

Abstract
Motivation: Combinatorial therapies have been under intensive research for cancer treatment. However, due to the large number of possible combinations among candidate compounds, exhaustive screening is prohibitive. Hence, it is important to develop computational tools that can predict compound combination effects, prioritize combinations and limit the search space to facilitate and accelerate the development of combinatorial therapies.
Results: In this manuscript we consider the NCI-DREAM Drug Synergy Prediction Challenge dataset to identify features informative about combination effects. Through systematic exploration of differential expression profiles after single compound treatments and comparison of molecular structures of compounds, we found that synergistic levels of combinations are statistically significantly associated with compounds’ dissimilarity in structure and similarity in induced gene expression changes. These two types of features offer complementary information in predicting experimentally measured combination effects of compound pairs. Our findings offer insights on the mechanisms underlying different combination effects and may help prioritize promising combinations in the very large search space.
Availability and Implementation: The R code for the analysis is available on https://github.com/YiyiLiu1/DrugCombination.
Contact: hongyu.zhao@yale.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Designing effective therapies for cancer treatment is an important yet challenging task. Although monotherapies are commonly used to treat cancer (Joensuu et al., 2001; Vogel et al., 2002), they suffer from many problems, such as acquired resistance and poor safety (LoRusso et al., 2012). Combinatorial therapies, by utilizing compounds impacting multiple biological processes simultaneously, have great potentials to overcome these problems and are attracting growing interest in drug development (Al-Lazikani et al., 2012; Jia et al., 2009). Indeed, there are many effective combinations used in practice. For example, the combination of Lapatinib and Capecitabine can achieve improved efficacy in breast cancer treatment (Geyer et al., 2006). In this study we focus on two-way combinations because most studies and available data to date are on two-way combinations and yet all existing methods are still limited in their performance in predicting the effectiveness of two-way combinations. Generally, when two compounds act together, their combination effect can be categorized into three main types: additive, if the effect of combination is equivalent to the sum of the effects of two compounds acting individually; synergistic, if the combination effect is greater than additive; and antagonistic, if the combination effect is less than additive. Typically compound combination effects are inferred through cell culture experiments, where cell viabilities under treatment with compounds in combination and treatment with compounds individually are measured and compared. However, since the number of possible combinations grows rapidly with the number of compounds under consideration, exhaustive experimental screening of all these combinations is prohibitively costly. Therefore, it is very useful to develop computational methods that can prioritize different combinations in order to reduce the search space for screening experiments. There are inspiring attempts made towards this goal (Guimera and Sales-Pardo, 2013; Huang et al., 2013, 2014; Li et al., 2015; Pang et al., 2014; Zhao et al., 2011). However, while it is known combination effects can be cell-line specific (Held et al., 2013), most existing computational methods were trained using only the specific types of combination effects (synergistic/additive/antagonistic) collected from literature review or databases, without considering the cell lines they were tested on or other experimental contexts. Besides, many of these methods rely heavily on prior knowledge of drug combination mechanisms, which is far from complete and accurate.
In this article, we aim to identify features informative on combination effects of compound pairs using the dataset from the NCI-DREAM Drug Synergy Prediction Challenge (Bansal et al., 2014). This DREAM challenge provided gene expression profiles of OCL-LY3 diffuse large B-cell lymphoma cells pre- and post- 14 single compound treatments, and experimentally-evaluated the combination effects of 91 pairwise combinations of these 14 compounds. In the original challenge, participants proposed different methods to predict combination effects using gene expression profiles, but no unanimous conclusion could be drawn on the relation between synergistic effects and transcriptome changes induced by single compounds (Bansal et al., 2014). Although a few participants considered compound chemical/molecular features such as structure, the relevance of these features to combination effects was not demonstrated. In this article, we adopt the structural similarity of compounds defined in PubChem (Kim et al., 2016) and consider four similarity measures for gene expression changes resulting from single compound treatments. We systematically explore whether the structural similarity and expression similarity is associated with combination effects using PC-index and resampled Spearman correlation (Bansal et al., 2014). Because we are interested in prioritizing compound pairs likely having synergistic effects, we also evaluate the area under the receiver operation characteristic curve (AUC) for synergistic combination prediction using structural similarity and expression similarity individually and jointly. For the NCI-DREAM challenge data set, we find that synergistic effect prediction using gene expression similarity defined with one of the four measures outperforms the best method reported in the original challenge, and that prediction using structural similarity also outperforms most of the methods proposed for the original challenge (Bansal et al., 2014). Moreover, combining gene expression and structure information together further improves predictive power for synergistic effects. Our findings in this paper may lead to better computational methods to prioritize compound combinations and facilitate future drug development.
2 Methods
2.1 Data preprocessing
We analyzed the data distributed for the NCI-DREAM Drug Synergy Prediction Challenge (Bansal et al., 2014; Goswami et al., 2015; Yang et al., 2015). In this project, the OCL-LY3 cell line was perturbed by 14 single compounds, each with 2 concentrations (IC20’s of 24 and 48 h), and gene expression profiles of untreated samples, DMSO-treated (DMSO was used as a control media) and single compound-treated samples were generated at three time points (6, 12 and 24 h after treatment). All expression profiles were measured using the Human Genome U219 96-Array plate (Affymetrix), in triplicate except that DMSO-treated ones were in octuplicate. Quality-controlled and RMA-normalized (Irizarry et al., 2003) data (in log2 scale) were provided by the Challenge. All 91 pairwise combinations of the 14 compounds () were tested with each compound concentrated at its IC20 of 60 h. Combination effects of these compound pairs were assessed using the Bliss independence model (Bliss, 1939). Mean Excess over Bliss (EOB) values (estimated from five replicates) were provided as a measurement of the synergy levels and standard errors (SE) were also given to quantify the uncertainties. We used these experimentally measured combination effects as ‘gold standard’ in our analysis.
We first applied RUVr (Risso et al., 2014) to remove potential batch effects in gene expression measurements. We considered compounds, concentrations and time points as treatment status (‘wanted variation’) and varied the number of hidden factors (‘unwanted variation’) from 1 to 40. Based on the relative log expression (RLE) plots (Supplementary Figs S1 and S2), we set in our analysis. We also reported results with other values of in supplementary information (Supplementary Figs S3–S10 and Supplementary Tables S1–S5), which suggest that our results are quite robust to the specific choice of k.
2.2 Structural similarity measure
We used substructure key-based 2D Tanimoto similarity score from the PubChem database (Kim et al., 2016) to quantify the structural similarity between two compounds. PubChem generates a binary fingerprint (an ordered list of binary bits) to represent the presence or absence of specific chemical substructure for each compound. It then defines the similarity score between two compounds as
where is the count of bits shared by the two compounds, and and are the counts of bits in the two compounds, respectively.
It can be seen from the above formula that the Tanimoto score is always between 0 and 1. According to PubChem, a Tanimoto score of or greater is considered statistically significant at the 95% significance level (Kim et al., 2016).
2.3 Gene expression similarity measures
We used the average of three/eight replicates to represent gene expression levels under each experimental condition. We then calculated the fold changes between treatments (single compounds) and control (DMSO). The following metrics were adopted to evaluate the global differential expression similarities between two drugs.
Direction-based
This metric considers the concordance of the direction of gene expression changes (up or down) following single-compound treatments. It was motivated by the second best performing method in the original challenge (Goswami et al., 2015). However, unlike what was done there, we did not assume a common core gene set for all compounds since different compounds may impact different genes and pathways. Instead, we selected out genes with large expression changes for each compound and measured the concordance of two compounds on their corresponding ‘signature’ genes. Specifically, we discretized expression changes into three levels: for probes with fold changes within a certain range around and thus viewed as unaffected by a compound; and for probes with fold changes outside the range and considered down-regulated and up-regulated, respectively. We then define the expression similarity score between compound and compound as
where is the total number of probes (48 789 in this dataset) and is the status (, or ) for probe treated with compound .
GSEA-based
This metric was proposed in the context of constructing drug similarity network with transcriptome data (Iorio et al., 2009, 2010). The original method (Iorio et al., 2009, 2010) first ranks probes from the most up-regulated to the most down-regulated in each of the six datasets measured at three time points and two concentrations, and then merges the six lists using pair-wise Spearman’s Footrule, Borda Merging Method and Kruskal Algorithm (Diaconis and Graham, 1977; Lin, 2010) into a unified ranked list hierarchically. Then for compounds and , it calculates four Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) scores: and , the enrichments of compound ’s top probes and bottom probes in compound ’s expression list; and , the enrichments of compound ’s top probes and bottom probes in compound ’s expression list. A distance between and is defined as:
Since the values of ’s are between and , is between and . In our analyses, we define the corresponding similarity score as:
which is between to . We also calculated without merging the six lists for each compound.
Pearson correlation-based
Under each treatment condition, we ranked all the probes according to their absolute log2-transformed intensity differences, and then selected top probes as signatures. We calculated the Pearson correlation between two compounds’ expression profiles on all 48 789 probes as well as on the union of their signature gene sets.
Spearman correlation-based
We also calculated the Spearman correlation between two compounds’ expression profiles on all 48 789 probes as well as on the union of their signature gene sets.
2.4 Evaluation of concordance between similarity-based scores and synergistic levels of compound pairs
We used probabilistic concordance index (PC-index) and resampled Spearman correlation to quantify the concordance between the similarity-based scores and synergistic levels of compound pairs (Bansal et al., 2014).
2.4.1 PC-index
Suppose that there are a total of compound pairs. Let the similarity-based scores for compound pair be , and the experimentally measured EOB and its SE be and , then for two compound pairs and , we first compute
where
PC-index is defined as
PC-index takes experimental errors into account when evaluating the concordance between the predictions and the ‘gold standard’. It is symmetric around , i.e. if a prediction has pc-index , then flipping it over (exactly opposite order) gives pc-index . The largest value (PCmax) is reached when the predictions are entirely concordant with the ‘gold standard’. Due to noise terms, PCmax will not be generally. In this study,
2.4.2 Resampled spearman correlation
Similar to PC-index, resampled Spearman correlation accounts for uncertainties of the experiment. It assumes that the experimental measurement of mean EOB is noisy and follows a normal distribution, , with and equal to the mean EOB and SE obtained from the experiment. For each compound pair , we randomly sample a new from , and then calculated the spearman correlation between and . We repeat the process for 10 000 times and use the mean of the 10 000 Spearman correlations as a final resampled Spearman correlation score .
2.4.3 Statistical significance estimation
We simulated 10 000 independent random predictions (10 000 random permutations of the 91 pairs) and calculated their PC-indices and resampled Spearman correlations. We use these scores from random predictions as the empirical null distributions for PC-index and resampled Spearman correlation. Then we estimated the P-value of PC-index and for each similarity-based prediction as
2.5 Synergistic combination prediction
We treated compound pairs with experimentally measured synergistic combination as positive and all others as negative, and adopted logistic regression models to predict synergistic combinations using gene expression similarity and structural similarity, individually and jointly. We performed 100 rounds of 3-fold cross validation. In each round, we estimated the cross-validation AUC using trapezoidal method to integrate the ROC curve. We reported the mean AUC over 100 rounds for each classifier as an evaluation of its performance. To assess the statistical significance of improvement brought by structural information compared to using gene expression only, we randomly permutated the structural similarity scores of the 91 pairs 1000 times, and for each permutation, we calculated the mean AUC (over 100 rounds of cross validation) of the classifier combining the permutated structural similarity score and the original gene expression similarity score. We calculated the P-value of AUC improvement as
Similarly, we calculated the P-value of AUC improvement by adding gene expression information as
3 Results
We evaluated the structural similarity of the 14 compounds and also compared their effects on the transcriptome of the OCL-LY3 cell line. Following the performance evaluation methods of the original DREAM challenge (Bansal et al., 2014), we used PC-index and resampled Spearman correlation coefficient to measure the associations between these similarity scores and the synergistic levels. Additionally, we evaluated these scores’ ability to predict synergistic combinations using AUC from cross validation.
3.1 Structural similarity negatively correlates with synergism
A negative correlation can be observed between EOB for two compounds against their structural similarity scores (Fig. 1). The PC-index between EOB and the negative structural similarity score was (P-value ), and the resampled Spearman correlation between them was (P-value ). This result outperforms most of the methods in the original challenge (Supplementary Table S6). It indicates that synergistic effect is significantly negatively correlated with structural similarity.
Fig. 1.
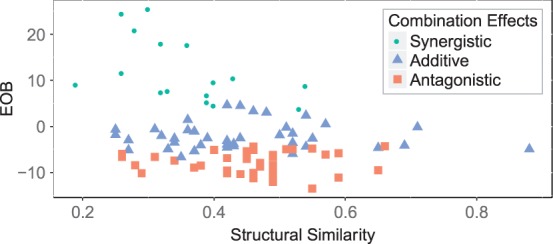
EOB against structural similarity of compounds. Activities of compound pairs are discretized into three states as defined in the original challenge: synergistic (circle), additive (triangle) and antagonistic (square)
In addition, there is an interesting pattern behind this overall negative correlation. In this dataset, compound pairs with EOB and EOB/SE were classified as having synergistic effects; similarly, those with EOB and EOB/SE were defined to be antagonistic; and the rest of the combinations were classified as additive (Bansal et al., 2014). We observed that none of the compound pairs with large structural similarity scores (e.g.>0.6) have synergistic effects, while the combination effects of the compound pairs with small structural similarity scores spread across all the three classes. We will discuss possible mechanisms for this phenomenon in Section 4.
3.2 Gene expression similarity positively correlates with synergism
We considered four measures for the similarity of gene expression changes brought by two compounds individually and all the similarity scores showed positive correlations with EOB.
We defined the first similarity score based on the directions (up or down) of gene expression changes. We set different thresholds on fold changes to define signatures and calculated the PC-indices and resampled Spearman correlations between EOB and the direction-based similarity measurements (Fig. 2(a, b)). In all situations we considered, the PC-indices were greater than and the resampled Spearman correlations were greater than , which indicate positive correlations between synergistic level and gene expression similarity. Taking all genes into account (first bar of each data set in Fig. 2), only weak correlations could be observed; by excluding genes with small changes, signals in most of the six datasets (measured at three time points and two concentrations) became strong. The largest PC-index and resampled Spearman correlation were (P-value ) and (P-value ), respectively.
Fig. 2.
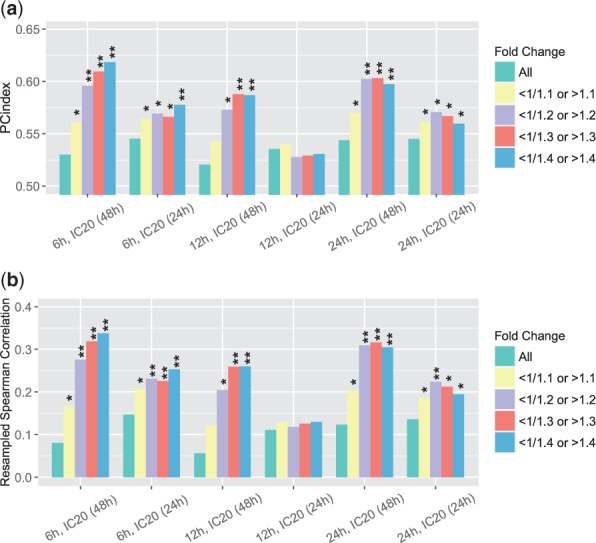
(a) PC-indices of direction-based gene expression similarity scores. (b) Resampled Spearman correlations of direction-based gene expression similarity scores. When calculating the similarity scores, we considered all probes as well as probes with fold changes beyond certain ranges only. *P-value <0; **P-value <
The second gene expression similarity score we considered was based on GSEA. We set different cutoffs to define top and bottom signature genes and the resulting PC-indices and resampled Spearman correlations between these expression similarities and EOB were shown in Figure 3(a, b). Again, we could observe positive correlations between EOB and these expression similarities in all cases. Here the largest PC-index and resampled Spearman correlation we got were (P-value ) and (P-value ), respectively.
Fig. 3.
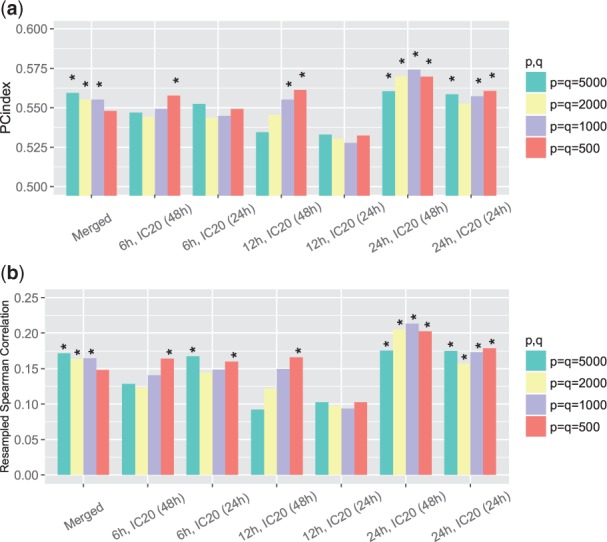
(a) PC-indices of GSEA-based gene expression similarity scores. (b) Resampled Spearman correlations of GSEA-based gene expression similarity scores. When calculating the similarity scores, we considered different numbers of top () and bottom () signatures; we included the results for a merged data set and six unmerged data sets separately. *P-value <; **P-value <
The third and fourth expression similarities we considered were based on the more conventional Pearson and Spearman correlations. Similar to what we did earlier, besides considering all the probes, we also selected strong signals (probes with the largest expression changes under the treatment of each single compound) and calculated the correlations only on these probes. We presented the PC-indices and resampled Spearman correlations between EOB and these two correlation-based scores in Figures 4(a, b) and 5(a, b), respectively. These two expression similarity measures also had positive correlations with synergistic levels. The largest PC-index and the largest resampled Spearman correlation we got for the Pearson correlation-based similarity were (P-value ) and (P-value ), respectively. The largest PC-index and resampled Spearman correlation we got for Spearman correlation-based similarity were (P-value ) and (P-value ), respectively.
Fig. 4.
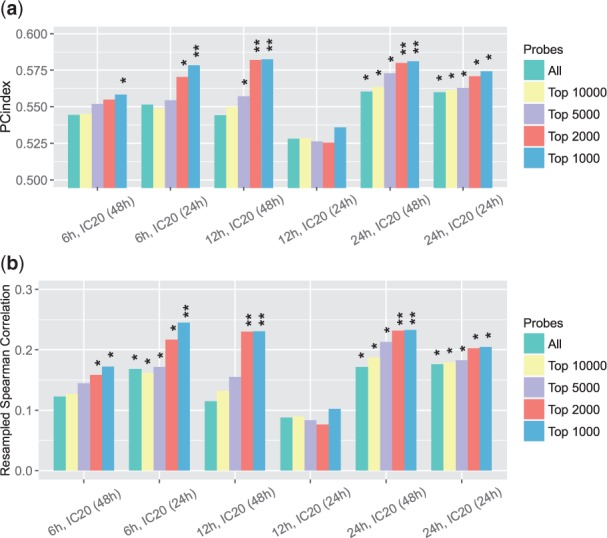
(a) PC-indices of Pearson correlation-based gene expression similarity scores. (b) Resampled Spearman correlations of Pearson correlation-based gene expression similarity scores. When calculating the similarity scores, we considered all probes as well as probes with largest expression changes only. *P-value <; **P-value <0.01
Fig. 5.
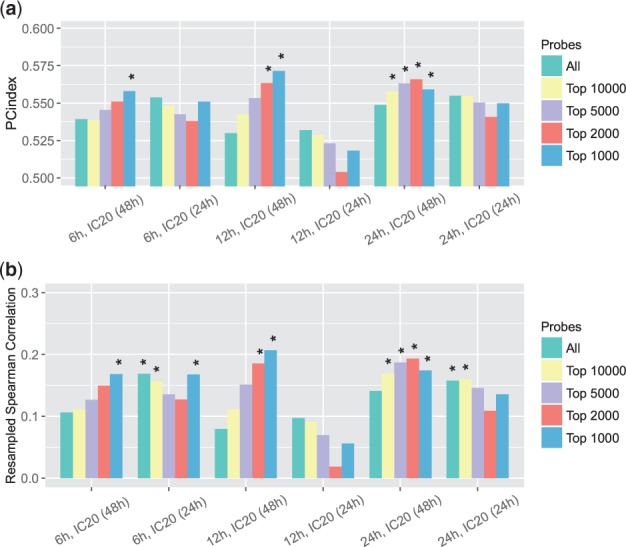
(a) PC-indices of Spearman correlation-based gene expression similarity scores. (b) Resampled Spearman correlations of Spearman correlation-based gene expression similarity scores. When calculating the similarity scores, we considered all probes as well as probes with largest expression changes only. *P-value <; **P-value <
Similar to structural similarity, we observed (Supplementary Fig. S11) that it is less likely for the compound pairs with relatively small gene expression similarity scores to have synergistic effects, while for the compound pairs with relatively large gene expression similarity scores, their combination effects vary case by case.
In addition, we also note the following two observations. First, according to our results, all six datasets have certain information that could be utilized, and no dosage or exposure time is ‘optimal’ in terms of transcriptome concordance with experimentally measured synergistic levels. Second, we found that signals can usually be strengthened when we focus on a subset of signature genes influenced most by the perturbations rather than considering all of them together. This is reasonable as many genes may not be impacted by individual drug perturbations and introduce noises.
3.3 Structural similarity and gene expression similarity provide complementary information on synergistic combination prediction
Since compound pairs with synergistic effects are most worthy of further investigation, we trained logistic regression models to predict synergistic combinations using structural similarity score, each of the four gene expression similarity scores, individually and jointly. The AUCs using gene expression similarity alone and jointly with structural similarity, estimated from 100 rounds 3-fold cross-validations, are listed in Tables 1–4. The AUC using structural similarity alone is 0.73.
Table 1.
AUC of logistic regression model to predict synergistic combinations: direction-based expression similarity scores
| 6 h, IC20 (48 h) | 6 h, IC20 (24 h) | 12 h, IC20 (48 h) | 12 h, IC20 (24 h) | 24 h, IC20 (48 h) | 24 h, IC20 (24 h) | |
|---|---|---|---|---|---|---|
| All | 0.43 [0.70] | 0.70 [0.77] | 0.47 [0.71] | 0.67 [0.76] | 0.56 [0.73] | 0.56 [0.71] |
| FC > 1.1 or < 1/1.1 | 0.55 [0.71] | 0.77 [0.80] | 0.43 [0.70] | 0.69 [0.77] | 0.66 [0.76] | 0.68 [0.74] |
| FC > 1.2 or < 1/1.2 | 0.69 [0.75] | 0.81 [0.83] | 0.62 [0.73] | 0.74 [0.78] | 0.79 [0.82] | 0.78 [0.79] |
| FC > 1.3 or < 1/1.3 | 0.75 [0.77] | 0.81 [0.81] | 0.74 [0.78] | 0.74 [0.79] | 0.84 [0.85] | 0.79 [0.82] |
| FC > 1.4 or < 1/1.4 | 0.77 [0.77] | 0.82 [0.80] | 0.76 [0.80] | 0.73 [0.78] | 0.85 [0.84] | 0.79 [0.81] |
AUCs are shown using gene expression similarity only and combining gene expression similarity with structural similarity (in brackets). FC, fold change.
Table 2.
AUC of logistic regression model to predict synergistic combinations: GSEA-based expression similarity scores
| Merged | 6 h, IC20 (48 h) | 6 h, IC20 (24 h) | 12 h, IC20 (48 h) | 12 h, IC20 (24 h) | 24 h, IC20 (48 h) | 24 h, IC20 (24 h) | |
|---|---|---|---|---|---|---|---|
| p=q=5000 | 0.65 [0.73] | 0.51 [0.72] | 0.74 [0.78] | 0.46 [0.71] | 0.66 [0.76] | 0.64 [0.74] | 0.65 [0.73] |
| p=q=2000 | 0.66 [0.74] | 0.52 [0.72] | 0.75 [0.79] | 0.43 [0.71] | 0.65 [0.76] | 0.67 [0.75] | 0.65 [0.73] |
| p=q=1000 | 0.66 [0.74] | 0.55 [0.72] | 0.76 [0.79] | 0.42 [0.71] | 0.65 [0.75] | 0.68 [0.75] | 0.66 [0.74] |
| p=q=500 | 0.65 [0.74] | 0.57 [0.72] | 0.77 [0.79] | 0.42 [0.71] | 0.65 [0.75] | 0.68 [0.75] | 0.66 [0.74] |
AUCs are shown using gene expression similarity only and combining gene expression similarity with structural similarity (in brackets).
Table 3.
AUC of logistic regression model to predict synergistic combinations: Pearson correlation-based expression similarity scores
| 6 h, IC20 (48 h) | 6 h, IC20 (24 h) | 12 h, IC20 (48 h) | 12 h, IC20 (24 h) | 24 h, IC20 (48 h) | 24 h, IC20 (24 h) | |
|---|---|---|---|---|---|---|
| All | 0.48 [0.71] | 0.74 [0.78] | 0.42 [0.71] | 0.63 [0.75] | 0.62 [0.74] | 0.64 [0.73] |
| Top 10000 | 0.50 [0.71] | 0.75 [0.78] | 0.42 [0.71] | 0.62 [0.74] | 0.64 [0.75] | 0.64 [0.72] |
| Top 5000 | 0.54 [0.72] | 0.77 [0.79] | 0.42 [0.71] | 0.61 [0.74] | 0.67 [0.76] | 0.64 [0.73] |
| Top 2000 | 0.57 [0.72] | 0.79 [0.79] | 0.46 [0.71] | 0.58 [0.73] | 0.68 [0.76] | 0.62 [0.73] |
| Top 1000 | 0.58 [0.72] | 0.81 [0.80] | 0.46 [0.71] | 0.59 [0.73] | 0.69 [0.75] | 0.60 [0.72] |
AUCs are shown using gene expression similarity only and combining gene expression similarity with structural similarity (in brackets).
Table 4.
AUC of logistic regression model to predict synergistic combinations: Spearman correlation-based expression similarity scores
| 6 h, IC20 (48 h) | 6 h, IC20 (24 h) | 12 h, IC20 (48 h) | 12 h, IC20 (24 h) | 24 h, IC20 (48 h) | 24 h, IC20 (24 h) | |
|---|---|---|---|---|---|---|
| All | 0.45 [0.71] | 0.72 [0.78] | 0.45 [0.71] | 0.65 [0.76] | 0.59 [0.73] | 0.59 [0.71] |
| Top 10000 | 0.48 [0.71] | 0.75 [0.78] | 0.42 [0.71] | 0.63 [0.75] | 0.63 [0.75] | 0.63 [0.72] |
| Top 5000 | 0.54 [0.72] | 0.75 [0.79] | 0.43 [0.71] | 0.59 [0.74] | 0.66 [0.75] | 0.62 [0.72] |
| Top 2000 | 0.60 [0.73] | 0.74 [0.79] | 0.46 [0.71] | 0.50 [0.73] | 0.68 [0.75] | 0.51 [0.71] |
| Top 1000 | 0.61 [0.73] | 0.76 [0.79] | 0.49 [0.71] | 0.48 [0.72] | 0.67 [0.75] | 0.45 [0.69] |
AUCs are shown using gene expression similarity only and combining gene expression similarity with structural similarity (in brackets).
It can be seen that most classifiers achieved satisfactory results in identifying synergistic combinations. Moreover, using structural similarity and gene expression similarity together does lead to greater predictive power. Specifically, for direction-based gene expression similarity scores, combining them together with structural similarity, in all the 30 cases (6 datasets 5 thresholds), 28 had larger AUCs compared with using expression similarity alone (all with P-values <), and 23 had larger AUCs compared with using structural similarity alone (22 with P-values <); for GSEA-based gene expression similarity scores, among the 28 cases (7 datasets 4 thresholds), combining them together with structural similarity outperformed using expression similarity alone and using structural similarity alone in 28 cases (all with P-value <) and 19 cases (8 with P-values <), respectively; for Pearson correlation-based gene expression similarity scores, when using two features, 29 out of 30 cases (6 datasets × 5 thresholds) had larger AUCs than using expression similarity alone (all with P-values <) and 15 had larger AUCs than using structural similarity alone (7 with P-value <); for Spearman correlation-based gene expression similarity scores, AUCs in all the 30 cases (6 datasets × 5 thresholds) were larger when using both features than using expression similarity alone (all with P-values <), and AUCs in 13 cases were larger than using structural similarity alone (6 with P-value <). Hence, we concluded for most of these gene expression similarity scores, their predictions for synergistic combinations were significantly improved when incorporating structural information. Especially in the cases where expression similarity scores were less informative (Figs 2–5 and Tables 1–5), the increments were remarkable; as it is not always easy to identify an ‘optimal’ dosage/duration in transcriptome effect measurements, we believe including structural information would benefit synergistic combination prediction greatly. Besides, for most cases where gene expression similarity demonstrated certain power to predict synergistic combinations, combining them and structural similarity together did lead to better performance over using structural similarity alone, too. Additionally, we found that the dataset measured at 6 h post-treatment with IC20’s of 24 h was most useful here compared to others, a phenomenon not observed when examining the scores’ correlations to EOBs (Section 3.2). We investigated the expression similarity scores on this dataset and found that most of them indeed could separate out synergistic pairs from the rest more effectively, as compared to measurements obtained from other datasets, synergistic pairs tend to be more concentrated on the relatively large gene expression similarity parts using these scores; while PC-index and resampled Spearman correlation emphasize more on the global concordance between the similarity score and EOBs, such signal was diluted.
In summary, we observed that both structural similarity and gene expression similarity are informative to predict synergistic effects; and these two types of features provide complementary information.
4 Discussion and conclusion
Developing combinatorial therapies for cancer treatment has attracted increasing attention due to their great potentials as compared to monotherapies. One major challenge to develop combinatorial therapies is the large search space of possible combinations, therefore computationally predicting combination effects and prioritizing combinations is vitally important. In this paper, we have analyzed the experimentally measured 91 combinations of 14 compounds from the NCI-DREAM Drug Synergy Prediction Challenge to identify features that are informative on combination effects.
We observed that structural similarity is statistically significantly negatively correlated with synergism. There has been previous work utilizing structural similarity to model compound combination effects, based on the assumption that if the combination of compound A and compound B has a specific effect, compounds having similar structure to compound A (B) tend to have the same interaction with compound B (A) (e.g. Vilar et al., 2012). Compared with these work and several methods in the original challenge, we systematically investigated the relation between structural similarity and synergism. We hypothesize that the negative correlation we observed may be caused by ‘competitive binding’ (Cokol et al., 2011; Jia et al., 2009), i.e. when two compounds bind to similar targets they tend to have antagonistic or additive effects due to competing and interfering interactions between them. Specifically, when two compounds have similar structures (i.e. many share substructures), they more likely interact with the same protein sites or perturb the same biological processes, where competition exists, and reduces the chance of synergistic effects. On the other hand, compound pairs with distinct structures (lower structural similarity scores) can interact with different proteins or have varied functions, where there is no strong competition for ‘resources’; depending on the biological processes they influence and genomic context of the cell line they work on, their combination effects can be synergistic, additive or antagonistic.
We also found a statistically significant positive association between synergism and the similarity of gene expression changes caused by single compounds. This association is robust to the four different similarity measures we considered on all six gene expression profiles. We hypothesize that two compounds leading to different changes on gene expressions may offset each other’s effects when applied together, thus are less likely able to ‘collaborate’ to generate synergistic effects. On the other hand, if two compounds lead to similar gene expression changes, they may not have such ‘neutralizing’ problem and hence are more likely to be synergistic; yet, the actual effects in such cases can vary depending on the interactions of different biological processes affected by the two compounds.
In addition, we found both gene expression similarity and structural similarity are predictive of synergistic combinations. More importantly, these two distinct similarity scores provided complementary information on synergistic effect predictions; when utilized together, the performance can be further improved. Besides classifying compound combinations as synergistic/non-synergistic, similarly we also observed complementary informativeness of gene expression and structural similarity in the three-class (synergistic, additive and antagonistic) classification as well (Supplementary Tables S3–S5). Our findings in this paper should be useful for prioritizing compound combinations. They also provide insights for us to further investigate the mechanisms behind various combination effects.
There are several directions worth explorations in future studies. First, for compound structural similarity scores, we used 2D information here as it is robust and often generates superior results in activity prediction (Maggiora et al., 2014). In the future, when more accurate descriptions of 3D compound structures and reliable metrics of 3D structural similarity are available, we can carry out similar analysis based on 3D information. Second, we considered gene expression similarity defined on individual genes in this paper. Since genes are not isolated in biological systems, better measures may be defined by taking gene–gene interactions into account. Finally, in addition to structure and expressions, other types of molecular and biological features may be investigated and prove useful in predicting combination effects.
Supplementary Material
Acknowledgements
We acknowledge Dr David Stern of Yale Department of Pathology and Dr Kuang-Yao Lee of Zhao Lab for their helpful discussions.
Funding
This work was supported by National Institutes of Health (NIH) grants GM59507, CA154295, and CA196530.
Conflict of Interest: none declared.
References
- Al-Lazikani B. et al. (2012) Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol., 30, 679–691. [DOI] [PubMed] [Google Scholar]
- Bansal M. et al. (2014) A community computational challenge to predict the activity of pairs of compounds. Nat. Biotechnol., 32, 1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss C.I. (1939) The toxicity of poisons applied jointly. Ann. Appl. Biol., 26, 585–615. [Google Scholar]
- Cokol M. et al. (2011) Systematic exploration of synergistic drug pairs. Mol. Syst. Biol., 7, 544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaconis P., Graham R.L. (1977) Spearman's footrule as a measure of disarray. J. R. Stat. Soc.. Ser. B, 262–268. [Google Scholar]
- Geyer C.E. et al. (2006) Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med., 355, 2733–2743. [DOI] [PubMed] [Google Scholar]
- Goswami C.P. et al. (2015) A new drug combinatory effect prediction algorithm on the cancer cell based on gene expression and dose–response curve. CPT Pharmacomet. Syst. Pharmacol., 4, e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimera R., Sales-Pardo M. (2013) A network inference method for large-scale unsupervised identification of novel drug–drug interactions. PLoS Comput. Biol., 9, e1003374.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held M.A. et al. (2013) Genotype-selective combination therapies for melanoma identified by high-throughput drug screening. Cancer Discov., 3, 52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.L. et al. (2013) Systematic prediction of pharmacodynamic drug–drug interactions through protein–protein-interaction network. PLoS Comput. Biol., 9, e1002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L. et al. (2014) DrugComboRanker: drug combination discovery based on target network analysis. Bioinformatics, 30, 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F. et al. (2009) Identifying network of drug mode of action by gene expression profiling. J. Comput. Biol., 16, 241–251. [DOI] [PubMed] [Google Scholar]
- Iorio F. et al. (2010) Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA, 107, 14621–14626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry R.A. et al. (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics, 4, 249–264. [DOI] [PubMed] [Google Scholar]
- Jia J. et al. (2009) Mechanisms of drug combinations: interaction and network perspectives. Nat. Rev. Drug Discov., 8, 111–128. [DOI] [PubMed] [Google Scholar]
- Joensuu H. et al. (2001) Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N. Engl. J. Med., 344, 1052–1056. [DOI] [PubMed] [Google Scholar]
- Kim S. et al. (2016) PubChem substance and compound databases. Nucleic Acids Res., 44, D1202–D1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P. et al. (2015) Large-scale exploration and analysis of drug combinations. Bioinformatics, 31, 2007–2016. [DOI] [PubMed] [Google Scholar]
- Lin S. (2010) Space oriented rank-based data integration. Stat. Appl. Genet. Mol. Biol., 9, Article20 ISSN (Online), 1544-6115. [DOI] [PubMed] [Google Scholar]
- LoRusso P.M. et al. (2012) Accelerating cancer therapy development: the importance of combination strategies and collaboration. Summary of an Institute of Medicine Workshop. Clin. Cancer Res., 18, 6101–6109. [DOI] [PubMed] [Google Scholar]
- Maggiora G. et al. (2014) Molecular similarity in medicinal chemistry. J. Med. Chem., 57, 3186–3204. [DOI] [PubMed] [Google Scholar]
- Pang K.F. et al. (2014) Combinatorial therapy discovery using mixed integer linear programming. Bioinformatics, 30, 1456–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D. et al. (2014) Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol., 32, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A. et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar S. et al. (2012) Drug–drug interaction through molecular structure similarity analysis. J. Am. Med. Inform. Assoc., 19, 1066–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C.L. et al. (2002) Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol., 20, 719–726. [DOI] [PubMed] [Google Scholar]
- Yang J. et al. (2015) DIGRE: drug-induced genomic residual effect model for successful prediction of multidrug effects. CPT Pharmacomet. Syst. Pharmacol., 4, e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.M. et al. (2011) Prediction of drug combinations by integrating molecular and pharmacological data. PLoS Comput. Biol., 7, e1002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.