

Abstract
Coiled-coils refer to a bundle of helices coiled together like strands of a rope. It has been estimated that nearly 3% of protein-encoding regions of genes harbour coiled-coil domains (CCDs). Experimental studies have confirmed that CCDs play a fundamental role in subcellular infrastructure and controlling trafficking of eukaryotic cells. Given the importance of coiled-coils, multiple bioinformatics tools have been developed to facilitate the systematic and high-throughput prediction of CCDs in proteins. In this article, we review and compare 12 sequence-based bioinformatics approaches and tools for coiled-coil prediction. These approaches can be categorized into two classes: coiled-coil detection and coiled-coil oligomeric state prediction. We evaluated and compared these methods in terms of their input/output, algorithm, prediction performance, validation methods and software utility. All the independent testing data sets are available at http://lightning.med.monash.edu/coiledcoil/. In addition, we conducted a case study of nine human polyglutamine (PolyQ) disease-related proteins and predicted CCDs and oligomeric states using various predictors. Prediction results for CCDs were highly variable among different predictors. Only two peptides from two proteins were confirmed to be CCDs by majority voting. Both domains were predicted to form dimeric coiled-coils using oligomeric state prediction. We anticipate that this comprehensive analysis will be an insightful resource for structural biologists with limited prior experience in bioinformatics tools, and for bioinformaticians who are interested in designing novel approaches for coiled-coil and its oligomeric state prediction.
Keywords: coiled-coil, prediction, oligomeric state, polyglutamine
Introduction
First described in 1953 by Pauling and Crick [1], the proliferation of studies of coiled-coil domains (CCDs) in proteins has driven continued computational prediction in the past few decades. CCDs can be summarized as at least two or more helices that wrap around each other, which can be defined as a repeat Xn of residues, where X can be denoted as (a-b-c-d-e-f-g) and n can be described as the number of helices. It is estimated that nearly 10% of eukaryotic proteins harbour CCDs [2, 3]. Depending on the value of n, CCDs can be categorized into several groups, including antiparallel dimer, parallel dimer, trimer and tetramer (Figure 1). The colour scheme in Figure 1 is based on the B-factor values using PyMOL. CCDs exhibit a preference for hydrophobic residues at positions a and d, charged residues at positions e and g and hydrophilic residues at positions b, c and f [8, 9], which serve to stabilize helix oligomerization according to the ‘Peptide Velcro’ hypothesis [10]. This repeating Xn motif enables the prediction of CCDs and their oligomeric states based on protein sequences.
Figure 1.
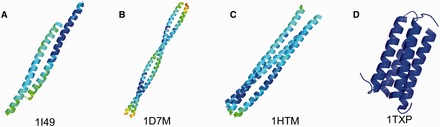
Examples of coiled-coil oligomeric states. (A) Antiparallel dimer (PDB Accession: 1I49 [4]). (B) Parallel dimer (PDB Accession: 1D7M [5]). (C) Trimer (PDB Accession: 1HTM [6]). (D) Tetramer (PDB Accession: 1TXP [7]). A colour version of this figure is available at BIB online: http://bib.oxfordjournals.org.
Experimental studies have confirmed that CCDs play a fundamental role in subcellular infrastructure and controlling trafficking of eukaryotic cells [11, 12]. The relatively high stability of CCDs has led to their promising use as delivery systems for a range of molecules. For example, cartilage oligomeric matrix protein (COMP) [13, 14] and right-handed protein [15] from Staphylothermus marinus have been used as drug delivery systems in anticancer therapies [3, 16, 17]. The five α-helix CCDs in COMP are capable of binding and carrying some important signalling molecules, including vitamins A and D3. Other successful applications of CCDs, peptides and motifs used in drug delivery systems have also been reported [18–22].
Sequence and structural analysis of CCDs have enabled the development of computational approaches for the prediction of CCDs from sequence alone [8–10, 23]. For example, Vincent et al. performed coiled-coil prediction for proteins from tenascins and thrombospondins families, analysed the motif conservation of different coiled-coil oligomeric states and revealed that sequence conservation allows trimers and pentamers of CCDs to be distinguished, providing useful insights for future coiled-coil prediction [23]. However, the rapid growth in prediction approaches since the last comprehensive comparison, which was reported almost a decade ago [24], creates an urgent need to critically assess and compare the now-large and diverse prediction methods. In this article, therefore, we present a comprehensive review of 12 sequence-based methods for coiled-coil prediction, offering insights into the nature of different predictors and facilitating potential improvement of CCD prediction. All predictors are critically reviewed in terms of input, model construction and outcome (i.e. prediction performance) [25, 26]. To evaluate the performance of coiled-coil predictors, independent tests were conducted with new test data sets (http://lightning.med.monash.edu/coiledcoil/) carefully collected and curated from different resources. Finally, as CCDs have been extensively found in disease-associated human polyglutamine (PolyQ) proteins [27], we applied various predictors to a data set of nine human proteins containing PolyQ repeats and discussed our findings.
Materials and methods
Predictors reviewed in this study
Table 1 summarizes the details of the tools of coiled-coil and its oligomeric state prediction that are evaluated in this article. These are COILS [28], PCOILS [29], Paircoil2 [30], SOSUIcoil [31], MARCOIL [32], CCHMM_PROF [33], SpiriCoil [34], SCORER 2.0 [35], LOGICOIL [36], PrOCoil [37], RFCoil [38] and Multicoil2 [39].
Table 1.
A comprehensive list of coiled-coil and oligomeric state predictors reviewed in this study
| Task | Tool publication date | Input format | Model highlighta | Evaluation |
Service |
||||
|---|---|---|---|---|---|---|---|---|---|
| Strategy | Output format | Web serviceb | Availability | Speedc | Reliabilityd | ||||
| Coiled-coil region prediction | COILS [28] | Raw sequence or SwissProt IDs | Pairwise residue probabilities | Algorithm tested with sequences of known globular proteins, randomly generated sequences and all the sequences in GenBank | Residue score and probability located in CCD | Yes | Yes (with third-party implementations) | Fast | Consistent |
| 1997 | |||||||||
| PCOILS [29] | Raw sequence/FASTA sequence | Pairwise profile comparison using protein evolution profile | Case study | Residue score and probability to located in CCD | Yes | Yes | Moderate | Results vary depending on BLAST database | |
| 2005 | |||||||||
| Paircoil2 [30] | FASTA sequence | Pairwise residue probabilities | Leave-family-out cross-validation | Residue score and probability to located in CCD | Yes | Yes | Fast | Unknowne | |
| 2006 | |||||||||
| MARCOIL [32] | FASTA Sequence | HMM based on MTIDK and other matrices | 150-fold cross-validation | Residue score and probability to located in CCD | Yes | Yes | Fast | Consistent | |
| 2002 | |||||||||
| CCHMM_PROF [33] | Raw sequence/FASTA sequence | HMM based on multiple sequence alignment | Overall accuracy, Segment overlap and case study | Overall probability of containing CCDs and Binary decision to (not) be in CCD | Yes | Yes | Moderate | Results vary depending on the BLAST database | |
| 2009 | |||||||||
| SpiriCoilf [34] | FASTA sequence | Structurally informed homology-based multiple HMMs | Independent test | Binary decision to (not) be in CCD | Yes | No | Fast | – | |
| 2010 | |||||||||
| SOSUIcoil [31] | One-letter symbol or multiple FASTA sequences | Canonical discriminant analysis | Independent test and case study | – | No | No | – | – | |
| 2008 | |||||||||
| Coiled-coil oligomeric state prediction | SCORER 2.0 [35] | Raw sequence and/or heptad register | Log-likelihood ratio with new defined score function | Independent test | Predicted scorer to be parallel dimeric and trimeric coiled-coil | Yes | Yes | Fast | Consistent |
| 2011 | |||||||||
| LOGICOIL [36] | Raw sequence and/or heptad register | Bayesian variable selection and multinomial probit regression | 10-fold cross-validation and leave-one-out cross-validation | Predicted score to be parallel dimer, antiparallel dimer, trimer and tetramer | Yes | Yes | Fast | Consistent | |
| 2013 | |||||||||
| PrOCoil [37] | Raw sequence and/or heptad register | SVM and coiled-coil Kernel | 10-fold cross-validation, nested cross-validation and case study | Predicted scorer to be parallel dimeric and trimeric coiled-coil | Yes | Yes | Fast | Consistent | |
| 2011 | |||||||||
| RFCoil [38] | Raw sequence and heptad register | Random forest with effective amino acid indices | 10-fold cross-validation and independent tests | Predicted probability to be parallel dimeric and trimeric coiled-coil | Yes | Yes | Fast | Consistent | |
| 2014 | |||||||||
| Coiled-coil region and oligomeric state prediction | Multicoil2 [39] | FASTA sequence | Pairwise residue correlation and HMM | Leave-family-out cross-validation | Residue probability to be located in non-coiled-coil, dimer or trimer | Yes | Yes | Fast | Consistent |
| 2011 | |||||||||
Note. aHMM—Hidden Markov Model; SVM—Support Vector Machines.
bThe URLs of predictors listed are: COILS—http://embnet.vital-it.ch/software/COILS_form.html; PCOILS—http://toolkit.tuebingen.mpg.de/pcoils; PairCoil2—http://groups.csail.mit.edu/cb/paircoil2/paircoil2.html; MARCOIL—http://toolkit.tuebingen.mpg.de/marcoil; SOSUIcoil—http://harrier.nagahama-i-bio.ac.jp/sosui/coil/submit.html (not available); CCHMM_PROF—http://gpcr.biocomp.unibo.it/cgi/predictors/cchmmprof/pred_cchmmprof.cgi; SpiriCoil – http://supfam.cs.bris.ac.uk/SUPERFAMILY/spiricoil/; SCORER 2.0—http://coiledcoils.chm.bris.ac.uk/Scorer/; LOGICOIL—http://coiledcoils.chm.bris.ac.uk/LOGICOIL/; PrOCoil—http://www.bioinf.jku.at/software/procoil/; RFCoil—http://protein.cau.edu.cn/RFCoil/index.php?page=introduction; Multicoil2—http://groups.csail.mit.edu/cb/multicoil2/cgi-bin/multicoil2.cgi.
cSpeed refers to the response time after submitting the sequence to the web server.
dReliability refers to whether the outputs of the predictor’s web server and its local executable are consistent.
ePaircoil2 is not runnable on our local machine.
fIn [34], SpiriCoil was also applied for oligomeric state prediction. The prediction performance was comparable with that of MULTICOIL, which is the previous version of Multicoil2.
Model input
The training data set is used to build a computational model to learn potential patterns hidden in the data set. Before model construction, data collection and preprocessing of the training data set were performed. Data sets with too much noise or imbalanced distribution may lead to unsatisfactory prediction performance of the model. There are two main ways to collect the CCD data to build the model. In some studies, the CCDs were extracted with SCOP [40] and SOCKET [41], while other studies extracted the data directly from a publicly available database regarding experimentally verified CCDs, for example, CC+ [42]. The CCDs in the CC+ database were annotated manually and with SOCKET, which has been widely used to extract reliable CCDs from protein structures. A cut-off value of 7.0Å was usually used for extracting coiled-coils from protein structures. Removal of sequence redundancy, an important step before model construction, was performed using CD-HIT [43].
Models construction and development
Relatively simple classification methods predict whether a protein sequence contains a CCD. More sophisticated predictors perform multiclass classifications that categorize coiled-coil regions into different forms of α-helical assembly, such as dimer, trimer and tetramer. We discuss below the different algorithms used in the predictors (Table 1).
COILS, the first reported algorithm for CCD prediction, is a statistically controlled predictor based on the amino-acid profile-based method. The similarity of a protein sequence with a structurally known protein is computed using a sliding window. The recommended window length for COILS is 28 to help remove false positives. PCOILS is an updated version of COILS that predicts coiled-coils through comparing pairwise protein evolution profiles based on user-provided multiple sequence alignment or PSI-BLAST [44]. Paircoil2 is the latest development of PAIRCOIL [45]. These predictors use pairwise residue correlations or probabilities to detect the coiled-coil motif in a protein sequence. The training data set of Paircoil2 is larger than that used for training PAIRCOIL because of the dramatically increased number of known coiled-coil sequences. SOSUIcoil uses amino acid physical properties to help determine an appropriate heptad register, followed by canonical discriminant analysis to discriminate coiled-coils.
Hidden Markov Models (HMM) has been used in a number of coiled-coil predictors. These include MARCOIL, CCHMM_PROF and SpiriCoil. CCHMM_PROF is an improved version of CCHMM [46], which used multiple sequence alignments instead of single sequence-based HMM. MARCOIL also uses single sequence-based HMMs, whereas SpiriCoil uses a large library of HMMs to predict coiled-coils that fall into known superfamilies. The application of SpiriCoil is limited to sequences that have reasonably high similarity to known families because of the use of the training data set for constructing SpiriCoil. On the other hand, MARCOIL, which uses explicit knowledge of existing coiled-coils to train a single HMM, possesses a more complicated algorithm to efficiently search for a variable length subsequence of high probability for coiled-coil formation. According to the HMM parameter t, MARCOIL model has two variations, MARCOIL-L (t = 0.001) and MARCOIL-H (t = 0.01).
MultiCoil [47], a predictor developed based on the PAIRCOIL algorithm, extends the dimeric coiled-coil prediction in PAIRCOIL to trimeric coiled-coils, using a multidimensional scoring approach. Multicoil2 further extends the algorithm to include pairwise correlations with HMM in a Markov Random Field. Multicoil2 also contains eight sequence-based features (including dimer probability, trimer probability, non-coiled probability, dimer correlations at distance 1–7, trimer correlations at distance 1–7, non-coiled correlations at distance 1–7, the hydrophobicity at the a and d positions) that are used to train the model (pairwise correlation HMM). The resulting algorithm integrated the sequence features and the pairwise interactions into a multinomial logistic regression to formulate an optimized scoring function for the classification of coiled-coil oligomeric state.
SCORER [48] uses a log-odd-based scoring system for the classification of coiled-coil sequences into parallel dimeric and trimeric coiled-coils. SCORER 2.0 combines an expanded and updated training set and a Bayes factor method, which takes into consideration the possible uncertainty in the profile tables. LOGICOIL is a predictor based on the combined and concurrent application of Bayesian variable selection and multinomial probit regression. The application of Bayesian paradigm can provide informative posterior distributions on the selected parameters, as well as offering a framework to apply this useful information based on biological data and expert knowledge. Traditional machine learning techniques, including support vector machine (SVM) [49] and random forest [50], have also been applied to coiled-coil oligomeric state prediction. For example, PrOCoil adopts an SVM based on identified rules converted into weighted amino-acid patterns. In addition to PrOCoil, PrOCoil-BA (PrOCoil-Balanced Accuracy) is an alternative model, which is optimized for balanced accuracy, i.e. the average of sensitivity and specificity. RFCoil uses random forest combined with effective amino-acid indices selected by Gini (a decision tree split function) decrease [51] and Kendall rank correlation coefficient [52].
Model evaluation
A variety of methods were used to assess the prediction performance of coiled-coil predictors listed in Table 1, including cross-validation, leave-one-out cross-validation, leave-family-out cross-validation, independent test and case study. Normally, cross-validation can avoid over-fitting caused by the training data set. The nature of cross-validation is to split the data set into N folds and combine N − 1 folds as the training data set, leaving the remaining fold as the test data set. Leave-one-out cross-validation and leave-family-out cross-validation are variations of cross-validation. Given a data set with D data samples, leave-one-out cross-validation combines D − 1 samples as the training data set and leaves the remaining one sample as the test sample. In this cross-validation, all samples in the data set are treated as a test sample once. If the data set is collected from different species/families, each subset from the same species/family is regarded as test data sets once, and other subsets from other families/species will be combined to form the training data set. The final performance for cross-validation is often averaged from the results of different combinations of the training data sets. The independent test is another method to assess the performance of bioinformatics tools. To test the performance of an algorithm on a new data set with a different data distribution, it is important to ensure that there is no overlap between the training data set and the independent test data set. Finally, the case study is as an effective way to test the performance of a method in real-world applications, providing useful insights into the method scalability and usefulness with unknown data.
Predictor utility
An important aspect of predictors in the biological research community is to provide a user-friendly web interface or a local tool to enable non-bioinformaticians to apply the model directly to their research. The usefulness of bioinformatics tools depends on three factors, i.e. the web interface, the output and interpretation of prediction results and the availability of locally runnable software. A user-friendly interface can provide appropriate guidance and instructions to avoid potential mistakes when using the web server. This is especially important when parameter settings are required before conducting prediction tasks. Among the predictors we tested, those predictors aimed at discriminating coiled-coils from non-coiled-coils (e.g. COILS, PCOILS, Paircoil2 and MARCOIL) require parameter settings before sequence submission. Documents are available online regarding the description of the parameters and their potential effect on the prediction performance. On the other hand, the predictors for coiled-coil oligomeric states are mostly parameter-free. For coiled-coil oligomeric state prediction, only sequence and its heptad register are required as the input (for example, SCORER 2.0, PrOCoil, RFCoil and LOGICOIL). Furthermore, SCORER 2.0, PrOCoil and LOGICOIL are also able to predict sequences without the prerequisite of knowing the coiled-coils/heptad registers by combing coiled-coil prediction and extracting heptad register from MARCOIL, without the necessity of performing a two-stage prediction.
Stand-alone software allows users to perform predictions for a large amount of sequences on local machines, offering an advantage over web servers. Among the coiled-coil predictors reviewed in this article, SpiriCoil and SOSUIcoil do not have available locally runnable tools. The local versions of SCORER 2.0, PrOCoil, RFCoil and LOGICOIL were written using the R package (http://www.r-project.org/). PrOCoil has been integrated with R so it can be downloaded and installed with the R console. Users should be aware of the difference in the length of the coiled-coils in the training data sets of different frameworks especially for the oligomeric state prediction. For SCORER 2.0, MultiCoil2, PrOCoil, RFCoil and LOGICOIL, the minimum lengths of their training coiled-coils are 15, 21, 8, 8 and 15, respectively. This means that one should take into consideration the length of the sequence when choosing appropriate predictors to obtain better prediction results. Although coiled-coil predictors recommend the preferable sequence lengths of coiled-coils, they can still predict the oligomeric state of the coiled-coils shorter than the specified length thresholds. Under such circumstance, it is the users’ responsibility to choose an appropriate predictor according to the length of query sequence before its submission.
Understandable and visualizable interpretation of the output is also important for better understanding the prediction results and their significance. The output of the coiled-coil predictors we reviewed is often organized in two ways, based on either a residue or a sequence basis. Most of the predictors for discrimination of coiled-coils from non-coiled-coils provide prediction outputs on a residue basis, which allows users to gain a detailed insight into each amino acid and its predicted score/probability. Moreover, COILS, PCOILS, Paircoil2 and MARCOIL also provide the visible plots of predicted score/probability for each amino acid and enable users to obtain an overview of predicted scores for the entire sequence. On the other hand, the predictors of coiled-coil oligomeric state (including SCORER 2.0 and LOGICOIL) provide only a final decision and an overall prediction score. These scores are not easy to interpret and understand. PrOCoil provides both prediction scores and visible plots for each amino acid. RFCoil, on the other hand, provides a matrix showing the probability of the query sequence forming a dimeric coiled-coil or a trimeric coiled-coil, which is relatively easy to understand.
A case study of coiled-coil prediction for human PolyQ proteins
As an extended test of the reviewed coiled-coil predictors, we examined the prediction consistency for nine disease-associated PolyQ proteins. We submitted their sequences to the corresponding web servers and obtained the prediction results. PolyQ proteins contain a stretch of repeated glutamine residues (termed the ‘PolyQ tract’). PolyQ repeats with more than seven residues are abundant in 128 proteins in the human proteome [53]. These repeats have important biological functions especially in transcription regulation, and proteins harbouring expanded PolyQ repeats are involved in neurodegenerative diseases [54]. The PolyQ diseases are caused in part by a gain-of-function mechanism of neuronal toxicity involving protein conformational changes that result in the formation and deposition of β-sheet rich aggregates [55]. Because PolyQ repeats are highly aggregation-prone [55], it is difficult to determine their structure by X-ray crystallography [56]. The widely accepted model of β-sheet-mediated aggregation has been recently challenged by experimental and bioinformatics studies showing that disease-associated PolyQ proteins contain CCDs largely overlapping with their PolyQ repeats [27]. We therefore investigated the prediction of CCDs in human proteins containing PolyQ repeats, using the data set containing the most updated nine disease-associated PolyQ proteins from UniProt database studied by Fiumara et al. [27], which is also available in the PolyQ database [53] (http://pxgrid.med.monash.edu.au/polyq/; Table 2).
Table 2.
The list of nine human disease-related PolyQ proteins
| Protein | Protein length | PolyQ tract | UniProt identifier | Associated disease |
|---|---|---|---|---|
| TATA binding protein | 339 | 58–95 | P20226 | Spinocerebellar ataxia 17 [57–59] |
| Huntingtin | 3142 | 18–38 | P42858 | Huntington disease [60] |
| Ataxin-1 | 815 | 197–208 | P54253 | Spinocerebellar ataxia 1 [61, 62] |
| 212–225 | ||||
| Ataxin-2 | 1313 | 166–188 | Q99700 | Spinocerebellar ataxia 2 [63–65] and Amyotrophic lateral sclerosis 13 [66] |
| Voltage-dependent P/Q-type calcium channel subunit alpha-1A (Brain calcium channel I) | 2505 | 2314–2324 | O00555 | Spinocerebellar ataxia 6 [67–70] |
| Atrophin-1 | 1190 | 484–502 | P54259 | Dentatorubro-pallidoluysian atrophy [71] |
| Ataxin 7 | 892 | 30–39 | O15265 | Spinocerebellar ataxia 7 [72] |
| Androgen receptor | 919 | 58–78 | P10275 | Spinocerebellar muscular atrophy or Kennedy disease [73] |
| Ataxin-3 | 364 | 296–305 | P54252 | Spinocerebellar ataxia 3 or Machado-Joseph disease [74] |
Results and discussion
Independent test and performance evaluation
In this section, to assess the prediction performance of the reviewed coiled-coil tools in an objective and fair manner, we assembled two independent test data sets (discussed below) and measured the performance [in terms of area under curve (AUC)] of all tested tools on these two data sets. In particular, as the previous versions of CCHMM, SCORER and MultiCoil have been upgraded as CCHMM_PROF, SCORER 2.0 and Multicoil2, respectively, we only evaluated the advanced versions in the independent test. In addition, as SOSUIcoil and SpiriCoil did not provide local executables, and it was not possible to run Paircoil2 without execution errors, these three predictors were not included in this test. According to the nature of the prediction tasks, we performed independent tests for two different types of tasks, namely, coiled-coil oligomeric state prediction and CCD prediction. Coiled-coil oligomeric state prediction usually requires CCDs and their heptad registers (i.e. a-g) as the input, while CCD prediction often takes protein sequences as input. For the first type, we evaluated the performance of coiled-coil oligomeric state predictors, including RFCoil, PrOCoil, SCORER 2.0, LOGICOIL and Multicoil2. For the second type, we compared the prediction performance of COILS, PCOILS, MARCOIL, CCHMM_PROF and Multicoil2.
Coiled-coil oligomeric state prediction
Test data set construction
We carefully prepared two different test data sets. For the first data set, CCDs and their respective heptad assignments were extracted from the PDB using SOCKET [41]. Only X-ray crystal structures were selected to ensure the quality of the data set (downloaded on 6 May 2014). SOCKET was applied to annotate the coiled-coils in a given structure with a default packing cut-off of 7.0Å, which was the same as that specified in the data set collection procedure of previous studies [37, 38]. In addition, to improve the quality of the data set, we further removed those structures with a resolution of worse than 4.0Å. Meanwhile, the structures with unnatural residues were also removed. For the second data set, we first culled coiled-coil class (h class) proteins from SCOPe [75] (the extended version of SCOP) and then verified the CCDs with SOCKET. Only the consensus sequences assigned by both SCOPe and SOCKET analysis that contained coiled-coils were retained to constitute the second data set, whereas the coiled-coil and heptad annotations were obtained by SOCKET. We subsequently examined the overlap between the second data set and the training data sets of RFCoil, PrOCoil, SCORER 2.0 and LOGICOIL. Our analysis showed that the majority of entries in the second data set were covered by the training data sets of the four predictors, suggesting that the second data set was not sufficiently large enough to be an independent test data set. Therefore, to address this, we first removed all the training data of investigated predictors from our data sets and then combined the first, second and other training data sets of the four predictors, and used CD-HIT to reduce the sequence redundancy of the resulting data set to ensure that the sequence identity of any two sequences in the data set was no more than 50%. For each cluster generated by CD-HIT, if all sequences in this cluster were from our first and second data sets, the representative sequence was collected. Although sequence redundancy can be reduced by other alternative ways, 50% has been commonly used as the preferred threshold for CCDs, as any threshold lower than 50% is deemed to be too strict for coiled-coil oligomeric state prediction [36]. Finally, the independent test data set contained 509 antiparallel dimers, 88 parallel dimers, 94 trimers and 36 tetramers (Supplementary Table S1; Additional file 1—http://lightning.med.monash.edu/coiledcoil/).
Performance comparison
Among the four reviewed predictors, RFCoil and PrOCoil were trained using coiled-coils with length ≥8 amino acids, while SCORER 2.0 and LOGICOIL were developed using coiled-coils with length >14 residues. In addition, RFCoil, PrOCoil and SCORER 2.0 were designed to classify parallel dimeric and trimeric coiled-coils. LOGICOIL is the only currently available predictor that can be used to predict four types of coiled-coil oligomeric states, including parallel/antiparallel dimers, trimers and tetramers. Therefore, to comprehensively evaluate the performance of these tools for predicting the two different types of coiled-coils, we first split the independent test data set into two subsets, one with coiled-coils >7 residues and the other with coiled-coils >14 amino acids. For each subset, we evaluated the prediction performance using AUC values. This included the performance comparison of parallel dimer and parallel trimer between the four predictors, as well as pairwise performance comparison of LOGICOIL. The receiver operating characteristic (ROC) curves of these different predictors are shown in Figure 2. We also notice that certain heptad registers for CCDs from SOCKET are non-canonical, which means that the heptad registers (i.e. a-g) are interrupted according to SOCKET annotations. In view of this, we further removed the coiled-coils with non-canonical heptad assignments and repeated our tests (Additional file 2 downloadable at http://lightning.med.monash.edu/coiledcoil/). The corresponding ROC curves of all predictors for predicting these coiled-coils without non-canonical heptad registers are shown in Figure 3. For Figures 2A, B, 3A and B, ‘positive’ and ‘negative’ indicate parallel dimeric and trimeric coiled-coils, respectively.
Figure 2.
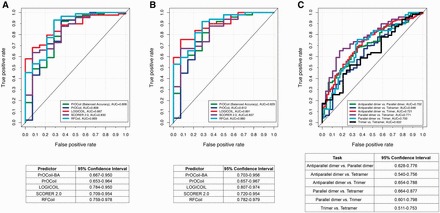
Performance comparison of coiled-coils with non-canonical heptad registers between RFCoil, SCORER 2.0, PrOCoil and LOGICOIL on the independent test. (A) ROC curves and the 95% confidence intervals for parallel dimeric and trimeric coiled-coils with length ≥8 amino acids. (B) ROC curves and the 95% confidence intervals for parallel dimeric and trimeric coiled-coils with length ≥15 amino acids. (C) ROC curves and the 95% confidence intervals of LOGICOIL for pairwise oligomeric state prediction with coiled-coils with length ≥15 residues. A colour version of this figure is available at BIB online: http://bib.oxfordjournals.org.
Figure 3.
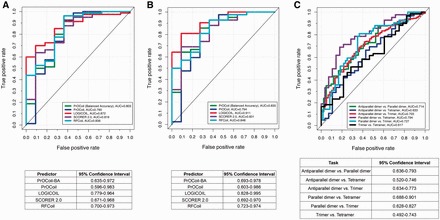
Performance comparison of coiled-coils without non-canonical heptad registers between RFCoil, SCORER 2.0, PrOCoil and LOGICOIL on the independent test. (A) ROC curves and the 95% confidence intervals for parallel dimeric and trimeric coiled-coils with length ≥8 amino acids. (B) ROC curves and the 95% confidence intervals for parallel dimeric and trimeric coiled-coils with length ≥15 amino acids. (C) ROC curves and the 95% confidence intervals of LOGICOIL for pairwise oligomeric state prediction with coiled-coils with length ≥15 residues. A colour version of this figure is available at BIB online: http://bib.oxfordjournals.org.
We note that generally, when testing with parallel dimeric and trimeric coiled-coils, LOGICOIL and RFCoil achieved the highest AUC values (see Figures 2A, B, 3A and B). Although LOGICOIL was trained using longer coiled-coil sequences, most of which contained canonical heptads, it was able to predict shorter coiled-coils with non-canonical heptads. Pairwise AUC values can be observed in Figures 2C and 3C, where LOGICOIL achieved the highest AUC values when predicting parallel dimer and tetramer (with AUC values of 0.771 and 0.794, respectively). However, distinguishing tetramer from trimer appears to be the most challenging task. PrOCoil-BA performed constantly better than PrOCoil when tested with both short and long coiled-coils (see Figures 2A, B, 3A and B). In addition to AUC values, we also computed the 95% confidence interval using the ‘pROC’ package [76]. The 95% confidence intervals are shown for each ROC curve in the corresponding tables in Figures 2 and 3. It can be seen that most of the 95% confidence intervals are overlapped. This suggests that even though the compared predictors achieved different AUC values, it is difficult to determine which predictor is the ‘statistically significant’ best model. For each of the parallel dimeric and trimeric testing samples, we also applied majority voting to generate consensus results and compared the performance of majority voting with other individual predictors (Supplementary Tables S2 and Supplementary Data). It is clear that majority voting could indeed improve the prediction accuracy when testing oligomeric state prediction of coiled-coils with length ≥15 amino acids that contained both canonical and non-canonical heptad registers. Because dimeric coiled-coils are more prevalent than trimer and tetramer, all these predictors were trained with imbalanced training data sets. Accordingly, some predictors are highly biased. For example, when testing RFCoil, we noticed that RFCoil could readily predict dimeric coiled-coils with high confidence, but often wrongly predicted many trimers as dimers. This is probably because of the limited number of trimers included in the training data set, and hence the trained RFCoil model did not generalize and perform well on trimer prediction. Therefore, to address this problem in future work, we recommend that certain techniques for imbalanced data processing and mining be applied (e.g. oversampling or undersampling) to enrich the imbalanced samples. Oversampling and undersampling [77] are both basic (opposite but equivalent) methodologies for sampling the data with imbalanced class distribution. Oversampling is a technique that randomly selects samples from the class where the number of samples is quite small to enrich the samples in this class, while undersampling randomly selects samples from the class where the number of samples in this class is large to reduce the number of samples in this class. These two techniques are basic and easy to implement. More complex and advanced techniques for imbalanced biological/medical data mining tasks also exist [78–80].
We next compared the prediction performance of Multicoil2 and other predictors. Multicoil2 accepts the full-length protein sequences as the input rather than coiled-coil sequences and their respective heptad registers. Instead of providing an overall score for the input sequence, Multicoil2 generates predicted probabilities for each individual residue in the sequence to form parallel dimers, parallel trimers or non-coiled-coils. Here, to compare with other methods, we calculated the average of the predicted probabilities by Multicoil2, normalized the value into the range of [0, 1] and removed the predicted non-coiled-coils from the results (the prediction threshold was set as 0.5). We combined the parallel dimeric and trimeric coiled-coils with length >= 21 amino acids (given that Multicoil2 can only predict CCDs with length >= 21 amino acids) in the data set used in our independent test with the dimers and trimers sequences in the Multicoil2 training data set and applied CD-HIT to remove the sequence redundancy, ensuring that the identity between any two sequences in the resulting data set was no more than 50%. As a result, only 22 CCDs remained in the resulting data set. For the remaining CCDs, we downloaded their complete protein sequences so that we could use them as the input to Multicoil2. Multicoil2 predicted only 11 of 22 (50.0%) sequences that contained CCDs that overlapped with SOCKET annotation. Therefore, we compared only the prediction performance of different predictors on these 11 ‘valid’ CCDs (Figure 4; Additional file 3—http://lightning.med.monash.edu/coiledcoil/). In Figure 4, ‘positive’ and ‘negative’ represent parallel dimeric and trimeric coiled-coils, respectively. LOGICOIL correctly classified all the parallel dimeric and trimeric coiled-coils, while Multicoil2 and PrOCoil obtained the lowest AUC value. Consistent with the results in Figures 2 and 3, PrOCoil-BA performed better than PrOCoil (greater by 0.2), followed by RFCoil and SCORER 2.0. In addition, the 95% confidence intervals suggest that LOGICOIL was the best predictor based on this independent testing data set. Consistent with the AUC values shown in Figure 4, LOGICOIL correctly classified all the test samples. It is noteworthy that the majority voting strategy achieved an accuracy of 90.9%, which was ranked as the second best accuracy according to the accuracies of other individual predictors (Supplementary Table S4).
Figure 4.
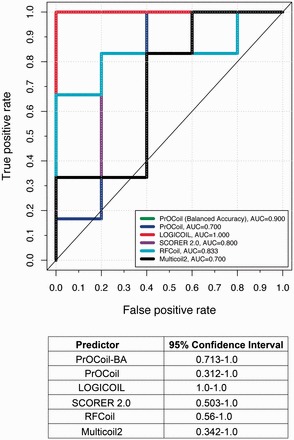
ROC curves and the 95% confidence intervals of Multcoil2 and other predictors for parallel dimeric and trimeric coiled-coil prediction. A colour version of this figure is available at BIB online: http://bib.oxfordjournals.org.
CCD prediction
Testing data set construction
The positive data set comprised protein sequences containing annotated CCDs based on SOCKET. For the negative data set, we extracted protein entries of alpha and beta classes (a/b; i.e. c class) from the SCOPe database, except for superfamilies c.37.1, c.49.2, c.67.1 and c.93.1, which are annotated to contain CCDs [24]. Protein sequences were extracted from PDB, and those sequences that contain unnatural amino acids were removed. These sequences were further validated by SOCKET with a loosened threshold of 7.4Å [33] to ensure they did not contain any CCDs. After removing all the available training data of investigated predictors from our testing data set, we combined our testing data sets with the available training data sets of CCHMM_PROF, MARCOIL and Multicoil2. We then applied CD-HIT to remove the sequence redundancy, so that the sequence identity between any two sequences was not >30%. Similar to the construction process of the independent test data set for CCD oligomeric state prediction, for each cluster generated by CD-HIT, only representative sequences from the clusters where there were no samples from the training data sets of the compared predictors in this cluster were collected. After this procedure, the final data set included a total of 1643 sequences, 601 of which did not contain any CCDs and 1042 containing 2176 CCDs (Additional files 4 and 5—http://lightning.med.monash.edu/coiledcoil/). CCHMM_PROF and PCOILS require the position-specific scoring matrix (PSSM) generated by PSI-BLAST as the input to make the prediction. Accordingly, we used the Uniref90 database to generate the PSSM profiles of all the tested sequences and conduct the comparison, which was also used as the search database by CCHMM_PROF [33]. The parameters for PSI-BLAST was preliminarily set by the PCOILS program; for CCHMM_PROF, we used the same parameters described in [33].
Performance comparison
Firstly, we evaluated the effectiveness of different predictors for identifying CCDs by calculating the averaged probability score for each protein. If a protein was predicted to contain coiled-coil residues, the probability was calculated as the averaged score of all predicted coiled-coil residues; otherwise, if a protein was not predicted to have CCDs, then the calculated probability was the averaged score of all residues of the whole protein. The ROC curves and corresponding AUC values of the compared predictors are shown in Figure 5A, where ‘positive’ represents the sequences containing CCDs, while ‘negative’ indicates the sequences without CCDs. Because Multicoil2 can only predict protein sequences with CCDs >21 amino acids, we provided the results of Multicoil2 on both the entire test data set (termed ‘Multicoil2-all’) and a subset that only contained proteins with coiled-coils >= 21 amino acids (termed ‘Multicoil2-21’). It is apparent that Multicoil2-21 identified the majority of coiled-coils and achieved the highest AUC value of 0.898, followed by CCHMM_PROF (AUC = 0.811). The AUC value of PCOILS was higher than COILS by 0.017, presumably owing to the incorporation of evolutionary information in the form of PSSM generated by PSI-BLAST. Next, we examined whether the identified CCDs were identical to those annotated by SOCKET. To do so, we compared all 2176 CCDs and their corresponding prediction scores of all reviewed predictors. A domain was predicted as a CCD if its probability was >0.5. For the negative protein (i.e. proteins without CCDs), if it was predicted to have a CCD, the average score would be calculated; otherwise, the average prediction score for each residue in this protein would be calculated. The results are shown in Figure 5B, where the ‘positive’ denotes CCDs while the ‘negative’ indicates the sequences without CCDs. Similar to Figure 5A, CCHMM_PROF and Multicoil2-21 again achieved the highest and second highest AUC values (AUC = 0.906 and 0.863, respectively), suggesting that the majority of their predicted CCDs were consistent with the SOCKET assignment. COILS obtained the lowest performance with an AUC score of only 0.607. We also note that Multicoil2-all achieved a lower AUC score, possibly owing to its restriction of having a length requirement of coiled-coils during the model training. The performance comparison results between individual predictors and majority voting are shown in Supplementary Table S5. Because the minimum length of coiled-coils used for training Multicoil2 is 21, we further filtered the testing data set with different thresholds of coiled-coil lengths to perform the CCD coverage test. Although majority voting did not improve the overall prediction accuracy, the performance of majority voting was still competitive compared with individual predictors (Supplementary Table S5).
Figure 5.
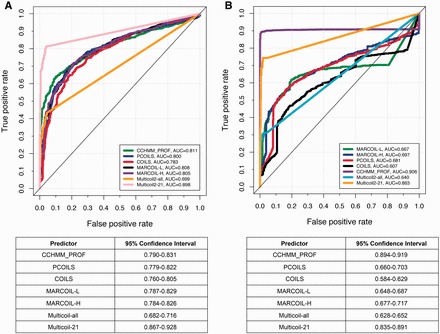
Performance comparison of CCD predictors. (A) ROC curves and the 95% confidence intervals of different predictors for identifying coiled-coil domains. (B) ROC curves and the 95% confidence intervals of different predictors, showing the consistency between the predicted CCDs and those annotated by SOCKET based on the protein structures. A colour version of this figure is available at BIB online: http://bib.oxfordjournals.org.
CCD and CCD oligomeric state prediction for human PolyQ proteins
Identification of CCDs
We first made a consensus-based decision for CCD prediction based on the predictors that are capable of discriminating coiled-coils from non-coiled-coils. The predictors used in this step were COILS, PCOILS, Paircoil2 (the p-score version with different window sizes and probability score version), MARCOIL, CCHMM_PROF, SpiriCoil and Multicoil2. Strikingly, the results are largely inconsistent between different predictors (Supplementary Tables S6–S13), making it difficult to generate a consensus prediction. Only a small portion of the proteins was predicted to harbour CCDs according to the prediction results of PCOILS, Paircoil2 (both p-score and probability score versions), SpiriCoil and Multicoil2. In contrast, COILS, MARCOIL and CCHMM_PROF predicted several CCDs within the nine PolyQ proteins. Most of the predicted coiled-coils overlapped or flanked the PolyQ tract. Based on the prediction results, the final decisions of predicted CCDs were made through majority voting (i.e. the CCD peptides need to be predicted by at least four predictors; the results are listed in Table 3). In the prediction of CCDs in nine disease-associated PolyQ proteins by Fiumara et al. [27], only two relatively old CCDs predictors were used (COILS and Paircoil2). We note that the results of Fiumara et al. are inconsistent with our predictions in this several state-of-the-art predictors. This discrepancy highlights that it remains a challenging task to develop reliable and consistent CCD prediction methods, and that attention should be paid when only a few specific methods are used to make the prediction, especially when these methods are used to guide and interpret experimental investigations such as the studies by Fiumara et al. [27].
Table 3.
The consensus CCDs predicted by at least four predictors
| Protein | Predicted coiled-coils | Protein structure | Sequence | Overlapping PolyQ tract | Agreed by |
|---|---|---|---|---|---|
| Voltage-dependent P/Q-type calcium channel subunit alpha-1A (Brain calcium channel I) | 720–747 | 3BXK (B/D = 1955–1975) | AQELTKDEQEEEEAANQKLALQKAKEVA | No | COILS, PCOILS, Paircoil2 (P-score version), CCHMM_PROF, Multicoil2 and MARCOIL |
| Atrophin-1 | 793–819 | – | AKKRADLVEKVRREAEQRAREEKERER | No | COILS, PCOILS, Paircoil2 (P-score version), CCHMM_PROF, Multicoil2 (cut-off = 0.5) and MARCOIL |
Prediction of oligomeric state of PolyQ proteins
To examine the potential oligomeric states of the peptides listed in Table 3, we performed the prediction using RFCoil, SCORER 2.0, PrOCoil and LOGICOIL (Supplementary Tables S14 and Supplementary Data). Because COILS, MARCOIL, PCOILS, Paircoil2 and Multicoil2 all provided heptad registers, we used these heptads to facilitate the oligomeric state prediction. As we can see, with different heptad registers, RFCoil, SCORER 2.0 and PrOCoil produced consistent prediction results (dimer formation), while the oligomeric state predictions from LOGICOIL were variable.
Conclusion
Given the functional significance of CCDs, computational biologists are motivated to develop more accurate and reliable predictors for CCD prediction. Aiming at providing a comprehensive review of coiled-coil predictors to non-bioinformaticians, this article describes and compares a number of widely used coiled-coil predictors in terms of their input, model construction and model evaluation. Independent tests reveal that LOICOIL achieved the overall highest AUC value when used to predict parallel dimeric and trimeric coiled-coils. For CCD prediction, Multicoil2 achieved the highest AUC value when detecting long CCDs in proteins, while CCHMM_PROF achieved the highest AUC value for the coverage of detected CCDs without the length limitation of CCDs. A case study of nine PolyQ proteins demonstrated that coiled-coil predictions were quite different among different predictors, which could further confound the consensus prediction analysis. We conclude that coiled-coil prediction is still a challenging task, and we expect that more powerful algorithms with improved prediction performance will emerge with the increasing availability of coiled-coil data.
Key Points
This article provides a comprehensive review on the current progress of computational approaches for coiled-coil domain (CCD) prediction and coiled-coil oligomeric state prediction.
Independent tests using rigorously prepared data sets highlight that Multicoil2 (tested with long coiled-coils) and CCHMM_PROF achieved the highest area under curve (AUC) values for coiled-coil domain prediction, while LOGICOIL achieved the highest AUC value for parallel dimeric and trimeric prediction.
The CCD prediction results on nine PolyQ proteins show inconsistencies of CCD prediction, which should be borne in mind when using prediction methods to make meaningful and reliable biological inferences.
This review serves as a useful guide for researchers who want to gain a better understanding of state-of-the-art approaches in this area and aim to develop their own methods with improved performance.
Supplementary Material
Acknowledgements
The authors would like to thank Derek N. Woolfson, Mauro Delorenzi and Piero Fariselli for providing the training data sets for LOGICOIL, MARCOIL and CCHMM_PROF, respectively.
Funding
China Scholarship Council (CSC) and Monash University Joint PhD Student Scholarship (to C.L.); Higher Degree by Research Scholarship (HDR) awarded by Monash University, Malaysia (to C.C.H.C); JSPS, Japan (grant-in-aid #26240034 to T.A.); Hundred Talents Program of the Chinese Academy of Sciences (CAS) (J.S.); NIH R01 (AI111990 to J.S.); Monash University Major Inter-Disciplinary Research Project grant (201402 to J.S.); and A.M.B. is an NHMRC Senior Research Fellow (1022688).
References
- 1. Lupas A. Coiled coils: new structures and new functions. Trends Biochem Sci 1996;21:375–82. [PubMed] [Google Scholar]
- 2. Grigoryan G, Keating AE. Structural specificity in coiled-coil interactions. Curr Opin Struct Biol 2008;18:477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McFarlane AA, Orriss GL, Stetefeld J. The use of coiled-coil proteins in drug delivery systems. Eur J Pharmacol 2009;625:101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tarricone C, Xiao B, Justin N, et al. The structural basis of Arfaptin-mediated cross-talk between Rac and Arf signalling pathways. Nature 2001;411:215–19. [DOI] [PubMed] [Google Scholar]
- 5. Burkhard P, Kammerer RA, Steinmetz MO, et al. The coiled-coil trigger site of the rod domain of cortexillin I unveils a distinct network of interhelical and intrahelical salt bridges. Structure 2000;8:223–30. [DOI] [PubMed] [Google Scholar]
- 6. Bullough PA, Hughson FM, Skehel JJ, et al. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994;371:37–43. [DOI] [PubMed] [Google Scholar]
- 7. Whitson SR, LeStourgeon WM, Krezel AM. Solution structure of the symmetric coiled coil tetramer formed by the oligomerization domain of hnRNP C: implications for biological function. J Mol Biol 2005;350:319–37. [DOI] [PubMed] [Google Scholar]
- 8. Gromiha MM, Parry DA. Characteristic features of amino acid residues in coiled-coil protein structures. Biophys Chem 2004;111:95–103. [DOI] [PubMed] [Google Scholar]
- 9. Mason JM, Arndt KM. Coiled coil domains: stability, specificity, and biological implications. Chembiochem 2004;5:170–6. [DOI] [PubMed] [Google Scholar]
- 10. Arndt KM, Pelletier JN, Muller KM, et al. Comparison of in vivo selection and rational design of heterodimeric coiled coils. Structure 2002;10:1235–48. [DOI] [PubMed] [Google Scholar]
- 11. Gillingham AK, Munro S. Long coiled-coil proteins and membrane traffic. Biochim Biophys Acta 2003;1641:71–85. [DOI] [PubMed] [Google Scholar]
- 12. Rose A, Schraegle SJ, Stahlberg EA, et al. Coiled-coil protein composition of 22 proteomes–differences and common themes in subcellular infrastructure and traffic control. BMC Evol Biol 2005;5:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo Y, Bozic D, Malashkevich VN, et al. All-trans retinol, vitamin D and other hydrophobic compounds bind in the axial pore of the five-stranded coiled-coil domain of cartilage oligomeric matrix protein. EMBO J 1998;17:5265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ozbek S, Engel J, Stetefeld J. Storage function of cartilage oligomeric matrix protein: the crystal structure of the coiled-coil domain in complex with vitamin D(3). EMBO J 2002;21:5960–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stetefeld J, Jenny M, Schulthess T, et al. Crystal structure of a naturally occurring parallel right-handed coiled coil tetramer. Nat Struct Biol 2000;7:772–76. [DOI] [PubMed] [Google Scholar]
- 16. Eriksson M, Hassan S, Larsson R, et al. Utilization of a right-handed coiled-coil protein from archaebacterium Staphylothermus marinus as a carrier for cisplatin. Anticancer Res 2009;29:11–18. [PubMed] [Google Scholar]
- 17. Boulikas T, Vougiouka M. Recent clinical trials using cisplatin, carboplatin and their combination chemotherapy drugs (review). Oncol Rep 2004;11:559–95. [PubMed] [Google Scholar]
- 18. Deacon SP, Apostolovic B, Carbajo RJ, et al. Polymer coiled-coil conjugates: potential for development as a new class of therapeutic “molecular switch". Biomacromolecules 2011;12:19–27. [DOI] [PubMed] [Google Scholar]
- 19. Hodges RS. Boehringer Mannheim award lecture 1995. La conference Boehringer Mannheim 1995. De novo design of alpha-helical proteins: basic research to medical applications. Biochem Cell Biol 1996;74:133–54. [DOI] [PubMed] [Google Scholar]
- 20. Kakizawa Y, Furukawa S, Ishii A, et al. Organic-inorganic hybrid-nanocarrier of siRNA constructing through the self-assembly of calcium phosphate and PEG-based block aniomer. J Control Release 2006;111:368–70. [DOI] [PubMed] [Google Scholar]
- 21. Wu K, Liu J, Johnson RN, et al. Drug-free macromolecular therapeutics: induction of apoptosis by coiled-coil-mediated cross-linking of antigens on the cell surface. Angew Chem Int Ed Engl 2010;49:1451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pechar M, Pola R. The coiled coil motif in polymer drug delivery systems. Biotechnol Adv 2013;31:90–6. [DOI] [PubMed] [Google Scholar]
- 23. Vincent TL, Woolfson DN, Adams JC. Prediction and analysis of higher-order coiled-coils: insights from proteins of the extracellular matrix, tenascins and thrombospondins. Int J Biochem Cell Biol 2013;45:2392–401. [DOI] [PubMed] [Google Scholar]
- 24. Gruber M, Soding J, Lupas AN. Comparative analysis of coiled-coil prediction methods. J Struct Biol 2006;155:140–5. [DOI] [PubMed] [Google Scholar]
- 25. Chang CC, Song J, Tey BT, et al. Bioinformatics approaches for improved recombinant protein production in Escherichia coli: protein solubility prediction. Brief Bioinform 2014;15:953–62. [DOI] [PubMed] [Google Scholar]
- 26. Chang CC, Tey BT, Song J, et al. Towards more accurate prediction of protein folding rates: a review of the existing web-based bioinformatics approaches. Brief Bioinform 2014;16(2):314–24. [DOI] [PubMed] [Google Scholar]
- 27. Fiumara F, Fioriti L, Kandel ER, et al. Essential role of coiled coils for aggregation and activity of Q/N-rich prions and PolyQ proteins. Cell 2010;143:1121–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lupas A. Predicting coiled-coil regions in proteins. Curr Opin Struct Biol 1997;7:388–93. [DOI] [PubMed] [Google Scholar]
- 29. Gruber M, Soding J, Lupas AN. REPPER–repeats and their periodicities in fibrous proteins. Nucleic Acids Res 2005;33:W239–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McDonnell AV, Jiang T, Keating AE, et al. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics 2006;22:356–58. [DOI] [PubMed] [Google Scholar]
- 31. Tanizawa H, Ghimire GD, Mitaku S. A hight performance prediction system of coiled coil domains containing heptad breaks: SOSUIcoil. Chem-Bio Inform J 2008;8:16. [Google Scholar]
- 32. Delorenzi M, Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 2002;18:617–25. [DOI] [PubMed] [Google Scholar]
- 33. Bartoli L, Fariselli P, Krogh A, et al. CCHMM_PROF: a HMM-based coiled-coil predictor with evolutionary information. Bioinformatics 2009;25:2757–63. [DOI] [PubMed] [Google Scholar]
- 34. Rackham OJ, Madera M, Armstrong CT, et al. The evolution and structure prediction of coiled coils across all genomes. J Mol Biol 2010;403:480–93. [DOI] [PubMed] [Google Scholar]
- 35. Armstrong CT, Vincent TL, Green PJ, et al. SCORER 2.0: an algorithm for distinguishing parallel dimeric and trimeric coiled-coil sequences. Bioinformatics 2011;27:1908–14. [DOI] [PubMed] [Google Scholar]
- 36. Vincent TL, Green PJ, Woolfson DN. LOGICOIL–multi-state prediction of coiled-coil oligomeric state. Bioinformatics 2013;29:69–76. [DOI] [PubMed] [Google Scholar]
- 37. Mahrenholz CC, Abfalter IG, Bodenhofer U, et al. Complex networks govern coiled-coil oligomerization–predicting and profiling by means of a machine learning approach. Mol Cell Proteomics 2011;10:M110.004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li C, Wang XF, Chen Z, et al. Computational characterization of parallel dimeric and trimeric coiled-coils using effective amino acid indices. Mol Biosyst 2015;1:354–60. [DOI] [PubMed] [Google Scholar]
- 39. Trigg J, Gutwin K, Keating AE, et al. Multicoil2: predicting coiled coils and their oligomerization states from sequence in the twilight zone. PLoS One 2011;6:e23519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Andreeva A, Howorth D, Chandonia JM, et al. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res 2008;36:D419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Walshaw J, Woolfson DN. Socket: a program for identifying and analysing coiled-coil motifs within protein structures. J Mol Biol 2001;307:1427–50. [DOI] [PubMed] [Google Scholar]
- 42. Testa OD, Moutevelis E, Woolfson DN. CC+: a relational database of coiled-coil structures. Nucleic Acids Res 2009;37:D315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fu L, Niu B, Zhu Z, et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 2012;28:3150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Altschul SF, Madden TL, Schaffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Berger B, Wilson DB, Wolf E, et al. Predicting coiled coils by use of pairwise residue correlations. Proc Natl Acad Sci USA 1995;92:8259–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fariselli P, Molinini D, Casadio R, et al. Prediction of structurally-determined coiled-coil domains with hidden Markov models. Bioinform Res Dev Proc 2007;4414:292–302. [Google Scholar]
- 47. Wolf E, Kim PS, Berger B. MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci 1997;6:1179–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Woolfson DN, Alber T. Predicting oligomerization states of coiled coils. Protein Sci 1995;4:1596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cortes C, Vapnik V. Support-vector networks. Mach Learn 1995;20:273–97. [Google Scholar]
- 50. Breiman L. Random forests. Mach Learn 2001;45:5–32. [Google Scholar]
- 51. Raileanu LE, Stoffel K. Theoretical comparison between the Gini Index and Information Gain criteria. Anna Math Artif Intell 2004;41:77–93. [Google Scholar]
- 52. Kendall M. A new measure of rank correlation. Biometrika 1938;30:9. [Google Scholar]
- 53. Robertson AL, Bate MA, Androulakis SG, et al. PolyQ: a database describing the sequence and domain context of polyglutamine repeats in proteins. Nucleic Acids Res 2011;39:D272–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bilen J, Bonini NM. Drosophila as a model for human neurodegenerative disease. Annu Rev Genet 2005;39:153–71. [DOI] [PubMed] [Google Scholar]
- 55. Saunders HM, Bottomley SP. Multi-domain misfolding: understanding the aggregation pathway of polyglutamine proteins. Protein Eng Des Sel 2009;22:447–51. [DOI] [PubMed] [Google Scholar]
- 56. Kim MW, Chelliah Y, Kim SW, et al. Secondary structure of Huntingtin amino-terminal region. Structure 2009;17:1205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zuhlke C, Hellenbroich Y, Dalski A, et al. Different types of repeat expansion in the TATA-binding protein gene are associated with a new form of inherited ataxia. Eur J Hum Genet 2001;9:160–4. [DOI] [PubMed] [Google Scholar]
- 58. Nakamura K, Jeong SY, Uchihara T, et al. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 2001;10:1441–8. [DOI] [PubMed] [Google Scholar]
- 59. Silveira I, Miranda C, Guimaraes L, et al. Trinucleotide repeats in 202 families with ataxia: a small expanded (CAG)n allele at the SCA17 locus. Arch Neurol 2002;59:623–9. [DOI] [PubMed] [Google Scholar]
- 60. Lin B, Nasir J, MacDonald H, et al. Sequence of the murine Huntington disease gene: evidence for conservation, alternate splicing and polymorphism in a triplet (CCG) repeat [corrected]. Hum Mol Genet 1994;3:85–92. [DOI] [PubMed] [Google Scholar]
- 61. Banfi S, Servadio A, Chung MY, et al. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat Genet 1994;7:513–20. [DOI] [PubMed] [Google Scholar]
- 62. Quan F, Janas J, Popovich BW. A novel CAG repeat configuration in the SCA1 gene: implications for the molecular diagnostics of spinocerebellar ataxia type 1. Hum Mol Genet 1995;4:2411–13. [DOI] [PubMed] [Google Scholar]
- 63. Pulst SM, Nechiporuk A, Nechiporuk T, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 1996;14:269–76. [DOI] [PubMed] [Google Scholar]
- 64. Sanpei K, Takano H, Igarashi S, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 1996;14:277–84. [DOI] [PubMed] [Google Scholar]
- 65. Imbert G, Saudou F, Yvert G, et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet 1996;14:285–91. [DOI] [PubMed] [Google Scholar]
- 66. Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010;466:1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhuchenko O, Bailey J, Bonnen P, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet 1997;15:62–9. [DOI] [PubMed] [Google Scholar]
- 68. Jodice C, Mantuano E, Veneziano L, et al. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum Mol Genet 1997;6:1973–8. [DOI] [PubMed] [Google Scholar]
- 69. Tonelli A, D'Angelo MG, Salati R, et al. Early onset, non fluctuating spinocerebellar ataxia and a novel missense mutation in CACNA1A gene. J Neurol Sci 2006;241:13–17. [DOI] [PubMed] [Google Scholar]
- 70. Romaniello R, Zucca C, Tonelli A, et al. A wide spectrum of clinical, neurophysiological and neuroradiological abnormalities in a family with a novel CACNA1A mutation. J Neurol Neurosurg Psychiatry 2010;81:840–3. [DOI] [PubMed] [Google Scholar]
- 71. Nagafuchi S, Yanagisawa H, Ohsaki E, et al. Structure and expression of the gene responsible for the triplet repeat disorder, dentatorubral and pallidoluysian atrophy (DRPLA). Nat Genet 1994;8:177–82. [DOI] [PubMed] [Google Scholar]
- 72. David G, Abbas N, Stevanin G, et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 1997;17:65–70. [DOI] [PubMed] [Google Scholar]
- 73. Echaniz-Laguna A, Rousso E, Anheim M, et al. A family with early-onset and rapidly progressive X-linked spinal and bulbar muscular atrophy. Neurology 2005;64:1458–60. [DOI] [PubMed] [Google Scholar]
- 74. Kawaguchi Y, Okamoto T, Taniwaki M, et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 1994;8:221–8. [DOI] [PubMed] [Google Scholar]
- 75. Fox NK, Brenner SE, Chandonia JM. SCOPe: Structural Classification of Proteins–extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res 2014;42:D304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chawla NV. Data Mining for Imbalanced Datasets: An Overview, Data Mining and Knowledge Discovery Handbook, 2nd edn, Springer, United States of America, 2010:875–86. [Google Scholar]
- 78. Munkhdalai T, Namsrai OE, Ryu K. Self-training in significance space of support vectors for imbalanced biomedical event data. BMC Bioinformatics 2015;16(Suppl 7):S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wu K, Edwards A, Fan W, et al. Classifying imbalanced data streams via dynamic feature group weighting with importance sampling. Proc SIAM Int Conf Data Min 2014;2014:722–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yang P, Xu L, Zhou BB, et al. A particle swarm based hybrid system for imbalanced medical data sampling. BMC Genomics 2009;10(Suppl 3):S34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.