

Abstract
Aim/hypothesis
Here, our aim was to examine whether VLDL and apolipoprotein (apo) CIII induce endoplasmic reticulum (ER) stress, inflammation and insulin resistance in skeletal muscle.
Methods
Studies were conducted in mouse C2C12 myotubes, isolated skeletal muscle and skeletal muscle from transgenic mice overexpressing apoCIII.
Results
C2C12 myotubes exposed to VLDL showed increased levels of ER stress and inflammatory markers whereas peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) and AMP-activated protein kinase (AMPK) levels were reduced and the insulin signalling pathway was attenuated. The effects of VLDL were also observed in isolated skeletal muscle incubated with VLDL. The changes caused by VLDL were dependent on extracellular signal-regulated kinase (ERK) 1/2 since they were prevented by the ERK1/2 inhibitor U0126 or by knockdown of this kinase by siRNA transfection. ApoCIII mimicked the effects of VLDL and its effects were also blocked by ERK1/2 inhibition, suggesting that this apolipoprotein was responsible for the effects of VLDL. Skeletal muscle from transgenic mice overexpressing apoCIII showed increased levels of some ER stress and in-flammatory markers and increased phosphorylated ERK1/2 levels, whereas PGC-1α levels were reduced, confirming apoCIII effects in vivo. Finally, incubation of myotubes with a neutralising antibody against Toll-like receptor 2 abolished the effects of apoCIII on ER stress, inflammation and insulin resistance, indicating that the effects of apoCIII were mediated by this receptor.
Conclusions/interpretation
These results imply that elevated VLDL in diabetic states can contribute to the exacerbation of insulin resistance by activating ERK1/2 through Toll-like receptor 2.
Keywords: AMPK, apoCIII, ERK1/2, TLR2, VLDL
Introduction
Insulin resistance and type 2 diabetes mellitus are characterised by the presence of atherogenic dyslipidaemia, which includes the following cluster of abnormalities: high levels of triacylglycerols, low levels of HDL-cholesterol and the appearance of small, dense LDLs [1]. Atherogenic dyslipidaemia frequently precedes type 2 diabetes mellitus by several years, indicating that derangement of lipid metabolism is an early event in the development of this disease [2]. It is now well accepted that the different components of atherogenic dyslipidaemia are closely linked and are initiated by insulin resistance through overproduction of triacylglycerolrich VLDL [1, 2]. In addition to triacylglycerols, VLDLs also contain apolipoproteins, of which apolipoprotein (apo) CIII is one of the most abundant [3] with levels that are closely correlated with serum triacylglycerol levels [4]. Plasma apoCIII increases plasma triacylglycerols predominantly through the inhibition of VLDL hydrolysis by lipoprotein lipase and by inhibiting chylomicron and VLDL clearance by the liver [5], but it also causes inflammation in endothelial cells [6]. Furthermore, some studies have associated elevated circulating apoCIII with insulin resistance [7], although others did not find a relationship [8].
Whereas the effects of insulin resistance on lipoprotein metabolism have been studied extensively [1, 2], little is known about the effects of elevated VLDL and apoCIII on the molecular mechanism of insulin resistance in skeletal muscle cells. This is important, since the primary site of insulin-stimulated glucose disposal is skeletal muscle and this can account for up to 90% of glucose clearance [9]. As a result, loss of skeletal muscle insulin sensitivity is believed to be critical in the pathogenesis of type 2 diabetes [10]. The mechanisms involved in the development of insulin resistance are currently unclear, but accumulating evidence points to the presence of a chronic low-level inflammatory process [11]. Among other mechanisms, endoplasmic reticulum (ER) stress [12] and Toll-like receptors (TLRs) [13] can activate prion-flammatory signalling pathways, including inhibitor of κB (IκΒ) kinase β (ΙΚΚ-β)–NF-κB. Thus, IKK-β phosphorylates pathway whereas, once activated, NF-κB regulates the expression of multiple inflammatory mediators, which also contribute to insulin resistance [11].
In the present study, we examined whether VLDL and apoCIII induce ER stress, inflammation and insulin resistance in skeletal muscle cells.
Methods
Materials
Escherichia coli (K12 strain) lipopolysaccharide (ultrapure) and PAM3CSK4 (tripalmitoylated cysteine-, serine- and lysine-containing peptide) were purchased from InvivoGen (San Diego, CA, USA). LDH Cytotoxicity Assay Kit (88953) was from Thermo Scientific (Waltham, MA, USA) and the Elisa kit for measuring IL-6 secretion (Novex, KMC0061) was from Life Technologies (Carlsbad, CA, USA).
Plasma VLDL isolation
VLDL particles (< 1.006 g/ml) were isolated by ultracentrifugation at 100,000g for 24 h from normolipidaemic human plasma obtained in EDTA-containing vacutainer tubes (total cholesterol ≤ 5.2 mmol/l, triacylglycerols ≤ 1 mmol/l). To obtain VLDL particles containing low or high amounts of apoCIII, we further isolated light VLDL (Svedberg flotation units 60–400) from normolipidaemic and hypertriacylglycerolaemic (triacylglycerols ≥ 2.5 mmol/l) human plasma by ultracentrifugation at 56,000g for 1 h. VLDL preparations were extensively dialysed in PBS and then triacylglycerol and apoB concentrations were measured using a commercial kit adapted to a COBAS c501 autoanalyser (Roche Diagnostics, Rotkreuz, Switzerland). ApoB/triacylglycerol ratios were similar in both light VLDL preparations. ApoCIII levels were determined using a nephelometric commercial kit (Kamiya Biomedical Company, Seattle, WA, USA) adapted to COBAS c501 autoanalyser. Cells were treated with 300 μg/ml of filtered VLDL, based on triacylglycerol concentration, as previously described [14].
Cell culture
Mouse mycoplasma free C2C12 cells (ATCC, Manassas, VA, USA) were maintained, grown and differentiated to myotubes as previously described [15]. ATCC provided authentication of the cells. Where indicated, cells were treated with 10 μmol/l U0126, 100 μg/ml apoCIII (purity > 95%) (Abcam, Cambridge, UK), 50 μg/ml TLR2 neutralising antibody (InvivoGen) or control non-immune IgG for 24 h. Cells were transiently transfected with 50 nmol/l siRNA against extracellular signal-regulated kinase (ERK) 1/2 (Santa Cruz, Dallas, TX, USA) and siRNA control using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions.
Animals
Skeletal muscle (gastrocnemius) samples from male wild-type and transgenic mice overexpressing human apoCIII (apoCIII Tg; C57BL/6J background) were frozen in liquid nitrogen and then stored at − 80°C. For ex vivo experiments, skeletal muscles were isolated from male C57BL/6J mice (6– 8 weeks old) and mounted in an incubation bath as previously described [16] in the presence or absence of 500 μg/ml VLDL. Experimenters were not blind to group assignment or outcome assessment. For further details, please refer to the electronic supplementary material (ESM) Methods.
RNA preparation and quantitative RT-PCR
Relative levels of specific mRNAs were assessed by real-time PCR, as previously described [15]. For details, see ESM Methods. The primer sequences used are shown in ESM Table 1.
Immunoblotting
Isolation of total and nuclear protein extracts was performed as described elsewhere [15]. Western blot analysis was performed using antibodies against total (1:1000, 9272) and phospho-Akt (Ser473) (1:1000, 9271), glucose-regulated protein78 (GRP78)/binding immunoglobulin protein (BiP) (1:1000, 3177), insulin receptor β-subunit (IRβ) (1:1000, 3020), CCAT-enhancer-binding protein homologous protein (CHOP) (1:1000, 5554), total eukaryotic initiation factor 2α (eIF2α) (1:1000, 9722) and phospho-eIF2α (Ser51) (1:1000, 9721S), total signal transducer and activator of transcription 3 (STAT3) (1:1000, 9132) and phospho-STAT3 (Tyr705) (1:1000, 9131), total extracellular signal-regulated kinase (ERK) 1/2 (1:1000, 9102) and phospho-ERK1/2 (Thr202/Tyr204) (1:1000, 9101), total acetyl-CoA carboxylase (ACC) (1:1000, 3662) and phospho-ACC (Ser79) (1:1000, 3661), NQO1 (1:500, 62,262), nuclear respiratory factor 1 (NRF1) (1:500, 12,381), nuclear factor-E2-related factor 2 (NRF2) (1:500, 4399), phospho-IRS-1 (Ser307) (1:500, 2381), IκΒα (1:500, 9242), p65 (1:500, 3034), total AMP-activated protein kinase (AMPK) (1:1000, 2532) and phospho-AMPK (Thr172) (1:1000, 2531) (all from Cell Signaling Technology, Danvers, MA, USA; numbers indicate catalogue number), oxidative phosphorylation (1:1000, ab110413) (OXPHOS), peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α; (1:1000, ab54481) (Abcam), OCT-1 ( 1:500, sc-8024 X), peroxisome proliferator-activated receptor (PPAR)β/δ (1:500, sc-7197), prohibitin (1:500, sc-377037), suppressor of cytokine signalling 3 (SOCS3) (1:500, sc-51699), Tribbles 3 (TRB3) (1:500,sc-365842), glyceraldehyde 3-phosphate dehydrogenase (1:500, sc-32233), total IRS-1 (1:500, sc-560) and β-actin (1:500, sc-47778) (all from Santa Cruz; numbers indicate catalogue number). Detection was achieved using the Western Lightning Plus-ECL chemiluminescence kit (PerkinElmer, Waltham, MA, USA). The equal loading of proteins was assessed by Ponceau S staining. For validation, we used a protein marker (Precision Plus Protein Dual Color Standards 1610374; Bio-Rad, Hercules, CA, USA), on the same blots. All of these commercially available antibodies showed a single distinct band at the molecular weight indicated in the datasheets.
Electrophoretic mobility shift assay
The electrophoretic mobility shift assay (EMSA) was performed as described in ESM Methods.
2-Deoxy-D-(1,2-[3H]N)glucose uptake
Glucose uptake experiments were performed as described in ESM Methods.
Image analysis
The chemiluminescent blots were imaged using the ChemiDoc MP imager (Bio-Rad). Image acquisition and subsequent densitometric analysis of the corresponding blots were performed using ImageLab software version 4.1 (Bio-Rad). For further details, see ESM Methods.
Statistical analyses
Results were normalised to levels in control groups and are expressed as mean ± SD. Significant differences were established by either Student’s t test or two-way ANOVA, according to the number of groups compared, using GraphPad Prism V4.03 software (GraphPad Software, San Diego, CA, USA). When significant variations were found by two-way ANOVA, the Tukey–Kramer multiple comparison post hoc test was performed. Differences were considered significant at p < 0.05.
Results
VLDL induces ER stress, inflammation and insulin resistance in myotubes
VLDL exposure significantly increased expression of the ER stress markers Bip (also known as Hspa5), Chop (Ddit3) and Nqo1, the latter being an NRF2-target gene activated by ER stress (Fig. 1a). Consistent with the presence of VLDL-induced ER stress, the protein levels of BiP, phospho-eIF2α, CHOP and TRB3, a pseudokinase that mediates ER stress-induced insulin resistance in myotubes [17], were increased by VLDL (Fig. 1b). VLDL exposure also increased the mRNA levels of inflammatory genes such as Il6, Mcp1 (also known as Ccl2) and Tnfα (Tnf), whereas the mRNA expression of the NF-κB inhibitor IκBα (Nfkbia) was reduced (Fig. 1c). IL-6 induces insulin resistance by activating STAT3, which in turn upregulates the transcription of SOCS3. SOCS3 inhibits insulin signalling through several distinct mechanisms, including IRS degradation [18]. In agreement with the increase in IL-6 expression, the mRNA levels of Socs3 were also increased after VLDL exposure (Fig. 1c). The potential activation of the NF-κB pathway by VLDL was confirmed by the presence of reduced protein levels of IκBα and enhanced levels of the p65 subunit of NF-κB (Fig. 1d). Similarly, increased protein levels of phospho-STAT3 (phosphorylated at Tyr705) and SOCS3 demonstrated the activation of the STAT3–SOCS3 pathway by VLDL (Fig. 1e).
Fig. 1.
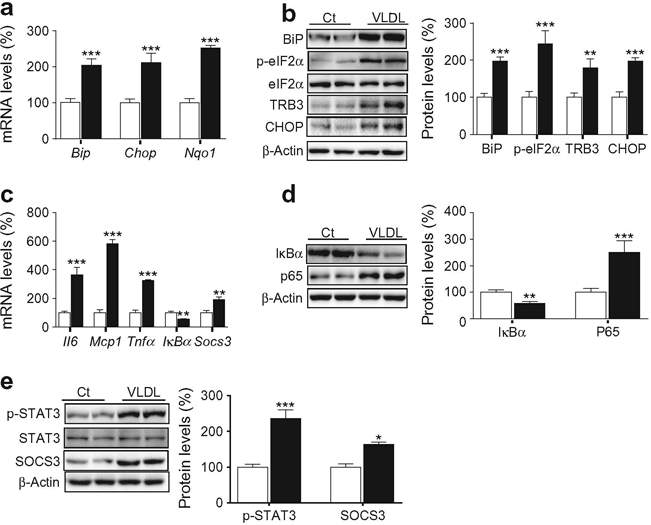
VLDL induces ER stress and inflammation. Mouse C2C12 myotubes were incubated in the presence (black bars) or absence (control, Ct, white bars) of 300 μg/ml VLDL for 24 h. (a) mRNA abundance of Bip, Chop and Nqo1. mRNA levels are normalised to Aprt (n = 8–10, five independent C2C12 cultures were used). (b) BiP, phospho-eIF2α (Ser51), TRB3, CHOP and β-actin protein levels. (c), mRNA abundance of Il6, Mcp1, Tnfα, IκBα and Socs3. (d) IκBα, p65 and β-actin protein levels. (e) Phospho-STAT3 (Tyr705), SOCS3 and β-actin protein levels. The graphs show quantification expressed as a percentage of control samples. Data are means ± SD of five independent experiments and were compared by Student’s t test. *p < 0.05, **p < 0.01 and ***p < 0.001 vs control
Mitochondrial function is transcriptionally controlled by PGC-1α [19], which plays a critical role in skeletal muscle metabolic function. In fact, some studies indicate that the reported reduction in PGC-1α expression and/or function in the skeletal muscle of individuals who have diabetes or are at risk for diabetes [20, 21] induces insulin resistance by reducing oxidative phosphorylation and lipid oxidation, leading to accumulation of lipid derivatives in skeletal muscle [22]. Myotubes exposed to VLDL showed a reduction in the mRNA expression of Pgc1α (also known as Ppargc1a) (Fig. 2a). This transcriptional co-activator regulates the activity of several transcription factors, including PPARα and PPARβ/δ, which control the expression/function of genes involved in fatty acid oxidation (FAO) [23]. The expression of these transcription factors and that of their target genes involved in FAO, such as those encoding acyl-coA oxidase (Acox, also known as Acox1) and medium chain acyl-CoA dehydrogenase (Mcad, also known as Acadm), was also decreased by VLDL (Fig. 2a). In addition, PGC-1α protein levels were downregulated by VLDL and, consistent with this reduction, the protein levels of its downstream transcription factor NRF1 [24] were also reduced (Fig. 2b). FAO is also under the control of AMPK, whose activation exerts multiple protective effects, including inhibition of inflammation and insulin resistance [25]. Activation of this kinase upregulates PGC-1α levels and increases FAO by phosphorylating ACC at Ser79, leading to inhibition of ACC’s activity and decreased malonyl-CoA content, which inhibits carnitine palmitoyltransferase (CPT-1), the rate-limiting step in FAO in mitochondria [25]. VLDL reduced the levels of both phospho-AMPK and phospho-ACC in myotubes (Fig. 2b), whereas it increased the protein levels of the redox transcription factor NRF2 and the protein encoded by its target gene Nqo1 (Fig. 2c).
Fig. 2.
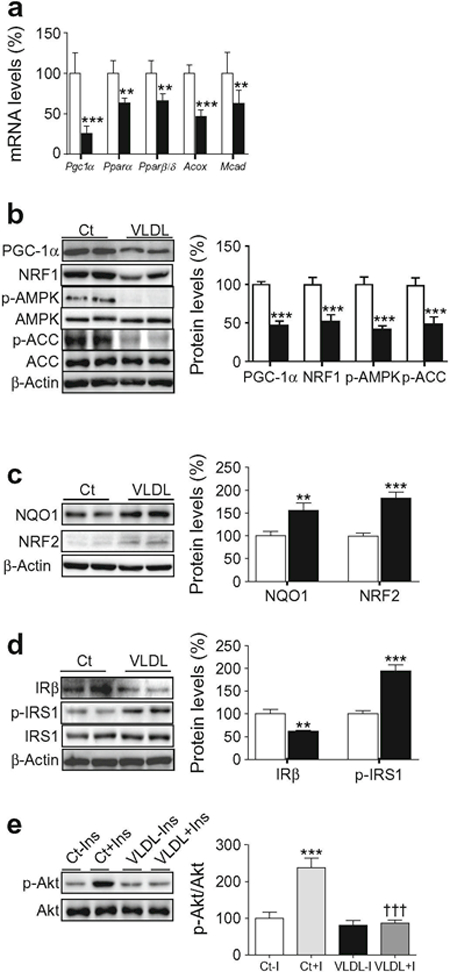
VLDL reduces PGC-1α and AMPK levels and induces insulin resistance. Mouse C2C12 myotubes were incubated in the presence (black bars) or absence (control, Ct, white bars) of 300 μg/ml VLDL for 24 h. (a) Pgc1α, Pparα (Ppara), Pparβ/δ (Pparb/Ppard), Acox and Mcad mRNA levels (n = 8–10, five independent C2C12 cultures were used). (b) PGC-1α, NRF1, phospho-AMPK (Thr172), phospho-ACC (Ser79) and β-actin protein levels. (c) NQO1, NRF2 and β-actin protein levels. (d) IRβ, phospho-IRS-1 (Ser307), and β-actin protein levels. (e) Phospho-Akt (Ser473) protein levels. Where indicated, cells were incubated with 100 nmol/l insulin (Ins, I) for the last 10 min. The graphs show quantification expressed as a percentage of control samples. Data are means ± SD of five independent experiments and compared by Student’s t test (a–d) or two-way ANOVA followed by Tukey post hoc test (e). **p < 0.01 and ***p < 0.001 vs control; †††p < 0.001 vs control cells incubated with insulin
When we examined proteins involved in the insulin signalling pathway, we observed that in agreement with a previous study reporting that ER stress reduced insulin receptor levels in adipocytes [26], protein levels of IRβ were reduced in VLDL-exposed cells (Fig. 2d). In addition, VLDL increased IRS-1 phosphorylation at Ser307 (Fig. 2d) and blunted insulin-stimulated Akt phosphorylation (Fig. 2e).
VLDL increases ER stress, mitochondrial dysfunction and inflammation in isolated skeletal muscle
Next, we examined the effects of VLDL on skeletal muscle. Gastrocnemius muscles isolated from mice were incubated with VLDL for 6 h, which resulted in an increase in the mRNA expression and protein levels of BiP and phospho-eIF2α, whereas no changes were observed in CHOP (Fig. 3a, b). Muscles exposed to VLDL also showed a significant increase in the mRNA levels of Il6, Mcp1 and Tnfα (Fig. 3c), consistent with the reduction in IκBα (Fig. 3d). VLDL also reduced IRβ protein levels and increased IRS phosphorylation at Ser307 (Fig. 3d). Similar to what we observed in vitro, VLDL caused a marked reduction in the expression of Pgc1α, Pparα, Pparβ/δ, and their target genes involved in FAO (Fig. 3e). Consistent with the reported regulation of mitochondrial OXPHOS genes [27] and NRF1 [24] by PGC-1α, the reduction in the protein levels of this transcriptional co-activator caused by VLDL was accompanied by a reduction in NRF1 and the different OXPHOS complexes (Fig. 3f). In addition, a reduction was detected in phospho-AMPK and phospho-ACC in muscles exposed to VLDL (Fig. 3g).
Fig. 3.
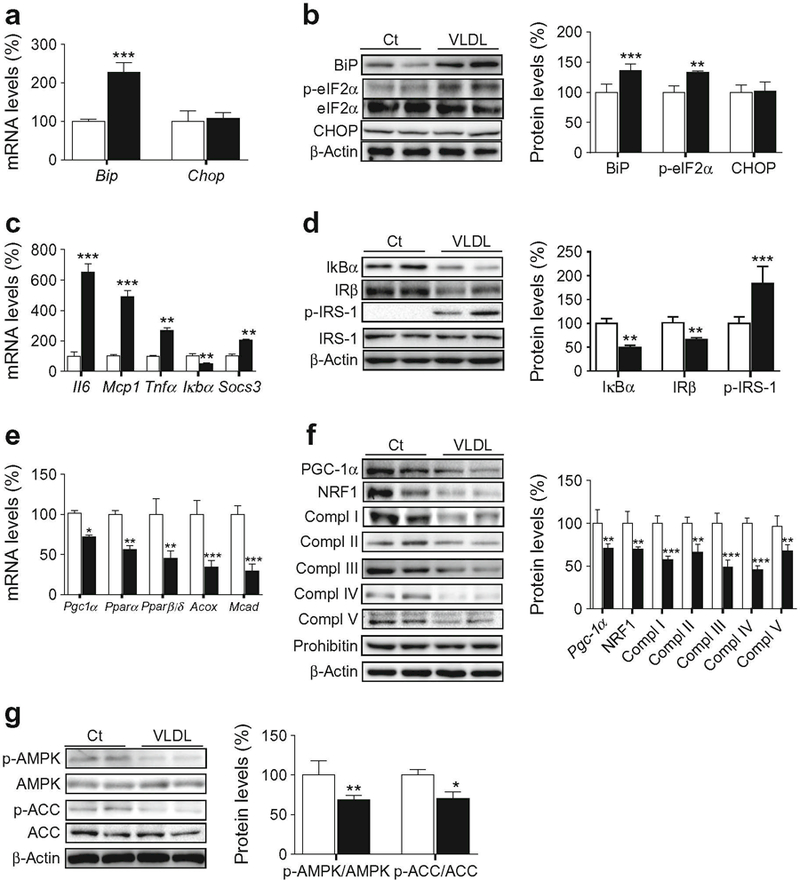
VLDL induces ER stress and inflammation, reduces the levels of mitochondrial proteins and attenuates the insulin signalling pathway in isolated skeletal muscle. Mouse gastrocnemius muscles were incubated in the presence (black bars) or absence (control, Ct, white bars) of 500 μg/ml VLDL for 6 h. (a) mRNA abundance of Bip and Chop. (b) BiP, PhosphoeIF2α (Ser51), CHOP and β-actin protein levels. (c) mRNA abundance of Il6, Mcp1, Tnfα, Iκbα and Socs3. (d) IκBα, IRβ, phospho-IRS-1 (Ser307), and β-actin protein levels. (e) Pgc1α, Pparα, Pparβ/δ, Acox and Mcad mRNA levels. (f) PGC-1α, NRF1, OXPHOS complexes (Compl), prohibitin and β-actin protein levels. (g) Phospho-AMPK (Thr172), phospho-ACC (Ser79) and β-actin protein levels. The graphs show quantification expressed as a percentage of control. Data are means ± SD of five independent experiments and were compared by Student’s t test. *p < 0.05, **p < 0.01 and ***p < 0.001 and vs control
ERK1/2 inhibition prevents the effects of VLDL
Interestingly, TLR-mediated NF-κB activation requires mitogen-activated protein kinase (MAPK)–ERK (MEK) 1/2 [28] and activation of both MEK1/2 and NF-κB results in the downregulation of PGC-1α in myotubes [29]. Similarly, an inhibitory crosstalk between AMPK and ERK1/2 has been reported and inhibition of ERK1/2 was found to improve AMPK and Akt pathways and to reverse ER stress-induced insulin resistance in myotubes [30]. These data prompted us to investigate whether the ERK–MAPK cascade was involved in the effects mediated by VLDL. This possibility was supported by the fact that VLDL increased phospho-ERK1/2 levels in both cultured myotubes and isolated muscle (Fig. 4a). Next, we used U0126, a potent and specific ERK1/2 inhibitor that binds to MEK, thereby inhibiting its catalytic activity and phosphorylation of ERK1/2, to investigate whether inhibition of this kinase prevented the effects caused by VLDL. U0126 prevented the increase in the expression of ER stress and inflammatory markers (Fig. 4b) and the reduction in genes involved in FAO (Fig. 4c). Knockdown of ERK1/2 by siRNA transfection (ESM Fig. 1) confirmed that this kinase was responsible for the effects of VLDL on ER stress and inflammation (Fig. 4d) and the reduction in genes involved in FAO (Fig. 4e).
Fig. 4.
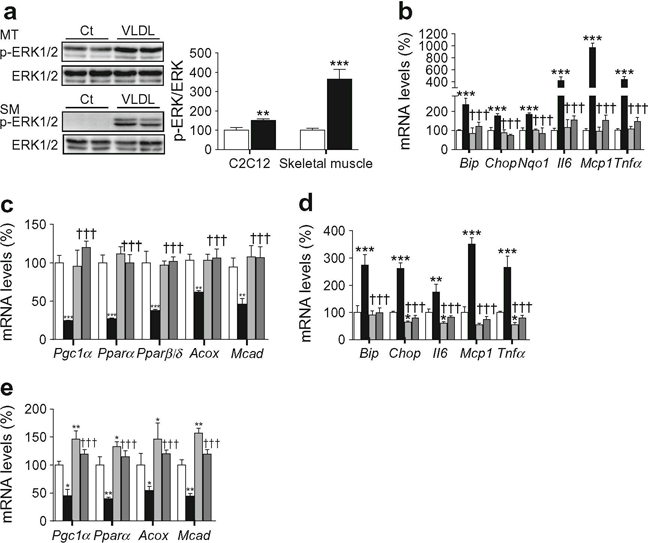
ERK1/2 inhibition and knockdown prevents the effects of VLDL. (a) C2C12 myotubes (MT) and isolated skeletal muscles (SM) were in-cubated in the presence (black bars) or absence (control, Ct, white bars) of 300 μg/ml VLDL (myotubes) or 500 μg/ml VLDL (muscle) and the protein levels of phospho-ERK1/2 (Thr202/Tyr204) were analysed. (b, c) C2C12 myotubes were incubated in the presence (black bars) or absence (control, white bars) of 300 μg/ml VLDL for 24 h; 10 μmol/l U0126 was added to control (light grey bars) or VLDL-treated (dark grey bars) myotubes and the mRNA abundance of Bip, Chop, Nqo1, Il6, Mcp1 and Tnfα (b) and Pgc1α, Pparα, Pparβ/δ, Acox and Mcad (c) was evaluated. (d, e) C2C12 cells were transfected with control siRNA or ERK1/2siRNA and incubated in the presence or absence of 300 μg/ml VLDL. The mRNA abundance of Bip, Chop, Il6, Mcp1 and Tnfα (d) and Pgc1α, Pparα, Acox and Mcad (e) was evaluated. White bars, control siRNA; light grey bars ERK1/2 siRNA; black bars VLDL + control siRNA; dark grey bars VLDL+ERK1/2 siRNA. The graphs show quantification expressed as a percentage of control. Data are means ± SD of five independent experiments and were compared by Student’s t test (a) or two-way ANOVA followed by Tukey post hoc test (b–e). *p < 0.05, **p < 0.01 and ***p < 0.001 vs control; †††p < 0.001 vs VLDL-exposed cells
ApoCIII mimics the effects of VLDL through TLR2
Given that apoCIII is the most abundant apolipoprotein in VLDL in individuals with diabetes [3], we next investigated whether this apolipoprotein was responsible for the effects of VLDL in myotubes. Exposure of myotubes to light VLDL with high or low apoCIII content isolated from plasma of hypertriacylglycerolaemia or normolipidaemic individuals, respectively, showed that light VLDL with low levels of apoCIII did not cause the effects observed with VLDL with high apoCIII content (ESM Fig. 2a,b). Incubation of myotubes with apoCIII did not cause toxicity (ESM Fig. 2c) and led to a significant increase in phospho-ERK1/2 (Fig. 5a), TRB3, phospho-eIF2α and BiP protein levels (Fig. 5b), as well as secretion of IL-6 (ESM Fig. 2d), indicating that this apolipoprotein induces ER stress and inflammation. In contrast, apoCIII exposure reduced the expression of Pgc1α, Pparα and Pparβ/δ (Fig. 5c) and reduced the protein levels of PGC-1α and NRF1 (Fig. 5d). In agreement with this, apoCIII reduced the DNA-binding activity of PPARβ/δ (Fig. 5e). Moreover, the effects of apoCIII were concentration dependent (ESM Fig. 3). The induction of ER stress caused by apoCIII was accompanied by a reduction in the protein levels of IRβ and an increase in IRS-1 phosphorylated at Ser307 (Fig. 5f), whereas insulin-stimulated Akt phosphorylation was mitigated (Fig. 5g). No changes were observed in ER stress and inflammatory markers or in the protein levels of PGC-1α and phospho-ERK1/2 when cells were incubated with apoCI, indicating that the effects of apoCIII were specific (ESM Fig. 4a,b). In addition, VLDL and apoCIII reduced insulin-stimulated glucose uptake, whereas light VLDL low in apoCIII did not (ESM Fig. 4c). Moreover, apoCIII intensified the effects of the saturated fatty acid palmitate on the levels of ER stress markers, ERK1/2 phosphorylation, PGC-1α and insulin signalling pathway (ESM Fig. 4d), indicating that the increase in apoCIII might exacerbate the effects of lipids on insulin resistance.
Fig. 5.
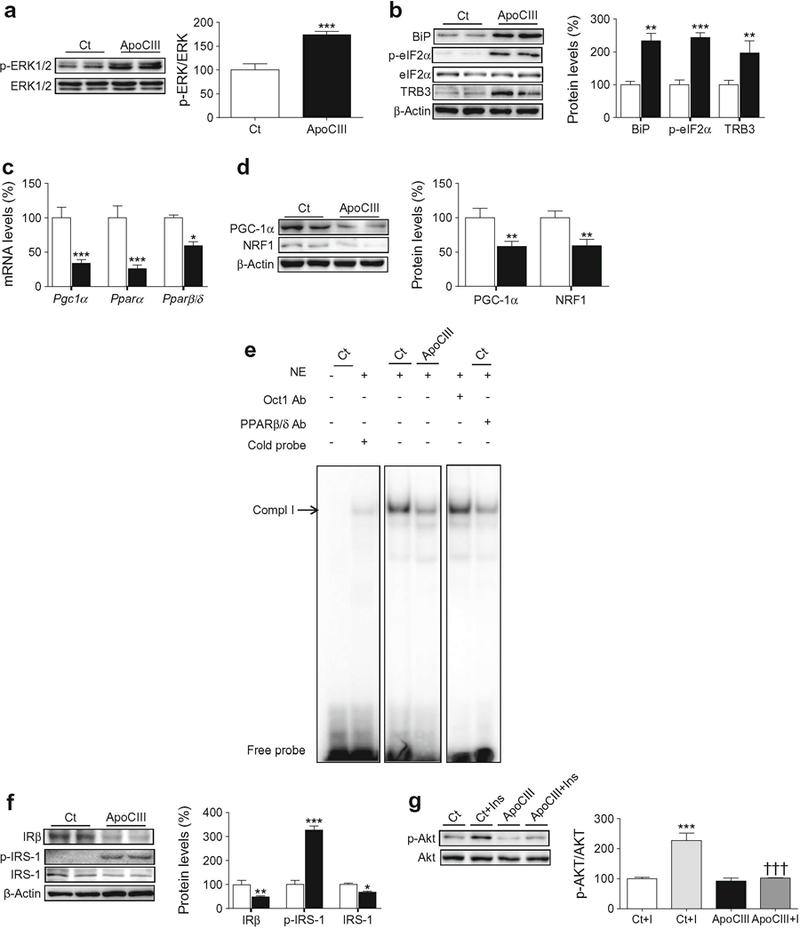
ApoCIII activates ERK1/2 and induces ER stress, inflammation and insulin resistance. C2C12 myotubes were incubated in the presence (black bars) or absence (control, Ct, white bars) of 100 μg/ml apoCIII for 24 h. (a) Phospho-ERK1/2 (Thr202/Tyr204) protein levels. (b) BiP, phospho-eIF2α (Ser51), TRB3 and β-actin protein levels. (c) mRNA abundance of Pgc1α, Pparα and Pparβ/δ. (d) PGC-1α, NRF1 and β-actin protein levels. (e) Autoradiograph of EMSA performed with a 32P-labelled PPAR nucleotide and crude nuclear protein extract (NE) from C2C12 myotubes. One main specific complex (Compl I) based on competition with a molar excess of unlabelled probe is shown. The supershift assay performed by incubating NE with an antibody (Ab) directed against PPARβ/δ shows a reduction in the band, whereas the band is unchanged by an unrelated antibody against Oct1. (f) IRβ, phospho-IRS-1 (Ser307) and β-actin protein levels. (g) Phosphorylated Akt (Ser473) protein levels. Where indicated, cells were incubated with 100 nmol/l insulin (Ins, I) for the last 10 min. The graphs show quantification expressed as a percentage of control. Data are means ± SD of five independent experiments and were compared by Student’s t test (a–f) or two-way ANOVA followed by Tukey post hoc test (g). *p < 0.05, **p < 0.01 and***p < 0.001 vs control; †††p < 0.001 vs control cells incubated with insulin
The increase in the expression of ER stress and inflammatory markers caused by apoCIII was blunted by co-incubation with U0126 (Fig. 6a). Likewise, U0126 prevented the increase in the protein levels of BiP, phospho-eIF2α and phospho-ERK1/2 caused by apoCIII (Fig. 6b). Inhibition of the MAPK–ERK1/2 pathway also prevented the reduction in IκBα and the increase in the DNA-binding activity of NF-κB (ESM Fig. 5a) and the increase in NRF2 and phospho-STAT3 (Tyr705) (Fig. 6c) observed in cells exposed only to apoCIII. ApoCIII also reduced the expression of Pgc1α, Pparα and Pparβ/δ and their target genes involved in FAO–changes that were abolished by U0126 (Fig. 6d). Additionally, the reduction in the protein levels of PGC-1α, phospho-AMPK and phospho-ACC was reversed by U0126 (Fig. 6e). siRNA knockdown of ERK1/2 confirmed that this kinase was responsible for the increase in ER stress and inflammation (Fig. 6f) and the reduction in FAO genes (Fig. 6g) caused by apoCIII.
Fig. 6.
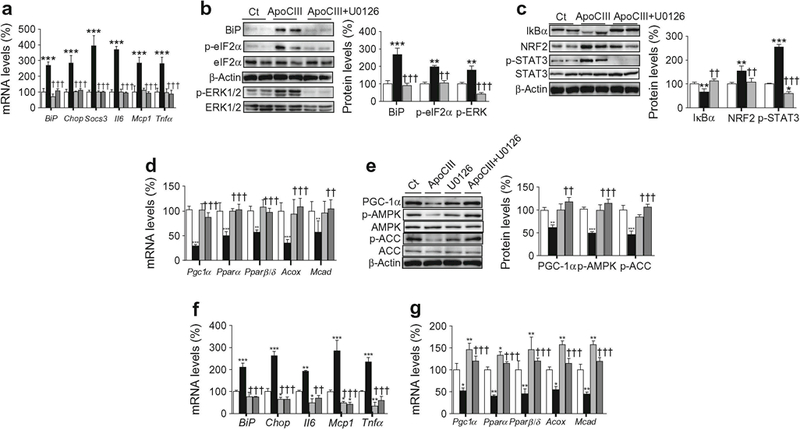
ERK1/2 inhibition prevents the effects of apoCIII on ER stress and inflammation. (a–e) C2C12 myotubes were incubated in the presence (black bars) or absence (control, Ct, white bars) of 100 μg/ml apoCIII for 24 h; 10 μmol/l U0126 was added to control myotubes (light grey bars) or apoCIII-treated myotubes (dark grey bars). (a) mRNA abundance of Bip, Chop, Socs3, Il6, Mcp1 and Tnfα. (b) BiP, phospho-eIF2α (Ser51), phospho-ERK1/2 (Thr202/Tyr204) and β-actin protein levels. (c) IκBα, NRF2, phospho-STAT3 (Tyr705) and β-actin protein levels. (d) mRNA abundance of Pgc1α, Pparα, Pparβ/δ, Acox and Mcad. (e) PGC-1α, phospho-AMPK (Thr172), phospho-ACC (Ser79) and β-actin protein levels. (f, g) C2C12 myotubes were transfected with control or ERK1/2 siRNA and incubated in the presence or absence of 100 μg/ml apoCIII for 24 h. The mRNA abundance of Bip, Chop, Il6, Mcp1 and Tnfα (f) and Pgc-1α, Pparα, Pparβ/δ, Acox and Mcad (g) was evaluated. White bars, control siRNA; light grey bars ERK1/2 siRNA; black bars, apoCIII+ control siRNA; dark grey bars, apoCIII+ERK1/2 siRNA The graphs show quantification expressed as a percentage of control. Data are means ± SD of five independent experiments and were compared by two-way ANOVA followed by Tukey post hoc test. *p < 0.05, **p < 0.01 and ***p < 0.001 vs control; ††p < 0.01 and †††p < 0.001 vs apoCIII-exposed cells
Next, we examined whether some of the changes caused by apoCIII in vitro were observed in skeletal muscle of transgenic mice with human apoCIII overexpression (apoCIII Tg) (Fig. 7a). These mice have marked elevations in plasma triacylglycerols but no impairment of glucose tolerance [31]. However, apoCIII Tg mice fed a high-fat diet show hepatic insulin resistance [7] and are more susceptible to development of diabetes [32]. In skeletal muscle of apoCIII Tg mice fed a standard diet, increased Chop, Il6 and Tnfα expression was detected when these mice were compared with non-transgenic littermates, whereas no changes were observed in Bip mRNA levels (Fig. 7b). Moreover, the marked increase in the protein levels of phospho-ERK1/2 in skeletal muscle from apoCIII Tg mice was accompanied by a reduction in PGC-1α protein levels (Fig. 7c, d).
Fig. 7.
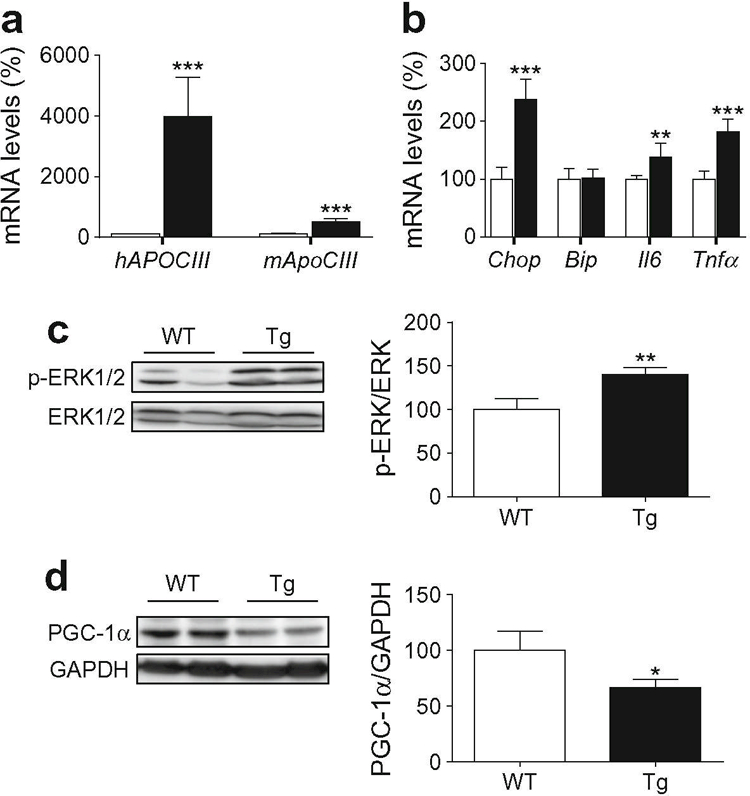
Skeletal muscle from apoCIII Tg mice shows increased levels of phospho-ERK1/2. Skeletal muscle from male non-transgenic (WT, white bars) and apoCIII Tg mice (Tg, black bars) was used. (a) mRNA abundance of human APOCIII and mouse ApoCIII (Apoc3). (b) mRNA abundance of Chop, Bip, Il6 and Tnfα. (c) Phospho-ERK1/2 (Thr202/Tyr204) protein level. (d) PGC-1α protein levels. The graphs show quantification expressed as a percentage of WT value. Data are means ± SD (n = 5 per group) and were compared by Student’s t test. *p < 0.05, **p < 0.01 and ***p < 0.001 vs WT mice
Since TLRs activate ERK1/2 and cause inflammation [28], we examined whether apoCIII acted through these receptors. We incubated mouse C2C12 myotubes exposed to apoCIII with a selective neutralising antibody against either TLR2 or IgG (ESM Fig. 5b). In the presence of this neutralising antibody, the increase in phospho-ERK1/2 levels caused by apoCIII alone was blunted (Fig. 8a). Consistent with a crucial role for ERK1/2 in the effects caused by apoCIII, the TLR2 neutralising antibody prevented the apoCIII-induced changes in the mRNA (Fig. 8b) and protein (Fig. 8c) levels of ER stress and inflammatory markers. Likewise, TLR2 neutralisation partially reversed the reduction in the protein levels of IRβ 8d) (Fig. 8d), blunted the increase in phospho-IRS-1 (Ser307) and prevented the reduction in PGC-1α and NRF1 (Fig. 8e). Blocking TLR2 also prevented the apoCIII-induced reduction in the expression of genes involved in FAO (Fig. 8f).
Fig. 8.
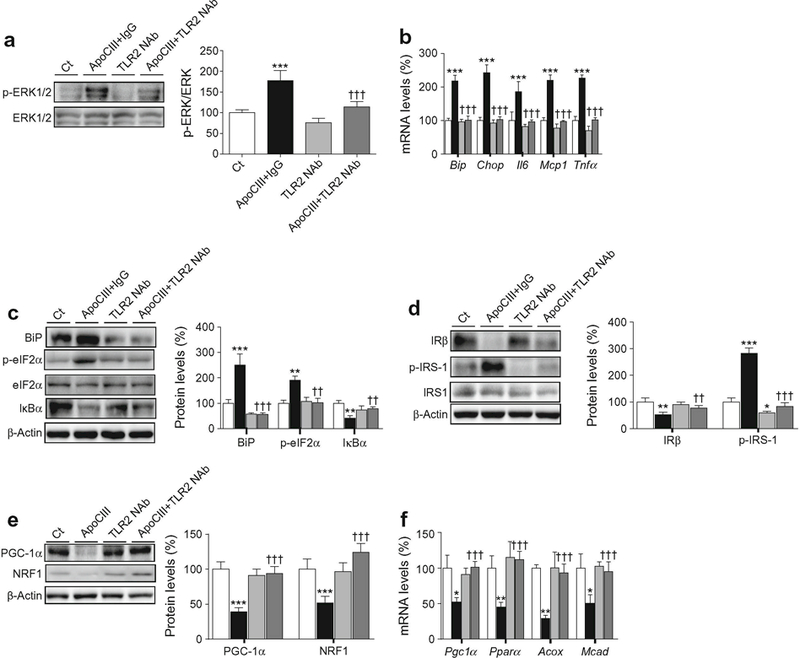
TLR2 mediates the effects of apoCIII on ERK1/2, ER stress and inflammation. Mouse C2C12 myotubes were incubated in the presence or absence (control, Ct, white bars) of 100 μg/ml apoCIII for 24 h; 50 μg/ml of IgG was added to apoCIII-treated myotubes (black bars) or 50 μg/ml of the neutralising antibody against TLR2 (TLR2NAb) was added to the control (light grey bars) or apoCIII-treated (dark grey bars) myotubes. (a) Phospho-ERK1/2 (Thr202/Tyr204) protein levels. (b) mRNA abundance of Bip, Chop, Il6, Mcp1 and Tnfα. (c) BiP, phospho-eIF2α (Ser51), IκBα, and β-actin protein levels. (d) IRβ, phospho-IRS-1 (Ser307) and β-actin protein levels. (e) PGC-1α, NRF1 and β-actin protein levels. (f) mRNA abundance of Pgc1α, Pparα, Acox and Mcad mRNA. The graphs show quantification expressed as a percentage of control. Data are means ± SD of five independent experiments and were compared by two-way ANOVA followed by Tukey post hoc test. *p < 0.05, **p < 0.01 and ***p < 0.001 vs control; ††p < 0.01 and †††p < 0.001 vs apoCIII-exposed cells
Discussion
Although it is well established that insulin resistance drives atherogenic dyslipidaemia, there is little evidence on whether the increase in VLDL particles associated with insulin-resistant states exacerbates the insulin resistance. Our findings demonstrate that exposure of myotubes and isolated skeletal muscle to VLDL increases the levels of ER stress and inflammatory markers and attenuates the insulin signalling pathway. These data indicate that increased levels of VLDL particles may contribute towards exacerbation of insulin resistance. Our findings also demonstrate that apoCIII may be the VLDL component responsible for the changes caused by VLDL exposure. This is interesting, since apoCIII expression is increased by insulin deficiency, insulin resistance [33, 34] and hyperglycaemia [35], converting apoCIII into the most abundant VLDL apolipoprotein in individuals with diabetes [3], suggesting that the increase in apoCIII levels in diabetic states may contribute to exacerbation of these conditions. In this regard, it is interesting to note that humans with a mutation in the APOCIII gene (also known as APOC3) that results in a reduction in the half-life of apoCIII, show a favourable lipoprotein pattern, increased insulin sensitivity and longevity and protection against cardiovascular diseases [36, 37]. Recent evidence seems to confirm that apoCIII plays a key role in diabetes. Thus, decreasing apoCIII in mice results in improved glucose tolerance [38]. In agreement with this, antisense-mediated lowering of plasma apoCIII improves dyslipidaemia and insulin sensitivity in humans with type 2 diabetes [39] and a null mutation in human APOCIII confers a favourable plasma lipid profile, although it does not improve insulin sensitivity [8].
The mechanism by which VLDL and apoCIII increase ER stress and inflammation and attenuate insulin signalling in myotubes seems to involve ERK1/2 activation. This kinase has been implicated in the development of insulin resistance associated with obesity and type 2 diabetes [40]. In fact, Erk1−/− mice (also known as Mapk3−/− mice) challenged with a high-fat diet are resistant to obesity and are protected from insulin resistance [41]. In addition, hyperinsulinaemic–euglycaemic clamp studies have demonstrated an increase in whole-body insulin sensitivity in ob/ob-Erk1−/− mice associated with an increase in both insulin-stimulated glucose disposal in skeletal muscles and adipose tissue insulin sensitivity [42].
In the present study, apoCIII-induced ERK1/2 activation was accompanied by a reduction in AMPK activity. An inhibitory crosstalk exists between AMPK and ERK1/2 and activation of ERK1/2 inhibits AMPK and promotes ER stress-induced insulin resistance in skeletal muscle cells [14, 29]. Hence, VLDL and apoCIII-induced ER stress might be a result of the reduction in AMPK activity. In fact, AMPK activation inhibits ER stress [14, 43], whereas the reduction in its activity promotes ER stress [44]. Moreover, VLDL- and apoCIII-induced ER stress ultimately results in activation of the IKKβ–NF-κΒ pathway, which attenuates the insulin signalling pathway by phosphorylating IRS-1 in serine residues and increases the transcription of inflammatory genes. In agreement with this, we found that ERK1/2 inhibition or knockdown prevented the changes in ER stress and inflammation and the attenuation of the insulin signalling pathway caused by VLDL. Moreover, ERK1/2 inhibition prevented the reduction in AMPK caused by apoCIII, confirming the negative crosstalk between ERK1/2 and AMPK.
Similarly, the reduction in AMPK caused by apoCIII-induced ERK1/2 activation may contribute to reduced PGC-1α levels, since PGC-1α is an important mediator of AMPK-induced gene expression and AMPK activation regulates PGC-1α transcription [45]. Given the key role of PGC-1α in regulating the activity of transcription factors involved in FAO, such as PPARs [22], the reduction in PGC-1α following treatment with VLDL or apoCIII leads to a decrease in the expression of genes involved in FAO, suggesting that it can promote the deleterious effects of saturated fatty acids [11].
VLDLs also bind to the VLDL receptor, which is a determinant factor in adipose tissue inflammation and adipocyte macrophage infiltration when stimulated with VLDL from hyperlipidaemic mice [13]. Although we cannot discount a role for this receptor, the fact that the effects of VLDL from normolipidaemic individuals are mimicked by apoCIII seems to suggest that most of the effects of these lipoproteins are caused by the presence of apoCIII in these particles.
Interestingly, our findings indicate that the effects of apoCIII are mediated by TLR2. TLR2 not only recognises numerous lipid-containing molecules but also it recognises endogenous proteins [46]. It is expressed in skeletal muscle cells and is involved in fatty acid-induced insulin resistance [47]. Moreover, activation of the TLR2 pathway ultimately leads to NF-κB and ERK1/2 activation [48]. Likewise, TLR2 deficiency improves insulin sensitivity and attenuates cytokine expression [49]. Our findings confirm the importance of TLR2 in insulin resistance and indicate that its activation by VLDL and apoCIII induces ER stress, inflammation and insulin resistance.
In conclusion, our findings show that VLDL- and apoCIII-induced TLR2 activation results in ER stress, in-flammation and insulin resistance by activating ERK1/2 in skeletal muscle cells. These results imply that elevated VLDL in diabetic states can contribute to the exacerbation of insulin resistance.
Supplementary Material
Acknowledgements
We thank the University of Barcelona’s Language Advisory Service for revising the manuscript.
Funding This study was partly supported by funds from the Spanish Ministerio de Economía y Competitividad (SAF2012–30708 and SAF2015–64146-R to MVC), the Generalitat de Catalunya (2014SGR-0013 to MVC), NIH NIDDK (DK101663 to ABK), USDA NIFA (11874590 to ABK) and USDA NIFA Hatch Formula Funds (2015–31200-06009 to ABK), an Instituto de Salud Carlos III grant (PI16–00139 to JCE-G) and European Union ERDF funds. CIBER de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM) is an Instituto de Salud Carlos III project (Grant CB07/08/0003 to MVC). GB was supported by an FPI grant from the Spanish Ministerio de Economía y Competitividad.
Abbreviations
- ACC
Acetyl-CoA carboxylase
- AMPK
AMP-activated protein kinase
- Apo
Apolipoprotein
- apoCIII Tg
Transgenic mice overexpressing human apoCIII
- BiP
Binding immunoglobulin protein
- CPT-1
Carnitine palmitoyltransferase 1
- CHOP
CCAAT-enhancer-binding protein homologous protein
- eIF2
Εukaryotic initiation factor 2α
- EMSA
Electrophoretic mobility shift assay
- ER
Endoplasmic reticulum
- ERK
Extracellular signal-regulated kinase
- FAO
Fatty acid oxidation
- GRP78
Glucose-regulated protein 78
- IκB
Inhibitor of κB
- IKK
IκΒ kinase β
- IR
Insulin receptor β-subunit
- IRE-1
Inositol-requiring 1 transmembrane kinase/endonuclease-1α
- MAPK
Mitogen-activated protein kinase
- MCAD
Medium chain acyl-CoA dehydrogenase
- MCP-1
Monocyte chemoattractant protein 1
- MEK
MAPK–ERK
- NRF1
Nuclear respiratory factor 1
- NRF2
Nuclear factor-E2-related factor 2
- OXPHOS
Oxidative phosphorylation
- PERK
Eukaryotic translation initiation factor-2α kinase 3
- PGC-1
Peroxisome proliferator-activated receptor γ coactivator 1α
- PPAR
Peroxisome proliferator-activated receptor
- SOCS
Suppressor of cytokine signalling 3
- STAT3
Signal transducer and activator of transcription 3
- TLR
Toll-like receptor
- TRB3
Tribbles 3
- UPR
Unfolded protein response
- XBP1
X-box binding protein-1
Footnotes
Data availability Data are available on request from the authors.
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
References
- 1.Xiao C, Dash S, Morgantini C, Hegele RA, Lewis GF (2016) Pharmacological targeting of the atherogenic dyslipidemia complex: the next frontier in CVD prevention beyond lowering LDL cholesterol. Diabetes 65:1767–1778 [DOI] [PubMed] [Google Scholar]
- 2.Adiels M, Olofsson SO, Taskinen MR, Borén J (2008) Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol 28:1225–1236 [DOI] [PubMed] [Google Scholar]
- 3.Hiukka A, Fruchart-Najib J, Leinonen E, Hilden H, Fruchart JC, Taskinen MR (2005) Alterations of lipids and apolipoprotein CIII in very low density lipoprotein subspecies in type 2 diabetes. Diabetologia 48:1207–1215 [DOI] [PubMed] [Google Scholar]
- 4.Campos H, Perlov D, Khoo C, Sacks FM (2001) Distinct patterns of lipoproteins with apoB defined by presence of apoE or apoC-III in hypercholesterolemia and hypertriglyceridemia. J Lipid Res 42: 1239–1249 [PubMed] [Google Scholar]
- 5.Aalto-Setälä K, Fisher EA, Chen X et al. (1992) Mechanism of hypertriglyceridemia in human apolipoprotein (apo) CIII transgenic mice. Diminished very low density lipoprotein fractional catabolic rate associated with increased apo CIII and reduced apo E on the particles. J Clin Invest 90:1889–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawakami A, Aikawa M, Alcaide P, Luscinskas FW, Libby P, Sacks FM (2006) Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation 114:681–687 [DOI] [PubMed] [Google Scholar]
- 7.Lee HY, Birkenfeld AL, Jornayvaz FR et al. (2011) Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatology 54:1650–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pollin TI, Damcott CM, Shen H et al. (2008) A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 332:1702–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeFronzo RA, Gunnarsson R, Björkman O, Olsson M, Wahren J (1985) Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J Clin Invest 76:149–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdul-Ghani MA, DeFronzo RA (2010) Pathogenesis of insulin resistance in skeletal muscle. J Biomed Biotechnol 2010:476279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schenk S, Saberi M, Olefsky JM (2008) Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest 118:2992–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salvadó L, Palomer X, Barroso E, Vázquez-Carrera M (2015) Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol Metab 26:438–448 [DOI] [PubMed] [Google Scholar]
- 13.Könner AC, Brüning JC (2011) Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol Metab 22:16–23 [DOI] [PubMed] [Google Scholar]
- 14.Nguyen A, Tao H, Metrione M, Hajri T (2014) Very low density lipoprotein receptor (VLDLR) expression is a determinant factor in adipose tissue inflammation and adipocyte-macrophage interaction. J Biol Chem 289:1688–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salvadó L, Barroso E, Gómez-Foix AM et al. (2014) PPARβ/δ prevents endoplasmic reticulum stress-associated inflammation and insulin resistance in skeletal muscle cells through an AMPK-dependent mechanism. Diabetologia 57:2126–2135 [DOI] [PubMed] [Google Scholar]
- 16.Alkhateeb H, Chabowski A, Bonen A (2006) Viability of the isolated soleus muscle during long-term incubation. Appl Physiol Nutr Metab 31:467–476 [DOI] [PubMed] [Google Scholar]
- 17.Koh HJ, Toyoda T, Didesch MM et al. (2013) Tribbles 3 mediates endoplasmic reticulum stress-induced insulin resistance in skeletal muscle. Nat Commun 4:1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howard JK, Flier JS (2006) Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab 17:365–371 [DOI] [PubMed] [Google Scholar]
- 19.Handschin C, Spiegelman BM (2006) Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev 27:728–735 [DOI] [PubMed] [Google Scholar]
- 20.Patti ME, Butte AJ, Crunkhorn S et al. (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A 100:8466–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mootha VK, Lindgren CM, Eriksson KF et al. (2003) PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34:267–273 [DOI] [PubMed] [Google Scholar]
- 22.Miura S, Kai Y, Ono M, Ezaki O (2003) Overexpression of peroxisome proliferator-activated receptor γ coactivator-1α down-regulates GLUT4 mRNA in skeletal muscles. J Biol Chem 278:31385–31390 [DOI] [PubMed] [Google Scholar]
- 23.Vega RB, Huss JM, Kelly DP (2000) The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol 20:1868–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Z, Puigserver P, Andersson U et al. (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98:115–124 [DOI] [PubMed] [Google Scholar]
- 25.Zhang BB, Zhou G, Li C (2009) AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab 9:407–416 [DOI] [PubMed] [Google Scholar]
- 26.Zhou L, Zhang J, Fang Q et al. (2009) Autophagy-mediated insulin receptor down-regulation contributes to endoplasmic reticulum stress-induced insulin resistance. Mol Pharmacol 76:596–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT (2009) Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci U S A 106:20405–20410 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Chung S, Lapoint K, Martinez K et al. (2006) Preadipocytes mediate lipopolysaccharide-induced inflammation and insulin resistance in primary cultures of newly differentiated human adipocytes. Endocrinology 147:5340–5351 [DOI] [PubMed] [Google Scholar]
- 29.Coll T, Jové M, Rodríguez-Calvo R et al. (2006) Palmitate-mediated downregulation of peroxisome proliferator-activated receptor-gamma coactivator 1α in skeletal muscle cells involves MEK1/2 and nuclear factor-κB activation. Diabetes 55:2779–2787 [DOI] [PubMed] [Google Scholar]
- 30.Hwang SL, Jeong YT, Li X et al. (2013) Inhibitory cross-talk between the AMPK and ERK pathways mediates endoplasmic reticulum stress-induced insulin resistance in skeletal muscle. Br J Pharmacol 169:69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reaven GM, Mondon CE, Chen YD, Breslow JL (1994) Hypertriglyceridemic mice transgenic for the human apolipoprotein C-III gene are neither insulin resistant nor hyperinsulinemic. J Lipid Res 35:820–824 [PubMed] [Google Scholar]
- 32.Salerno AG, Silva TR, Amaral ME et al. (2007) Overexpression of apolipoprotein CIII increases and CETP reverses diet-induced obesity in transgenic mice. Int J Obes 31:1586–1595 [DOI] [PubMed] [Google Scholar]
- 33.Chen M, Breslow JL, Li W, Leff T (1994) Transcriptional regulation of the apoC-III gene by insulin in diabetic mice: correlation with changes in plasma triglyceride levels. J Lipid Res 35:1918– 1924 [PubMed] [Google Scholar]
- 34.Altomonte J, Cong L, Harbaran S et al. (2004) Foxo1 mediates insulin action on apoC-III and triglyceride metabolism. J Clin Invest 114:1493–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caron S, Verrijken A, Mertens I et al. (2011) Transcriptional activation of apolipoprotein CIII expression by glucose may contribute to diabetic dyslipidemia. Arterioscler Thromb Vasc Biol 31:513–519 [DOI] [PubMed] [Google Scholar]
- 36.Atzmon G, Rincon M, Schechter CB et al. (2006) Lipoprotein genotype and conserved pathway for exceptional longevity in humans. PLoS Biol 4:e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A (2014) Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 371:32–41 [DOI] [PubMed] [Google Scholar]
- 38.Åvall K, Ali Y, Leibiger IB et al. (2015) Apolipoprotein CIII links islet insulin resistance to β-cell failure in diabetes. Proc Natl Acad Sci U S A A112:E2611–E2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Digenio A, Dunbar RL, Alexander VJ et al. (2016) Antisense-mediated lowering of plasma apolipoprotein C-III by volanesorsen improves dyslipidemia and insulin sensitivity in type 2 diabetes. Diabetes Care 39:1408–1415 [DOI] [PubMed] [Google Scholar]
- 40.Ozaki KI, Awazu M, Tamiya M et al. (2016) Targeting the ERK signaling pathway as a potential treatment for insulin resistance and type 2 diabetes. Am J Physiol Endocrinol Metab 310:E643–E651 [DOI] [PubMed] [Google Scholar]
- 41.Bost F, Aouadi M, Caron L et al. (2005) The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes 54:402–411 [DOI] [PubMed] [Google Scholar]
- 42.Jager J, Corcelle V, Grémeaux T et al. (2011) Deficiency in the extracellular signal-regulated kinase 1 (ERK1) protects leptin-deficient mice from insulin resistance without affecting obesity. Diabetologia 54:180–189 [DOI] [PubMed] [Google Scholar]
- 43.Dong Y, Zhang M, Wang S et al. (2010) Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes 59:1386–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dong Y, Zhang M, Liang B et al. (2010) Reduction of AMP-activated protein kinase α2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 121:792–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cantó C, Auwerx J (2009) PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol 20:98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rubartelli A, Lotze MT (2007) Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol 28:429–436 [DOI] [PubMed] [Google Scholar]
- 47.Senn JJ (2006) Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J Biol Chem 281:26865–26875 [DOI] [PubMed] [Google Scholar]
- 48.Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K (1999) The kinase TAK1 can activate the NIK-IκB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 398:252–256 [DOI] [PubMed] [Google Scholar]
- 49.Kuo LH, Tsai PJ, Jiang MJ et al. (2011) Toll-like receptor 2 deficiency improves insulin sensitivity and hepatic insulin signalling in the mouse. Diabetologia 54:168–179 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.