

Abstract
Purpose:
To describe inflammatory ocular findings in patients with autoimmune lymphoproliferative syndrome (ALPS).
Methods:
Retrospective review of medical records for ALPS patients seen at National Eye Institute between 2003 and 2013.
Results:
Twenty-nine ALPS patients previously referred for ocular or visual symptoms or history of prolonged corticosteroid use were identified. Mean age was 20 years (range, 4–66 years). The majority were male (n = 21, 72.4%) and Caucasian (n = 24, 82.8%). Ten (34.5%) had abnormal ocular findings, the most common of which was an ocular inflammatory disorder (n = 4, 13.8%). Uveitis was seen in two patients with ALPS-FAS and one with ALPS-U, all of whom required long-term systemic immunosuppression. One patient with ALPS-FAS had history of optic neuritis.
Conclusions:
ALPS can have intraocular inflammatory manifestations that require routine follow-up to ensure appropriate and timely treatment of intraocular disease. Long-term immunosuppression may be needed for patients with ALPS-associated uveitis.
Keywords: ALPS, autoimmune lymphoproliferative syndrome, eye findings, ocular inflammation, uveitis
INTRODUCTION
Autoimmune lymphoproliferative syndrome (ALPS) is a rare inherited disorder that manifests early in life with generalized lymphadenopathy, splenomegaly, hepatomegaly, multi-lineage autoimmune cytopenias, and the expansion of CD4-CD8- double negative T cells in the circulation.1 Mutations in genes that mediate lymphocyte apoptosis have been shown to result in inappropriate persistence and accumulation of autoreactive and potentially oncogenic lymphocytes and have become the basis of ALPS diagnostic classification.1, 2 Patients with germline homozygous or heterozygous mutations in FAS are classified as ALPS-FAS, patients with somatic mutations in FAS are classified as ALPS-sFAS, patients with germline mutations in FAS ligand (FASLG) are classified as ALPS-FASLG, and patients with germline mutations in caspase 10 are classified as ALPS-CASP10.3 Patients who meet diagnostic criteria but do not have known genetic mutations in FAS, FASLG, or CASP10 are classified as ALPS-U (undetermined).
Mutations in the FAS gene are most frequently found in patients with ALPS.4 Fas is a 48-kDa type I transmembrane protein of the tumor necrosis factor (TNF) superfamily, and Fas ligand (FasL) is a 40-kDa type II transmembrane protein also of the TNF superfamily that binds Fas to induce programmed cell death.2, 5 Their normal interaction is important for maintaining self-tolerance, while their dysfunction plays a key role in the pathogenesis of various autoimmune diseases, including Hashimoto’s thyroiditis, multiple sclerosis, Sjögren’s syndrome, and type 1 diabetes mellitus.6–8 Autoimmunity frequently occurs in ALPS and usually presents 2 to 3 years after lymphoproliferation. The most common autoimmune manifestations of ALPS are autoimmune hemolytic anemia (AIHA), immune-mediated thrombocytopenia (ITP), and autoimmune neutropenia (AIN); autoimmune liver and kidney disease are less common.1, 9 ALPS patients are also at increased risk of both Hodgkin’s and non-Hodgkin’s lymphoma and therefore require close surveillance.10
The interaction between FAS and FASLG is particularly important for maintaining cell homeostasis in immune-privileged organs, such as the testes, brain, placenta, and the eye.2 In the eye, Fas/FasL binding triggers apoptosis in invading Fas-positive inflammatory cells, thereby preventing inflammation and maintaining immune privilege within the eye.11 While autoimmune diseases are well known to be associated with ALPS, only two cases of anterior uveitis and one case of panuveitis have been previously reported in association with ALPS.4, 12 The purpose of this study was to describe the ocular features, primarily those that are inflammatory in nature, in patients with ALPS.
PATIENTS AND METHODS
All patients with a diagnosis of ALPS were seen under the ALPS Natural History Study protocol (NCT00001350) at the National Institute of Allergy and Infectious Diseases (NIAID). Patients who reported ocular or visual symptoms during a study physician’s review of systems were referred to the National Eye Institute (NEI) for further evaluation. Patients with a history of prolonged corticosteroid use were also referred for ophthalmologic evaluation due to higher risk of developing cataracts and elevated intraocular pressure. Those referred between 2003 and 2013 were identified using a database search of NEI electronic medical records. Patients were seen at the NEI under clinical research protocols NCT00655096 or NCT00708955. All study protocols (NCT00001350, NCT00655096 and NCT00708955) were approved by Institutional Review Boards at the National Institutes of Health (NIH) and adhered to the tenets of the Declaration of Helsinski.
All patients underwent a complete ophthalmologic examination with dilation. Demographic and clinical data, including age, race/ethnicity, sex, clinical history, best-corrected visual acuity (BCVA), ocular findings, and ophthalmic imaging findings, were collected by retrospective chart review. For patients with ocular manifestations, the nature and anatomic location of ocular findings (e.g., anterior segment, posterior segment) were noted. For those with manifestations of intraocular inflammation, the location and activity of inflammation were determined by using Standardization of Uveitis Nomenclature criteria.13 All results are reported with descriptive statistics.
RESULTS
Twenty-nine patients with a genetic diagnosis of ALPS were identified. Mean age at diagnosis was 20 years (range, 4–66 years), and the majority of these patients were male (n = 21, 72.4%) and Caucasian (n = 24, 82.8%). At the time of referral, median BCVA was 20/20 in both eyes (range, 20/16–20/32 in right eye, 20/16–20/40 in left eye), intraocular pressure was within normal limits, and 10 (34.5%) patients had one or more findings on ophthalmologic examination. Ocular findings were variable and are summarized in Table 1. Because ALPS is known to be associated with various autoimmune diseases, we mainly focused on evaluating the presence of ocular inflammatory disease in this cohort. Of 29 patients, four (13.8%) had evidence of intraocular inflammation, including uveitis (n = 3; 10.3%) and history of optic neuritis (n = 1; 3.4%).
Table 1.
Ophthalmic Findings in 10 ALPS Patients Referred for Ocular or Visual Complaints
| No. | Gender | Age (years) | ALPS Diagnosis | Finding(s) on Ophthalmologic Examination |
|---|---|---|---|---|
| 1 | F | 4 | ALPS-FAS | Panuveitis (bilateral)1 |
| 2 | M | 21 | ALPS-U | Posterior uveitis with choroidal infiltrates (bilateral)1 |
| 3 | M | 36 | ALPS-FAS | Severe diabetic retinopathy treated with panretinal photocoagulation (type 1 diabetes mellitus) |
| 4 | F | 48 | ALPS-FAS | Inactive chorioretinal scar, mild optic nerve pallor (secondary to prior episode of optic neuritis)1 |
| 5 | F | 19 | ALPS-FAS | Megalopapillae2 |
| 6 | M | 15 | ALPS-FAS | Strabismus |
| 7 | F | 66 | ALPS-FAS | Bilateral lacrimal gland and extraocular muscle enlargement |
| 8 | M | 16 | ALPS-FAS | Anterior-intermediate uveitis (bilateral)1 |
| 9 | M | 23 | ALPS-FASLG | Trichiasis, peripheral corneal scar |
| 10 | M | 24 | ALPS-FAS | Megalopapillae2 |
Evidence of past or currently active intraocular inflammation
No evidence of ocular hypertension or glaucomatous changes, likely congenital
Abbreviation: ALPS, autoimmune lymphoproliferative syndrome; FASLG, FAS ligand; U, undetermined Of 29 ALPS patients referred to the National Eye Institute for ocular or visual symptoms or for history of prolonged corticosteroid use, 10 patients had findings on ophthalmologic examination. These findings were varied, and a few patients had multiple findings on examination. Three patients had active intraocular inflammation in both eyes. One patient had remote history of optic neuritis and vitreous hemorrhage secondary to immune-mediated thrombocytopenia with an inactive chorioretinal scar.
Case 1
Anterior-intermediate uveitis was observed in a 16-year-old Hispanic male with ALPS-FAS, who developed uveitis 4 years after his ALPS diagnosis. The patient also had a history of thrombocytopenia, lymphadenopathy, and splenomegaly, for which he previously received corticosteroids, intravenous immunoglobulin (IVIG), and rituximab. At the time of examination, he had excellent visual acuity (VA) at 20/16 in both eyes. Ophthalmologic examination showed in both eyes: trace AC cells with no flare, posterior synechiae, trace vitreous cells with snowballs in the left eye but no haze, and mild peripheral vascular sheathing (Fig. 1A-D). Fluorescein angiography (FA) also revealed peripheral vascular leakage in both eyes (Fig. 1E,F). The patient was started on mycophenolate mofetil (MMF) with good response, but he required additional topical corticosteroids for mild recurrences of AC inflammation and occasional periocular corticosteroid injections for poor compliance with systemic medications. After 4.5 years of follow-up, his VA remained at 20/16 and 20/20 in his right and left eyes, respectively.
Figure 1:
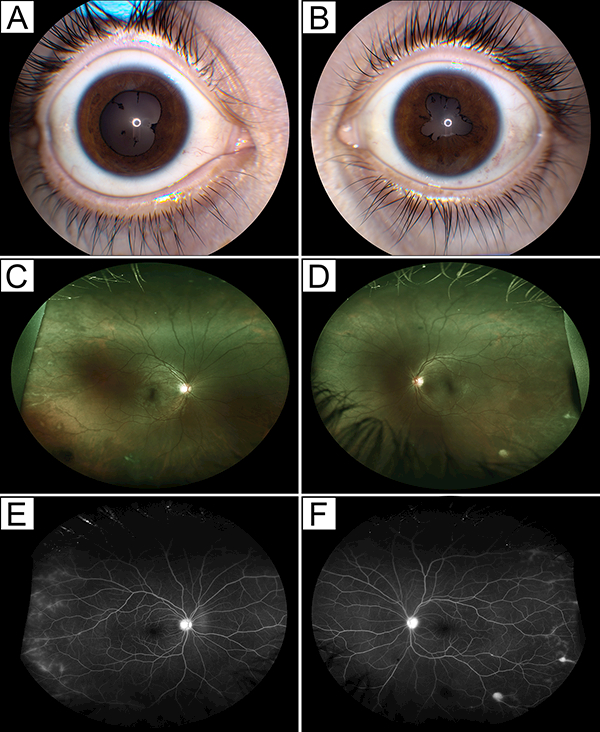
Bilateral anterior-intermediate uveitis in a 16-year-old male with ALPS-FAS. (A,B) Clinical photographs show posterior synechiae in both eyes (A, right eye; B, left eye) at presentation. (C,D) Wide-field color fundus photographs show few peripheral chorioretinal lesions and peripheral vascular sheathing (E,F) that stain on wide-field fundus angiography. Peripheral retinal vessels also show late leakage in both eyes (C,E, right eye; D,F, left eye).
Case 2
Panuveitis developed in a 4-year-old Caucasian female, who was diagnosed with ALPS-FAS earlier that year.12 She also had a history of thrombocytopenia, neutropenia, and splenomegaly, for which she received IVIG and rituximab and later underwent splenectomy. Her initial examination showed BCVA of 20/20 and 20/40 in her right and left eyes, respectively. Keratic precipitates and 3+ AC cell and flare with posterior synechiae were noted in the left eye (Fig. 2A,B). Chorioretinal scars were also visualized in the inferior periphery of the left eye. The patient was initially treated with topical corticosteroids with immediate symptomatic improvement despite the absence of changes in VA or clinical examination. In the same year, the patient developed intraocular inflammation in her right eye and was started on topical corticosteroids in that eye. Over the next year, the patient had a waxing and waning course without complete resolution of AC inflammation and with development of vitreous haze. She was treated with methotrexate, but was later switched to cyclosporine following elevation of her liver enzymes. For approximately 6 months, the patient’s uveitis resolved transiently following weekly rituximab treatment for 4 weeks to manage her ITP. She eventually required both cyclosporine and MMF for adequate control of intraocular inflammation (Fig. 2C) as well as occasional adjunct periocular corticosteroid injections for cystoid macular edema (CME). She also developed band keratopathy, posterior synechiae, and posterior subcapsular cataracts in both eyes. After 14 years of follow-up, she maintained good VA at 20/16 and 20/40 in her right and left eyes, respectively, with mild cataracts, no uveitic activity, and no CME in both eyes.
Figure 2:
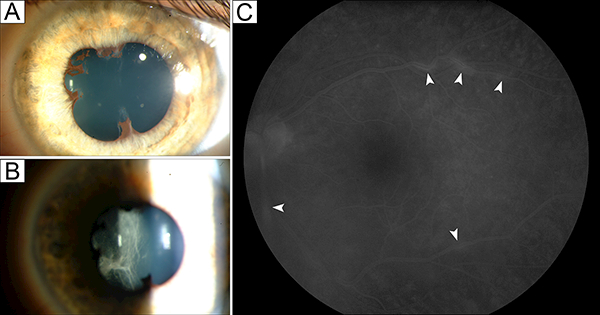
Bilateral panuveitis in a 4-year-old female with ALPS-FAS. Clinical photographs from early in her disease course show posterior synechiae (A) and anterior vitreous cells and condensation (B), with unremarkable findings on fluorescein angiography (FA). (C) A few years later, late-phase FA shows staining of the vascular wall at multiple points in the posterior pole of the left eye (arrowheads), which resolved with cyclosporine and mycophenolate mofetil.
Case 3
Posterior uveitis was seen in a 21-year-old Caucasian male, who was diagnosed with ALPS-U at 16 years of age. He had a history of thrombocytopenia, lymphadenopathy, and splenomegaly, was treated with IVIG, and underwent splenectomy. He initially presented to the NEI with acute vision loss in his left eye and was already on MMF for approximately 3 years as a steroid-sparing agent to manage his ALPS-associated autoimmune cytopenias and infiltrative lung lesions. He had good VA at 20/16 and 20/32 in his right and left eyes, respectively. Ophthalmologic examination showed trace AC cells in his left eye and deep white choroidal infiltrates in all four quadrants in both eyes, though greater in the left (Fig. 3A,B). Central macular serous detachment was also noted in the left eye and corresponded to a central visual field defect on Humphrey visual field testing. Optical coherence tomography confirmed the presence of macular subretinal fluid in the left eye (Fig. 3C). Indocyanine green angiography showed evidence of deep chorioretinal lesions, which also stained on FA (Fig. 3D,E). AC paracentesis was performed to exclude any lymphoproliferative process and showed undetectable levels of interleukin (IL)-10 and IL-6. An extensive work-up, including brain magnetic resonance imaging (MRI) and lumbar puncture, was unrevealing for any known infectious or malignant process. The patient was started on high-dose pulse corticosteroids followed by an oral prednisone taper, and his MMF dose was increased from 1250 mg daily to 2000 mg daily. His submacular fluid improved rapidly (Fig. 3F), and his deep white chorioretinal lesions resolved over the following 8 months, during which oral prednisone was slowly tapered and then stopped. Three years later, he developed idiopathic bilateral disc swelling without any signs of intraocular inflammation. His VA, visual fields, and color vision remained unaffected. Repeat MRI was unremarkable, and his disc swelling resolved spontaneously. A few months later, however, the patient developed leukemia and received an allogeneic hematopoietic stem cell transplant. He has since remained in remission. Five years following his initial diagnosis of uveitis, his eyes were quiet off of all immunosuppression with essentially normal visual fields and good VA at 20/20 in both eyes.
Figure 3:
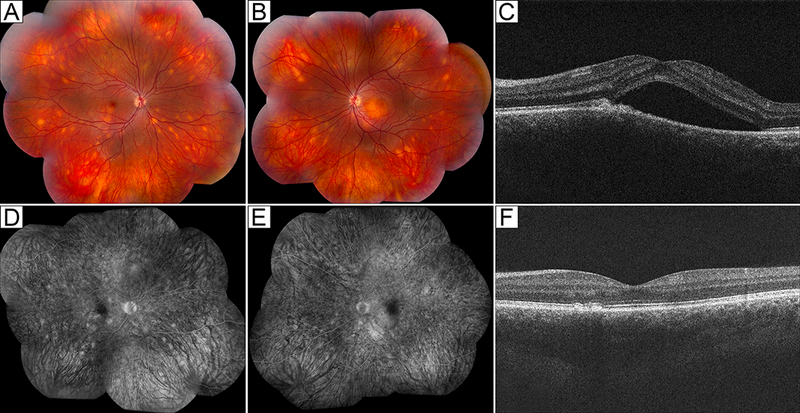
Posterior uveitis in a 21-year-old male with ALPS-U (ALPS with undetermined genetic defect). (A,B) Color fundus photographs show scattered deep white choroidal lesions in both eyes (A, right eye; B, left eye). (C) Optical coherence tomography shows submacular fluid in the right eye. (D,E) Fluorescein angiography shows staining of deep choroidal lesions in both eyes and mild perifoveal leakage in the left eye (D, right eye; E, left eye). (F) With treatment, the patient’s chorioretinal lesions (not shown) and submacular fluid resolved.
Case 4
A 48-year-old Caucasian female with ALPS-FAS had one episode of optic neuritis treated with oral corticosteroids as well as vitreous hemorrhage secondary to ITP approximately 24 years after her ALPS diagnosis. At the time of referral, BCVA was 20/32 and 20/25 in the right and left eyes, respectively, with full color vision and no afferent pupillary defect. A chorioretinal scar was visualized temporal to the macula in the right eye, and the optic nerve of the right eye was mildly pallid temporally. Approximately 4 years later, BCVA remained essentially the same at 20/40 and 20/20 in the right and left eyes, respectively, with no changes on ophthalmologic examination.
DISCUSSION
Autoimmune cytopenias (e.g., AIHA, ITP, AIN) are most commonly associated with ALPS, but other autoimmune disorders, such as hepatitis, glomerulonephritis, primary biliary cirrhosis, and Guillain-Barré syndrome, can also be seen in ALPS patients.9 To our knowledge, only three cases of ALPS-associated uveitis have been reported in the literature to date.4, 12 This study aimed to describe the ocular manifestations of ALPS in patients who were referred to the NEI for ocular or visual symptoms while being seen for the ALPS Natural History Study at the NIAID.
In our cohort, four (13.8% of ocular cohort, 1.7% of entire ALPS cohort) patients developed ocular inflammatory manifestations 1 to 24 years after their initial ALPS diagnosis, with uveitis typically presenting within 1 to 5 years. Although not every patient in the ALPS Natural History Study was examined by an ophthalmologist, it is readily known that ocular inflammatory disorders are not common in ALPS; only a few cases of ALPS-associated uveitis have been previously described.4, 12 Rieux-Laucat et al. reported two cases of anterior uveitis among 16 ALPS patients with heterozygous FAS mutations; the first case was bilateral, and the second was presumably unilateral panuveitis—albeit predominantly anterior—given that retinal vasculitis was also seen in the same eye.4 Lim et al. from our group also characterized a case of bilateral panuveitis,12 and an update to this case was included within this case series. Notably, in this study, all three cases of uveitis were bilateral, involved the posterior segment, and required long-term systemic immunosuppression for adequate disease control. The types of treatment used in the two uveitis cases reported by Rieux-Laucat et al. are not clearly described,4 but approximately 50 to 60% of ALPS patients require long-term systemic immunosuppression to control ALPS-associated autoimmune cytopenias and typically do not respond to short-term corticosteroids alone.14 This becomes an important consideration when trying to balance the intended therapeutic effects of immunomodulation to treat the underlying autoimmune disease and the seemingly paradoxical immune compromise seen with ALPS-related multi-lineage cytopenias. Even so, prompt but long-term systemic immunosuppression may be necessary to treat potentially vision-threatening intraocular inflammation in ALPS patients, many of whom are quite young and already carry a large burden of autoimmune disease. Fortunately, VA outcomes in our case series were good with appropriate and timely treatment.
In addition to potentially complex treatment considerations, differentiating between an autoimmune and a neoplastic etiology of intraocular inflammatory changes seen in ALPS patients can be challenging. The 21-year-old ALPS patient with posterior uveitis in this case series was already on chronic steroid-sparing immunosuppressive treatment with MMF for management of his ALPS-associated cytopenias and pulmonary infiltrates prior to his initial presentation to the NEI. A comprehensive work-up at the time was negative for malignancy, and thus, his deep, widespread, white choroidal infiltrates were presumed to be inflammatory in nature and were treated accordingly. Although the patient developed leukemia 3 years later, the patient’s clinical response to corticosteroids and the lack of recurrence of choroidal lesions with acute leukemia suggest that his choroidal lesions were in fact inflammatory and not malignant at presentation. Nonetheless, his case demonstrates the importance of regular follow-up and close observation in ALPS patients.
As with all retrospective studies, our findings must be interpreted with caution. Referral bias, or bias in seeking ophthalmologic consultation, may have affected the true proportion of ocular findings in ALPS patients seen at the NIAID. A total of 232 ALPS patients were seen by the NIAID from 2003 to 2013, and 29 of these patients reported ocular or visual symptoms on a physician’s review of systems or had a history of prolonged corticosteroid use. Thus, it is possible that there were other patients enrolled in the natural history protocol who could have had ocular findings on ophthalmologic examination, but were unfortunately never examined because they did not report any symptoms. We therefore cannot provide a precise estimation of the occurrence of intraocular inflammation in ALPS. However, our study suggests that the minimum frequency of ocular inflammatory disease is approximately 2% in ALPS patients and approximately 14% among those reporting ocular or visual symptoms.
Although ALPS is exceedingly rare, it may be underdiagnosed because of its variable phenotypic expression and clinical overlap with other lymphoproliferative disorders. Nonetheless, it is highly unlikely that a patient presenting to a uveitis specialist will have ALPS in the absence of such findings. However, history of generalized lymphadenopathy, splenomegaly, hepatomegaly, multi-lineage autoimmune cytopenias, other autoimmune diseases, and/or lymphoma, especially in a child, adolescent, or young adult, should prompt the clinician to broaden the differential diagnosis to include ALPS.
In this study, we present the ophthalmologic findings of ALPS patients who were referred to the NEI for ocular or visual complaints. In addition to other autoimmune disorders, at least 1.7% of ALPS patients, or at least 13.8% of ALPS patients reporting symptoms, can have potentially vision-threatening ocular inflammatory disease that can manifest at any time during their disease course. Long-term immunosuppression may be required to adequately treat ALPS-associated intraocular inflammation, as illustrated by the long-term follow-up of all three patients with uveitis in our ALPS cohort. Early diagnosis and prompt intervention are likely to improve visual outcomes. Thus, routine ophthalmologic examination of ALPS patients with any ocular or visual symptoms is recommended.
Acknowledgments
FUNDING
This study was supported by the NEI and NIAID Intramural Research Programs. Dr. Ucar’s work was also supported by a grant from The Scientific and Technological Research Council of Turkey (TUBITAK). Miss Kim’s research was supported by the NIH Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and generous contributions to the Foundation for the NIH from Pfizer, Inc., Doris Duke Charitable Foundation, Newport Foundation, American Association for Dental Research, Howard Hughes Medical Institute, Colgate-Palmolive Company as well as other private donors. For a complete list, please visit the Foundation website at http://fnih.org/work/education-training-0/medical-research-scholars-program.
Footnotes
DECLARATION OF INTEREST
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Color versions of one or more of the figures in the article can be found online at www.tandfonline.com/ioii.
REFERENCES
- 1.Rao VK, Oliveira JB. How I treat autoimmune lymphoproliferative syndrome. Blood. 2011;118: 5741–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee HO, Ferguson TA. Biology of FasL. Cytokine Growth Factor Rev. 2003;14: 325–335. [DOI] [PubMed] [Google Scholar]
- 3.Oliveira JB, Bleesing JJ, Dianzani U, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010;116: e35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rieux-Laucat F, Blachere S, Danielan S, et al. Lymphoproliferative syndrome with autoimmunity: A possible genetic basis for dominant expression of the clinical manifestations. Blood. 1999;94: 2575–2582. [PubMed] [Google Scholar]
- 5.Szabo I, Gulbins E, Apfel H, et al. Tyrosine phosphorylation-dependent suppression of a voltage-gated K+ channel in T lymphocytes upon Fas stimulation. J Biol Chem. 1996;271: 20465–20469. [DOI] [PubMed] [Google Scholar]
- 6.Kong L, Ogawa N, Nakabayashi T, et al. Fas and Fas ligand expression in the salivary glands of patients with primary Sjogren’s syndrome. Arthritis Rheum. 1997;40: 87–97. [DOI] [PubMed] [Google Scholar]
- 7.De Maria R, Testi R. Fas-FasL interactions: a common pathogenetic mechanism in organ-specific autoimmunity. Immunol Today. 1998;19: 121–125. [DOI] [PubMed] [Google Scholar]
- 8.DeFranco S, Bonissoni S, Cerutti F, et al. Defective function of Fas in patients with type 1 diabetes associated with other autoimmune diseases. Diabetes. 2001;50: 483–488. [DOI] [PubMed] [Google Scholar]
- 9.Sneller MC, Dale JK, Straus SE. Autoimmune lymphoproliferative syndrome. Curr Opin Rheumatol. 2003;15: 417–421. [DOI] [PubMed] [Google Scholar]
- 10.Straus SE, Jaffe ES, Puck JM, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood. 2001;98: 194–200. [DOI] [PubMed] [Google Scholar]
- 11.Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270: 1189–1192. [DOI] [PubMed] [Google Scholar]
- 12.Lim WK, Ursea R, Rao K, et al. Bilateral uveitis in a patient with autoimmune lymphoproliferative syndrome. Am J Ophthalmol. 2005;139: 562–563. [DOI] [PubMed] [Google Scholar]
- 13.Jabs DA, Nussenblatt RB, Rosenbaum JT, Standardization of Uveitis Nomenclature Working G. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005;140: 509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Price S, Shaw PA, Seitz A, et al. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood. 2014;123: 1989–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]