

Abstract
Background
Doxorubicin (DOX) is the most widely used chemotherapeutic agent that has multimodal cytotoxicity. The main cytotoxic actions of DOX occur in the nucleus. The emergence of drug-resistant cancer cells that have the ability to actively efflux DOX out of the nucleus, and the cytoplasm has led to the search for a more effective derivative of this drug.
Materials and methods
We created a new derivative of DOX that was derived via simple conjugation of the 3′ amino group of DOX to the dexamethasone molecule.
Results
Despite having a lower cytotoxic activity in MCF-7 cells, the conjugated product, DexDOX, exerted its actions in a manner that was different to that of DOX. DexDOX rapidly induced MCF-7 cell apoptosis without entering the nucleus. Further analysis showed that Dex-DOX increased cytosolic oxidative stress and did not interfere with the cell cycle. In addition, the conjugated product retained its cytotoxicity in multidrug resistance-1-overexpressing MCF-7 cells that had an approximately 16-fold higher resistance to DOX.
Conclusion
We have synthesized a new derivative of DOX, which has the ability to overcome multidrug resistance-1-induced resistance. This molecule may have potential as a future chemotherapeutic agent.
Keywords: doxorubicin, dexamethasone, drug-resistant tumor, bioconjugation, multidrug resistance, reactive oxygen species
Introduction
Doxorubicin (DOX), a member of the anthracycline antibiotic family, was originally extracted from Streptomyces peucetius var. caesius.1 Rather than displaying antimicrobial properties, DOX and other anthracyclines have been shown to have strong cytotoxicity, and therefore, DOX derivatives are generally used as chemotherapeutic agents. To date, DOX is the most widely used drug in standard chemotherapeutic regimens and is used to treat a wide range of solid and hematologic malignancies.2,3 Several studies have suggested that DOX has complex cytotoxic activities that are not fully understood. DOX directly diffuses into the cell cytoplasm and binds to the cytoplasmic proteasome’s 20S subunit.4 Once the DOX-proteasome complex has entered the nucleus via nuclear pores, it then inhibits topoisomerase II, the key enzyme that maintains DNA tension.5,6 In addition, DOX also intercalates to the DNA strand, preferably at cytosine-guanine nucleotide pair.7–9 These intranuclear processes are considered to be the main mechanisms by which DOX cytotoxicity leads to apoptosis.10 DOX can generate cytotoxicity by inducing oxidative stress. The reductive activation of the aglycone structure of DOX leads to the formation of semiquinone radicals, which are powerful reactive oxygen species (ROS) that can cause ROS-mediated cell death.11–13 Mitochondrial dysfunction, P53, and AMP-activated protein kinase activation are also involved in DOX-induced apoptosis.14–16 Recently, DOX was proposed to cause cell death through autophagy and necrosis via poly (ADP-ribose) polymerase-1 (PARP-1)-induced DNA damage.17 Interestingly, inhibition of one of these pathways did not circumvent DOX-mediated cell death. This observation suggests that these cytotoxic effects may act together.
Tumor drug-resistance is widely recognized as being one of the most important clinical issues currently being faced by oncologists. Upregulation of the multidrug resistance (MDR) gene is one of the main mechanisms of resistance employed by numerous anti-cancer drugs, including DOX.18–21 MDR encodes P-glycoprotein (P-gp), which is an ATP-binding cassette pump that is responsible for the efflux of DOX out of the cells. A previous study demonstrated that P-gp actively promotes the efflux of DOX from the nucleus, where the drugs bind to both TOPO II and DNA.22 Pharmacological blockage as well as gene-targeted downregulation of the P-gp pump have been shown to reverse DOX sensitivity. Therefore, many research groups have focused on developing methods to downregulate P-gp in an attempt to overcome DOX-resistance in cancer cells.23–28
A number of DOX derivatives have been constructed to date, and those derivatives have shown additional properties compared to DOX.29–33 We believe that modification of DOX is another possible approach that might overcome P-gp efflux. In this study, we took simple bio-conjugation to covalently conjugate DOX to dexamethasone, a potent, synthetic, lipophilic hormone. Our results demonstrate that the conjugated molecule, designated as DexDOX, has potent cytotoxic effects. However, these effects appear to be different to those of DOX. Furthermore, the new molecule was also able to escape MDR-1 overexpression-induced resistance, and therefore, this derivative may have potential for use as a future therapeutic agent.
Materials and methods
Cell lines and cell culture
The human breast cancer cell line, MCF-7, was obtained from the American Type Cell Collection (ATCC; Manassas, VA, USA). The cells were cultured to 70%–80% confluence in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco®, Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (FBS) (Gibco®) at 37°C and 5% CO2.
MDR-1 overexpression
To stimulate drug-resistance in MCF-7, we transfected MCF-7 cells with an ABCB1 pCMV GFP-tagged plasmid (OriGene Technologies Inc., Rockville, MD, USA). One day prior to transfection, the cells were plated at 10,000 cells per cm2 in a 25-mL flask. Transfection was performed by mixing 2 μg of ABCB1 pCMV GFP-tagged plasmid with Xfect transfection reagent (Clontech, Takara Bio USA Inc., Mountain View, CA, USA) to a final volume of 100 μL. Next, 2.5 μL of Xfect polymer was added into the tube containing the plasmid. The tube was then vortexed and incubated for 10 min at room temperature (RT). The contents of the tube were then added to the cells. Our preliminary analysis showed that approximately 85% of the cells were expressing GFP at day 5 following transfection.
One-pot, two-step conjugation of dexamethasone to DOX
One-pot, two-step reactions were performed as follows. In the first step of the reaction, equal moles of DOX (Selleck Chemicals, Houston, TX, USA) and 2-iminothiolane (Sigma-Aldrich Co., St Louis, MO, USA) were mixed together in DMSO (Sigma-Aldrich Co.) for 48 h at RT. Equal moles of dexamethasone (Selleck Chemicals) comparing to the moles of DOX in DMSO were then added dropwise to the first reaction, and then mixed, and incubated for 96 h at RT. The products were further purified by flash chromatography using a pre-cast silica column (StarFlash™ Column, Starlab Scientific Co Ltd, Xi’an City, Shaanxi Province, People’s Republic of China). In some experiments, the products were dialyzed in deionized water with a molecular weight cutoff of 1,000 Dalton (Pur-A-Lyzer™ Mega 1000 Dialysis Kit, Sigma-Aldrich, St Louis, MO, USA) before carrying out the flash chromatography. The chromatography fractions were dried via negative pressure evaporation. The product was kept dry by placing it into a desiccator.
Thin-layer chromatography (TLC)
TLC was performed at each step of the reaction and purification process. Samples were dropped onto and dried in pre-cast silica gel (Miles Scientific, Newark, DE, USA). TLC was run using a 15:1.5:5 ratio of chloroform:acetic acid:methanol (V/V). The gels were then either directly visualized or visualized under UV light.
Mass spectroscopy
Molecular weight of DexDOX was determined by atmospheric pressure chemical ionization (APCI) microTOF mass spectroscopy (Bruker Biosciences Corporation, Billerica, MA, USA). The sample was dissolved in 0.05% formic acid in methanol (v/v). The acquisition parameters of operating were positive ion polarity, 5,000 V capillary, 1.6 Bar nebulizer, 200°C dry heater, and 8.0 l/min dry gas. The spectrum was scanned from 100 m/z to 1,800 m/z.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay
MCF-7 cells and MDR-1-transfected MCF-7 cells were plated overnight in a 96-well plate (4,000 cells per well). Cells were treated with either DOX at a final concentration of 0–12.5 μg/mL or DexDOX at a final concentration 0–120 μg/mL in triplicate for 24 h. Next, 10 μL/well of WST-1/ECS solution from the Quick Cell Proliferation Colorimetric Assay Kit (BioVision, Inc., Milpitas, CA, USA) was added, and the solution was incubated for 2 h at 37°C and 5% CO2. Absorbance was measured at 480 nm with the Biochrom Anthos 2010 microplate reader (Biochrom Ltd, Cambourne, Cambridge, UK). IC50s were calculated from linear regression from the absorbance OD of each triplicate concentration.34
Video confocal fluorescence microscopy
Confocal microscopy was performed to allow analysis of the intracellular distribution and cytopathic effects. Microscopy was performed using an LSM 800 confocal microscope (Carl Zeiss Meditech AG, Jena, Germany). Low numbers of MCF-7 cells were seeded into 24-well plates (3,000 cells/well) overnight. On the day of the experiment, culture media was replaced with 10% FBS DMEM added as a supplement, together with either 100 μg/mL DexDOX or 3 μg/mL DOX (approximately IC60). Videos were recorded using ZEN software version 2.1, every 20 min for a total duration of 12 h.
Flow cytometry for cell cycle analysis and apoptosis detection
MCF-7 cells were treated with either 100 μg/mL of DexDOX or 3 μg/mL of DOX for 48 h. Both adherent and floating cells were collected and washed twice with cold PBS. The cell pellet was resuspended in 200 μL of PBS, and then, 10 μL of PI staining mixture was added to each of the samples. The samples were then incubated for 30 min at RT. Analysis was carried out using FACs Calibur (BD Biosciences, San Jose, CA, USA). For analysis of apoptosis, cells were stained with FITC Annexin V Apoptosis kit and PI (BioLegend®, San Diego, CA, USA) according to the manufacturer’s protocol.
Fluorescent microscopy of dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay
MCF-7 cells were seeded overnight into 8-well chamber slides at 4,000 cells per well. Cells were washed with HBSS buffer. Pre-warmed DCFH-DA solution was then added to a final concentration 100 μM. Next, the cells were incubated for 30 min at 37°C, 5% CO2. The DCFH-DA solution was then removed. Fresh media containing either 3 μg/mL of DOX or 100 μg/mL of DexDOX was then added. The expression of dichlorofluorescin (DCF) in treated cells was observed at 0, 20, 40, and 60 min by fluorescent microscopy (Olympus BX51, Tokyo, Japan) and images taken using the Olympus DP71 system.35
Statistical analysis
Numerical data are expressed as mean ± SD. Comparison between two groups was assessed by paired t-tests (SPSS version 13). A P-value <0.05 was considered to be statistically significant.
Results
Purification and characterization of DexDOX product
The conjugation process was carried out in DMSO at RT. The product was purified by flash chromatography. The fractions collected from the chromatography were then subjected to vacuum drying, resulting in a dried purple material. This material could not be dissolved in water, methanol, or ethanol, suggesting that the molecule is strongly non-polar (Figure 1A). TLC analysis showed the presence of a single band that had a different retardation factor (Rf) to the original molecule and to unpurified products (Figure 1B). The solution of purified product in DMSO showed a purple color and had a peak absorbance wavelength at 505 nm as compared to 484 nm for DOX (Figure 1C). APCI mass spectroscopy revealed that the molecule had a mass of 1,037 Dalton (Figure 1D). Based on these findings, we postulated the most likely chemical structure of the conjugated molecule in Figure 1A and designated the molecule as DexDOX.
Figure 1.

Conjugation scheme and characterization of DexDOX.
Notes: (A) The one-pot, two-step conjugation process, first step: a reaction of DOX and 2-iminothiolane at 1:1 mole, second step: equal molarity of dexamethasone was added. The reactions were performed in DMSO at RT. (B) Representative image of TLC showing lane 1: DOX, lane 2: dexamethasone, lane 3: 2-iminothiolane; lane 4: reactions products; fraction A targeted products, fraction B, lane 5: DexDOX (purified fraction a), lane 6: DOX + dexamethasone. (C) Representative color of DexDOX and DOX in DMSO solution. (D) APCI mass-spectroscopy analysis of DexDOX.
Abbreviations: RT, room temperature; TLC, thin-layer chromatography; APCI, atmospheric pressure chemical ionization; DOX, doxorubicin.
Cytotoxicity of DexDOX in MCF-7 cells
We evaluated the antiproliferative effect of DexDOX by MTT assay. Following a 24 h exposure to varying concentrations of DOX and DexDOX, the number of viable MCF-7 cells were found to decrease. An average of calculated IC50s for DexDOX from four, independent experiments was 90.31±7.3 μg/mL. In contrast, the average IC50s for DOX under the same conditions was 2.8±0.9 μg/mL. This result clearly indicates that DexDOX has less cytotoxicity to MCF-7 cells than DOX.
Differences in cellular distribution of DexDOX and DOX
Cellular entry of DexDOX was evaluated by flow cytometry analysis. MCF-7 cells were treated with DexDOX or DOX for 2 h prior to flow cytometry analysis. The results indicate that about 70% of DexDOX-treated MCF cells and almost all of the DOX-treated cells emitted a fluorescence signal at the time of flow cytometry analysis (Figure 2). As both DOX and DexDOX emitted fluorescent signals, we employed live cell confocal video microscopy to study the intracellular distribution of both molecules. The fluorescence signals of DOX were found to increase gradually over time during the observation period. The signals were observed in both the cytoplasm and nucleus, but predominately inside the nucleus (Figure 3). Interestingly, the number of pyknotic cells (arrow), which indicate the presence of apoptotic cells, gradually increased with increased intranuclear accumulation of DOX. The intracellular distribution and the accumulation of DexDOX observed were different from that of DOX. Fluorescence signals of DexDOX peaked at 3 min after exposure, suggesting that this molecule was more efficient at entering MCF-7 cells than DOX. The DexDOX fluorescence signal rapidly decreased. Notably, apoptotic cells were observed at the early phase of the exposure to DexDOX, and their numbers increased further as the fluorescence signal decreased. Interestingly, fluorescence signals from DexDOX were observed only in the cytoplasm, suggesting that this molecule could not enter the nucleus. Taken together, these differences in the patterns of cellular diffusion and distribution suggest that the mechanisms of cytotoxicity employed by DexDOX and DOX are not identical.
Figure 2.
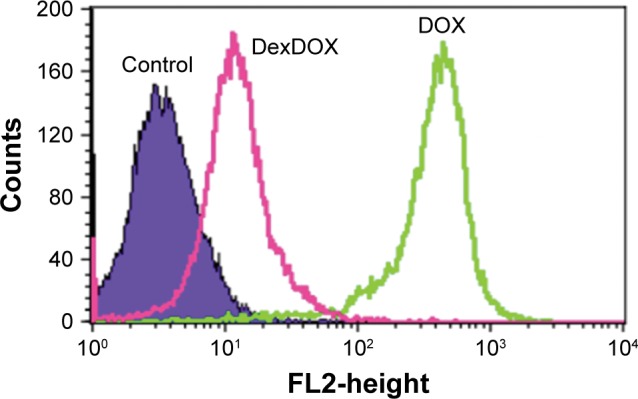
Cellular penetration and retention of DexDOX.
Note: Flow cytometry of MCF-7 cells 2 h following incubation with DexDOX and DOX.
Abbreviation: DOX, doxorubicin.
Figure 3.

Cellular penetration and subcellular distribution of DexDOX.
Notes: Representative images from video-assisted confocal microscopy showed that DexDOX entered MCF-7 more rapidly than DOX. The fluorescence signal was rapidly lost without entering the nucleus. Interestingly, pyknotic cells, which are indicative of apoptosis, were observed (arrow) after the fluorescence signal was lost. In contrast, DOX gradually diffused into cells and accumulated in the nucleus. The apoptotic cells (arrow) were observed in the later phase.
Abbreviation: DOX, doxorubicin.
DexDOX-induced cell death via ROS generation, independent of the cell cycle interruption
Flow cytometry analysis of the cell cycle showed that under the conditions used, the majority of the MCF-7 treated with DOX, 55.0%±1.3% of the cells were in G0/G1 phase and approximately 27.4%±0.7% of the cells were in G2/M phase. In contrast, 48 h treatment of the cells with DexDOX did not affect cell cycle progression, about 75.4%±2.2% of the cells were in G0/G1 and 11.5%±1.4% were in G2/M phase as compared to the untreated cells in which 77.4%±2.4% and 10.4%±1.8% of the cells were in G0/G1 and G2/M, respectively (Figure 4A). Flow cytometry of Annexin V showed that following a 48 h exposure to DOX, approximately 70% of the cells were Annexin+ PI−, indicating that those cells were in an early apoptotic phase/state. In contrast, exposure of DexDOX showed the presence of double-positive Annexin V and PI, indicating that the cells were in late apoptosis (Figure 4B). These findings supported the results of video confocal microscopy, which showed that an early apoptosis occurred following exposure to DexDOX. To further understand the mechanisms leading to cell death, we indirectly evaluated intracellular ROS generation in DexDOX-treated cells. The expression of intracellular DCF, an oxidized product of DCFH-DA, was subsequently analyzed. Our results demonstrate that DexDOX increased DCF expression during the early phase of the cell cycle (20–60 min) (Figure 4C). The increase in DCF signals during the first 60 min suggested that cellular ROS increased during this period.
Figure 4.

DexDOX-induced apoptosis independent of the cell cycle interruption.
Notes: (A) Cell cycle analysis demonstrated that treatment with DexDOX did not induce cell cycle arrest, whereas DOX induced cycle arrest in the G2/M phase. (B) Representative analysis of Annexin V and PI showed that the majority of the MCF-7 cells were double-positive for Anexin V and PI following 48 h of treatment with DexDOX. In contrast, MCF-7 induced MCF-7-expressed Annexin V. (C) Representative images of DCFH-DA-treated cells showing sequential expression of DCF (green fluorescence) were generated from oxidization of DCFH-DA following exposure to DexDOX.
Abbreviations: DCFH-DA, dichloro-dihydro-fluorescein diacetate; DOX, doxorubicin.
DexDOX can overcome MDR-1-induced resistance in MCF-7 cells
We tested the effect of DexDOX in MDR-1-overexpressing cells. Five days following transfection, approximately 87% of MCF-7 cells displayed a green fluorescent signal. The MTT assay showed that MDR-1-transfected cells could be defined as being resistant to DOX. The IC50s of DOX in MDR-1-transfected cells were 45.15±7.1 μg/mL, or approximately 16-fold of the IC50s of DOX in MCF-7 cells. In contrast, the IC50s of DexDOX in MDR-1-overexpressing cells were 101.82±14.0 μg/mL, which did not differ to those of the control, MCF-7, indicating that overexpression of MDR-1 did not lead to a decrease in the cytotoxicity of DexDOX (Figure 5).
Figure 5.

IC50 of DexDOX and DOX in MDR-1-overexpressing cells.
Notes: (A) Bar graph comparing IC50s of DOX and DexDOX in control MCF-7. (B) MDR-1-overexpressing MCF-7 cells (*P<0.05).
Abbreviations: MDR, multidrug resistance; DOX, doxorubicin.
Discussion
Bio-conjugation, a process used to attach a bioactive molecule to another molecule via a covalent bond, leads to the formation of a novel chemical structure with may have enhanced properties compared to those of the original molecule. In this study, we synthesized a novel derivative of DOX by simple conjugation with dexamethasone. The chemical reaction is simple and does not require any catalyst or exogenous energy. We have demonstrated that the new derivative has considerable cytotoxicity, but acts via a different mechanism to that of DOX.
Dexamethasone is a potent glucocorticoid analog, synthetic, lipophilic, and hormone-based drug that is widely used to treat a variety of medical conditions and is included in many standard chemotherapeutic regimens. The 3′ amino group in the sugar moiety of DOX which was the conjugation site is known to be a crucial site allowing the DOX molecule to intercalate to the cytosine molecule in the DNA strand. Therefore, it is hypothesized that modification at this residue is likely to interfere with DNA intercalation and topoisomerase inhibition, both of which are known to be key cytotoxic mechanisms employed by DOX. Our video-assisted microscopy study clearly demonstrated that DexDOX did not enter the nucleus showing an obvious difference with DOX in this respect.
Interestingly, transferrin-conjugated DOX that was modified at the 3′ amino group, was also found to be unable to enter the nucleus.33 Taken together with our results, the data suggest that this amino group of the sugar moiety may be crucial for giving these molecules the ability to enter the nucleus.
Despite a loss of ability to enter the nucleus, DexDOX elicited potent and rapid cytotoxicity. This molecule showed a greater efficiency in cellular penetration than DOX. This may be due to the lipophilic property of dexamethasone. Peak cytoplasmic fluorescence emission occurred within the first 3 minutes of exposure to DexDOX and was followed by a rapid loss of fluorescence signal. Interestingly, initiation of apoptosis was found to coincide with loss of DexDOX’s fluorescence signal. It is known that the fluorescence signal of DOX and other anthracyclins originates from a free electron traveling along the aglycone structure. However, fluorescence signal loss in our experiment is too rapid to be the result of energy consumption efflux pump function. Therefore, we speculated that a breakdown of the aglycone structure of DexDOX had occurred in the cytoplasm. Redox-cycling of aglycone breaks the aglycone structure down, forming semiquinone radicals which are a potent ROS, and this is the core process of DOX-mediated oxidative injuries.12 Our results demonstrate evidence of increased ROS in MCF-7 cells treated with DexDOX in parallel with the observation of a reduction in the cytoplasmic signal of DEXDOX. This suggests that the loss of DexDOX fluorescence may contribute to cellular ROS generation. The presence of ROS was detected within 15 min, a relatively early stage, following exposure to DOX.36 It was previously reported that potentiating oxidative stress of DOX can overcome the effect of efflux pump-induced resistance.13 DexDOX-induced cellular cell cycle independent apoptosis after entering cells. We speculated that the rapid action of DexDOX in the generation of ROS may be crucial for overcoming the P-gp efflux pump, which needs sufficient time to extrude the drug from cells. Therefore, the overexpression of P-gp had little effect on DexDOX cytotoxicity.
DOX is best known for its cardiotoxicity. Again, the molecular mechanisms of DOX-induced myocardial damage remain unclear. A line of evidence suggests that ROS generation contribute to myocardial injury.3 However, antioxidant treatments failed to improve myocardial damage. Of interest, a recent study showed that mice lacking the topoisomerase 2β in cardiomyocyte were resistant to DOX-induced myocardial damage.37 Since DexDOX generated oxidative stress without entering the nucleus, this molecule should be useful for further understanding the mechanism of DOX-induced cardiotoxicity.
Conclusion
We have synthesized a new derivative of DOX by conjugating the dexamethasone molecule to DOX. This molecule showed potent cytotoxicity to MCF-7 cells in a manner that is different to that of DOX. Furthermore, we have shown that this molecule has the ability to escape the effect of P-gp overexpression on drug resistance and that, therefore, it may be useful for the treatment of drug-resistant tumor cells.
Acknowledgments
We thank Dr Jatupol Lingsakul, Scientific and Technological Research Equipment Centre, Chulalongkorn University for his technical assistance and advice. We are grateful to Dr Ryoji Sassa (Okasaki, Nagoya) for his generous support. This work was supported by Lerdsin Foundation and SS Manufacturing. Kamanotip Chaikomon was supported by The 90th and 100th Anniversary of Chulalongkorn University Funds.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Arcamone F, Cassinelli G, Fantini G, et al. Adriamycin, 14-hydroxy-daunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Reprinted from Biotechnology and Bioengineering, Vol. XI, Issue 6, Pages 1101–1110 (1969) Biotechnol Bioeng. 2000;67(6):704–713. doi: 10.1002/(sici)1097-0290(20000320)67:6<704::aid-bit8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 2.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56(2):185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 3.Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52(6):1213–1225. doi: 10.1016/j.yjmcc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Kiyomiya K, Matsuo S, Kurebe M. Mechanism of specific nuclear transport of adriamycin: the mode of nuclear translocation of adriamycin-proteasome complex. Cancer Res. 2001;61(6):2467–2471. [PubMed] [Google Scholar]
- 5.Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984;226(4673):466–468. doi: 10.1126/science.6093249. [DOI] [PubMed] [Google Scholar]
- 6.Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 2009;9(5):338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agudelo D, Bourassa P, Bérubé G, Tajmir-Riahi HA. Intercalation of antitumor drug doxorubicin and its analogue by DNA duplex: structural features and biological implications. Int J Biol Macromol. 2014;66:144–150. doi: 10.1016/j.ijbiomac.2014.02.028. [DOI] [PubMed] [Google Scholar]
- 8.Swift LP, Rephaeli A, Nudelman A, Phillips DR, Cutts SM. Doxorubicin-DNA adducts induce a non-topoisomerase II-mediated form of cell death. Cancer Res. 2006;66(9):4863–4871. doi: 10.1158/0008-5472.CAN-05-3410. [DOI] [PubMed] [Google Scholar]
- 9.Yang F, Teves SS, Kemp CJ, Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys Acta. 2014;1845(1):84–89. doi: 10.1016/j.bbcan.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cutts SM, Nudelman A, Rephaeli A, Phillips DR. The power and potential of doxorubicin-DNA adducts. IUBMB Life. 2005;57(2):73–81. doi: 10.1080/15216540500079093. [DOI] [PubMed] [Google Scholar]
- 11.Berlin V, Haseltine WA. Reduction of adriamycin to a semiquinone-free radical by NADPH cytochrome P-450 reductase produces DNA cleavage in a reaction mediated by molecular oxygen. J Biol Chem. 1981;256(10):4747–4756. [PubMed] [Google Scholar]
- 12.Malisza KL, McIntosh AR, Sveinson SE, Hasinoff BB. Semiquinone free radical formation by daunorubicin aglycone incorporated into the cellular membranes of intact Chinese hamster ovary cells. Free Radic Res. 1996;24(1):9–18. doi: 10.3109/10715769609087995. [DOI] [PubMed] [Google Scholar]
- 13.Kostrzewa-Nowak D, Paine MJ, Wolf CR, Tarasiuk J. The role of bioreductive activation of doxorubicin in cytotoxic activity against leukaemia HL60-sensitive cell line and its multidrug-resistant sublines. Br J Cancer. 2005;93(1):89–97. doi: 10.1038/sj.bjc.6602639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huigsloot M, Tijdens IB, Mulder GJ, van de Water B. Differential regulation of doxorubicin-induced mitochondrial dysfunction and apoptosis by Bcl-2 in mammary adenocarcinoma (MTLn3) cells. J Biol Chem. 2002;277(39):35869–35879. doi: 10.1074/jbc.M200378200. [DOI] [PubMed] [Google Scholar]
- 15.Chen MB, Wu XY, Gu JH, Guo QT, Shen WX, Lu PH. Activation of AMP-activated protein kinase contributes to doxorubicin-induced cell death and apoptosis in cultured myocardial H9c2 cells. Cell Biochem Biophys. 2011;60(3):311–322. doi: 10.1007/s12013-011-9153-0. [DOI] [PubMed] [Google Scholar]
- 16.Wang S, Konorev EA, Kotamraju S, Joseph J, Kalivendi S, Kalyanaraman B. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. Intermediacy of H(2)O(2)- and p53-dependent pathways. J Biol Chem. 2004;279(24):25535–25543. doi: 10.1074/jbc.M400944200. [DOI] [PubMed] [Google Scholar]
- 17.Shin HJ, Kwon HK, Lee JH, et al. Doxorubicin-induced necrosis is mediated by poly-(ADP-ribose) polymerase 1 (PARP1) but is independent of p53. Sci Rep. 2015;5:15798. doi: 10.1038/srep15798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueda K, Cardarelli C, Gottesman MM, Pastan I. Expression of a full-length cDNA for the human “MDR1” gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Acad Sci U S A. 1987;84(9):3004–3008. doi: 10.1073/pnas.84.9.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zaman GJR, Flens MJ, van Leusden MR, et al. The human multidrug resistance-associated protein MRP is a plasma membrane drug-efflux pump. Proc Natl Acad Sci U S A. 1994;91(19):8822–8826. doi: 10.1073/pnas.91.19.8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marbeuf-Gueye C, Broxterman HJ, Dubru F, Priebe W, Garnier-Suillerot A. Kinetics of anthracycline efflux from multidrug resistance protein-expressing cancer cells compared with P-glycoprotein-expressing cancer cells. Mol Pharmacol. 1998;53(1):141–147. doi: 10.1124/mol.53.1.141. [DOI] [PubMed] [Google Scholar]
- 21.Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. 2003;22(47):7468–7485. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- 22.Shen F, Chu S, Bence AK, et al. Quantitation of doxorubicin uptake, efflux, and modulation of multidrug resistance (MDR) in MDR human cancer cells. J Pharmacol Exp Ther. 2008;324(1):95–102. doi: 10.1124/jpet.107.127704. [DOI] [PubMed] [Google Scholar]
- 23.Tabe Y, Konopleva M, Contractor R, et al. Up-regulation of MDR1 and induction of doxorubicin resistance by histone deacetylase inhibitor depsipeptide (FK228) and ATRA in acute promyelocytic leukemia cells. Blood. 2006;107(4):1546–1554. doi: 10.1182/blood-2004-10-4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim DW, Kim KO, Shin MJ, et al. siRNA-based targeting of antiapoptotic genes can reverse chemoresistance in P-glycoprotein expressing chondrosarcoma cells. Mol Cancer. 2009;8:28. doi: 10.1186/1476-4598-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferry DR, Traunecker H, Kerr DJ. Clinical trials of P-glycoprotein reversal in solid tumours. Eur J Cancer. 1996;32A(6):1070–1081. doi: 10.1016/0959-8049(96)00091-3. [DOI] [PubMed] [Google Scholar]
- 26.Teodori E, Dei S, Martelli C, Scapecchi S, Gualtieri F. The functions and structure of ABC transporters: implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance (MDR) Curr Drug Targets. 2006;7(7):893–909. doi: 10.2174/138945006777709520. [DOI] [PubMed] [Google Scholar]
- 27.Sims JT, Ganguly SS, Bennett H, Friend JW, Tepe J, Plattner R. Imatinib reverses doxorubicin resistance by affecting activation of STAT3-dependent NF-κB and HSP27/p38/AKT pathways and by inhibiting ABCB1. PLoS One. 2013;8(1):e55509. doi: 10.1371/journal.pone.0055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ha JS, Byun J, Ahn DR. Overcoming doxorubicin resistance of cancer cells by Cas9-mediated gene disruption. Sci Rep. 2016;6:22847. doi: 10.1038/srep22847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiss RB. The anthracyclines: will we ever find a better doxorubicin? Semin Oncol. 1992;19(6):670–686. [PubMed] [Google Scholar]
- 30.Chhikara BS, Mandal D, Parang K. Synthesis, anticancer activities, and cellular uptake studies of lipophilic derivatives of doxorubicin succinate. J Med Chem. 2012;55(4):1500–1510. doi: 10.1021/jm201653u. [DOI] [PubMed] [Google Scholar]
- 31.Yu S, Zhang G, Zhang W, et al. Synthesis and biological activities of a 3′-azido analogue of doxorubicin against drug-resistant cancer cells. Int J Mol Sci. 2012;13(3):3671–3684. doi: 10.3390/ijms13033671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piorecka K, Stanczyk W, Florczak M. NMR analysis of antitumor drugs: doxorubicin, daunorubicin and their functionalized derivatives. Tetrahedron Lett. 2017;58(2):152–155. [Google Scholar]
- 33.Kratz F, Beyer U, Roth T, et al. Transferrin conjugates of doxorubicin: synthesis, characterization, cellular uptake, and in vitro efficacy. J Pharm Sci. 1998;87(3):338–346. doi: 10.1021/js970246a. [DOI] [PubMed] [Google Scholar]
- 34.Kalyanaraman B, Darley-Usmar V, Davies KJ, et al. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med. 2012;52(1):1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tupchiangmai W, Choksakulporn S, Tewtrakul S, Pianwanit S, Sritanaanant Y. Use of a hexasubstituted benzene scaffold in the development of multivalent HIV-1 integrase inhibitors. Chem Pharm Bull (Tokyo) 2014;62(8):754–763. doi: 10.1248/cpb.c14-00074. [DOI] [PubMed] [Google Scholar]
- 36.Ciftci O, Ullrich O, Schmidt CA, Diestel A, Hass R. Regulation of the nuclear proteasome activity in myelomonocytic human leukemia cells after adriamycin treatment. Blood. 2001;97(9):2830–2838. doi: 10.1182/blood.v97.9.2830. [DOI] [PubMed] [Google Scholar]
- 37.Zhang S, Liu X, Bawa-Khalfe T, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nature Med. 2012;18(11):1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]