

Abstract
Philadelphia (Ph)-like acute lymphoblastic leukemia (ALL) is a molecular subtype of high-risk B-cell ALL characterized by formation of abnormal gene fusions involving tyrosine kinase (TK) and cytokine receptor genes and activation of TK signaling. Because of the diversity of associated genetic changes, the detection of Ph-like ALL cases currently requires multiple cytogenetic and molecular assays; thus, our goal was to develop a consolidated workflow for detecting genetic abnormalities in Ph-like ALL. We found that total and targeted RNA sequencing (RNAseq)-based approach allowed the detection of abnormal fusion transcripts (EBF1-PDGFRB, P2RY8-CRLF2, RCSD1-ABL1, and RCSD1-ABL2). The bioinformatics algorithm accurately detected the fusion transcripts without prior input about possible events. Additionally, we showed that RNAseq analysis enabled evaluation for disease-associated sequence variants in expressed transcripts. While total RNAseq can be a second tier approach allowing discovery of novel genetic alterations, the targeted RNAseq workflow offers a clinically applicable method for the detection of fusion transcripts.
Keywords: Ph-like ALL, RNAseq, fusion transcripts, expression, sequence variant
Introduction
The reciprocal translocation between chromosomes 9 and 22 resulting in a formation of the oncogenic BCR-ABL1 fusion on the derivative chromosome 22 (also known as the Philadelphia (Ph) chromosome) is considered a high-risk feature in B acute lymphoblastic leukemia (B-ALL) with an increased risk of relapse.[1,2] However, the addition of tyrosine kinase inhibitors (TKIs) to the treatment protocols has significantly improved the outcomes for the patients with Ph-positive B-ALL [3] highlighting the power of therapies that target key oncogenic mutations in leukemia cells. Recent genomic profiling of ALL has identified an ALL subtype with a gene expression signature resembling that of Ph-positive ALL, but without the presence of Ph-chromosome. This molecular subtype of ALL is typically referred to as ‘Ph-like’, and is characterized by genetic alterations affecting a myriad of kinase, cytokine or cytokine receptor (CR) genes.[4–8] Similar to Ph-positive cases prior to the introduction of TKI therapies, the ‘Ph-like’ leukemias have a high probability of relapse and carry a poor prognosis with standard treatment regiments.[4] Since they are also associated with activated kinase signaling pathways, there is a growing interest in investigating the efficacy of adding TKIs to the standard chemotherapy treatment for patients with this disease subtype. Numerous in vitro and in vivo studies, coupled with an increasing number of case reports, have documented that Ph-like ALLs can be successfully targeted by TKIs.[9–11] However, further investigation of the efficacy of TKIs and their implementation as part of standard treatment will require the development of robust and reliable assays for the accurate genetic diagnosis of Ph-like ALL cases. Due to the variety of genetic abnormalities in Ph-like ALL, thorough diagnostic evaluation for this subtype requires methods that can simultaneously test for a large number of abnormal gene fusions, along with other types of mutations including sequence base changes. Current clinical diagnostic methods for Ph-like ALL involve a laborious ‘candidate abnormality’ approach and stepwise series of cytogenetic and molecular assays to evaluate for possible genetic lesions that are implicated in the disease.[6] Due to the prohibitive costs, lack of sequencing and computing infrastructure, and shortage of specialized bioinformatics personnel, clinical diagnostic laboratories have lagged behind in adopting comprehensive genomic assays like exome, whole genome and whole transcriptome sequencing, which were used in the research setting to study genetic alterations in Ph-like ALL. Current advances in next-generation sequencing (NGS) technology have provided efficient methods to identify abnormal gene fusions either at the DNA or RNA level,[12,13] and these technologies are becoming increasingly accessible for clinical use due to continued improvements in technical and analytical performance and considerable reduction in sequencing costs.
Transcriptome or RNA sequencing (RNAseq) has been successfully used to identify known or novel oncogenic fusions in a variety of cancers,[12,14] including Ph-like ALL.[6] We hypothesized that RNAseq-based approaches can also be effectively applied in routine diagnostics. We hence conducted a retrospective study to test the feasibility and clinical applicability of using total RNAseq and targeted RNAseq for the identification of genetic alterations associated with Ph-like ALL in a subset of known samples. We found both approaches reliably identified expected gene fusions in four cases of Ph-like ALL which were previously characterized by a combination of traditional cytogenetic and molecular methods. Furthermore, as the clinically relevant genetic aberrations of Ph-like ALL also include a limited set of previously characterized point mutations, we were able to utilize total RNAseq data to evaluate the expressed transcripts for acquired oncogenic sequence variants.
Materials and methods
Specimen selection and procurement
After approval by the University of Chicago Institutional Review Board, four cases were identified by searching available clinical and research databases in which previous conventional karyotype analysis or Chromosome Microarray (CMA) analysis identified abnormalities suggestive of Ph-like ALL (Supplementary Figure S1). Case A was an 11-year-old female with newly diagnosed B-ALL, who had an induction failure after a four-drug induction chemotherapy regimen. CMA analysis showed an interstitial deletion on chromosome 5, with breakpoints within the EBF1 and PDGFRB genes. Upon addition of TKIs to the chemotherapy backbone based on the presence of an EBF1–PDGFRB fusion in leukemia cells, the patient achieved complete remission and proceeded to hematopoietic stem cell transplantation (HSCT). Case B was a 35-year-old female with relapsed B-ALL whose disease was refractory to several lines of salvage treatment, and she expired due to disease progression and chemotherapy complications. CMA analysis of a relapse sample showed a deletion within the pseudoautosomal region of the X-chromosome with the breakpoints in the P2RY8 and CRLF2 genes. Case C was a 15-year-old female diagnosed with B-ALL with the t(1;9)(q24;q34.1) shown previously to result in a fusion between the RCSD1 gene (1q24) and the ABL1 gene (9q34.1).[11,15] The patient achieved complete remission upon addition of TKIs to the post-induction chemotherapy regimen, but developed fatal complications after HSCT. Case D was previously described by Raca et al. [16] The patient was a 20-year-old male diagnosed with refractory precursor B-ALL with a normal karyotype, in whom CMA analysis revealed a complex rearrangement affecting the long arm of chromosome 1 (1q24.2), with two of the breakpoints occurring within the RCSD1 and ABL2 genes. The patient received HSCT in the presence of low-level minimal residual disease (MRD), and achieved complete remission post-transplant.
The patients were treated at the University of Chicago Medicine between January 2014 and August 2015. Cryopreserved leukemia cells were obtained from the University of Chicago Cancer Cytogenetics Laboratory Tissue Repository (IRB#13465), where they were deposited after obtaining consent for banking and future research use from the patients or their parents.
Total and targeted RNA sequencing
RNAseq libraries were constructed using the Illumina TruSeq RNA Sample Prep Kit v2 (Illumina Inc., San Diego, CA), following the manufacturer’s recommendations. For targeted transcriptome sequencing, the completed cDNA libraries were subjected to an additional enrichment step using the IDT xGEN Lockdown probes according to the manufacturer’s protocol (Integrated DNA Technologies Inc., Coralville, IA). A total of 13 kinase genes were enriched with DNA probe sets designed at 1 × tiling against the coding regions for targeted RNAseq. A detailed description of the procedures for library preparation, sequencing and data analysis is provided in the Supplementary methods.
RNAseq data analysis and fusion detection
The detection of fusion transcripts and the mapping of the specific breakpoints were accomplished using the FusionCatcher algorithm.[17] A detailed description of the analysis workflow is provided in the Supplementary methods. The sample processing and data analysis workflow for total RNAseq and targeted RNAseq are outlined in Figure 1.
Figure 1.

Flowchart of RNAseq procedures and workflow of analysis.
Detection of nucleotide variants from total RNAseq data
The sequencing output files in FastQ format were first subjected to quality control metrics (see Supplementary methods) and matching read pairs were retained (cmpfastq) before alignment to the genomic reference (hg19) using the RNAseq alignment algorithm STAR (Spliced Transcripts Alignment to a Reference).[18] Using the .bam output generated from STAR, additional data processing was conducted using picard tools (AddOrReplaceReadGroups and MarkDuplicates) to prepare the RNAseq data for variant analysis using the Genome Analysis Tool Kit (GATK).[19] Further GATK preprocessing involved the use of the SplitNCigarReads function to reformat the RNAseq data, before proceeding with variant calling using the HaploTypeCaller according to GATK Best Practices recommendation.[20,21] Variant calling was conducted by comparing the mapped sequence data to the hg19 reference genome to generate vcf files. The sequence changes were further annotated using ANNOVAR,[22] and filtered to exclude variants that are frequently observed in the general population. [23–25] Nonsynonymous variants were considered and reported with the associated rs numbers (dbSNP138) [26] if they were previously described as being mutated in Ph-like ALL.[6] (Supplementary Table S3).
Results
Detailed results of the morphologic evaluation, immunotyping, and cytogenetic testing for all four cases are presented in the Supplementary Table S1 and Supplementary Figure S1(a–c).
Fusion detection and breakpoint mapping from total RNAseq
The whole transcriptome sequencing yielded an average of approximately 163 million reads per sample (Supplementary Table S4). Using the FusionCatcher algorithm, the output included a list of candidate gene fusions for each sample, together with the number of read pairs and unique reads spanning the fusion junctions and thus supporting each candidate fusion (see Additional data: Excel files 1 and 2). For each of the four samples the abnormality that was expected based on previous CMA results or karyotype analysis was identified by FusionCatcher either as the first or the second candidate fusion (Table 1). The other detected fusion candidates were mostly designated as out-of-frame and read-through transcripts and disregarded for further evaluation. On a high performance-computing environment (128 Gb ram cluster), the total RNAseq data took approximately 20 h of analyses. The FusionCatcher output is available as Supplemental data (Excel file 1 and 2), demonstrating the candidate fusion genes detected for each sample and the final summary output file.
Table 1.
Number of read pairs spanning fusion junction as computed by FusionCatcher.
| Total RNAseq |
Targeted RNAseq |
|||
|---|---|---|---|---|
| Case | Spanning read pairs | Comments | Spanning read pairs | Comments |
| A | 63 | EBF1-PDGFRB as top hit (total detected: 69 fusions) | 345 | Detected 3 fusions with EBF1-PDGFRB as top hit |
| B | 13 | P2RY8-CRLF2 as 2nd hit (total detected: 7 fusions) | 604 | Only detected P2RY8-CRLF2 |
| C | 98 | RCSD1-ABL1 as top hit (total detected: 4 fusions) | 149 | Only detected RCSD1-ABL1 |
| D | 5 | RCSD1-ABL2 as 2nd hit (total detected: 9 fusions) | 129 | Only detected RCSD1-ABL2 |
| Average | 44.75 | – | 306.75 | – |
RNAseq allowed us to accurately delineate fusion junctions for all detected abnormal gene fusions, as the exact junction points were sequenced through, and the resulting sequences included in the output from the FusionCatcher data analyses. For Case A, RNAseq analysis showed that the fusion event occurred between exon 15 of the EBF1 gene and exon 11 of the PDGFRB gene (Figure 2(a)), resulting in the formation of an in-frame fusion transcript, expected to produce a chimeric EBF1-PDGFRB protein [9,10] (Figure 2(a) and Additional data: Excel files 1 and 2). For Case B, the fusion transcripts were shown to contain the 5′-UTR of P2RY8 joined to exon 1 of CRLF2. This chromosomal rearrangement resulted in juxtaposition of the promoter region of P2RY8 upstream of the coding sequences of CRLF2 (Figure 2(b)), with overexpression of CRLF2 acting as the main driver of the disease.[27] For Case C, RNAseq data were consistent with a fusion between the exon 3 of RCSD1 and exon 4 of ABL1 [11] (Figure 2(c)), resulting in the formation of an in-frame RCSD1-ABL1 fusion transcript. Finally for Case D, the fusion event resulted from a complex rearrangement of chromosome 1 [16] and led to expression of an abnormal transcript which joined RCSD1 exon 3 with exon 4 or 5 (based on alternative variant transcripts NM_001136001and NM_001136000, respectively) of the ABL2 gene (Figure 2(d)). The transcript was shown to be in-frame, likely coding for a functional RCSD1-ABL2 fusion protein.
Figure 2.
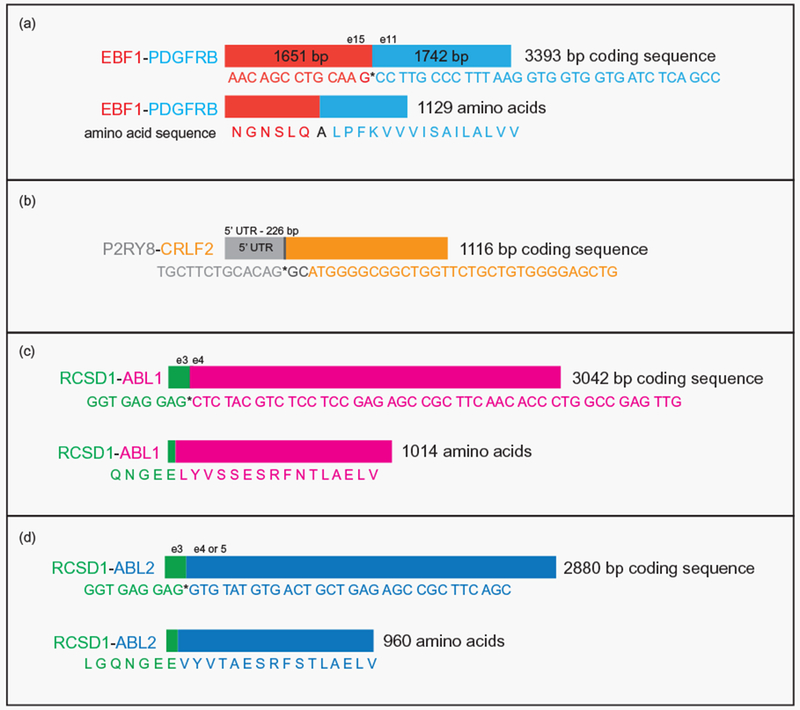
Schematic representation of the fusion transcripts detected by FusionCatcher analysis. Fusion transcripts as detected by the algorithm in the studied patients: (a) EBF1-PDGFRB, (b) P2RY8-CRLF2, (c) RCSD1-ABL1, and (d) RCSD1-ABL2.
In each of the four cases, whole transcriptome sequencing also allowed us to detect prominent up-regulation of the implicated kinase gene (Supplementary Figure S2 and Supplementary Table S6). The results of expression analysis demonstrate that RNAseq-based approaches will be able to detect those cases of Ph-like ALL where up-regulation of target kinase genes occurs through mechanisms (including point mutations or translocations with immunoglobulin heavy and light chain loci), which do not create fusion transcripts.
Analysis of sequence variants using total RNAseq data
Using data from total RNAseq, we were able to conduct a targeted analysis for sequence variants in the genes that have been shown to be mutated in Ph-like ALL.[6] (Supplementary Table S3) As Ph-like ALL cases found to have CRLF2 overexpression are frequently associated with mutations in the genes coding for the members of the JAK-family kinase,[6,8] it is valuable during genetic diagnostics of Ph-like ALL to test for sequence variants and gene fusions in a single assay. Our patient Case B was verified by RNAseq to have a P2RY8–CRLF2 fusion that causes overexpression of the CRLF2 gene. By sequence variant analysis using the same RNAseq dataset, we were able to confirm that JAK1, JAK2, and JAK3 gene mutations were not present in this patient’s sample. Additionally, we screened other genes that have been previously shown to be mutated in Ph-like ALL [6] for acquired, disease-associated sequence variants in all four patient samples. Analysis of the selected 27 genes detected nonsynonymous sequence variants in the NOTCH1 gene and the NOTCH1 and PAX5 genes (Table 2) in Samples A and D respectively. Both variants were present at around 44–53% variant allele frequency. These variants were documented in the dbSNP138 database, but were found to have less than 1% minimum allele frequency in the ExAC, ESP, and 1000g project database. Without sequencing corresponding germ-line samples, we are unable to determine if these variants are constitutional, or represent acquired somatic mutations in the leukemic cells.
Table 2.
Sequence variants detected in the studied Ph-like ALL cases.
| Sample | Fusion detected | Mutations detected | Coding sequence change | Amino acid change | dbSNP138 | Allele frequency (%) | ExAC MAF (%) |
|---|---|---|---|---|---|---|---|
| A | EBF1-PDGFRB | NOTCH1 | c.G4049T | p.R1350L | rs150343794 | 45.1 | 0.07 |
| B | P2RY8-CRLF2 | – | – | – | – | – | – |
| C | RCSD1-ABL1 | – | – | – | – | – | – |
| D | RCSD1-ABL2 | NOTCH1 | c.G3788A | p.R1263H | rs377594681 | 44.4 | 0.00097 |
| PAX5 | c.T6A | p.D2E | rs139701864 | 53.2 | 0.32 |
Fusion detection from targeted RNAseq
In addition to whole transcriptome sequencing, the samples were concurrently analyzed by targeted RNAseq, to ascertain the efficiency and clinical applicability of this approach for detecting gene fusions associated with Ph-like ALL. Targeted transcriptome sequencing required significantly reduced sequencing output, and approximately 370,000 reads were obtained per sample (Supplementary Table S5). The targeted RNAseq analyses using FusionCatcher were completed in less than 20 min. Furthermore, we observed an enrichment of read pairs that span the fusion junction using targeted sequencing (mean spanning read pairs =306.75 vs. 44.75 for total transcriptome sequencing) (Table 1).
The fusion transcripts detected using total RNAseq were also effectively identified using targeted RNAseq. Although the enrichment strategy only utilized probes targeting the coding regions of 13 Ph-like ALL associated kinase genes, the reads mapping to the variable non-targeted fusion partners were easily identified in all four cases from the targeted RNAseq data (Additional data, Excel file 2). This is because the junction fragments containing sequences from both fusion partners could be effectively enriched during library preparation, through partial hybridization to the kinase-partner specific probes within the enrichment probe set. Sequencing of these chimeric fragments subsequently allowed easy identification of the unknown partner genes fused to the targeted kinases. This strategy of targeting only the kinase genes reduces the cost of the highly purified oligo probe sets and proves that chimeric fragments can be efficiently captured for sequencing and readily detected by using probes against only one partner.
Importantly, we verified that expression analysis for the genes of interest which was successfully performed in total RNAseq experiments can also be carried out using targeted RNAseq datasets. Specifically, we observed 10–20× more reads aligning to each studied kinase gene in a sample where that particular kinase was involved in an abnormal fusion (data not shown). Both targeted and total RNAseq therefore provide valuable information about expression levels of the genes of interest.
Discussion
The presence of activating kinase mutations in Ph-like ALL signifies high-risk disease,[16] but has been shown to cause leukemia cells to be vulnerable to TKIs and other targeted therapies.[6] In spite of their clear prognostic and therapeutic significance, diagnostic evaluation for genetic abnormalities associated with Ph-like ALL is still not widely available. The main challenge in reliable detection of Ph-like ALL in the clinical setting is the extreme diversity of the underlying genetic abnormalities,[6] which consist of a variety of chromosomal rearrangements (translocations, inversions, deletions, and duplications) and intragenic deletions and point mutations, affecting more than 15 TK and CR genes and more than 30 genes with other cellular functions.[6,8] This genetic heterogeneity precludes development of a robust, simple and affordable testing strategy to detect Ph-like ALL in the clinical setting. The majority of the gene fusions associated with Ph-like ALL are caused by cryptic chromosomal rearrangements which cannot be detected by conventional cytogenetic analysis, prompting clinical and research laboratories to develop alternative cytogenetic and molecular assay for this disease type (Supplementary Table S7 and Supplementary Figure S3). However, detection of Ph-like ALL cannot be efficiently accomplished by targeted, low-throughput methods like fluorescence in situ hybridization (FISH) and real-time polymerase chain reaction (RT-PCR). Testing strategies that are based on detecting the typical expression signature rather than specific disease-associated genetic alterations [28] are more appropriate as first-line screens; they have to be combined with additional molecular methods to detect exact abnormal fusions or point mutations in leukemia cells, so that appropriate targeted agents can be incorporated into treatment.
The performance of NGS-based approaches for detection of abnormal gene fusions has significantly improved over time. Faulted initially by low specificity and sensitivity due to technical issues (artifacts during library preparation, sequencing errors, poor alignment of short reads) and biological factors (false positives due to read-through transcripts, low expression level of certain fusions), these methods are now more reliable due to development of sophisticated bioinformatics tools which incorporate multi-layered filtering strategies to eliminate false-positive calls without sacrificing the sensitivity.[12,29]
Stadt et al. have recently described NGS-based detection of gene fusions and other genetic abnormalities in Ph-like ALL, using capture-based target enrichment of genomic DNA.[30] However, this requires the sequencing of large intronic regions where translocations breakpoints may occur, resulting high costs for assay development and optimization due to the extensive enrichment-probes synthesized for specific coverage requirements. Furthermore, commercially available RNA-based assays using anchored multiplex PCR (AMP) library preparation followed by NGS [13] require the use of proprietary reagents and technologies that are costly.
A possible shortcoming of our proposed RNA-based method is that it cannot directly identify Ph-like ALL cases caused by CRLF2 and EPOR translocations with the IGH locus, which do not result in production of abnormal fusion transcripts.[31] However, this limitation is compensated by the advantage of possible gene expression analysis. We demonstrated this by conducting expression analysis in our samples, and observing up-regulation of the TK genes involved in the fusion events (Supplementary Figure S2 and Supplementary Table S6). In addition to conducting expression analysis of the RNAseq data, screening for Ph-like ALL cases associated with CRLF2 rearrangements in the clinical setting can be accomplished by flow-cytometry,[32] while a simple FISH assay with the IGH break-apart probe can be used together with NGS testing to screen for translocations involving the EPOR and CRLF2 loci with the IGH locus.
Although the use of RNAseq data for gene expression analysis and even fusion detection has become well-established, reliable detection of sequence variants using RNAseq remains challenging due to the nature of transcriptome assembly that calls for technically difficult computational solutions.[33] However, we hypothesized that even if detection of unknown sequence variants may be unreliable, RNAseq could be adopted to search for a limited number of well characterized hot-spot mutations. The generation of sequence variant information without the need to conduct additional DNA-based targeted or exome sequencing can be highly valuable in both research and clinical settings, thus prompting efforts to develop bioinformatics computational tools that can overcome the issues of detecting variants using RNAseq data.[33,34] From our datasets, we could evaluate the leukemia samples for the presence of sequence variants in genes associated with Ph-like ALL (Supplementary Table S3), without additional cost incurred from DNA-based sequencing that would otherwise have to be conducted in parallel. This additional use of the RNAseq dataset is therefore a cost-effective method to sieve cases of Ph-like ALL for relevant genetic variants for further validation.
Since total RNAseq is associated with substantial reagent, sequencing and analysis costs and cannot be easily implemented in clinical laboratories, we propose the use of targeted RNAseq for detecting Ph-like ALL abnormalities. We found that such targeted panels can also be used to evaluate expression levels of the targeted kinase genes and screen for hot-spot mutations in the targeted expressed transcripts. The sequencing can be accomplished by the more commonly available lower throughput sequencer like the Illumina Miseq instrument. In our workflow (Figure 1), the targeted assay based on enrichment of transcripts corresponding to 13 kinase genes commonly implicated in Ph-like ALL reliably detected the expected abnormal fusion transcripts, with a substantial increase in the number of detected supporting reads as compared to whole transcriptome analysis. The targeted approach also allowed for a significant decrease in sequencing output, analysis time, and computing power.
Hematological malignancies like Ph-like ALL are amenable to the use of RNAseq in the clinical setting due to the relative ease of obtaining fresh unfixed tissue for the extraction of RNA. Budgeting approximately 1 million reads as being sufficient for our targeted RNAseq assay to detect fusion events in Ph-like ALL, the estimated reagent and sequencing cost for the targeted assay is less than $200 per sample, and will continue to decrease as sequencing technologies advance in the future. Additionally, the workflow could be accommodated easily in any clinical NGS laboratory and the RNAseq libraries could be batched for the sequencing runs with other clinical samples processed for other NGS panels. The assay is easily scalable, since probes for additional targets can be added to the enrichment panel as new genes get implicated in the pathogenesis of the disease of interest.
With inclusion of the Ph-like ALL in the 2016 revision to the World Health Organization classification of acute leukemia [35] and growing availability of improved treatment options, the need for reliable detection of Ph-like ALL at the time of diagnosis will keep increasing. Fortunately, NGS-based methods for efficient detection of genetic abnormalities in Ph-like ALL are becoming more readily available.[13,30] Our data show that total and targeted RNAseq combined with a specialized bioinformatics analysis represent additional effective approaches in identification of abnormal gene fusions that are the most frequent mutations responsible for pathogenesis of Ph-like ALL. While whole transcriptome analysis appears optimal for research studies allowing discovery of novel genetic alterations, we propose that the targeted RNAseq approach as a tier one test with the advantage of cost and time savings, can easily be adopted by clinical laboratories, and be used to identify cases of Ph-like ALL in the clinical setting, thus enabling the selection of appropriate targeted therapies.
Supplementary Material
Abbreviations:
- ALL:
acute lymphoblastic leukemia
- Ph:
Philadelphia
- TK:
tyrosine kinase
- TKI:
tyrosine kinase inhibitor
- CR:
cytokine receptor
- NGS:
next-generation sequencing
- RNAseq:
RNA sequencing
- SNP:
single nucleotide polymorphism
- RT-PCR:
real-time polymerase chain reaction
- FISH:
fluorescence in situ hybridization
Footnotes
Potential conflict of interest: Disclosure forms provided by the authors are available with the full text of this article at http://dx.doi.org/10.1080/10428194.2016.1219902.
References
- [1].Jones LK, Saha V. Philadelphia positive acute lymphoblastic leukemia of childhood. Br J Haematol 2005;130:489–500. [DOI] [PubMed] [Google Scholar]
- [2].Fielding AK, Rowe JM, Richards SM, et al. Prospective outcome data on 267 unselected adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia confirms superiority of allogeneic transplantation over chemotherapy in the pre-imatinib era: results from the International ALL Trial MRC UKALLXII/ECOG2993. Blood. 2009;113:4489–4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schultz KR, Bowman WP, Aledo A, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. J Clin Oncol 2009;27:5175–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009;360:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 2009;10:125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 2014;371:1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Roberts K, Payne-Turner D, McCastlain KK, et al. High frequency and poor outcome of Ph-like acute lymphoblastic leukemia in adults. Blood. 2015;126:2618. [Google Scholar]
- [8].Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lengline E, Beldjord K, Dombret H, et al. Successful tyrosine kinase inhibitor therapy in a refractory B-cell precursor acute lymphoblastic leukemia with EBF1-PDGFRB fusion. Haematologica. 2013;98:e146–e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Weston BW, Hayden MA, Roberts KG, et al. Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1-PDGFRB-positive acute lymphoblastic leukemia. J Clin Oncol 2013;31:e413–e416. [DOI] [PubMed] [Google Scholar]
- [11].Mustjoki S, Hernesniemi S, Rauhala A, et al. A novel dasatinib-sensitive RCSD1-ABL1 fusion transcript in chemotherapy-refractory adult pre-B lymphoblastic leukemia with t(1;9)(q24;q34). Haematologica. 2009;94:1469–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Maher CA, Kumar-Sinha C, Cao X, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med 2014;20:1479–1484. [DOI] [PubMed] [Google Scholar]
- [14].Zhao Q, Caballero OL, Levy S, et al. Transcriptome-guided characterization of genomic rearrangements in a breast cancer cell line. Proc Natl Acad Sci USA. 2009;106:1886–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kamran S, Raca G, Nazir K. RCSD1-ABL1 translocation associated with IKZF1 gene deletion in B-cell acute lymphoblastic leukemia. Case Rep Hematol 2015;2015:353247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Raca G, Gurbuxani S, Zhang Z, et al. RCSD1-ABL2 fusion resulting from a complex chromosomal rearrangement in high-risk B-cell acute lymphoblastic leukemia. Leuk Lymphoma. 2015;56:1145–1147. [DOI] [PubMed] [Google Scholar]
- [17].Nicorici D, Satalan M, Edgren H, et al. FusionCatcher – a tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv 2014;doi: 10.1101/011650. [DOI]
- [18].Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc 2015;10:1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lek M, Karczewski K, Minikel E, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tennessen JA, Bigham AW, O’Connor TD, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Abecasis GR, Auton A, Genomes Project C, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chiaretti S, Brugnoletti F, Messina M, et al. CRLF2 overexpression identifies an unfavorable subgroup of adult B-cell precursor acute lymphoblastic leukemia lacking recurrent genetic abnormalities. Leuk Res 2016;41:36–42. [DOI] [PubMed] [Google Scholar]
- [28].Harvey RC, Kang H, Roberts KG, et al. Development and validation of a highly sensitive and specific gene expression classifier to prospectively screen and identify B-precursor acute lymphoblastic leukemia (ALL) patients with a Philadelphia chromosome-like (“Ph-like” or “BCR-ABL1-Like”) signature for therapeutic targeting and clinical intervention. Blood. 2013;122:826. [Google Scholar]
- [29].Stransky N, Cerami E, Schalm S, et al. The landscape of kinase fusions in cancer. Nat Commun 2014;5:4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Stadt UZ, Escherich G, Indenbirken D, et al. Rapid capture next-generation sequencing in clinical diagnostics of kinase pathway aberrations in B-cell precursor ALL. Pediatr Blood Cancer. 2016;63:1283–1286. [DOI] [PubMed] [Google Scholar]
- [31].Moorman AV, Schwab C, Ensor HM, et al. IGH@ translocations, CRLF2 deregulation, and microdeletions in adolescents and adults with acute lymphoblastic leukemia. J Clin Oncol 2012;30:3100–3108. [DOI] [PubMed] [Google Scholar]
- [32].Russell LJ, Capasso M, Vater I, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114:2688–2698. [DOI] [PubMed] [Google Scholar]
- [33].Piskol R, Ramaswami G, Li JB. Reliable identification of genomic variants from RNA-seq data. Am J Hum Genet 2013;93:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cirulli ET, Singh A, Shianna KV, et al. Screening the human exome: a comparison of whole genome and whole transcriptome sequencing. Genome Biol 2010;11:R57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.