

Abstract
Motivation
Bacterial resistance to antibiotics, particularly plasmid-encoded resistance to beta lactam drugs, poses an increasing threat to human health. Point mutations to beta-lactamase enzymes can greatly alter the level of resistance conferred, but predicting the effects of such mutations has been challenging due to the large combinatorial space involved and the subtle relationships of distant residues to catalytic function. Therefore we desire an information-theoretic metric to sensitively and robustly detect both local and distant residues that affect substrate conformation and catalytic activity.
Results
Here, we report the use of positional mutual information in multiple microsecond-length molecular dynamics (MD) simulations to predict residues linked to catalytic activity of the CTX-M9 beta lactamase. We find that motions of the bound drug are relatively isolated from motions of the protein as a whole, which we interpret in the context of prior theories of catalysis. In order to robustly identify residues that are weakly coupled to drug motions but nonetheless affect catalysis, we utilize an excess mutual information metric. We predict 31 such residues for the cephalosporin antibiotic cefotaxime. Nine of these have previously been tested experimentally, and all decrease both enzyme rate constants and empirical drug resistance. We prospectively validate our method by testing eight high-scoring mutations and eight low-scoring controls in bacteria. Six of eight predicted mutations decrease cefotaxime resistance greater than 2-fold, while only one control shows such an effect. The ability to prospectively predict new variants affecting bacterial drug resistance is of great interest to clinical and epidemiological surveillance.
Availability and implementation
Excess mutual information code is available at https://github.com/kassonlab/positionalmi
Contact
kasson@virginia.edu
1 Introduction
Beta lactamases are enzymes produced by bacteria that confer resistance to a broad range of clinically used antibiotics that share a common chemical core structure (Llarrull et al., 2010; Matagne et al., 1998). Use of these drugs has led to the emergence of beta lactamases with a broad spectrum of activity that render many classes of antibiotics ineffective (Boucher et al., 2013a). In the past, this has spurred the development of new beta lactam antibiotics, but that pipeline has slowed in recent decades, creating a clinical problem of highly resistant infections (Boucher et al., 2013b; Levy and Marshall, 2004). Understanding the determinants of activity and specificity in beta lactamases is key to both monitoring the emergence of further resistance and the targeted design of new antibacterial agents. Here, we focus on the CTX-M family, a group of extended spectrum beta lactamases that have spread globally in recent years and utilize a catalytic mechanism roughly analogous to serine proteases (Shimamura et al., 2002). CTX-M enzymes are currently the most prevalent extended-spectrum beta lactamases in both hospital-associated and community-acquired infections and are thus of substantial clinical concern (Cantón and Coque, 2006; Valverde et al., 2004). Even when the resulting infections are treatable, presence of a CTX-M enzyme greatly increases the morbidity resulting from bacterial infection (Cantón and Coque, 2006; Livermore et al., 2007; Philippon et al., 1989).
Although the key catalytic residues in CTX-M enzymes are well known, we wish to understand the basis for modulation of activity and ligand specificity in these enzymes. Even though comprehensive mutagenesis of CTX-M has not yet been experimentally feasible, reports of individual mutations show that single point mutations can alter drug spectrum and catalytic activity (Aumeran et al., 2003; Gazouli et al., 1998; Pérez-Llarena et al., 2011, 2008; Sougakoff et al., 1988). Such point mutants have been identified in clinical isolates of bacteria (Bonnet et al., 2001, 2003; Delmas et al., 2006; Poirel et al., 2002) as well as laboratory mutagenesis experiments (Aumeran et al., 2003; Gazouli et al., 1998; Pérez-Llarena et al., 2011, 2008; Sougakoff et al., 1988).
A comprehensive experimental understanding of how mutations affect beta lactamase drug resistance has been hindered by the combinatorial magnitude of the problem. Even for point mutations on CTX-M enzymes alone, rigorous quantitation of how mutants affect activity against a large panel of drugs is extremely resource-intensive, entailing the screening of ∼5000 mutants (260 residues × 19 amino acid changes) against multiple antibiotics. Therefore, we wish to identify residues that may contribute to activity and specificity but are not absolutely essential to function. Prediction of such residues can guide a more targeted set of mutagenesis experiments. In designing such an approach, we wish to consider all residues in the protein, not simply those in direct contact with the drug or previously identified via serendipitous mutations.
We have chosen to predict residues modulating antibiotic resistance in CTX-M enzymes based on analysis of molecular dynamics (MD) simulations. We hypothesize that the conformational dynamics of the enzyme and its substrate will yield insight into catalytic activity even though we do not consider catalysis explicitly in classical MD simulations, unlike reactive methods (Hermann et al., 2009; Nichols et al., 2015; Xu et al., 2007). Classical MD simulations have been previously used to predict or explain mutations in a number of enzyme systems (Karplus and Kuriyan, 2005; Rod et al., 2003; Watney et al., 2003).
To predict individual residues to mutate, we seek to identify the influence of individual atoms on catalytic activity rather than overall conformational substates of the enzyme. MD simulations provide a means to quantify this ‘influence’ by measuring positional relatedness between protein atoms and a bound drug, based on the hypothesis that the conformation and orientation of the drug and its environment are related to catalytic activity. We sample conformations from MD simulations, which estimate a Boltzmann-weighted ensemble.
Positional mutual information provides a robust non-linear metric to quantify relatedness of positional displacement in MD simulations (Cover and Thomas, 2012; Kasson et al., 2009). Normalized covariance matrices have also been used for such measurements (Kamberaj and van der Vaart, 2009; Karplus and Kushick, 1981), but such approaches are restricted by a linear correlation approximation (Ichiye and Karplus, 1991; Lange and Grubmüller, 2006), which is less desirable for detecting subtle yet important motions. Mutual information quantifies how much knowledge of the probability distribution of positional displacement for one atom i affects the distribution for another atom j and thus provides a much more general means of detecting relatedness.
Although positional mutual information has been used to analyze large-scale movements in a manner analogous to principal components analysis (Brandman et al., 2012; Lange and Grubmüller, 2006), here we desire a more focused approach. To predict residues important to catalytic activity, we score by mutual information to drug conformation. This is corrected for bulk protein movement, yielding excess mutual information
| (1) |
where I(i,j) denotes the mutual information between residues i and j (Kasson et al., 2009). Such a metric encapsulates the precise question ‘how related is movement of atom i to the drug conformation’. This metric is designed to identify functionally important residues that meet this criterion; it is of course not designed to detect residues that might not be motionally correlated with the drug but still important to catalysis.
Simulations and analysis were performed on the CTX-M9 beta lactamase in complex with one of two antibiotics: cefotaxime and meropenem (Fig. 1). CTX-M enzymes hydrolyze antibiotics by way of an acyl-enzyme intermediate where the antibiotic is covalently bound to the enzyme (Chen et al., 2005). CTX-M9 is able to efficiently hydrolyze cefotaxime, whereas it only forms the acyl intermediate for meropenem without completing hydrolysis (Chen et al., 2005). The conformational dynamics of this intermediate may thus yield insight into the residues important for hydrolytic specificity in these enzymes. The positional mutual information matrices for both CTX-M9:meropenem and CTX-M9:cefotaxime complexes yield important insight regarding the organization of these enzymes. We then score residues that may affect catalytic activity of CTX-M9 using excess mutual information. These predictions yield a set of residues that have previously been identified as affecting catalytic activity and a set of novel, previously untested predictions. To validate our predictions, we tested the top-scoring residues for CTX-M9:meropenem and CTX-M9:cefotaxime via alanine mutagenesis, a common means to assess the effect of ablating a residue side chain. Six of these eight mutants had a >2-fold decrease in cefotaxime resistance while only one of the four lowest scoring residues for CTX-M9:meropenem and CTX-M9:cefotaxime similarly decreased resistance.
Fig. 1.
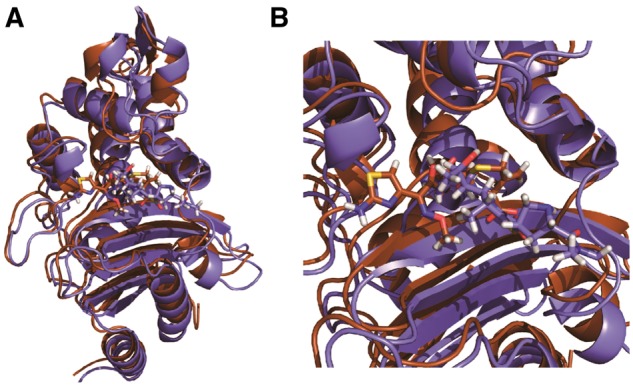
Structures of CTX-M9:drug complexes. The overlaid structures of CTX-M9 acylated to meropenem (brown online) and cefotaxime (violet online) are rendered in panel (A) with a close-up of the drug-binding pocket in panel (B). Protein is rendered in cartoon form and drug in sticks. These structures were used for MD simulations that served as the basis for positional mutual information calculations (Color version of this figure is available at Bioinformatics online.)
2 Methods
2.1 MD simulations
We obtained the apo crystal structure of CTX-M9 from the Protein Data Bank (PDB Code 2P74) (Chen et al., 2007). The acylated meropenem structure was generated by least-squares RMSD fitting of the acyl-meropenem intermediate of SHV-1 (PDB Code 2ZD8) (Nukaga et al., 2007) with missing atoms added via rigid-body fitting. The acylated CTX-M9:cefotaxime structure was generated via least squares rigid-body alignment of all common atoms on the beta lactam ring of meropenem and cefotaxime on a Thr71Ser CTX-M9 mutant. Meropenem and cefotaxime were parameterized using the Amber Antechamber program with AM1-BCC partial charges (Case et al., 2005). All simulations were run using Gromacs 4.5 with the AMBER99SB-ILDN force field and TIP3P explicit water (Hornak et al., 2006; Lindorff-Larsen et al., 2010; Pronk et al., 2013) in a periodic octahedral box with a minimum periodic image separation of 2 nm. The solvent consisted of approximately 24 000 water molecules and 150 mM NaCl. Simulations were run with a 2 fs time step and hydrogen bonds were constrained using LINCS (Hess et al., 1997). The temperature was maintained at 37°C using a velocity-rescaling thermostat (Bussi et al., 2007) and the pressure was maintained at 1 bar with a coupling constant of 10 ps. Short-range non-bonded and electrostatic interactions were truncated at 1.2 nm, and long-range electrostatics were treated with Particle Mesh Ewald (Darden et al., 1993).
CTX-M9:meropenem simulations were then run using the Folding@Home platform, and CTX-M9:cefotaxime simulations were run on a Cray XC30 or on NVidia GPGPUs. 200 independent simulations were run of CTX-M9:meropenem complexes; 22 of these were randomly selected for subsampled analysis, totaling 2460 ns with a median simulation length of 106 ns. The first 46 ns of each simulation were discarded and the remaining aggregate 2150 ns used for analysis. Three longer independent simulations CTX-M9:cefotaxime complexes were analyzed; two simulations had lengths of 954 ns and one of 289 ns, after truncation, totaling 2197 ns. Snapshots were recorded every 50 ps.
2.2 Mutual information analysis of CTX-M9 dynamics
We calculated displacements for all atoms of the CTX-M9:drug complex after rigid-body alignment of the binding pocket to the starting structure of each respective simulation. For this purpose, the binding pocket was defined as residues having at least one non-hydrogen atom within 1 nm of the acylated drug carbonyl in >90% of simulation snapshots and where the root-mean-squared positional fluctuation of the backbone atoms was <7 Å. Alignment using the binding pocket as a reference was chosen to minimize artifactual drug motion from alignment error; a comparison where alignment was performed on the whole protein is presented in the Supplementary Material and yields similar results. These displacements where is the index of each atom and t is the time from the initial alignment structure were then used to calculate mutual information and symmetric uncertainty in a fashion similar to that which we have reported previously (Kasson et al., 2009). Mutual information was calculated between two atoms , using
| (2) |
| (3) |
The probability density function and were estimated using 2-D histograms of and with 32 bins at even intervals min(,) to max(,) and min(,) to max(,).
Mutual information values were then normalized using symmetric uncertainty, which represents the relatedness of a pair of atoms independent of the motion undergone by each atom
| (4) |
Excess mutual information was calculated for each atom using
| (5) |
where k are atoms in the beta-lactam ring of the drug (C6, C7, C8, N10 and O9 for cefotaxime and C5, C6, C7, N4 and O71 for meropenem) and l are all atoms not in the binding pocket defined as above.
2.3 Sequence retrieval and processing
CTX-M family nucleotide sequences were retrieved using published accession numbers (Canton et al., 2012). These sequences were then translated and individually aligned to a protein sequence of CTX-M9 (ACR66304.1) using TBLASTN (Altschul et al., 1990; Leinberger et al., 2010). Resulting sequences were aligned using MUSCLE (Edgar, 2004). From this alignment, a consensus sequence was generated for all residues identical across the CTX-M9 family to identify conserved residues.
2.4 Creation and resistance measurements of mutants
We performed site-directed alanine mutagenesis on selected residues in CTX-M9 (Saladin et al., 2002). Mutants were tested for cefotaxime resistance using a Kirby–Bauer antibiotic disc assay (Bauer et al., 1959). Bacteria were grown to an optical density of 0.1 and then evenly spread on Mueller Hinton agar plates with a cefotaxime antibiotic disc placed in the center. The diameter of clearance was measured after 12–16 h of incubation. Resistance was measured as fold-change in apparent inhibitory concentration, calculated as the squared diameter of clearance for CTX wild-type divided by the square of the diameter of the mutant.
3 Results
We used positional mutual information to analyze the conformational dynamics of CTX-M9 with a bound antibiotic, either cefotaxime or meropenem, based on MD simulations. Multiple microsecond-length simulations were used to obtain good statistical sampling of positional motions of the enzyme-drug complex. Mutual information provides a non-linear analogue to measure correlated motions. This approach identifies pairs and networks of atoms that are dynamically related and thus statistically interact either directly or indirectly. When applied to the active site of an enzyme, it thus enables a unified analysis of short-range and long-range interactions that may influence catalysis and in this case drug resistance. Furthermore, by measuring the excess mutual information of protein atoms to the drug compared to the rest of the protein, we identified residues that may influence the dynamics of the bound beta lactam ring and potentially subsequent drug hydrolysis.
3.1 Pairwise symmetric uncertainty to analyze positional relationships in CTX-M9
To quantify pairwise relationships between all pairs of atoms in the CTX-M9:drug complex, we calculated a positional symmetric uncertainty matrix based on MD simulation trajectories. Symmetric uncertainty was used as a normalized information-theoretic metric of positional relatedness (calculated by dividing mutual information by the sum of entropies, see Section 2 for details). For cefotaxime, this yields a 3977 × 3977 matrix. Because the size of this matrix is N2 in the number of atoms, we selected the top 0.25% of interactions (5% squared) for analysis or the top 19 771 pairs. Similarly, symmetric uncertainty analysis of CTX-M9:meropenem yielded a 3992 × 3992 matrix where the top 19 920 pairs comprise the top 0.25% of interactions.
At a coarse level, the resulting symmetric uncertainty matrices show, as expected, strong relationships between directly interacting atoms as well as stabilization of secondary structure elements. Thresholded matrices for CTX-M9:cefotaxime and CTX-M9:meropenem complexes are plotted in Figures 2B and 3B, and the top-scoring interactions are rendered as dotted lines on the protein structure in Figures 2A and 3A. This analysis yields statistically coupled yet spatially distant atom pairs as well as a number of strong interactions from directly contacting atoms, as discussed below. The strong near-diagonal band reflects the expected high symmetric uncertainty for atoms that are directly connected by bonded interactions; in addition, salt bridges and van der Waals contacts between sequence-distant but spatially proximate atoms also resulted in pairs with high symmetric uncertainty. Secondary structural elements are typically strongly connected, except when such structures are so strongly stabilized that they are immobile over the multi-microsecond timescales sampled and thus have near-zero positional entropy. Also present in the high-scoring pairs are interactions between nearby secondary structural elements. For example, in the CTX-M9:cefotaxime simulations, a number of high-scoring pairs were measured between the alpha helix containing residues Ala28-Ser40 (Fig. 2A, atom indices 45–243) and the helix containing residues Arg276-Ala287 (atom indices 3752-3953).
Fig. 2.
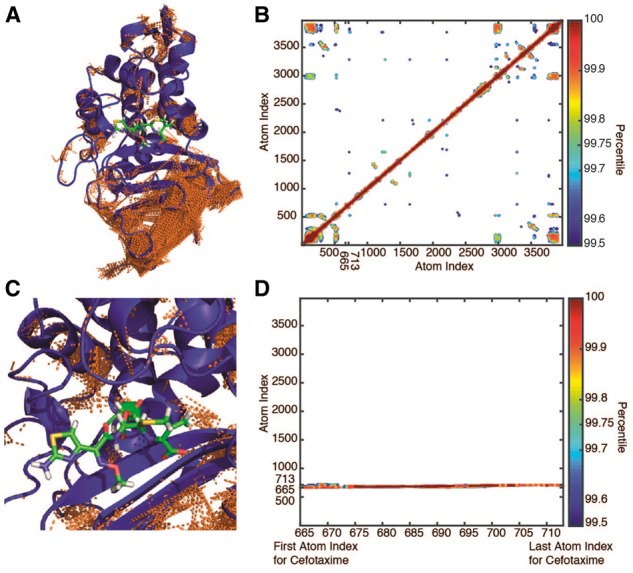
Pairwise positional symmetric uncertainty in CTX-M9:cefotaxime complexes. Symmetric uncertainty (normalized mutual information) is used to quantify the degree to which atom motions are associated. Rendered in panel (A) are the top 0.25% of symmetric uncertainty pairs shown as brown lines on the CTX-M9:cefotaxime structure. Panel (B) shows a contour plot of the atom–atom symmetric uncertainty matrix contoured at 0.1% intervals from the 99.5th to 100th percentiles. While numerous connections are identified between secondary structural elements, the drug has few high-ranking interactions with protein atoms, indicating a relative isolation of drug motion from protein motion. This is illustrated in the inset rendering in panel (C) and the portion of the symmetric uncertainty matrix corresponding to drug interactions in panel (D) that shows only self-interactions scoring above 99.5% (Color version of this figure is available at Bioinformatics online.)
Fig. 3.
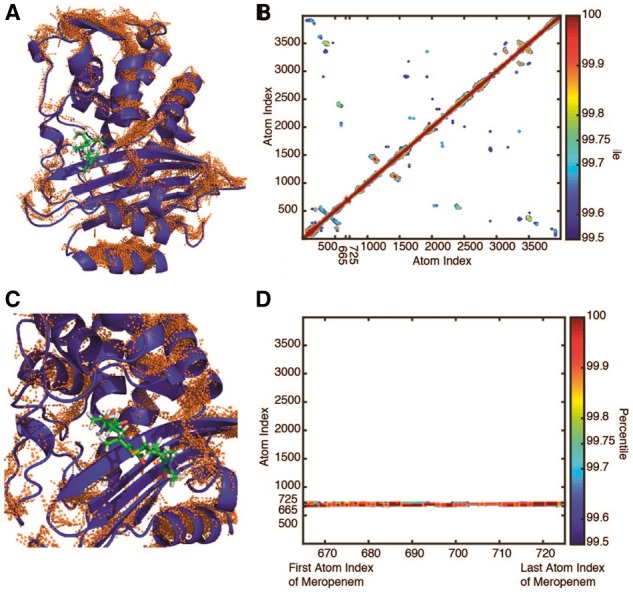
Pairwise positional symmetric uncertainty in CTX-M9:meropenem complexes. The top 0.25% of symmetric uncertainty pairs are rendered as brown lines on the protein structure in panel (A), with the matrix contoured at 0.1% intervals at 99.5% and above in panel (B) and insets showing drug interactions in panels (C) and (D). Similar to CTX-M9:cefotaxime, CTX-M9:meropenem simulations showed few interactions between the drug and enzyme or the drug-binding pocket and the rest of the enzyme (Color version of this figure is available at Bioinformatics online.)
One striking finding is the lack of strong relationships between the bound drug and the major catalytic residues and similarly between the drug-binding pocket and the rest of the enzyme in both sets of simulations. The highest-scoring drug-protein symmetric uncertainty value occurs below our 99.75% statistical cutoff, and such linkages remain sparse even at much lower cutoffs (Figs. 2D and 3D). This suggests that the drug and catalytic geometry are relatively isolated from conformational fluctuations of the rest of the protein. Such a finding makes sense in light of theories of catalytic preorganization in enzymes, which can be interpreted to state that optimal catalytic efficiency results from minimal fluctuations of the catalytic residues (Warshel, 1998, 2002). This is well supported by crystallographic studies of CTX-M9 alone and in complex with different transition-state analogues; in one such series of structures, key catalytic residues such as Ser130, Lys73, Glu166 and Ser237 shift only an average of 0.1 Å over a set of substrate analogues spanning the catalytic cycle (Chen et al., 2005).
Despite this relative isolation, symmetric uncertainty analysis of CTX-M9:cefotaxime yielded one potentially important network involving Asn104, Arg276 and Asn170. This network is connected by linkages at the 99.5th percentile, below our statistical cutoff, but stronger than any other networks of spatially distant residues. Although none of these residues is directly involved in catalysis, previous work has suggested they have a strong role in function. Asn170 is believed to be involved in establishing the hydrogen bond network (Delmas et al., 2008) with the catalytic water molecule while Asn104 directly interacts with acylamide side chain of cefotaxime (Shimamura et al., 2002). None of these interactions met the pre-defined threshold for significance in our analysis, but they constitute intriguing candidates for further testing.
3.2 Excess mutual information identifies residues linked to drug motion
While symmetric uncertainty enables a global analysis of statistical interaction networks in the protein–drug complex, a more targeted statistical metric is desired to identify residues associated with particular motions of the bound drug. Excess mutual information quantifies drug–protein positional coupling in a fashion corrected for protein motions and capable of robustly identifying even weak but physically significant coupling. Excess mutual information measures the symmetric uncertainty between a protein atom and the beta-lactam ring but corrects for bulk protein motion by subtracting the average symmetric uncertainty to the rest of the protein (see Section 2 for details). The top 5% of protein atoms as scored via excess mutual information to the beta-lactam ring were then selected for both CTX-M9:cefotaxime and CTX-M9:meropenem (Table 1 and Fig. 4). These top-scoring atoms yield a prediction of functionally important residues for beta-lactam hydrolysis for CTX-M9. The top 5% of atoms scored by excess mutual information in CTX-M9:cefotaxime simulations covered 35 residues, 31 after removing ‘singleton’ residues with only one atom selected (of 265 residues total).
Table 1.
Top-scoring residues via positional excess mutual information
| Rank | CTX-M9:cefotaxime | CTX-M9:meropenem |
|---|---|---|
| 1 | T235a,b | T235a,b |
| 2 | N132a | G236b,c |
| 3 | T71a,b,c | T216 |
| 4 | Y264b | T71a,b,c |
| 5 | N245a,b | R276a,c |
| 6 | D246a,b | S130b,c |
| 7 | N104a,b,c | A219a,b,c |
| 8 | Y234a | D246a,b |
| 9 | I221b | Y234a |
| 10 | N106c | Y73a,b,c |
| 11 | R222 | M68b |
| 12 | Y73a,b,c | N132a |
| 13 | D233b | A218a |
| 14 | Y105a,b | C69b |
| 15 | V103a,c | S237a,c |
| 16 | S220c | N214a |
| 17 | A219a,bc | L119 |
| 18 | Y60 | N245a,b |
| 19 | A218a | A125 |
| 20 | A263 | N104a,b,c |
| 21 | Q128a,b | E166c |
| 22 | D131a,b | T133 |
| 23 | V262b | V103a,c |
| 24 | L225 | Y105a,b |
| 25 | N214a | G217 |
| 26 | V46b | T215b |
| 27 | L33b | L127b |
| 28 | S237a,c | Q128a,b |
| 29 | A231 | L102b |
| 30 | R276a,c | D131a,b |
| 31 | S72b | P167 |
| 32 | E110b |
aShared between CTX-M9:cefotaxime and CTX-M9:meropenem.
bConserved residue.
cResidue mutated experimentally; altered drug resistance observed.
Fig. 4.
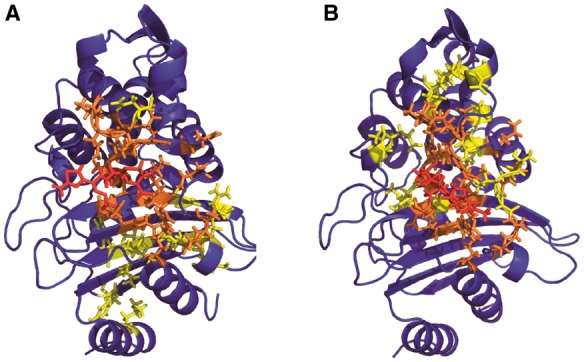
Residues linked to drug motion identified by excess positional mutual information. Residues corresponding to the top 5% of protein atoms scored by excess mutual information to the beta-lactam ring are rendered as sticks on the CTX-M9:cefotaxime structure in panel (A) and the CTX-M9:meropenem structure in panel (B). These residues constitute our predictions for sites where mutation will affect catalytic activity and drug resistance. The drug is shown in red online, residues identified in both enzyme:drug complexes in orange and residues identified in only one enzyme:drug complex in yellow (Color version of this figure is available at Bioinformatics online.)
Of these, nine have previously been tested via mutagenesis experiments, and in all nine mutations were confirmed to reduce catalytic activity, as assessed by a decrease in both kcat and minimum inhibitory concentration of drug to impede bacterial growth (Altschul et al., 1990; Aumeran et al., 2003; Edgar, 2004; Gazouli et al., 1998; Pérez-Llarena et al., 2011; Shimamura et al., 2002). In addition, 17 of the 31 residues are sequence-identical across the CTX-M family using the family definition provided in (Canton et al., 2012). Of the 10 top-scoring residues, 7 are sequence-identical across CTX-M9, and 3 have been previously tested and confirmed via experimental mutagenesis. This degree of sequence conservation further, although indirectly, supports a functional role for the residues thus identified.
Residues predicted by excess mutual information include amino acids both in the drug-binding pocket and distant from it (allosteric mutations). This second category (including Leu33, Val45, Ala245, Ala263 and Tyr264) is particularly interesting, as they are more difficult to identify via conventional methods. Previous experimental mutagenesis has shown that allosteric mutations can affect hydrolysis of cephalosporins via beta lactamase enzymes (Pérez-Llarena et al., 2011), and the prediction of novel allosteric mutations is a major goal of this work. Recent work based on evolutionary conservation has proposed distance to the active site as a means to score functional importance of enzymes (Jack et al., 2016). In our data, excess mutual information scores showed correlation values of (0.27 and 0.56) with distance to the catalytic residues, explaining 7.6 and 31% of the variance in MI respectively (see Supplementary Material), demonstrating that for this system excess mutual information contains information other than purely distance. In addition, high-scoring residues ranked moderately in positional mobility (top-scoring residues in CTX-M9:cefotaxime simulations have root-mean-squared fluctuation of 1.4–1.9 Å compared to an overall range of 0.53–7.72 Å and median 1.6 Å.
Corresponding analysis of the CTX-M9:meropenem complex yielded a number of residues in common with CTX-M9:cefotaxime (Fig. 4). The top 5% of atoms scored via excess mutual information mapped to 37 residues, 32 after singleton removal. Of these 32 residues, 17 were in common with CTX-M9:cefotaxime, and the remaining 15 diverged. Although it is tempting to analyze differential scoring between the CTX-M9:meropenem simulations and the CTX-M9:cefotaxime simulations as relating to the capacity of CTX-M9 to hydrolyze cefotaxime but not meropenem, we wish to remain conservative in this respect as the simulation sampling scheme differed slightly between CTX-M9:cefotaxime (fewer and longer simulations) and CTX-M9:meropenem (more simulations of 100–200 ns in length), and we cannot exclude a sampling bias in accounting for the different residues identified. Common residues included several involved in catalysis either directly or indirectly such as Ser237 and Arg276 (Chen et al., 2005; Pérez-Llarena et al., 2008). Top-scoring residues from CTX-M9:meropenem (but not CTX-M9:cefotaxime) included Ser130 and Glu166, two residues closely tied to catalytic function (Glu166 is believed by many to be the general base for cephalosporin hydrolysis in CTX-M enzymes) (Chen et al., 2005; Delmas et al., 2008; Tomanicek et al., 2011). The identification of these residues suggests that although CTX-M9 cannot fully hydrolyze meropenem, catalytic residues still interact with the drug in a coordinated manner.
3.3 Mutation at top-scoring sites decreases drug resistance
As an experimental test of our scoring method, we mutated each of the five top-scoring residues via excess mutual information singly to alanine. We used antibiotic resistance of transformed bacteria expressing each enzyme as a metric of enzymatic function. As a control, we also mutated the four lowest-scoring residues and four residues that scored closest to zero excess mutual information (relationship to drug motion equal to the average across the protein). Of the top-scoring residues, 6/8 had a greater than 2-fold reduction in cefotaxime antibiotic resistance (Fig. 5), with all mutants showing some reduction in resistance. Four of the most affected top-scoring residues were located near the drug-binding pocket while two were more distant from it, suggesting that excess mutual information can indeed identify both nearby and allosteric residues affecting function. Only one of the four lowest-scoring residues displayed a >2-fold reduction in cefotaxime antibiotic resistance (Fig. 6C). Similarly, of the four mutants scoring near zero (background correlation only), none showed a >2-fold decrease in drug resistance (Fig. 6C). These results demonstrate that excess mutual information can robustly identify (P < 0.05 via Fisher’s exact test or P < 0.01 via two-tailed Kolmogorv–Smirnov test) residues likely to be involved in enzyme function and consequent drug resistance.
Fig. 5.
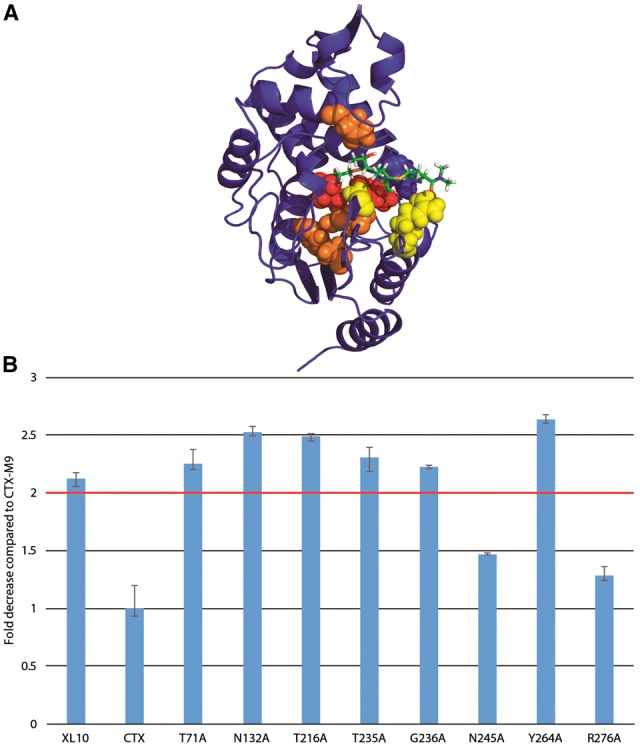
Mutation of top-ranking residues via positional mutual information greatly decreases cefotaxime drug resistance. The five highest-scoring residues for cefotaxime (orange online) and meropenem (yellow with residues in both sets as red) were selected for alanine mutagenesis. These were located both inside and distant from the drug pocket (A). Six of the eight mutants displayed a >2-fold drop in resistance (Color version of this figure is available at Bioinformatics online.)
Fig. 6.
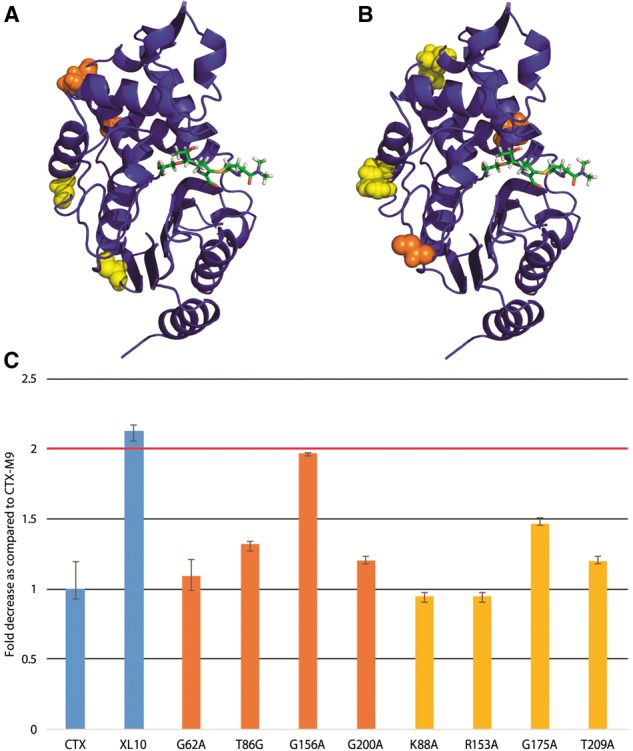
Mutation of lowest-scoring residues leaves cefotaxime drug resistance largely unaffected. As rendered in panel (A), the lowest-scoring residues and (B) the residues with closest to background mutual information in cefotaxime simulations (orange online) and meropenem simulations (yellow) were located outside the drug-binding pocket. As plotted in panel (C), none of near-background mutants (yellow) and one of the lowest scoring mutants (orange) showed a >2-fold decrease in drug resistance (Color version of this figure is available at Bioinformatics online.)
4 Discussion
We have developed an excess mutual information metric to predict residues important for drug hydrolysis in the CTX-M9 beta lactamase enzyme based on MD simulations. We initially developed excess positional mutual information to score protein residues on the influenza hemagglutinin glycoprotein that influence low-affinity binding of sialoglycans (Kasson et al., 2009). Here, we show how excess mutual information can be used in a much more sensitive and targeted fashion, detecting motions of an enzyme that are weakly but significantly coupled to dynamics of a bound substrate, and prospectively predicting and testing mutants that affect enzyme function. This marks a substantial expansion on the scope of problems for which excess mutual information can predict mutational effects: from initial work on relatively ‘floppy’ low-affinity ligand binding, it was not obvious that mutual information would successfully predict sites of mutation in a relatively rigid beta-lactamase enzyme. Although positional mutual information has been used very productively to identify conformational substates of a protein, use of excess mutual information allows a much more targeted measurement of how individual residues are related to ligand motion in a manner that is robust even in the presence of weak coupling. As we have shown, excess mutual information identifies residues where mutation alters kcat even though these residues are not strongly related to ligand or binding-pocket motions in the full N2 symmetric uncertainty matrix.
Our primary goal in developing excess mutual information is to obtain a metric that can predict both binding-pocket mutations and allosteric ones in a single integrated analysis. Since many structure-based approaches concentrate on the ligand-binding pocket, our prediction of both proximate and distant residues demonstrates the power of such an approach. Another motivation for the use of positional excess mutual information is that it provides an empirical measurement of positional relatedness that can be complementary to mutational analysis (Kasson et al., 2009) yet leverages state-of-the-art classical MD force fields for both force calculation and sampling of statistical ensembles of conformations. In contrast to methods that compute energy-based coupling of protein residues, analyses of MD trajectories naturally incorporate entropic terms and yield a free-energy-based coupling of protein residue motions.
Our analysis identified several previously untested mutations predicted to alter the catalytic activity and drug resistance conferred by CTX-M9. We tested a set of these prospective predictions utilizing bacterial drug resistance as a measure of enzyme function, comparing to low-scoring mutations. The predicted high-scoring mutations had a much greater effect on enzyme function than mid-scoring or low-scoring controls (6/8 showing a >2-fold drop in resistance versus 1/8 in the aggregate control groups; P < 0.01 via KS test). Furthermore, these mutations that alter drug resistance are located both within and outside the drug-binding pocket, with high-scoring mutations as far as 24 Å away, meeting our design criteria of a single global analysis to identify both local and allosteric mutants. The ability to anticipate altered drug-resistance of new variants is of great utility for clinical surveillance of CTX-M beta lactamases as well as drug development efforts.
Supplementary Material
Acknowledgements
We thank Folding@Home donors, Google Exacycle, and ORNL for computing time. We also thank J. Hays and M. Latallo for many helpful discussions.
Funding
This work was supported by the Hartwell Foundation; a Google Exacycle Award for Visiting Faculty; and the National Institutes of Health [5T32AI007046-39].
Conflict of Interest: none declared.
References
- Altschul S.F. et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Aumeran C. et al. (2003) Effects of Ser130Gly and Asp240Lys substitutions in extended-spectrum β-Lactamase CTX-M-9. Antimicrob. Agents Chemother., 47, 2958–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer A.W. et al. (1959) Single-disk antibiotic-sensitivity testing of staphylococci: an analysis of technique and results. AMA Arch. Intern. Med., 104, 208–216. [DOI] [PubMed] [Google Scholar]
- Bonnet R. et al. (2001) Novel cefotaximase (CTX-M-16) with increased catalytic efficiency due to substitution Asp-240–>Gly. Antimicrob. Agents Chemother., 45, 2269–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet R. et al. (2003) Effect of D240G substitution in a novel ESBL CTX-M-27. J. Antimicrob. Chemother., 52, 29–35. [DOI] [PubMed] [Google Scholar]
- Boucher H.W. et al. (2013a) 10 × ’20 progress—development of new drugs active against gram-negative bacilli: an update from the infectious diseases society of America. Clin. Infect. Dis., 56, 1685–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman R. et al. (2012) A-site residues move independently from P-site residues in all-atom molecular dynamics simulations of the 70S bacterial ribosome. PLoS One, 7, e29377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G. et al. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys., 126, 014101. [DOI] [PubMed] [Google Scholar]
- Canton R. et al. (2012) CTX-M enzymes: origin and diffusion. Antimicrob. Resist. Chemother, 3, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantón R., Coque T.M. (2006) The CTX-M β-lactamase pandemic. Curr. Opin. Microbiol., 9, 466–475. [DOI] [PubMed] [Google Scholar]
- Case D.A. et al. (2005) The Amber biomolecular simulation programs. J. Comput. Chem., 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. et al. (2005) Structure, function, and inhibition along the reaction coordinate of CTX-M beta-lactamases. J. Am. Chem. Soc., 127, 5423–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. et al. (2007) The acylation mechanism of CTX-M beta-lactamase at 0.88 a resolution. J. Am. Chem. Soc., 129, 5378–5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cover T.M., Thomas J.A. (2012) Elements of Information Theory. New York, John Wiley & Sons. [Google Scholar]
- Darden T. et al. (1993) Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys., 98, 10089–10092. [Google Scholar]
- Delmas J. et al. (2006) Prediction of the Evolution of Ceftazidime Resistance in Extended-Spectrum β-Lactamase CTX-M-9. Antimicrob. Agents Chemother., 50, 731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas J. et al. (2008) Structure and dynamics of CTX-M enzymes reveal insights into substrate accommodation by extended-spectrum β-Lactamases. J. Mol. Biol., 375, 192–201. [DOI] [PubMed] [Google Scholar]
- Edgar R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res., 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazouli M. et al. (1998) Sequence of the gene encoding a plasmid-mediated cefotaxime-hydrolyzing class A beta-lactamase (CTX-M-4): involvement of serine 237 in cephalosporin hydrolysis. Antimicrob. Agents Chemother., 42, 1259–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann J.C. et al. (2009) High level QM/MM modeling of the formation of the tetrahedral intermediate in the acylation of wild type and K73A mutant TEM-1 class A β-Lactamase†. J. Phys. Chem. A., 113, 11984–11994. [DOI] [PubMed] [Google Scholar]
- Hess B. et al. (1997) LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem., 18, 1463–1472. [DOI] [PubMed] [Google Scholar]
- Hornak V. et al. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiye T., Karplus M. (1991) Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins, 11, 205–217. [DOI] [PubMed] [Google Scholar]
- Jack B.R. et al. (2016) Functional sites induce long-range evolutionary constraints in enzymes. PLoS Biol., 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamberaj H., van der Vaart A. (2009) Extracting the causality of correlated motions from molecular dynamics simulations. Biophys. J., 97, 1747–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus M., Kuriyan J. (2005) Molecular dynamics and protein function. Proc. Natl. Acad. Sci. U S A., 102, 6679–6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus M., Kushick J.N. (1981) Method for estimating the configurational entropy of macromolecules. Macromolecules, 14, 325–332. [Google Scholar]
- Kasson P.M. et al. (2009) Combining molecular dynamics with bayesian analysis to predict and evaluate ligand-binding mutations in influenza hemagglutinin. J. Am. Chem. Soc., 131, 11338–11340., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange O.F., Grubmüller H. (2006) Generalized correlation for biomolecular dynamics. Proteins, 62, 1053–1061. [DOI] [PubMed] [Google Scholar]
- Leinberger D.M. et al. (2010) Integrated detection of extended-spectrum-beta-lactam resistance by DNA microarray-based genotyping of TEM, SHV, and CTX-M genes. J. Clin. Microbiol., 48, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S.B., Marshall B. (2004) Antibacterial resistance worldwide: causes, challenges and responses. Nat. Med., 10, S122–S129. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K. et al. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins., 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livermore D.M. et al. (2007) CTX-M: changing the face of ESBLs in Europe. J. Antimicrob. Chemother., 59, 165–174. [DOI] [PubMed] [Google Scholar]
- Llarrull L.I. et al. (2010) The future of the β-lactams. Curr. Opin. Microbiol., 13, 551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matagne A. et al. (1998) Catalytic properties of class A beta-lactamases: efficiency and diversity. Biochem. J., 330, 581–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols D.A. et al. (2015) Ligand-induced proton transfer and low-barrier hydrogen bond revealed by X-ray crystallography. J. Am. Chem. Soc., 137, 8086–8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nukaga M. et al. (2007) Inhibition of class A beta-lactamases by carbapenems: crystallographic observation of two conformations of meropenem in SHV-1. J. Am. Chem. Soc., 130, 12656–12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Llarena F.J. et al. (2008) Structure-function studies of arginine at position 276 in CTX-M beta-lactamases. J. Antimicrob. Chemother., 61, 792–797. [DOI] [PubMed] [Google Scholar]
- Pérez-Llarena F.J. et al. (2011) Distant and new mutations in CTX-M-1 β-Lactamase affect cefotaxime hydrolysis. Antimicrob. Agents Chemother., 55, 4361–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippon A. et al. (1989) Extended-spectrum beta-lactamases. Antimicrob. Agents Chemother., 33, 1131–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirel L. et al. (2002) Biochemical analysis of the ceftazidime-hydrolysing extended-spectrum beta-lactamase CTX-M-15 and of its structurally related beta-lactamase CTX-M-3. J. Antimicrob. Chemother., 50, 1031–1034. [DOI] [PubMed] [Google Scholar]
- Pronk S. et al. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics, 29, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rod T.H. et al. (2003) Correlated motion and the effect of distal mutations in dihydrofolate reductase. Proc. Natl. Acad. Sci. U S A., 100, 6980–6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saladin M. et al. (2002) Diversity of CTX-M beta-lactamases and their promoter regions from Enterobacteriaceae isolated in three Parisian hospitals. FEMS Microbiol. Lett., 209, 161–168. [DOI] [PubMed] [Google Scholar]
- Shimamura T. et al. (2002) Acyl-intermediate structures of the extended-spectrum class A beta-lactamase, Toho-1, in complex with cefotaxime, cephalothin, and benzylpenicillin. J. Biol. Chem., 277, 46601–46608. [DOI] [PubMed] [Google Scholar]
- Sougakoff W. et al. (1988) The TEM-3 β-lactamase, which hydrolyzes broad-spectrum cephalosporins, is derived from the TEM-2 penicillinase by two amino acid substitutions. FEMS Microbiol. Lett., 56, 343–348. [Google Scholar]
- Tomanicek S.J. et al. (2011) The active site protonation states of perdeuterated Toho-1 β-lactamase determined by neutron diffraction support a role for Glu166 as the general base in acylation. FEBS Lett., 585, 364–368. [DOI] [PubMed] [Google Scholar]
- Valverde A. et al. (2004) Dramatic increase in prevalence of fecal carriage of extended-spectrum β-Lactamase-producing enterobacteriaceae during nonoutbreak situations in Spain. J. Clin. Microbiol., 42, 4769–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A. (1998) Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J. Biol. Chem., 273, 27035–27038. [DOI] [PubMed] [Google Scholar]
- Warshel A. (2002) Molecular dynamics simulations of biological reactions. Acc. Chem. Res., 35, 385–395. [DOI] [PubMed] [Google Scholar]
- Watney J.B. et al. (2003) Effect of mutation on enzyme motion in dihydrofolate reductase. J. Am. Chem. Soc., 125, 3745–3750. [DOI] [PubMed] [Google Scholar]
- Xu D. et al. (2007) Antibiotic deactivation by a dizinc beta-lactamase: mechanistic insights from QM/MM and DFT studies. J. Am. Chem. Soc., 129, 10814–10822. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.