguay RL, Tal TL, Bai C, Tilton SC, Jin D, Yang D, Huang C, Dong Q. Early life perfluorooctanesulphonic acid (PFOS) exposure impairs zebrafish organogenesis. Aquatic toxicology (Amsterdam, Netherlands) 2014;150:124–132. doi: 10.1016/j.aquatox.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sant KE, Jacobs HM, Borofski KA, Moss JB, Timme-Laragy AR. Embryonic exposures to perfluorooctanesulfonic acid (PFOS) disrupt pancreatic organogenesis in the zebrafish, Danio rerio. Environmental Pollution. 2017;220(Part B):807–817. doi: 10.1016/j.envpol.2016.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jantzen CE, Annunziato KA, Bugel SM, Cooper KR. PFOS, PFNA, and PFOA Sub-Lethal Exposure to Embryonic Zebrafish Have Different Toxicity Profiles in Terms of Morphometrics, Behavior and Gene Expression. Aquatic toxicology (Amsterdam, Netherlands) 2016;175:160–170. doi: 10.1016/j.aquatox.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parolini M, Colombo G, Valsecchi S, Mazzoni M, Possenti CD, Caprioli M, Dalle-Donne I, Milzani A, Saino N, Rubolini D. Potential toxicity of environmentally relevant perfluorooctane sulfonate (PFOS) concentrations to yellow-legged gull Larus michahellis embryos. Environmental Science and Pollution Research. 2016;23:426–437. doi: 10.1007/s11356-015-5248-2. [DOI] [PubMed] [Google Scholar]
- 21.Xu J, Shimpi P, Armstrong L, Salter D, Slitt AL. PFOS induces adipogenesis and glucose uptake in association with activation of Nrf2 signaling pathway. Toxicology and Applied Pharmacology. 2016;290:21–30. doi: 10.1016/j.taap.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi X, Zhou B. The Role of Nrf2 and MAPK Pathways in PFOS-Induced Oxidative Stress in Zebrafish Embryos. Toxicological Sciences. 2010;115:391–400. doi: 10.1093/toxsci/kfq066. [DOI] [PubMed] [Google Scholar]
- 23.Cho HY, Gladwell W, Wang X, Chorley B, Bell D, Reddy SP, Kleeberger SR. Nrf2-regulated PPARγ Expression Is Critical to Protection against Acute Lung Injury in Mice. American Journal of Respiratory and Critical Care Medicine. 2010;182:170–182. doi: 10.1164/rccm.200907-1047OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhan L, Zhang H, Zhang Q, Woods CG, Chen Y, Xue P, Dong J, Tokar EJ, Xu Y, Hou Y, Fu J, Yarborough K, Wang A, Qu W, Waalkes MP, Andersen ME, Pi J. Regulatory Role of KEAP1 and NRF2 in PPARγ Expression and Chemoresistance in Human Non-small Cell Lung Carcinoma Cells. Free radical biology & medicine. 2012;53:758–768. doi: 10.1016/j.freeradbiomed.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rousseau ME, Sant KE, Borden LR, Franks DG, Hahn ME, Timme-Laragy AR. Regulation of Ahr signaling by Nrf2 during development: Effects of Nrf2a deficiency on PCB126 embryotoxicity in zebrafish (Danio rerio) Aquatic toxicology (Amsterdam, Netherlands) 2015;167:157–171. doi: 10.1016/j.aquatox.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukaigasa K, Nguyen LT, Li L, Nakajima H, Yamamoto M, Kobayashi M. Genetic evidence of an evolutionarily conserved role for Nrf2 in the protection against oxidative stress. Mol Cell Biol. 2012;32:4455–4461. doi: 10.1128/MCB.00481-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacobs HM, Sant KE, Basnet A, Williams LM, Moss JB, Timme-Laragy AR. Embryonic exposure to Mono(2-ethylhexyl) phthalate (MEHP) disrupts pancreatic organogenesis in zebrafish (Danio rerio) Chemosphere. 2018;195:498–507. doi: 10.1016/j.chemosphere.2017.12.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sundström M, Bogdanska J, Pham HV, Athanasios V, Nobel S, McAlees A, Eriksson J, DePierre JW, Bergman Å. Radiosynthesis of perfluorooctanesulfonate (PFOS) and perfluorobutanesulfonate (PFBS), including solubility, partition and adhesion studies. Chemosphere. 2012;87:865–871. doi: 10.1016/j.chemosphere.2012.01.027. [DOI] [PubMed] [Google Scholar]
- 29.Timme-Laragy AR, Goldstone JV, Imhoff BR, Stegeman JJ, Hahn ME, Hansen JM. Glutathione redox dynamics and expression of glutathione-related genes in the developing embryo. Free radical biology & medicine. 2013;65:89–101. doi: 10.1016/j.freeradbiomed.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones DP, Carlson JL, Samiec PS, Sternberg P, Jr, Mody VC, Jr, Reed RL, Brown LA. Glutathione measurement in human plasma. Evaluation of sample collection, storage and derivatization conditions for analysis of dansyl derivatives by HPLC. Clinica chimica acta; international journal of clinical chemistry. 1998;275:175–184. doi: 10.1016/s0009-8981(98)00089-8. [DOI] [PubMed] [Google Scholar]
- 31.Harris C, Hansen J. Oxidative Stress, Thiols, and Redox Profiles. In: Harris C, Hansen JM, editors. Developmental Toxicology. Humana Press; 2012. pp. 325–346. [DOI] [PubMed] [Google Scholar]
- 32.Harris C, Shuster DZ, Roman Gomez R, Sant KE, Reed MS, Pohl J, Hansen JM. Inhibition of glutathione biosynthesis alters compartmental redox status and the thiol proteome in organogenesis-stage rat conceptuses. Free radical biology & medicine. 2013;63:325–337. doi: 10.1016/j.freeradbiomed.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirlin WG, Cai J, Thompson SA, Diaz D, Kavanagh TJ, Jones DP. Glutathione redox potential in response to differentiation and enzyme inducers. Free radical biology & medicine. 1999;27:1208–1218. doi: 10.1016/s0891-5849(99)00145-8. [DOI] [PubMed] [Google Scholar]
- 34.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2–ΔΔCT Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 35.DeWitt JC, Shnyra A, Badr MZ, Loveless SE, Hoban D, Frame SR, Cunard R, Anderson SE, Meade BJ, Peden-Adams MM, Luebke RW, Luster MI. Immunotoxicity of Perfluorooctanoic Acid and Perfluorooctane Sulfonate and the Role of Peroxisome Proliferator-Activated Receptor Alpha. Critical Reviews in Toxicology. 2009;39:76–94. doi: 10.1080/10408440802209804. [DOI] [PubMed] [Google Scholar]
- 36.Takacs ML, Abbott BD. Activation of Mouse and Human Peroxisome Proliferator–Activated Receptors (α, β/δ, γ) by Perfluorooctanoic Acid and Perfluorooctane Sulfonate. Toxicological Sciences. 2007;95:108–117. doi: 10.1093/toxsci/kfl135. [DOI] [PubMed] [Google Scholar]
- 37.Laprairie RB, Denovan-Wright EM, Wright JM. Subfunctionalization of peroxisome proliferator response elements accounts for retention of duplicated fabp1 genes in zebrafish. BMC Evolutionary Biology. 2016;16:147. doi: 10.1186/s12862-016-0717-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sant KE, Timme-Laragy AR. Zebrafish as a Model for Toxicological Perturbation of Yolk and Nutrition in the Early Embryo. Current Environmental Health Reports. 2018 doi: 10.1007/s40572-018-0183-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park M, Helip-Wooley A, Thoene J. Lysosomal Cystine Storage Augments Apoptosis in Cultured Human Fibroblasts and Renal Tubular Epithelial Cells. Journal of the American Society of Nephrology. 2002;13:2878–2887. doi: 10.1097/01.asn.0000036867.49866.59. [DOI] [PubMed] [Google Scholar]
- 40.Timme-Laragy AR, Goldstone JV, Imhoff BR, Stegeman JJ, Hahn ME, Hansen JM. Glutathione redox dynamics and expression of glutathione-related genes in the developing embryo. Free Radical Biology and Medicine. 2013;65:89–101. doi: 10.1016/j.freeradbiomed.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams LM, Timme-Laragy AR, Goldstone JV, McArthur AG, Stegeman JJ, Smolowitz RM, Hahn ME. Developmental Expression of the Nfe2-Related Factor (Nrf) Transcription Factor Family in the Zebrafish, Danio rerio. PLoS ONE. 2013;8:e79574. doi: 10.1371/journal.pone.0079574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu Y, Sun Y, Zhu J, Yu L, Jiang X, Zhang J, Dong X, Ma B, Zhang Q. Oridonin exerts anticancer effect on osteosarcoma by activating PPAR-γ and inhibiting Nrf2 pathway. Cell Death & Disease. 2018;9:15. doi: 10.1038/s41419-017-0031-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee C. Collaborative Power of Nrf2 and PPARG Activators against Metabolic and Drug-Induced Oxidative Injury. Oxidative medicine and cellular longevity. 2017;2017:14. doi: 10.1155/2017/1378175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Evans BR, Karchner SI, Franks DG, Hahn ME. Duplicate aryl hydrocarbon receptor repressor genes (ahrr1 and ahrr2) in the zebrafish Danio rerio: Structure, function, evolution, and AHR-dependent regulation in vivo. Archives of Biochemistry and Biophysics. 2005;441:151–167. doi: 10.1016/j.abb.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 45.Timme-Laragy AR, Van Tiem LA, Linney EA, Di Giulio RT. Antioxidant responses and NRF2 in synergistic developmental toxicity of PAHs in zebrafish. Toxicological sciences : an official journal of the Society of Toxicology. 2009;109:217–227. doi: 10.1093/toxsci/kfp038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Richter CA, Garcia-Reyero N, Martyniuk C, Knoebl I, Pope M, Wright-Osment MK, Denslow ND, Tillitt DE. GENE EXPRESSION CHANGES IN FEMALE ZEBRAFISH (DANIO RERIO) BRAIN IN RESPONSE TO ACUTE EXPOSURE TO METHYLMERCURY. Environmental Toxicology and Chemistry/Setac. 2011;30:301–308. doi: 10.1002/etc.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fu YF, Du TT, Dong M, Zhu KY, Jing CB, Zhang Y, Wang L, Fan HB, Chen Y, Jin Y, Yue GP, Chen SJ, Chen Z, Huang QH, Jing Q, Deng M, Xi Liu T. Mir-144 selectively regulates embryonic α-hemoglobin synthesis during primitive erythropoiesis. Blood. 2009;113:1340–1349. doi: 10.1182/blood-2008-08-174854. [DOI] [PubMed] [Google Scholar]
- 48.Maradonna F, Evangelisti M, Gioacchini G, Migliarini B, Olivotto I, Carnevali O. Assay of vtg, ERs and PPARs as endpoint for the rapid in vitro screening of the harmful effect of Di-(2-ethylhexyl)-phthalate (DEHP) and phthalic acid (PA) in zebrafish primary hepatocyte cultures. Toxicology in Vitro. 2013;27:84–91. doi: 10.1016/j.tiv.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 49.Fai Tse WK, Li JW, Kwan Tse AC, Chan TF, Hin Ho JC, Sun Wu RS, Chu Wong CK, Lai KP. Fatty liver disease induced by perfluorooctane sulfonate: Novel insight from transcriptome analysis. Chemosphere. 2016;159:166–177. doi: 10.1016/j.chemosphere.2016.05.060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.