

Abstract
The interaction between the heart and brain is complex and integral to the maintenance of normal cardiovascular function. Even in the absence of coronary disease, acute neuronal injury can induce a variety of cardiac changes. Recent neuroimaging data revealed a network including the insular cortex, anterior cingulate gyrus, and amygdala playing a crucial role in the regulation of central autonomic nervous system. Damage in these areas has been associated with arrhythmia, myocardial injury, higher plasma levels of brain natriuretic peptide, catecholamines, and glucose. Some patients after brain injury may die due to occult cardiac damage and functional impairment in the acute phase. Heart failure adversely influences acute stroke mortality. Troponin and NT-proBNP are elevated in acute brain injury patients, in response to the activated renin–angiotensin–aldosterone system and other neurohumoral changes, as a protective mechanism for sympathoinhibitory activity. Such patients have been shown to be associated with higher short- and long-term mortality. While thrombolysis, neuroprotection, and other measures, alone or in combination, may limit the cerebral damage, attention should also be directed toward the myocardial protection. Early administration of cardioprotective medication aimed at reducing increased sympathetic tone may have a role in myocardial protection in stroke patients. For a full understanding of the brain–heart control, the consequences of disruption of this control, the true incidence of cardiac effects of stroke, and the evidence-based treatment options further research are needed.
Keywords: Acute neuronal injury, cardioprotection, insular cortex, receptor cross-talk, stress-induced cardiomyopathy
Introduction
Neurocardiology can be considered in categories such as the effects of the heart on the brain (i.e., embolic stroke of cardiac origin), the brain affecting the heart (i.e., neurogenic heart disease), and neurocardiac syndromes (i.e., Friedreich disease). This review will deal with various presentations of the neurogenic stress cardiomyopathy syndromes, their possible mechanisms, and their clinical management during the perioperative period.
History of Learning the Nature of the Brain–Heart Connection
Ivan Pavlov was the first to describe dysfunction of a visceral organ occurring due to a neurological insult. In 1942, Dr. Walter B. Cannon, Professor of Physiology at Harvard Medical School, published a paper entitled “'Voodoo’ Death,” in which he recounted anecdotal experiences, largely from the anthropology literature, of death from fright. He postulated that death was caused “by a lasting and intense action of the sympathico-adrenal system.”
Neurogenic Heart Disease
A wide variety of electrocardiographic changes are seen in the context of neurogenic heart disease. Dysrhythmias and repolarization changes are the two major categories of change that are regularly noted. It is likely that the life-threatening dysrhythmias found in patients with acute neurological diseases are due to repolarization change and results in ventricular tachycardia and/or ventricular fibrillation. Electrocardiography (ECG) changes are seen in the ST segment and T wave, which reflect abnormalities in repolarization. Most often, the changes are seen best in the anterolateral or inferolateral leads. In myofibrillar degeneration, the cells die in a hypercontracted state with prominent contraction bands. Many authors support the concept that the cause of the myocardial lesions is an autonomic storm with a contribution sympathetic overactivity (humoral arrival at the myocardium from the adrenal and by direct release into the cardiac muscle by intracardiac nerves) and in part caused by parasympathetic overactivity.[1,2]
Nervous System Stimulation
Lesions in the heart, indistinguishable histologically from stress and catecholamine-induced cardiac damage, can result due to neurological stimulation. Lateral hypothalamic stimulation results in hypertension and/or ECG changes similar to that seen in patients with central neurological damage. Other methods which induce these types of cardiac lesions include limbic cortical, mesencephalic reticular formation, stellate ganglion, and other region excitation. These cardiac and ECG abnormalities have been elicited in animals postadrenalectomy[2] and after vagotomy. Stellate ganglion block, beta blockers, and high spinal transections (C2 level) can inhibit the development of these cardiac lesions wherein sympathetic outflow/effect is blocked.[3]
Neurogenic stress cardiomyopathy
Hemorrhage into the subarachnoid space, traumatic brain injury, stroke, either hemorrhagic or ischemic in origin, infections of the central nervous system, acute stress, and epileptiform seizure activity results in a syndrome known as “neurogenic stunned myocardium,” or “neurogenic stress cardiomyopathy” (NSC). The cardiac involvement is manifested either in terms of ECG signs with Q-T interval prolongation, S-T-segment depression, T-wave inversion, and ventricular and supraventricular dysrhythmias, or in the form of left ventricular (LV) regional wall motion abnormalities, troponin release, and increased NT-proBNP.[4,5,6]
It is likely that patients with acute stroke die because of cardiac impairment which is occult in nature. The mechanism underlying could be an atheroma of the coronary artery since the risk factors for stroke and ischemic heart disease are similar, thereby increasing the risk of occult injury to the myocardium with elevated troponin values and impaired myocardial function as evidenced by elevated NT-proBNP. Elevated values of BNP are the most sensitive index and a measure of occult myocardial impairment.[7] Death due to impaired cardiac function may explain partly, failure of measures to reduce neurological damage not impacting mortality.
ECG changes are more commonly seen in patients with intracerebral (60%–70%) or subarachnoid haemorrhage (SAH) (40%–70%) than among those with ischemic stroke (15%–40%).[8] Unlike myocardial ischemia, cerebrogenic ECG changes evolve over several days and disappear in 2 weeks, but QT prolongation or U waves can be permanent. ECG abnormalities occur in 25%–75% of SAH patients and dysrhythmias are present in almost 100% of patients.[8] Serum markers of cardiac injury are increased in 20%–30% of patients with the most severe grades of SAH and wall motion abnormalities occur in 8%–13% of patients with regional or global kinetic patterns. Neurocardiogenic injury is associated with an increased risk of all-cause mortality, cardiac mortality, and heart failure. Despite high morbidity and mortality, NSC management is mainly supportive and symptomatic, based on the treatment of life-threatening events.
Pathophysiology/mechanisms for Neurogenic Stress Cardiomyopathy
The cardiac component of the problem
NSC is secondary to structural or functional brain damage and part of the stress-related cardiomyopathy syndrome spectrum including takotsubo syndrome with it's typical apical and midventricular myocardial dysfunction and significant overlap with NSC in clinical presentation, underlying pathophysiology, and reversibility.
The term NSC reflects the underlying pathophysiology of myocardial dysfunction related to the stress of catecholamine excess, triggered by an acute brain injury. The primary form of stress-related cardiomyopathy is likened to takotsubo cardiomyopathy. Stress-related cardiomyopathy syndrome has been ascribed to transient spasm of multiple coronary vessels, dysfunction of the microvasculature, spontaneous thrombolysis in the coronaries resulting in aborted myocardial infarction,[9] and the “catecholamine theory.”.[6] However, “catecholamine hypothesis,” consistent with catecholamine-mediated direct myocardial injury, is widely accepted.
The left ventricle contains apical-basal gradients of β-adrenergic receptors (β-ARs) and sympathetic innervation, with the apex characterized by highest βAR and lowest sympathetic nerve density. This pattern results in increased apical responsiveness to circulating catecholamines, predominantly epinephrine from the adrenal glands, as a compensatory mechanism for the sparse apical sympathetic innervation, to ensure optimal ventricular ejection during times of stress. High plasma levels of epinephrine can act as a negative inotrope through ligand-mediated trafficking of the AR β2AR from stimulatory G protein to inhibitory G protein subcellular signaling pathways. The β2AR is widely reported as being pleiotropic, having the potential to couple through Gs-adenylate cyclase-cAMP (like the β1 AR) and also through Giα, Gβγ, and non-G-protein pathways.
The process of ligand- or stimulus-directed trafficking or biased agonism explains that, at higher concentrations of epinephrine, the β2AR switches coupling to inhibitory Gi from Gs protein. This switch happening in favorable conditions of high catecholamine stress is because it depends on β2AR phosphorylation by both protein kinase A and G-protein receptor-coupled kinases (GRKs). Norepinephrine has a much lower affinity (20 times less) for the β2AR compared to the β1AR and also weaker β2AR stimulus trafficking to the Gi pathway. Although this negative inotropy is detrimental from a mechanical perspective, the Gs to Gi switch is potentially both antiapoptotic and antidysrhythmic and may represent a cardioprotective mechanism against β1AR catecholamine cardiotoxicity.
Polymorphisms of adrenergic receptors
Cardiac responsiveness to catecholamines is affected by genetic polymorphisms of the adrenoceptors. Single nucleotide polymorphisms of the adrenoreceptor have been associated with cardiac dysfunction of 3- to 5-fold increase, whereas combinations of two such polymorphisms after SAH showed a 10–15-fold increased risk for myocardial injury.[10] In patients with LV apical ballooning syndrome, a correlation was found with a polymorphism of G protein-coupled receptor kinase 5, a protein involved in postreceptor signal transduction.
Effects of high catecholamine level
High myocardial interstitial concentrations of norepinephrine result in myocyte calcium overload and cell death causing cardiac dysfunction, free radical production, and adenosine triphosphate depletion, with resulting ECG changes, failure of myocardial contraction, and possible cell death. The “myocardial contraction band” characterized by hypercontraction of sarcomeric myofibrils, eosinophilic transverse bands, and mononuclear infiltration of the interstitium has been associated with extreme sympathetic discharge. Cell death with ischemia results in cell relaxation, necrosis, and polymorphonuclear response in tissues with compromised vascular supply. In NSC, cell death results in hypercontracted state with the formation of contraction bands, early onset of calcification, and lesions myofibrillar in nature seen within minutes of onset, in proximity to cardiac innervation. Furthermore, regional wall motion abnormalities are reversible and not limited to single epicardial vessel distribution.
Role of inflammation
Inflammation has also been proposed as a contributory mechanism for myocardial injury in NSC. Release of pro-inflammatory cytokines is inhibited with acetylcholine, but dysfunction of the parasympathetic system can result in myocardial damage due to unmitigated inflammatory process. Patients with SAH have shown cerebrospinal fluid and serum levels of cytokinins being elevated contributing to enhancing neurocardiogenic injury. Comprehensive assessment of LV dysfunction may be performed by evaluation of LV systolic function, myocardial perfusion assessment, and test for sympathetic innervation of the heart.
The insular cortex and cardiovascular system
Literature on neurocardiology has focused mainly on the subcortical regions of the central autonomic nervous system. Recent studies support the thought that the cardiovascular system is regulated by modulation of the cortex.[11,12] Modern neuroimaging data using positron emission tomography and functional magnetic resonance revealed a network including the insular cortex, anterior cingulate gyrus, and amygdala playing a crucial role in the regulation of the central autonomic nervous system. Because the insular cortex is located in the region of the middle cerebral arteries, its structure tends to be exposed to a higher risk of cerebrovascular disease. The insular cortex damage has been associated with dysrhythmia, diurnal blood pressure variation disruption (e.g., a nondipper or riser pattern), myocardial injury, and sleep-disordered breathing, as well as higher plasma levels of brain natriuretic peptide, catecholamine, and glucose.[13]
The hypothalamic-pituitary-adrenocortical and sympathoadrenomedullary axes are the main biological systems activated during the stress response. Lateralization for cardiovascular function, with sympathetic tone predominantly regulated in the right insular region and parasympathetic effects situated in the left insula, is supported by several studies.[13,14] Bradycardia or hypotension was more frequent with stimulation of the left insular cortex, whereas tachycardia or hypertension was elicited if the right insula was stimulated.[14] The mechanism of NSC after acute brain injury may be related to disinhibition of the right insular cortex and a resulting enhancement of sympathetic tone.
Associations between heart rate variability and specific brain regions including the amygdala and ventromedial prefrontal cortex have been evidenced, further supporting a structural and functional link between the brain and the heart. Involvement of the insular cortex by cerebral infarction is associated with a nocturnal rise of blood pressure, QT prolongation, and cardiac dysrhythmias. In addition, the involvement of the insular cortex has a stronger association with the cardiovascular and autonomic disturbances than the infarction size and is an independent predictor of poor long-term outcome in patients with their first unilateral thromboembolic cerebral infarction. Amygdala is considered as a mediator of cardiovascular responses to emotional stimuli and particularly of negative stress responses.[15] Furthermore, damage to the hypothalamus is correlated with myocardial necrosis in SAH.[16] In addition, the brainstem is essential in mediating autonomic tone of the cardiovascular system.[17]
Neurogenic cardiac damage
Neurogenic cardiac damage after acute stroke and overactivation of β-ARs by catecholamine excess can lead to tonic opening of calcium channels, causing impaired sequestration of intracellular calcium ions, which is a necessary process for relaxation of cardiac muscles.[18] The prolonged contraction of cardiac muscles can lead to cell damage or death. Elevated troponin T is a poor prognostic sign after acute ischemic stroke.[19] Troponin elevations correlated with the severity of neurologic injury and cardiovascular abnormalities including LV dysfunction, pulmonary edema, and hypotension requiring vasopressors.
Monitoring for Neurogenic Stress Cardiomyopathy
All SAH patients should be screened on admission with a full cardiac evaluation including a complete clinical history, a chest X-ray, lipid profile, and electrolyte panel. A baseline assessment of cardiac function with troponin levels, serial enzymes, ECG, NT-proBNP level, and echocardiography may be beneficial, especially if any sign of myocardial dysfunction is present and cardiac output should be monitored in those patients with myocardial dysfunction or hemodynamic instability. An ejection fraction of <40% and troponin I <2.8 ng/ml are predictive of NSC rather than acute coronary syndromes. Elevated BNP levels also occur after SAH. A 2–3-fold increase in BNP levels in plasma but not in cerebrospinal fluid supports the heart as the source of increased BNP levels after SAH. Cardiac catheterization and cardiac MRI may be considered in patients to differentiate an acute coronary syndrome. Indices of sympathetic activity that can be used in the evaluation of NSC are tissue, plasma and urine catecholamine levels, heart rate variability, baroreflex sensitivity, cardiac metaiodobenzylguanidine (MIBG) scintigraphy, and microneurography.
Heart rate variability
It represents the beat-to-beat variability of heart rate. Power spectral analysis of sequence of 500 R–R intervals can detect two major bands: low frequency (LF) is the expression of baroreceptor-mediated regulation and occurs because of the contribution of parasympathetic and mainly sympathetic discharge, and high frequency (HF) reflects the modulation of vagus nerve discharge caused by respiration. Heart rate variability may detect the presence of autonomic neuropathy complicating acute brain injury. LF%, LF/HF, and Hunt and Hess Class are independent predictors of in-hospital mortality in patients with SAH.[20]
Baroreflex sensitivity
It is a measure of the reflex response (both vagal and sympathetic) to the stimulation of the baroreceptors induced by arterial pressure changes. It can be measured by infusion of vasoactive drugs or noninvasively through spontaneous variations in arterial pressure and R–R intervals.
Cardiac metaiodobenzylguanidine scintigraphy
This is the detection of the uptake of a specific tracer showing the activity of sympathetic postsynaptic fibers. MIBG bears a structural resemblance to norepinephrine, and the uptake of MIBG in various tissues closely parallels that of norepinephrine. MIBG is useful for the assessment of cardiac sympathetic nervous viability and function.
Microneurography MNSA, SSA, RNSA (muscle, skin, and renal sympathetic nerve activity)
These are direct recordings of the activity of the vasoconstrictive sympathetic fibers at muscle, skin, and renal levels.
Identification of high-risk patients
Identification of high-risk patients after acute stroke is important to arrange appropriate cardiac monitoring and effective management of dysrhythmias and to prevent cardiac morbidity and mortality. As higher National Institutes of Health Stroke Scale (NIHSS) correlates with impaired cardiovascular autonomic control, the NIHSS can be used to stratify risk of serious cardiac event. A history of heart failure,[21] high baseline serum creatinine, stroke severity, and long-corrected QT interval (QTc) or ventricular extrasystole, were independent risk factors for serious cardiac events. Prolonged and intensive cardiovascular monitoring is recommended in patients manifesting cerebrogenic cardiovascular disturbances and in high-risk patients with insular involvement, right-sided stroke, advancing age, coexisting hypertensive or coronary artery disease, or intense emotional stress. Severely affected patients should be evaluated by a cardiologist before the initiation of appropriate therapy.
Proposed potential treatments for neurogenic stress cardiomyopathy
Although the occurrence of NSC after acute brain injury is well recognized, anesthetic challenges of the neurologically critically ill patient presenting with NSC, the possible occurrence of myocardial stunning during anesthesia, and the impact of anesthesia on the autonomic nervous system during the perioperative period have not been fully explored. Hemodynamic instability, dysrhythmias, cardiogenic shock, pulmonary edema, and sudden cardiac death are the main concerns with a clinical presentation such as myocardial infarction. With high indices of overactivity of the sympathetic limb of the autonomic nervous system, there is a strong physiological role for beta blockade. Many studies have shown that β-blocker use was associated with less severe stroke on presentation and exerted its protective effect through sympatholytic action, inhibition of thrombin generation, reduced inflammation, and also protection of cardiac myocytes from norepinephrine-stimulated apoptosis.[22,23]
Although controversial, neurogenic stunned myocardium may benefit from inotropic medication to maintain equilibrium between myocardial oxygen supply and demand. Epinephrine-mediated stunning is recognized as a causative factor for NSC; administration of epinephrine may worsen negative inotropism further increasing the switch from Gs protein signaling to Gi protein signaling. Sympathomimetic drugs should be used with caution. Levosimendan theoretically may be the inotrope of choice in takotsubo cardiomyopathy-related shock; it increases myocardial systolic performance, improves coronary perfusion, has an antiapoptotic and an antistunning effect.[24] In patients with life-threatening acute LV failure, intra-aortic balloon pump and ventricular assist devices may also be necessary. In a retrospective analysis, SAH grading, increased plasma norepinephrine, and decreased plasma estradiol levels were independently associated with myocardial regional wall motion abnormalities.[25] Based on these results, the administration of estradiol to postmenopausal female patients with SAH complicated by NSC could be considered a possible therapeutic target. Furthermore, in patients with SAH after an aneurysmal bleed, changes in ECG such as prolongation of QTc interval, bradycardia, abnormalities of conduction, and changes in echocardiogram, can recover following aneurysmal clipping, on the 1st postoperative day.[26] Those with persistent changes in both ECG and ECHO have poor outcomes when followed up at 1 year. The presence of a thrombus in the LV may require anticoagulation to prevent systemic embolization if not contraindicated. Potential organ donors who have been treated aggressively with steroids, insulin, vasopressin, and thyroxine should be allowed to recover from NSC.
Cardioprotection in neurogenic stress cardiomyopathy
Occult cardiac impairment may account for stroke mortality, which opens a channel for cardioprotective intervention in NSC. Patients on beta blockers and angiotensin II receptor antagonists have shown mortality benefits in terms of stroke survival.[27,28] The role of drugs such as β-blockers, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, spironolactone, and other novel molecules with the potential of protecting the myocardium from adverse effects of the activated RAAS in the acute stroke phase have been studied. The benefit of using these drugs for acute stroke patients being the lack of need for neuroimaging and also widening the window period for therapy to a few days rather than a few hours. This may also improve cardiac functions such as contractility, which in turn can increase cerebral perfusion. This could be another potential benefit of protecting the heart for better perfusion of the brain.
Takotsubo cardiomyopathy
This is a stress-related cardiomyopathy syndrome mimicking an acute coronary event without demonstrable coronary artery stenosis or spasm, in which the heart takes on the appearance of a Japanese octopus fishing pot called a takotsubo [Figure 1].[29] Until now, there is no consensus on the diagnostic criteria for takotsubo cardiomyopathy. The modified Mayo clinic proposed diagnostic criteria includes: (1) transient hypokinesis, akinesis, or dyskinesis in the LV midsegments with or without apical involvement; regional wall motion abnormalities that extend beyond a single epicardial vascular distribution; and frequently, but not always, a stressful trigger; (2) the absence of obstructive coronary disease or angiographic evidence of acute plaque rupture; (3) new ECG abnormalities (ST-segment elevation and/or T-wave inversion) or modest elevation in cardiac troponin; and (4) the absence of pheochromocytoma and myocarditis.[30] This syndrome shares common pathophysiological mechanisms with NSC [Figure 2]. The switch of β2-receptors from Gs to Gi signaling causes stimulation of the cardioprotective PI3K/AKT signaling pathway, explaining the modest and patchy presence of myocardial cell death in takotsubo.[31]
Figure 1.
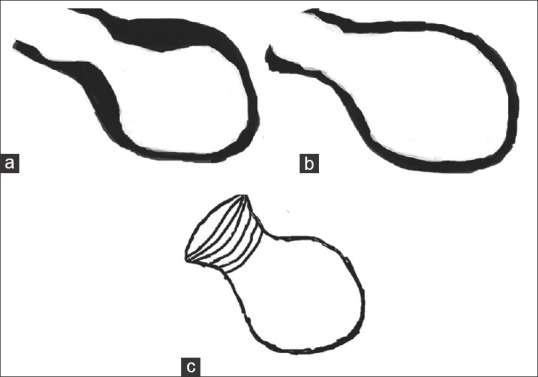
Schematic diagram of (a) end systolic and (b) end diastolic left ventricular silhouette with apical ballooning, (c) Japanese takotsubo octopus trap
Figure 2.
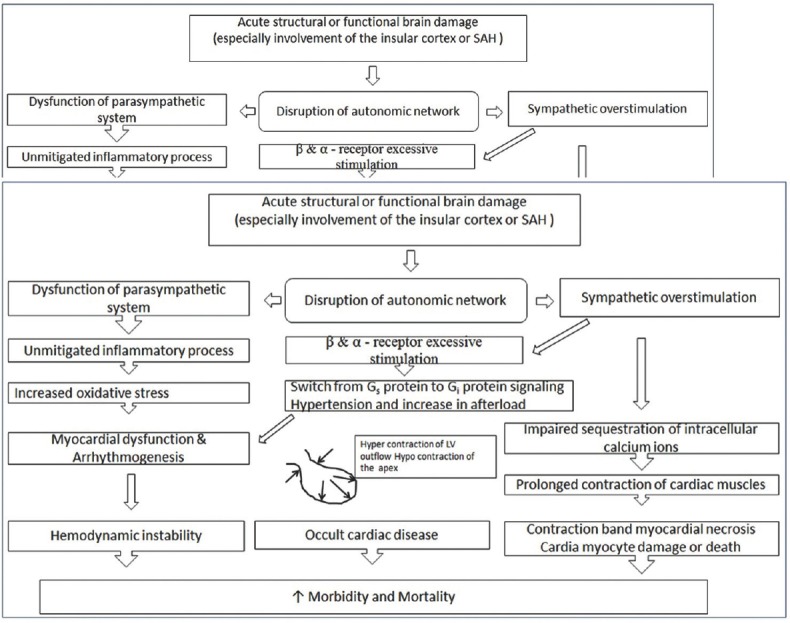
Pathophysiology of neurogenic stress cardiomyopathy
Reversibility of ventricular function occurs once the surge in epinephrine levels has cleared and the β2AR receptors switch back to Gs protein, enabling the inotropic function of the cardiomyocytes to recover. Under anesthesia, the stress of tracheal intubation and the consequent sympathetic reflex stimulation may explain the occurrence of takotsubo at induction. Myocardial dysfunction may also occur because of anaphylaxis, meperidine-induced histamine release, transfusion reaction as a result of histamine release, activation of the so-called histamine-adrenergic crosstalk, and the infusion of adrenergic drugs. Regional anesthesia provides optimal postoperative pain control but takotsubo syndrome has also been described during spinal anesthesia and in response to uncontrolled pain. Any potential triggering event that could result in a catecholamine surge and consequent cardiac dysfunction should be avoided during the entire perioperative period.
Opioids and the central α2-inhibitor, dexmedetomidine, both of which target the locus coeruleus, which regulates the reflex response to stress, may be the most appropriate drugs. Dexmedetomidine may also protect against psychological stress by depressing the activity of the amygdala related to anxiety. Magnesium, involved in several fundamental processes including gating of calcium channels, regulation of adenylate cyclase, cardiac excitability, control of vasomotor tone, and neurotransmitter release, has also been proposed as a potential drug to attenuate the stress response.
Conclusion
Intense brain–heart crosstalk is increasingly recognized in the acute phase after severe neurologic injury, NSC being the best known clinical life-threatening expression. Identifying patients at risk at an early stage of NSC and monitoring cardiovascular status at admission should be considered. A more complete understanding of the pathogenesis of the NSC requires further research. Vigilance and a high index of suspicion are essential to avoid misdiagnosis or delayed recognition, and the entire health-care team should be educated in the recognition of this potentially life-threatening syndrome. Much more clinical and basic research is needed to allow a full understanding of the brain–heart control, the consequences of disruption of this control, the true incidence of cardiac effects of stroke, and the evidence-based treatment strategies. The pathogenesis of myocyte damage in acute neuronal injury is thought to be neurally mediated through abnormal autonomic activity. However, coexistent acute coronary syndrome must also be considered.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Greenhoot JH, Reichenbach DD. Cardiac injury and subarachnoid hemorrhage. A clinical, pathological, and physiological correlation. J Neurosurg. 1969;30:521–31. doi: 10.3171/jns.1969.30.5.0521. [DOI] [PubMed] [Google Scholar]
- 2.Hawkins WE, Clower BR. Myocardial damage after head trauma and simulated intracranial haemorrhage in mice: The role of the autonomic nervous system. Cardiovasc Res. 1971;5:524–9. doi: 10.1093/cvr/5.4.524. [DOI] [PubMed] [Google Scholar]
- 3.Silvani A, Calandra-Buonaura G, Dampney RA, Cortelli P. Brain-heart interactions: Physiology and clinical implications. Philos Trans A Math Phys Eng Sci. 2016;374 doi: 10.1098/rsta.2015.0181. pii: 20150181. [DOI] [PubMed] [Google Scholar]
- 4.Mazzeo AT, Micalizzi A, Mascia L, Scicolone A, Siracusano L. Brain-heart crosstalk: The many faces of stress-related cardiomyopathy syndromes in anaesthesia and intensive care. Br J Anaesth. 2014;112:803–15. doi: 10.1093/bja/aeu046. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen H, Zaroff JG. Neurogenic stunned myocardium. Curr Neurol Neurosci Rep. 2009;9:486–91. doi: 10.1007/s11910-009-0071-0. [DOI] [PubMed] [Google Scholar]
- 6.Lee VH, Oh JK, Mulvagh SL, Wijdicks EF. Mechanisms in neurogenic stress cardiomyopathy after aneurysmal subarachnoid hemorrhage. Neurocrit Care. 2006;5:243–9. doi: 10.1385/NCC:5:3:243. [DOI] [PubMed] [Google Scholar]
- 7.Lehman R, Doust J, Glasziou P. Cardiac impairment or heart failure? BMJ. 2005;331:415–6. doi: 10.1136/bmj.331.7514.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheung RT, Hachinski V. Cardiac effects of stroke. Curr Treat Options Cardiovasc Med. 2004;6:199–207. doi: 10.1007/s11936-996-0014-x. [DOI] [PubMed] [Google Scholar]
- 9.Ibanez B, Choi BG, Navarro F, Farre J. Tako-tsubo syndrome: A form of spontaneous aborted myocardial infarction? Eur Heart J. 2006;27:1509–10. doi: 10.1093/eurheartj/ehl021. [DOI] [PubMed] [Google Scholar]
- 10.Zaroff JG, Pawlikowska L, Miss JC, Yarlagadda S, Ha C, Achrol A, et al. Adrenoceptor polymorphisms and the risk of cardiac injury and dysfunction after subarachnoid hemorrhage. Stroke. 2006;37:1680–5. doi: 10.1161/01.STR.0000226461.52423.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williamson JW, Fadel PJ, Mitchell JH. New insights into central cardiovascular control during exercise in humans: A central command update. Exp Physiol. 2006;91:51–8. doi: 10.1113/expphysiol.2005.032037. [DOI] [PubMed] [Google Scholar]
- 12.Nagai M, Hoshide S, Ishikawa J, Shimada K, Kario K. Insular cortex atrophy as an independent determinant of disrupted diurnal rhythm of ambulatory blood pressure in elderly hypertension. Am J Hypertens. 2009;22:723–9. doi: 10.1038/ajh.2009.71. [DOI] [PubMed] [Google Scholar]
- 13.Nagai M, Hoshide S, Kario K. The insular cortex and cardiovascular system: A new insight into the brain-heart axis. J Am Soc Hypertens. 2010;4:174–82. doi: 10.1016/j.jash.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Oppenheimer SM, Gelb A, Girvin JP, Hachinski VC. Cardiovascular effects of human insular cortex stimulation. Neurology. 1992;42:1727–32. doi: 10.1212/wnl.42.9.1727. [DOI] [PubMed] [Google Scholar]
- 15.Cheung RT, Hachinski VC, Cechetto DF. Cardiovascular response to stress after middle cerebral artery occlusion in rats. Brain Res. 1997;747:181–8. doi: 10.1016/s0006-8993(96)01137-7. [DOI] [PubMed] [Google Scholar]
- 16.Furlan JC, Fehlings MG. Cardiovascular complications after acute spinal cord injury: Pathophysiology, diagnosis, and management. Neurosurg Focus. 2008;25:E13. doi: 10.3171/FOC.2008.25.11.E13. [DOI] [PubMed] [Google Scholar]
- 17.Coote JH. Landmarks in understanding the central nervous control of the cardiovascular system. Exp Physiol. 2007;92:3–18. doi: 10.1113/expphysiol.2006.035378. [DOI] [PubMed] [Google Scholar]
- 18.Koppikar S, Baranchuk A, Guzmán JC, Morillo CA. Stroke and ventricular arrhythmias. Int J Cardiol. 2013;168:653–9. doi: 10.1016/j.ijcard.2013.03.058. [DOI] [PubMed] [Google Scholar]
- 19.James P, Ellis CJ, Whitlock RM, McNeil AR, Henley J, Anderson NE. Relation between troponin T concentration and mortality in patients presenting with an acute stroke: Observational study. BMJ. 2000;320:1502–4. doi: 10.1136/bmj.320.7248.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiu TF, Huang CC, Chen JH, Chen WL. Depressed sympathovagal balance predicts mortality in patients with subarachnoid hemorrhage. Am J Emerg Med. 2012;30:651–6. doi: 10.1016/j.ajem.2011.02.037. [DOI] [PubMed] [Google Scholar]
- 21.Zaroff JG, Leong J, Kim H, Young WL, Cullen SP, Rao VA, et al. Cardiovascular predictors of long-term outcomes after non-traumatic subarachnoid hemorrhage. Neurocrit Care. 2012;17:374–81. doi: 10.1007/s12028-011-9592-x. [DOI] [PubMed] [Google Scholar]
- 22.Laowattana S, Oppenheimer SM. Protective effects of beta-blockers in cerebrovascular disease. Neurology. 2007;68:509–14. doi: 10.1212/01.wnl.0000253186.23949.fd. [DOI] [PubMed] [Google Scholar]
- 23.Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation. 1998;98:1329–34. doi: 10.1161/01.cir.98.13.1329. [DOI] [PubMed] [Google Scholar]
- 24.Padayachee L. Levosimendan: The inotrope of choice in cardiogenic shock secondary to takotsubo cardiomyopathy? Heart Lung Circ. 2007;16(Suppl 3):S65–70. doi: 10.1016/j.hlc.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 25.Sugimoto K, Inamasu J, Hirose Y, Kato Y, Ito K, Iwase M, et al. The role of norepinephrine and estradiol in the pathogenesis of cardiac wall motion abnormality associated with subarachnoid hemorrhage. Stroke. 2012;43:1897–903. doi: 10.1161/STROKEAHA.111.646893. [DOI] [PubMed] [Google Scholar]
- 26.Jangra K, Grover VK, Bhagat H, Bhardwaj A, Tewari MK, Kumar B, et al. Evaluation of the effect of aneurysmal clipping on electrocardiography and echocardiographic changes in patients with subarachnoid hemorrhage: A Prospective observational study. J Neurosurg Anesthesiol. 2017;29:335–40. doi: 10.1097/ANA.0000000000000318. [DOI] [PubMed] [Google Scholar]
- 27.Barer DH, Cruickshank JM, Ebrahim SB, Mitchell JR. Low dose beta blockade in acute stroke (“BEST” trial): An evaluation. Br Med J (Clin Res Ed) 1988;296:737–41. doi: 10.1136/bmj.296.6624.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schrader J, Lüders S, Kulschewski A, Berger J, Zidek W, Treib J, et al. The ACCESS study: Evaluation of acute candesartan cilexetil therapy in stroke survivors. Stroke. 2003;34:1699–703. doi: 10.1161/01.STR.0000075777.18006.89. [DOI] [PubMed] [Google Scholar]
- 29.Bybee KA, Prasad A. Stress-related cardiomyopathy syndromes. Circulation. 2008;118:397–409. doi: 10.1161/CIRCULATIONAHA.106.677625. [DOI] [PubMed] [Google Scholar]
- 30.Prasad A, Lerman A, Rihal CS. Apical ballooning syndrome (Tako-tsubo or stress cardiomyopathy): A mimic of acute myocardial infarction. Am Heart J. 2008;155:408–17. doi: 10.1016/j.ahj.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 31.Paur H, Wright PT, Sikkel MB, Tranter MH, Mansfield C, O'Gara P, et al. High levels of circulating epinephrine trigger apical cardiodepression in a β2-adrenergic receptor/Gi-dependent manner: A new model of takotsubo cardiomyopathy. Circulation. 2012;126:697–706. doi: 10.1161/CIRCULATIONAHA.112.111591. [DOI] [PMC free article] [PubMed] [Google Scholar]