

Abstract
Background
Young children with medulloblastoma have inferior survival to older children. While the principal difference is due to radiation-sparing therapy, radiation is not an absolute requisite for survival of all. We aimed to estimate the event-free survival of young medulloblastoma patients with a robust sample size using different treatment strategies designed to defer, reduce, or delay radiation exposure. These strategies were deemed reasonable at the start of the trial by physicians and ethics review board. Additionally, we aimed to define the molecular characteristics associated with progression-free survival.
Methods
In this multicenter phase II trial we enrolled children younger than 3-years-old with newly diagnosed medulloblastoma, supratentorial primitive neuroectodermal tumor, pineoblastoma, atypical teratoid/rhabdoid tumor, high grade glioma, choroid plexus carcinoma, or ependymoma. Children ≥ 3 and < 5-years-old with newly diagnosed non-metastatic medulloblastoma without any high-risk features were also eligible. The study enrolled from 6 participating institutions. Patients were required to start therapy within 31 days from definitive surgery. Eligible patients had to have a Lansky performance score ≥ 30 (except for posterior fossa syndrome) and adequate hematopoietic, renal, and hepatic functions. Patients were ineligible if they had received any prior radiotherapy or chemotherapy. Patients were postoperatively stratified by clinical and histologic criteria into low-, intermediate-, and high-risk groups. All participants received 4 identical cycles of induction chemotherapy with high-risk patients also receiving vinblastine between induction cycles. Induction was followed by risk-adapted consolidation therapy: low-risk patients received a cyclophosphamide (1,500 mg/m2 IV day 1), etoposide (100mg/m2 IV day 1,2), carboplatin (AUC 5mg/mL/min IV day 2) regimen; intermediate-risk patients received focal radiation therapy (54 Gy with a clinical target volume of 5mm) to the tumor bed; and high-risk patients received chemotherapy with targeted IV topotecan (AUC 120-160 ng-hr/mL IV day 1-5) and cyclophosphamide (600mg/m2 IV day 1-5) or optional craniospinal radiation (23.4-39.6 Gy) for those who reach 3-years of age by the end of induction. After consolidation, all patients were to receive 6 cycles of oral maintenance chemotherapy with cyclophosphamide, topotecan and erlotinib. Per intent to treat all patients who received at least one dose of the first induction chemotherapy (methotrexate) were included in the analysis. Archived tumor specimens were sub-classified into molecular subdivisions based on DNA methylation profiles and overlaid with mutations and copy-number alterations. The co-primary endpoints for the study were (1) therapeutic: to estimate the event-free survival distribution of young medulloblastoma patients treated with risk-adapted therapy, and (2) biologic: to identify patterns of methylation profiling that are associated with progression-free survival among young patients with medulloblastoma treated with risk-adapted therapy. Medulloblastoma accrual drove the primary endpoints of the study it is the only entity discussed here. This trial is registered at ClinicalTrials.gov, number NCT00602667. The trial was permanently closed to accrual in April 2017.
Findings
Between November 27, 2007 and April 19, 2017, we enrolled 81 patients histopathologically diagnosed with medulloblastoma. All 81 started treatment and were included in the analysis. Accrual to the low-risk arm was suspended December 2, 2015 when the 1-year event-free survival was estimated to be 78.3%; a value below the 80% stopping rule boundary. The trial was closed to accrual once the specified accrual duration was reached. After a median follow up of 5.5 years (IQR 2.7-7.3) the 5-year event-free survival of the low-risk cohort was 55.3% (95% CI 33.3-77.3%); intermediate-risk was 24.6% (95% CI 3.6-45.6%); and high-risk was 16.7% (95% CI 3.4-30.0%) (p=0.0021). Survival by methylation subgroup showed 5-year progression-free survival of sonic hedgehog (SHH) was 51.1% (95% CI 34.6-67.6%) vs 8.3% (95% CI 0.0-24.0%) for group 3 (G3) (p<0.0001) vs 13.3% (95% CI 0.0-37.6%) for group 4 (G4) (p=0.095). The 5-year progression-free survival of two distinct methylation profiling patterns of SHH, medulloblastoma named subtype iSHH-I and iSHH-II, were 27.8% (95% CI 9.0-46.6%) and 75.4% (95% CI 55.0-95.8%), respectively (HR 4.17, 95% CI 1.51-11.53, p=0.0028). The 5-year progression-free survival of low-risk iSHH-II was 90.9% (95% CI 73.1-100.0%) vs 22.2% (95% CI 0.1-44.3%) for iSHH-I (HR 14.75, 95% CI 1.84-118.04, p=0.0007). All three risk-adapted therapies were well tolerated and no protocol-related deaths were observed. The most common adverse events were expected grade 3-4 toxicities including febrile neutropenia (48 patients [59%]), neutropenia (21 [26%]), infection with neutropenia (20 [25%]), leukopenia (15 [19%]), vomiting (15 [19%]) and anorexia (13 [16%]).
Interpretation
The risk-adapted approach failed to identify an acceptable benefit despite being well-tolerated. However, through the prespecified application of integrative genomics to this clinical trial cohort, we identified a good-responder SHH-subtype (iSHH-II) that exhibits excellent progression-free survival in the absence of radiation, intra-ventricular chemotherapy, or high-dose chemotherapy; a poor-responder SHH-subtype (iSHH-I); and very poor progression-free survival for G3/G4 medulloblastoma. These findings support the development of a molecularly-driven risk-adapted approach that will shape risk stratification and therapy on future medulloblastoma trials for young children.
Funding
American Lebanese Syrian Associated Charities, St Jude Children’s Research Hospital, NCI Cancer Center Grant (P30CA021765), Alexander and Margaret Stewart Trust, Sontag Foundation, and American Association for Cancer Research.
Introduction
Medulloblastoma is a malignant pediatric brain tumor that represents a leading cause of non-accidental death in children and adolescents.(1) Conventional treatment for medulloblastoma involves a combination of surgery, craniospinal irradiation and cytotoxic chemotherapy, curing 60-80% of patients often at the expense of debilitating side effects of therapy.(1–4) The severity of long-term neurocognitive consequences increases with dose of craniospinal irradiation and is inversely associated with the age at time of craniospinal irradiation. For children younger than 3-years at diagnosis (often called “infants”) this toxicity has come to be judged as unacceptable and front-line chemotherapy-based regimens have been prioritized over radiation. Despite limited success, the population of children treated with front-line chemotherapy-based regimens has been expanding up to 6-, and even 10-, years of age at diagnosis on some trials. The intent to decrease the risk of severe neurocognitive consequences that does not disappear in patients diagnosed after 3-years of age is laudable however survival consistently remains inferior to that of children treated with conventional radiation-centered therapies. Progression-free survival at 5-years of early childhood medulloblastoma from the most recently published trials that have employed radiation-sparing strategies range from 30-60% as compared to 70% in older children.(5–10) For those spared the debilitating neurologic and endocrine toxicities of craniospinal irradiation, the benefits are enormous,(11, 12) but this approach leaves about half of the population requiring salvage therapy or with terminal disease. Salvage therapy, which often includes craniospinal irradiation, is not always successful and, even when successful, may result in increased treatment-related morbidities from the successive therapies. Hence, improved methods to risk-stratify patients are needed to guide treatment decisions.
Evidence that the early childhood medulloblastoma population can be risk-stratified comes from the observation that different histopathological variants of medulloblastoma exhibit different outcomes. Categorically, all recently reported trials confirm better survival for patients with desmoplastic/nodular (DN) or closely related medulloblastoma with extensive nodularity (MBEN) tumors than for patients with tumors of other histologies [i.e. classic (CMB), large-cell/anaplastic (LCA)].(5, 8, 10, 11) The best reported 5-year progression-free survival was 90±7% for non-metastatic DN/MBEN patients on the German HITT 2000 trial that combined intra-ventricular and high-dose IV methotrexate with conventional chemotherapy.(13) Another trial, CCG-99703, reported a highly regarded 5-year progression-free survival of 78.6 ±11% for DN/MBEN patients administered conventional chemotherapy followed by repeated cycles of myeloablative chemotherapy.(10) However, risk of central nervous system (CNS) infection and leuko-encephalopathy with intraventricular therapy(9) and the toxic mortality rate of at least 2.6% with myeloablative regimens,(10) have led to attempts to eliminate the intra-ventricular requirement and/or reduce the intensity.
The successful treatment of some medulloblastomas with chemotherapy alone coupled with the desire to reduce intra-ventricular toxicity motivated the clinical trial designated, St. Jude Risk Adapted Therapy For Young Children with Embryonal Brain Tumors, High Grade Gliomas and Choroid Plexus Carcinoma or Ependymoma (SJYC07; NCT00602667). In this multicenter phase II trial, infants with medulloblastoma were postoperatively risk-stratified by clinical and histologic criteria into three groups: low, intermediate, and high (figure 1). In addition children ≥ 3 and < 5-years-old with newly diagnosed non-metastatic medulloblastoma without any high-risk features were also eligible for the intermediate risk group. The working hypothesis maintained that progression-free survival of young children with medulloblastoma could be improved with risk-adapted therapy. The primary therapeutic objective was to estimate the event-free survival in this population treated with risk-adapted therapy. All groups received induction chemotherapy that combined high-dose IV methotrexate with conventional chemotherapy followed by a consolidation phase unique to the risk-group. Low risk consolidation consisted of a familiar and well-tolerated carboplatin-based regimen.(14) Intermediate-risk patients received focal radiation therapy aimed at the tumor bed based on encouraging trial results for patients with localized disease.(7) High-risk patients were given a pharmacokinetically-targeted topotecan and cyclophosphamide regimen based on studies that suggested promising anti-medulloblastoma activity.(15) After consolidation, all risk groups received identical maintenance chemotherapy that consisted of alternating cycles of oral low-dose (metronomic) cyclophosphamide and topotecan with erlotinib to target tumor angiogenesis.(16, 17) None of the patients were prescribed intra-ventricular methotrexate or myeloablative regimens.
Figure 1. Trial profile.

*Enrolled on trial, to be reported on separately.
In addition, mindful of the emerging evidence that linked biologic heterogeneity with distinct clinical outcomes,(18, 19) another primary objective was to identify molecular patterns associated with progression-free survival in this cohort. Indeed, it is now widely accepted that medulloblastoma is a heterogeneous entity composed of at least four biologically and clinically distinct consensus subgroups: WNT, SHH, G3, and G4.(19) Moreover, a recent series of publications have reported on molecular heterogeneity within the consensus subgroups.(20–22) While the infant and young child medulloblastoma population has been included and explored as a subset of larger cohorts, it has never been systematically and rigorously explored on its own. Nor has molecular classification been applied to a uniformly treated clinical trial cohort of early childhood medulloblastoma and outcomes measured in the context of medulloblastoma subgroups and their associated subtypes.
Herein, we report on the primary outcomes of medulloblastoma patients treated on the SJYC07 trial and contextualize results with multidimensional molecular datasets, including a combined early childhood medulloblastoma series consisting of a large international cohort(20) and SJYC07 trial cases. These data de-clutter a complex entity and provide means for improved risk-stratification that can serve as a guideline for the next generation of early childhood medulloblastoma trials.
Methods
Study design and Participants
SJYC07 was a phase II study designed to evaluate risk-adapted multi-modality therapies in young children with brain tumors. The original design of this study was based on the St Jude historical cohort of 35 infant (0-3 years) medulloblastoma patients treated on various studies. Given the treatment heterogeneity as well as the small sample size in the historical cohort, SJYC07 aimed to estimate the outcome associated with the proposed treatment rather than to conduct a formal comparison. Using an ad hoc approach, the originally planned sample size was 140 infant medulloblastoma patients to be enrolled over 7 years with an estimated 73 events which was thought to be adequate to estimate the overall event-free survival curve as well as to conduct outcome associations with genomic subgroups. Following an amendment approved on September 17th 2010, eligibility was expanded to patients 3-5 years of age without clinically high-risk features to be treated on the intermediate risk arm where the intent was to estimate event-free survival separately in this cohort. On May 20th 2014, however, due to slow accrual as well as the emerging information regarding infant medulloblastoma outcomes from published studies, the study design was revised via an amendment to reduce the target sample size to 90 medulloblastoma subjects and to limit the time of enrollment to three additional years. Furthermore the design was revised to estimate and monitor outcome in each risk group separately. With the same amendment, 3-5 year-old subjects were combined with the <3 year old intermediate risk subjects since they received the same treatment. The following futility thresholds were introduced: 1-year event-free survival <80% in the low-risk cohort and 2-year event-free survival <20% in intermediate and high-risk cohorts. The monitoring was to be done periodically by comparing the observed event-free survival to these thresholds after the initial 10 patients in the low-risk group and the initial 15 patients in each of the intermediate and high risk groups reached the 1- and 2-year time point, respectively. Despite the drastic reduction in the sample size, the biologic primary objective was still deemed feasible since technology evolved, fresh-frozen tissue was no longer required, and >95% of patients had available FFPE tissue compared to initially estimated 50% with frozen tissue.
The study enrolled from 6 participating institutions (appendix p 3). Eligible participants were children younger than 3-years-old with newly diagnosed medulloblastoma, supratentorial primitive neuroectodermal tumor, pineoblastoma, atypical teratoid/rhabdoid tumor, high grade glioma, choroid plexus carcinoma, or ependymoma. Children ≥ 3 and < 5-years-old with newly diagnosed non-metastatic medulloblastoma without any high-risk features (defined below) were also eligible. All tumor samples were histologically confirmed and centrally reviewed by pathology prior to enrollment. Central neuropathology review was performed using standard histological preparations, established immunohistochemistry and interphase fluorescence in situ hybridization. MRI of brain with and without contrast and MRI spine with contrast and CSF cytology from lumbar spinal fluid (unless medically contraindicated) was required for disease evaluation. Patients were required to enroll and start therapy within 31 days from definitive surgery. Adequate hematopoietic (white blood cell [WBC] > 2,000/mm3, platelets > 50,000/mm3, hemoglobin > 8 g/dL, absolute neutrophil count [ANC] > 500/mm3), renal (serum creatinine < 3 times upper limit of normal [ULN]), and hepatic (alanine aminotransferase [ALT] < 5 times ULN, total bilirubin < 3 times ULN) functions and a Lansky performance status ≥ 30 (except for posterior fossa syndrome) were required. No prior radiotherapy or chemotherapy other than corticosteroids was allowed. No indication of estimated life expectancy of eligible patients was prespecified by the protocol.
Medulloblastoma accrual drove the primary endpoints of the study and it is the only entity discussed herein. Non-medulloblastoma entities are to be reported separately. Medulloblastomas were risk-stratified by clinical and histologic criteria into three treatment groups (figure 1).
Low-risk: < 3 years at time of diagnosis, no evidence of metastatic disease (M0), a surgical gross total resection (GTR) or near total resection (NTR) - defined as residual tumor or imaging abnormality [not definite for residual tumor] with a size of < 1 cm2 on postoperative imaging (R0), and DN/MBEN histology.
Intermediate-risk: < 3 years, M0, R0, with any histology other than DN/MBEN (i.e. CMB or LCA); OR < 3 years, M0, a subtotal resection (STR) – defined as residual tumor or imaging abnormality with a size of > 1 cm2 on postoperative imaging(R+), with DN/MBEN histology; OR patients 3-5 years, M0, R0, CMB or DN/MBEN histology without MYC or MYCN amplification.
High-risk: < 3 years old with evidence of metastatic disease (M+).
Patients aged 3-5 years with M+ disease, MYC or MYCN amplification, or LCA histology were not eligible and treatment on separate protocols with craniospinal irradiation was generally recommended.
The protocol was approved by the St Jude Children’s Research Hospital Institutional Review Board (IRB) and by local IRBs at participating institutions. The study was conducted in accordance with good clinical practice. Written informed consent was provided in all cases by the participant’s legal guardian.
A post-hoc analysis was performed using data sourced from 131 patients < 6-years old sourced from a published international cohort(20) added to 59 molecularly characterized SJYC07 samples. Histology and treatment information for the 131 patients were not collected.
Procedures
Treatment, for all risk groups (figure 1), consisted of 4 identical 28-day cycles of induction chemotherapy (16 weeks) which included high-dose (5 g/m2 or 2.5 g/m2 for patients ≤ 31-days-old at enrollment) intravenous (IV) methotrexate on day 1;, vincristine 1mg/m2 IV on day 8 and 15; cisplatin 75 mg/m2 IV on day 8; and cyclophosphamide 1.5 g/m2 IV on day 9. High-risk patients also received vinblastine 1 mg/m2 IV on day 17, 19, 22, 24, and 26 of each cycle.
Induction was followed by a consolidation phase unique to the risk groups. Low-risk patients received conventional doses of carboplatin, cyclophosphamide, and etoposide × 2 cycles (8 weeks). Intermediate-risk patients ≥ 12-months upon completion of induction received focal radiotherapy (RT) to the tumor bed (primary site dose of 54 Gy with a clinical target volume of 5mm over 6 weeks) while patients < 12-months-old received low-risk consolidation to delay focal RT until the age of 12-months. High-risk patients received targeted IV topotecan and cyclophosphamide (8 weeks) or, if ≥ 3-years-old upon completion of induction, could opt for craniospinal irradiation (23.4-39.6 Gy; dose determined by response after induction).(see attached protocol in appendix pp 19-258)
After consolidation, all risk groups received 6 cycles (24 weeks) of oral maintenance chemotherapy with cyclophosphamide, topotecan and erlotinib. The IV topotecan formulation was prepared with flavoring and given orally. For patients unable to swallow tablets, an oral elixir of cyclophosphamide was given and erlotinib tablets (25mg) were crushed.
Treatment continued until completion, disease progression, withdrawal of consent, or unacceptable toxicity. Disease progression was defined as > 25% increase in the size of any measurable lesion; the appearance of a new radiographically demonstrable lesion; or the conversion of negative CSF to positive, confirmed by two consecutive positive cytologic evaluations following two consecutive negative cytologic evaluations. Disease evaluations were performed every 2 months while on induction and consolidation therapy, every 3 months during maintenance and for the first year off therapy. From 12-24 months off therapy, disease evaluations occurred every 4 months with no MRI spine or LP required at 20 month off therapy. From 24-60 months off-therapy, disease evaluations occurred every 6 months with MRI spine only required annually and CSF cytology only required through 36 months off therapy. Laboratory studies were required prior to each therapy cycle and ANC> 500/mm3, platelets > 50,000/mm3 (without support), hemoglobin > 8 g/dL (with or without transfusion support) and total bilirubin < 3 times ULN were required prior to start each cycle. Auditory brainstem response (ABR) was required before first cisplatin dose, prior to induction cycle 3, at end of induction, prior to maintenance, at end of therapy, and annually thereafter. Dose reductions, delays, and omissions were permitted for: methotrexate with hepatotoxicity and nephrotoxicity; vincristine with seizure, neuropathy, and hepatotoxicity; cyclophosphamide for hematopoetic recovery; cisplatin for ototoxicty and nephrotoxicity; vinblastine for neuropathy, hepatotoxicity, myelosuppression. Carboplatin was permitted to be substituted for cisplatin for patients having grade 4 ototoxicity or bilateral hearing loss after prior cisplatin dose reduction. Toxicity was monitored and graded according to the Cancer Therapy Evaluation Program Common Terminology Criteria for Adverse Events version 3.0 (CTCAEv3.0). All adverse events grade 3, 4, and 5 (except those listed below) which occurred during treatment, for 30 days after the end of treatment, and which occurred later than 30 days after the end of treatment but felt to be at least possibly related to therapy were recorded. Exceptions included: Grade 3 elevation in ALT or AST which occurred within 7 days after methotrexate infusion; grade 3 and 4 hematologic toxicities that occurred from the beginning of induction therapy through the end of consolidation therapy for low and high risk patients; grade 3 and 4 hematologic toxicities that occurred from the beginning of induction therapy through the end of induction for the intermediate risk patients; grade 3 or 4 electrolyte abnormalities that occurred from the beginning of induction chemotherapy to the end of maintenance therapy were not recorded unless these resulted in hospitalization.
Tumor and matched blood samples were obtained with informed consent. DNA was extracted from formalin-fixed paraffin-embedded (FFPE) tissues and blood using the Maxwell® RSC DNA FFPE kit (#AS1450, Promega, Madison, WI), according to the manufacturer’s instructions, and quantified using Qubit (Thermo Fisher Scientific, Waltham, MA). DNA methylation-based classification was performed using methods previously described (Capper D, et al. Nature. In press). In brief, a machine learning approach based on the Random Forest algorithm was used to compare a given diagnostic sample to a cohort of > 2,000 reference samples; thus allowing us to confirm the diagnosis of medulloblastoma and assign molecular subgroups. For predicting SHH and Group 3/4 subtypes we performed t-SNE clustering combined with DBSCAN (20). DNA copy-number variants were inferred from DNA methylation arrays using the Conumee R package with default parameters (23). Medulloblastoma samples were exome sequenced alongside patient-matched blood samples. Tumor and germline DNA exomes were captured using the SureSelect Human All Exon V5 (Agilent Technologies) platform. All NGS data were harmonized and processed with the same analysis pipelines to ensure consistent germline and somatic mutation calling (20). All mutations and CNV calls were manually curated by inspecting sequence alignments and genome-wide copy-number plots. (appendix pp 16-18).
Outcomes
The co-primary endpoints for the study were (1) therapeutic: to estimate the event-free survival distribution of young medulloblastoma patients treated with risk-adapted therapy, and (2) biologic: to identify patterns of methylation profiling that are associated with progression-free survival among young patients with medulloblastoma treated with risk-adapted therapy.
Event-free survival was defined as the time interval from date of treatment initiation to date of relapsed or progressive disease, death, second malignancy, or to the date of last follow-up. Progression-free survival was defined as the time interval from date of treatment initiation to date of relapsed or progressive disease, or to the date of last follow-up. No second malignancies or death prior to relapse or progression had been observed in this cohort so event-free survival and progression-free survival are identical and hereon used interchangeably. Overall survival was defined as the time interval from date of treatment initiation to date of death from any cause or to date of last follow-up. Post-progression survival was defined as the time interval from date of progression to date of death from any cause or to date of last follow-up.
Secondary therapeutic endpoints were overall survival; event-free survival of non-medulloblastoma patients (to be reported separately); rates of local and distant disease progression in patients treated with focal RT (intermediate-risk); objective response rate to induction chemotherapy for patients with residual or metastatic disease (to be reported separately); the feasibility and toxicity of administering low dose IV vinblastine with induction chemotherapy to patients with metastatic disease; the feasibility and toxicity of administering cyclophosphamide and pharmacokinetically targeted topotecan to patients with metastatic disease, and to estimate the sustained objective response rate to such therapy in patients with measurable residual disease after induction (to be reported separately); the feasibility and toxicity of administering oral maintenance therapy in young children; to use quantitative magnetic resonance (MR) measures (volumetric, diffusion, and perfusion) of young brain tumor patients receiving chemotherapy including high-dose IV methotrexate to assess impact of treatment on developing brain (to be reported separately); the feasibility of using PET as an in-vivo dosimetric and distal edge verification system for patients treated with proton beam therapy (for participants enrolled at St Jude only; to be reported separately).
Secondary biological aims were to perform high resolution genome-wide analyses of chromosomal abnormalities and gene expression patterns, and evaluate the relationship of these to other clinicopathological variables; to evaluate specific tumor types for molecular abnormalities with suspected prognostic or therapeutic significance; to evaluate the feasibility of collecting frozen and fixed tumor samples for analysis using high-resolution molecular biology tools.
Secondary pharmacologic aims were to explain inter- and intra-patient pharmacokinetic and pharmacogenomic variability for methotrexate, cyclophosphamide, topotecan, and erlotinib in young children with brain tumors and to explore possible associations between pharmacokinetic parameters and patient specific covariates (e.g., age, sex, race, weight); to explore the relationship between clinical effect (toxicity and antitumor efficacy) and methotrexate pharmacokinetics; to assess the ability to achieve the target systemic exposure of IV topotecan in young patients with metastatic brain tumors.
Secondary cancer control aims were to explore possible associations between cerebrospinal fluid (CSF) neurotransmitter concentrations (e.g., dopamine and its metabolites) and the development of neurocognitive deficits; to explore the association between genetic polymorphisms affecting central dopaminergic transmission and specific phenotypes, including CNS neurotransmitter and neurocognitive performance phenotypes; to investigate changes in neuropsychological performance among patients enrolled in the study, and examine the impact of the proposed treatment regimen and other disease related factors (e.g., hydrocephalus) on neuropsychological performance; to assess the impact of changes in quantitative MR measures in the frontal lobe on neurocognitive performance in attention, working memory, and fluency; to assess the impact of changes in quantitative MR measures in the right frontal-parietal regions on neurocognitive performance on visual-spatial reasoning and processing speed; to assess the incidence of endocrinopathy after radiation therapy using photons or protons; to estimate the rate of longitudinal change in growth hormone secretion after conformal, intensity-modulated and proton beam radiation therapy.
These pharmacologic and cancer control aims are to be reported separately.
A post-hoc analysis was performed using data sourced from a published international cohort(20) that was added to molecularly characterized SJYC07 samples. The aim was to describe molecular characteristics particular to early childhood medulloblastoma such that recognizable patterns of methylation be applied to the trial cohort and outcomes measured.
Statistical analysis
This study was designed to estimate the event-free survival distribution associated with the proposed treatment approach. No specific statistical hypotheses were proposed rather the sample size was based on what was deemed feasible for patient accrual and tissue acquisition. Initially the trial proposed to enroll a total of 140 young medulloblastoma patients in 7 years, 50% of whom were expected to have fresh frozen tissue which was required for the planned molecular analyses. Based on historical data 73 events were expected after the planned follow-up of 1-year post accrual completion which was deemed adequate to describe event-free survival outcome and explore associations of molecular subtypes with progression-free survival. On May 30, 2014 after 7 years of accrual, due to slow accrual as well as the emerging information regarding infant medulloblastoma outcomes from published studies, the study design was revised to dramatically reduce the target sample size to 90 medulloblastoma subjects and to extend the time of enrollment to three additional years. Furthermore the design was revised to estimate and monitor outcome in each risk group separately. Based on available published data, the low-risk arm accrual was to be suspended if 1-year event-free survival estimate dropped below 80%. Monitoring was initiated after the first 10 patients reached the 1-year time point and was continued in a group sequential fashion. For the intermediate and high risk groups, a monitoring threshold of 2-year event-free survival <20% was established and monitoring was initiated after the first 15 patients in each cohort reached the 2-year time point. Once initiated, the monitoring was done periodically in approximately 6-month intervals in all three strata. Despite the reduction in the sample size, the biologic primary objective remained feasible due to the large percentage of patients (>95%) providing FFPE tissue which could now be used for the planned analyses instead of frozen tissue.
As per an intention-to-treat analysis all eligible medulloblastoma patients who received any methotrexate were included in the analyses for outcome and safety. All patients with adequate tissue for methylation profiling were included in the biologic analyses. Outcome distributions were estimated using the method of Kaplan and Meier; standard errors of outcome estimates were obtained using the method of Peto and Pike. Hazard ratios (HR) with associated 95% confidence intervals and p-values comparing outcome distributions were calculated using Cox regression using the likelihood ratio approach. In cases where the hazard ratio was not estimable due to small sample sizes or lack of events in an arm, the exact log-rank test was used to compare outcome distributions. Fisher’s exact test was used to compare distributions of categorical variables among patient groups (i.e. PTCH1, SUFU, SMO alterations, chromosome 2 gain between SHH subtypes). Statistical analyses were done using SAS v9.4. This trial is registered at ClinicalTrials.gov, number NCT NCT00602667 and was permanently closed to accrual in April 2017.
Role of the funding source
The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. GWR, CAB, AO-T, AG had full access to all the data. The corresponding author had final responsibility for the decision to submit for publication.
Results
Between November 27, 2007 and April 19, 2017, 293 children with newly diagnosed malignant CNS tumors were assessed for eligibility on SJYC07. Three patients were deemed ineligible: 2 for ineligible pathology diagnoses; 1 for failure to start treatment within the defined 31 day post-operative window. 290 eligible patients enrolled. 81 of the 290 were defined as having medulloblastoma (figure 1). All 81 medulloblastoma patients received methotrexate and were included in the analyses for the primary therapeutic aim. For the primary biologic aim 78 of the 81 clinical trial cases had available DNA methylation data for molecular classification. Three cases had no archival tissue available. 76 tumors molecularly classified as medulloblastoma and the remaining 2 cases identified as embryonal tumor with multilayered rosettes (ETMR) (figure 1).
Table 1 shows the general characteristics of the 81 medulloblastoma patients. Median age at enrollment was 2.3 years (IQR 1.3-2.9) and median follow-up was 5.5 years (IQR 2.7-7.3). 23 (28%) of 81 patients risk stratified as low-risk; 32 (40%) as intermediate-risk; and 26 (32%) as high-risk. The most common reason for not completing therapy was progression. Of the 47 patients who did not complete therapy, 37 (79%) progressed before therapy was complete, 8 (17%) withdrew, and 2 (4%) remained on maintenance therapy at the writing of this manuscript. Twenty-six (81%) intermediate-risk patients completed focal radiation therapy and only one high-risk patient opted for craniospinal irradiation instead of consolidation chemotherapy. 17 (74%) of 23 low-risk patients completed maintenance as compared to 12 (38%) out 32 intermediate risk patient and 5 (19%) out of 26 high-risk patients.
Table 1.
Clinical Characteristics of enrolled patients by clinical risk group
| Low (n=23) |
Intermediate (n=32) |
High (n=26) |
All (n=81) |
|
|---|---|---|---|---|
| Sex | ||||
|
| ||||
| Female | 10 (43%) | 12 (38%) | 14 (54%) | 36 (44%) |
| Male | 13 (57%) | 20 (62%) | 12 (46%) | 45 (56%) |
|
| ||||
| Age Group | ||||
|
| ||||
| <3 years | 23 (100%) | 16 (50%) | 26 (100%) | 65 (80%) |
| 3-5 years | – | 16 (50%) | – | 16 (20%) |
|
| ||||
| Metastatic Status | ||||
|
| ||||
| M0 | 23 (100%) | 32 (100%) | – | 55 (68%) |
| M+ | – | – | 26 (100%) | 26 (32%) |
| M1 | – | – | 1 (4%) | 1 (1%) |
| M2 | – | – | 7 (27%) | 7 (9%) |
| M3 | – | – | 18 (69%) | 18 (22%) |
|
| ||||
| Histology Group | ||||
|
| ||||
| MB-Classic | – | 25 (78%) | 10 (38%) | 35 (43%) |
| MB-LC/A | – | 1 (3%) | 5 (19%) | 6 (7%) |
| MB-DN/MBEN | 23 (100%) | 6 (19%) | 10 (38%) | 39 (48%) |
| DN | 17 (74%) | 5 (16%) | 8 (31%) | 30 (37%) |
| MBEN | 6 (26%) | 1 (3%) | 2 (8%) | 9 (11%) |
| MB-NOS | – | – | 1 (4%) | 1 (1%) |
|
| ||||
| R status | ||||
|
| ||||
| R0 | 20 (87%) | 30 (94%) | 18 (69%) | 68 (84%) |
| R+ | 3 (13%)* | 2 (6%) | 8 (31%) | 13 (16%) |
|
| ||||
| Extent of Resection | ||||
|
| ||||
| GTR | 19 (83%) | 30 (94%) | 13 (50%) | 62 (77%) |
| NTR | 1 (4%) | – | 5 (19%) | 6 (7%) |
| STR | 3 (13%) | 2 (6%) | 8 (31%) | 13 (16%) |
|
| ||||
| Methylation subgroup | ||||
|
| ||||
| SHH | 23 (100%) | 8 (25%) | 11 (42%) | 42 (52%) |
| G3 | – | 13 (41%) | 11 (42%) | 24 (30%) |
| G4 | – | 8 (25%) | 2 (8%) | 10 (12%) |
| ETMR | – | 1 (3%) | 1 (4%) | 2 (2%) |
| Unknown | – | 2 (6%) | 1 (4%) | 3 (4%) |
|
| ||||
| PFS Status | ||||
|
| ||||
| Not progressed | 13 (57%) | 9 (28%) | 5 (19%) | 27 (33%) |
| Progressed | 10 (43%) | 23(72%) | 21 (81%) | 54 (67%) |
|
| ||||
| Location of Relapse | ||||
|
| ||||
| None | 13 (57%) | 9 (28%) | 5 (19%) | 27 (33%) |
| Local | 6 (26%) | 1 (3%) | 4 (15%) | 11 (14%) |
| Distant | 3 (13%) | 19 (59%) | 10 (39%) | 32 (39%) |
| Local + Distant | 1 (4%) | 3 (9%) | 7 (27%) | 11 (14%) |
|
| ||||
| Survival Status | ||||
|
| ||||
| Alive | 20 (87%) | 19 (59%) | 12 (46%) | 51 (63%) |
| Dead | 3 (13%) | 13 (41%) | 14 (54%) | 30 (37%) |
Treated as low-risk because R0 at the end of induction prior to consolidation
On December 2, 2015 as part of routine interim monitoring the 1-year event-free survival rate in the low-risk arm was estimated to be 78.3%, which was below the protocol-specified stopping threshold of 80%. Thus the low-risk arm was closed to accrual and never re-opened. On April 19, 2017 the study was permanently closed after reaching the end of the proposed enrollment period.
The 5-year event-free survival and overall survival of the entire cohort at 5-years was 31.3% (95% CI 19.3-43.3%) and 59.4% (95% CI 45.7-73.1%), respectively (figure 2A; appendix p 6). Median time to progression/relapse was 8.4 months from start of treatment (IQR 5.4-12.0).
Figure 2. Event-free survival analysis.

Event-free survival of (a) all 81 participants and (b) by risk group as prespecifed in the protocol under the primary therapeutic aim. (c) Event-free survival of intermediate risk by age < 3 years (orange) and 3-5 years (green). (d) Event-free survival by histology. (e) Event-free survival of DN/MBEN samples by risk group. (f) Event-free survival of DN (black) and MBEN (grey). Panels c-f were not prespecified in the protocol and were performed to highlight event-free survival by major clinical characteristics. Hazard ratios with associated 95% confidence intervals and p-values comparing outcome distributions were calculated using Cox regression using the likelihood ratio approach. EFS = Event-free survival. HR = hazard ratio. Int = Intermediate-risk. NA = not applicable. DN = desmoplastic nodular. MBEN = medulloblastoma with extensive nodularity. DN/MBEN = desmoplastic nodular or medulloblastoma with extensive nodularity.
Event-free survival differed across risk groups (p=0.0021; figure 2B) mostly driven by the low-risk cohort. Event-free survival at 5-years for the low-risk cohort was 55.3% (95% CI 33.3-77.3%) compared to intermediate-risk (24.6% at 5 years, 95% CI 3.6-45.6%) and high-risk (16.7% at 5 years, 95% CI 3.4-30.0%). There was no significant difference between intermediate- and high-risk groups in event-free survival (HR 1.42, 95% CI 0.78-2.57; p=0.25) or in overall survival (HR 1.68, 95% CI 0.79-3.58; p=0.18). No significant difference was observed between the event-free survival of 3-5 year-olds, all treated in the intermediate-risk group, and the event-free survival of 0-3 year-old intermediate-risk patients (HR 1.22, 95% CI 0.54-2.76; p=0.64; figure 2C) (See appendix p 6 for overall survival estimates).
Most progressions/relapses occurred on therapy (40 [74%] of 54 progressions) with the majority (22 [55%] of the 40) occurring during or at the end of the maintenance phase. Table 1 shows the number of progression events and the survival status by risk-adapted group. The location of relapse in intermediate-risk patients was distant (outside the primary site) in the vast majority (19 [83%] of 23) and only one relapse was isolated to the primary site (table 1).
Of the 54 patients who progressed, 29 went on to receive craniospinal irradiation (median dose, 36 Gy [IQR 36-36]) at a median of 0.7 months (IQR 0.3-3.9) from the date of relapse/progression. Of these 29 patients, 18 (62%) are alive, compared to 6 (24%) of 25 who did not receive craniospinal irradiation (p<0.0001; appendix p 6).
Not unexpectedly, given that histology was part of the risk definition used in this study, histologic subtype was also significantly associated with outcome. (HRCMB vs. HRDN/MBEN 2.89, 95% CI 1.59-5.25; p=0.0005; HRLCA vs. HRDN/MBEN 10.14, 95% CI 3.79-27.16; p<0.0001; figure 2D; see appendix p 6 for overall survival). Comparison of risk group assignment to histology revealed that sixteen DN/MBEN patients were treated in the intermediate- or high-risk groups. No significant difference in event-free survival was seen between low-, intermediate-, or high-risk DN/MBEN (HRintermediate vs. HRlow 1.42, 95% CI 0.39-5.16; p=0.60; HRhigh vs. HRlow 1.48, 95% CI 0.51-4.34 p=0.48; figure 2E). Also, no significant difference was seen between MBEN and DN (HR 1.48, 95% CI 0.43-5.11; p=0.52; figure 2F).
The primary biologic aim was to identify molecular patterns associated with progression-free survival in medulloblastoma treated with risk-adapted therapy. Before undertaking the analyses we sought to describe and enrich our understanding of the molecular characteristics particular to early childhood medulloblastoma. A post-hoc analysis was performed on a larger cohort than what was collected on the SJYC07 trial. We collected data from 131 patients < 6-years old sourced from a published international cohort(20) and added 59 molecularly characterized SJYC07 samples. The age of 6 was selected to describe the full spectrum of early childhood medulloblastoma even though this inclusion would knowingly encompass more than what was captured on the SJYC07 trial. The combined cohort of 190 cases had both DNA methylation array data (derived from Illumina 450K or 850K methylation arrays) and matched germline and tumor sequencing (whole genome or whole exome) data (figure 3).
Figure 3. The genomic landscape of early childhood medulloblastoma.

(a) Establishing the molecular cohort (b) t-SNE plot of DNA methylation array data showing the distribution of consensus medulloblastoma subgroups (n = 208). (c) Oncoprint summarizing recurrently altered genes, frequent cytogenetic events, and sample annotations according to imedulloblastoma subgroup (n = 190).
Consensus molecular subgroup status was determined by a comparative analysis of tumor DNA methylation profiles against a large reference dataset. (Capper D, et al. Nature. In press). This divided samples of the molecular cohort into three subgroups: SHH (n=83; 44%), G3 (n=68; 36%), G4 (n=39; 20%). Consistent with prior reports, the WNT subgroup was absent from the entire cohort.(24, 25) In parallel, we applied t-distributed stochastic neighbor embedding (t-SNE) to molecularly-verified medulloblastomas. SHH tumors separated from non-SHH (G3 and G4) tumors conveying a highly distinct molecular profile (figure 3B). Conversely, G3 and G4 tumors clustered together and, as previously observed, exhibited some overlap, implying a close molecular relationship between these subgroups.(20, 22) The most recurrently mutated genes, copy number variants, and basic clinical characteristics (age, gender) were compared across methylation subgroups (figure 3C). Distribution of subgroups varied with age: for children < 3yrs (n=84), 73% were SHH (n=61), 25% were G3 (n=21), and 2% were G4 (n=2). For children 3-6 years (n=106), 21% were SHH (n=22), 44% were G3 (n=47), and 35% were G4 (n=37). t-SNE analysis performed exclusively on SHH methylation data (n=87) uncovered two subtypes that we designated as iSHH-I and iSHH-II (figure 4A). iSHH-I (n=37) was characterized by a median age of 2.0 years at diagnosis (range, 0.2 – 5.0 years) with only 5 of 37 (14%) patients falling between 3-6 years. The male: female ratio was 1.3:1. iSHH-II (n=46) was characterized by a median age of 2.2 years at diagnosis (range, 0.6 – 5.9 years). The age distribution for iSHH-II patients was broader than observed for iSHH-I, with 17 of 46 (37%) iSHH-II patients ≥3 years old. Gender distribution for iSHH-II showed a slight female predominance of 1.3:1 female: male. PTCH1 alterations (mutations and focal deletions) were more common in iSHH-II (48%) compared to iSHH-I (35%) (figure 4B,C). In contrast, deleterious SUFU alterations were significantly enriched in iSHH-I (32%) and rare in iSHH-II (4%) (p=0.0009). Similarly, chromosome 2 gain was significantly enriched in iSHH-I (35%) compared to iSHH-II (4%) (p=0.001; figure 4B,C; appendix p 10). Additional genetic alterations differentially distributed between iSHH subtypes included activating SMO mutations (13% of iSHH-II; absent in iSHH-I; p=0.03) and BCOR mutations (9% of iSHH-II; absent in iSHH-I; p=0.12), as well as chromosome 9q deletions (52% of iSHH-II; 24% in iSHH-I; p=0.01). To describe non-SHH tumors (iG3/iG4) relative to the eight methylation subtypes described among medulloblastomas of all ages,(20) we mapped their distribution on the published methylation-defined substructure (appendix p 15). Subtypes most commonly associated with G3 (II, III, IV) were more heavily represented, while subtypes V, VI, and VIII associated with G4 were under-represented compared to our recent study which included medulloblastoma patients of all age groups.(20) Consequently, prominent chromatin-modifier mutations associated with subtypes VI and VIII were virtually absent, whereas driver events such as MYC amplification and recently defined KBTBD4 mutations (associated mainly with subtypes II and III) were observed. The most abundant non-SHH subtype in patients <3-years was subtype IV; characterized by almost no known driver events but exhibiting widespread copy-number aberrations (appendix p 15).
Figure 4. Molecular features of early childhood SHH medulloblastoma subtypes.

(a) t-SNE plot showing separation of SHH medulloblastoma samples into two subtypes, SHH-I and SHH-II (n = 87). (b) Oncoprint summarizing recurrently altered genes, frequent cytogenetic events, and sample annotations for SHH medulloblastoma subtypes (n = 87). (c) Distribution of genetic events associated with the SHH pathway in SHH-I and SHH-II (n = 87).
With this improved understanding of the molecular patterning that occurs across early childhood medulloblastoma we analyzed the distribution and outcomes of the76 molecularly classified medulloblastoma tumors on SJYC07 in order to meet the primary biologic objective of the study. The majority of patients were SHH (42/76; 55%), followed by G3 (24/76; 32%), and G4 (10/76; 13%) (table 2). Evaluation of clinical risk groups according to molecular subgroup showed that the low-risk group was comprised solely of SHH patients (23/23; 100%), whereas intermediate- and high-risk groups were a mixture of SHH, G3, and G4 (table 1, 2).
Table 2.
Clinical characteristics of enrolled patients by molecular subgroup.
| SHH (n=42) |
G3 (n=24) |
G4 (n=10) |
All (n=76) |
|
|---|---|---|---|---|
| Sex | ||||
|
| ||||
| Female | 20 (48%) | 12 (50%) | 3 (30%) | 35 (46%) |
| Male | 22 (52%) | 12 (50%) | 7 (70%) | 41 (54%) |
|
| ||||
| Age Group | ||||
|
| ||||
| <3 years | 39 (93%) | 18 (75%) | 3 (30%) | 60 (79%) |
| 3-5 years | 3 (7%) | 6 (25%) | 7 (70%) | 16 (21%) |
|
| ||||
| Metastatic Status | ||||
|
| ||||
| M0 | 31 (74%) | 13 (54%) | 8 (80%) | 52 (68%) |
| M+ | 11 (26%) | 11 (46%) | 2 (20%) | 26 (32%) |
| M1 | – | 1 (4%) | – | 1 (1%) |
| M2 | 5 (12%) | 1 (4%) | 1 (10%) | 7 (9%) |
| M3 | 6 (14%) | 9 (38%) | 1 (10%) | 16 (21%) |
|
| ||||
| Histology Group | ||||
|
| ||||
| MB-Classic | 3 (7%) | 19 (79%) | 9 (90%) | 31 (41%) |
| MB-LC/A | – | 5 (21%) | 1 (10%) | 6 (8%) |
| MB-DN/MBEN | 39 (93%) | – | – | 39 (51%) |
| DN | 30 (72%) | – | – | 30 (39%) |
| MBEN | 9 (21%) | – | – | 9 (12%) |
|
| ||||
| R status | ||||
|
| ||||
| R0 | 34 (81%) | 21 (88%) | 9 (90%) | 64 (84%) |
| R+ | 8 (19%) | 3 (12%) | 1 (10%) | 12 (16%) |
|
| ||||
| Extent of Resection | ||||
| GTR | 32 (76%) | 17 (71%) | 9 (90%) | 58 (76%) |
| NTR | 2 (5%) | 4 (17%) | – | 6 (8%) |
| STR | 3 (19%) | 3 (12%) | 1 (31%) | 12 (16%) |
|
| ||||
| Risk subgroup | ||||
|
| ||||
| Low | 23 (55%) | – | – | 23 (30%) |
| Intermediate | 8 (19%) | 13 (54%) | 8 (80%) | 29 (38%) |
| High | 11 (26%) | 11 (46%) | 2 (20%) | 24 (32%) |
|
| ||||
| PFS Status | ||||
|
| ||||
| Not progressed | 22 (52%) | 2 (8%) | 5 (19%) | 27 (33%) |
| Progressed | 20 (48%) | 22 (92%) | 21 (81%) | 54 (67%) |
|
| ||||
| Location of Relapse | ||||
|
| ||||
| None | 22 (52%) | 2 (8%) | 2 (20%) | 26 (34%) |
| Local | 7 (17%) | 2 (8%) | 1 (10%) | 10 (13%) |
| Distant | 8 (19%) | 16 (67%) | 7 (70%) | 31 (41%) |
| Local + Distant | 5 (12%) | 4 (17%) | – | 9 (12%) |
|
| ||||
| Survival Status | ||||
|
| ||||
| Alive | 31 (74%) | 12 (50%) | 7 (70%) | 50 (66%) |
| Dead | 11 (26%) | 12 (50%) | 3 (30%) | 26 (34%) |
Survival by methylation subgroup revealed that progression-free survival of SHH was superior to that of G3 (HR 4.07, 95% CI 2.19-7.59; p<0.0001; figure 5A,) and G3/G4 combined (non-SHH) (HR 3.18, 95% CI 1.79-5.66; p<0.0001; appendix p 7). The difference in progression-free survival between SHH compared to G4 was not statistically significant (HR 2.02, 95% CI 0.89-4.62, p=0.095; figure 5A).
Figure 5. Progression-free survival analysis of SJYC07 medulloblastoma Subgroups and SHH medulloblastoma subtypes.
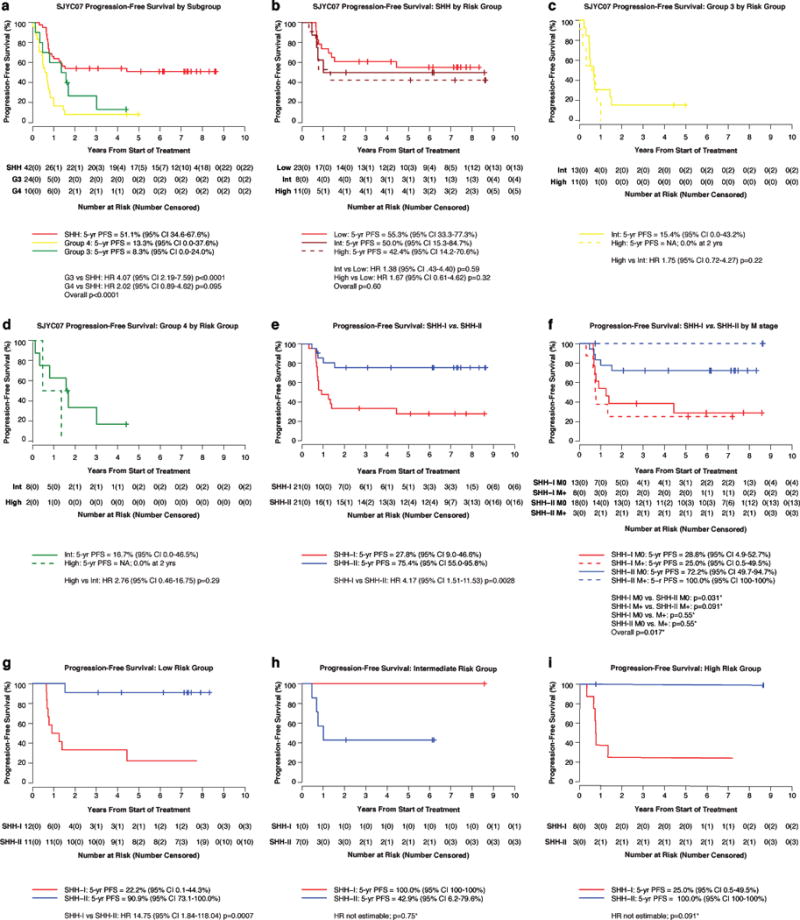
(a) Progression-free survival of SHH (red), Group 3 (yellow), Group 4 (green) patients. Progression-free survival of (b) SHH (c) Group 3 (d) Group 4 by low-, intermediate-, and high-risk groups. Progression-free survival plots showing outcome differences for SHH-I (red) and SHH-II (blue) subtypes by (e) entire cohort (f) metastatic disease (M0 = solid; M+ = dashed) (g) low- (h) intermediate- (i) high-risk groups. Hazard ratios with associated 95% confidence intervals and p-values comparing outcome distributions were calculated using Cox regression using the likelihood ratio approach. In f-i where the hazard ratio was not estimable due to small sample sizes or lack of events in an arm, the exact log-rank test was used to compare outcome distributions. All comparisons shown were prespecifed in the protocol under the primary biologic aim. PFS = Progression-free survival. HR = hazard ratio. SHH = Sonic hedgehog. G3 = Group 3. G4 = Group 4. Int = Intermediate-risk. NA = not applicable. M0 = non-metastatic. M+ = metastatic. *p values based on exact log-rank tests
No statistical difference in progression-free survival was observed between the three risk groups of SHH disease (HRintermediate vs. HRlow 1.38, 95% CI 0.43-4.40; p=0.59; HRhigh vs. HRlow 1.67, 95% CI 0.61-4.62; p=0.32; overall p=0.60; figure 5B), the two risk groups of G3 (HR 1.75, 95% CI 0.72-4.27; p=0.22; figure 5C), nor the two risk groups of G4 (HR 2.76, 95% CI 0.46-16.75; p=0.29; figure 5D)(See appendix pp 8-9 for overall survival and post-progression survival by subgroup).
For the SJYC07 SHH cohort only (iSHH-I n=21, iSHH-II n=21), progression-free survival of iSHH-II was significantly better than iSHH-I (HR 4.17, 95% CI 1.51-11.53; p=0.0028; figure5E). This trend persisted when adjusted for metastatic disease (5-year progression-free survival estimates were 100% (95% CI 100-100%) and 72.2% (95% CI 49.7-94.7%) for SHH-II M+ and M0 patients, compared to 25.0% (95% CI 0.5-49.5%) and 28.8% (95% CI 4.9-52.7%) for SHH-I M+ and M0 patients, respectively p = 0.017; figure 5F). The 5-year progression-free survival of clinically-defined low-risk iSHH-II (n=11) was 90.9% (95% CI 73.1-95.8%), whereas 5-year progression-free survival for low-risk iSHH-I (n=12) was 22.2% (95% CI 0.1-44.3%) (HR 14.75, 95% CI 1.84-118.04, p=0.0007; figure 5G). A separation in the progression-free survival curves was observed between iSHH-I and iSHH-II risk-stratified to the high-risk group but this was not statistically significant (5-year PFSiSHH-II 100%, 95% CI 100-100% vs 5-year PFSiSHH-I 25.0%, 95% CI 0.5-49.5%; p=0.091; figure 5I). Only 1 intermediate-risk iSHH-I patient was enrolled and thus no outcome comparison could be made with intermediate-risk iSHH-II (n=7) (figure 5H). Most of the relapses among iSHH-II (4 out of 5; 80%) occurred in patients treated on the intermediate-risk arm (table 2). No significant difference was observed in age, gender, extent of resection, histology, metastatic status, or assignment of risk group between iSHH-I and iSHH-II (appendix p 11). Differences in overall survival between iSHH-I and iSHH-II were also not significant (appendix p 12). Of the 15 relapsed iSHH-I patients, 8 (53%) died and 5 of 7 living patients received craniospinal irradiation.
Given that SUFU and chromosome 2 gain were significantly enriched in the SHH-I group we performed a post-hoc analysis and plotted the entire SHH cohort (n=42) according to SUFU mutation, chromosome 2 gain status, and combined chromosome 2 gain status and/or SUFU mutation (appendix p 13). Significant difference in progression-free survival was only seen in chromosome 2 gain status (HR 2.78, 95% CI 1.10-7.03; p=0.043). Survival of iSHH-I patients was no different for those with chromosome 2 alterations or SUFU mutations than those without (HR 1.00, 95% CI 0.35-2.81; p=0.99; appendix p 14)
Given the number of non-SHH subtypes and the modest number of non-SHH patients enrolled on SJYC07 (n=34; appendix p 15), no conclusions could be made about survival differences between subtypes I-VIII.
Therapy was well tolerated, no patients discontinued for drug related toxicity, and no protocol-related deaths were observed. The most common recorded grade 3 and 4 toxicities were hematologic, infectious, and gastrointestinal (table 3). 5 serious adverse events were reported (sepsis/bacteremia, pulmonary edema, viral respiratory infection requiring hospitalization, catheter related infection, and bone fracture) and only 2 (sepsis/bacteremia and pulmonary edema) were assessed as possibly, likely, or definitely related to study treatment. 14 (17%) of 81 patients had dose modifications due to toxicity (5[22%] of23 low risk, 6 [19%] of 32 intermediate risk, and 3 [12%] of 26 high risk).
Table 3.
Grade 3 and 4 adverse events deemed to be treatment-related
| Grade 3 | Grade 4 | |||||||
|---|---|---|---|---|---|---|---|---|
| LR (n=23) |
IR (n=32) |
HR (n=26) |
All (n=81) |
LR (n=23) |
IR (n=32) |
HR (n=26) |
All (n=81) |
|
| Hematologic | ||||||||
|
| ||||||||
| Febrile neutropenia | 15 (65%) | 15 (47%) | 18 (69%) | 48 (59%) | – | – | – | – |
| Neutropenia | 11 (47%) | 8 (25%) | 2 (8%) | 21 (26%) | 2 (9%) | 1 (3%) | 2 (8%) | 5 (6%) |
| Leukopenia (total WBC) | 7 (30%) | 6 (19%) | 2 (8%) | 15 (19%) | – | – | – | – |
| Anemia | 3 (13%) | – | 1 (4%) | 4 (5%) | 1 (4%) | – | – | 1 (1%) |
| Thrombocytopenia | – | 1 (3%) | 2 (8%) | 3 (4%) | 1 (4%) | – | 1 (4%) | 2 (3%) |
| Hemorrhage/Bleeding | 2 (9%) | 1 (3%) | – | 3 (4%) | – | – | – | – |
| Hematoma | 1 (4%) | – | – | 1 (1%) | – | – | – | – |
| Lymphopenia | – | 1 (3%) | – | 1 (1%) | – | – | – | – |
|
| ||||||||
| Infectious | ||||||||
|
| ||||||||
| Documented infection with Grade 3 or 4 neutropenia (blood, catheter-related, pneumonia, colitis, mucositis, otitis, cellulitis, cystitis) | 9 (39%) | 4 (13%) | 7 (26%) | 20 (25%) | – | – | 1 (4%) | 1 (1%) |
| Infection without Grade 3 or 4 neutropenia | 7 (30%) | 4 (13%) | 4 (15%) | 15 (19%) | – | 1 (3%) | – | 1 (1%) |
| Mucositis/stomatitis | 2 (9%) | 3 (9%) | 2 (8%) | 7 (9%) | – | – | 1 (4%) | 1 (1%) |
| Colitis, infectious | 2 (9%) | 1 (3%) | 3 (12%) | 6 (7%) | – | – | – | – |
| Infection - Other | 2 (9%) | 2 (6%) | 1 (4%) | 5 (6%) | – | – | – | – |
| Infection with unknown ANC | 1 (4%) | – | 1 (4%) | 2 (3%) | – | – | – | – |
| Cystitis | – | – | 1 (4%) | 1 (1%) | – | – | – | – |
| Hypotension/Sepsis | – | – | 1 (4%) | 1 (1%) | – | – | – | – |
|
| ||||||||
| Gastrointestinal | ||||||||
|
| ||||||||
| Vomiting | 5 (21%) | 3 (9%) | 7 (26%) | 15 (19%) | - | – | – | – |
| Anorexia | 3 (13%) | 4 (13%) | 6 (23%) | 13 (16%) | – | – | – | – |
| Diarrhea | 2 (9%) | 1 (3%) | 5 (19%) | 8 (10%) | – | – | – | – |
| Nausea | – | 3 (9%) | – | 3 (4%) | – | – | – | – |
| ALT elevation | – | – | 1 (4%) | 1 (1%) | – | 1 (3%) | – | 1 (1%) |
| AST elevation | – | – | – | – | – | 1 (3%) | – | 1 (1%) |
|
| ||||||||
| Constitutional | ||||||||
|
| ||||||||
| Dehydration | 1 (4%) | 2 (6%) | 1 (4%) | 4 (5%) | – | – | – | – |
| Ataxia (incoordination) | 1 (4%) | – | – | 1 (1%) | – | – | – | – |
| Irritability (children <3 years of age) | 1 (4%) | – | – | 1 (1%) | – | – | – | – |
| Fever (in the absence of neutropenia) | – | – | 1 (4%) | 1 (1%) | – | – | – | – |
| Constitutional Symptoms - Other | – | – | 1 (4%) | 1 (1%) | – | – | – | – |
| Fatigue (asthenia, lethargy, malaise) | – | – | 1 (4%) | 1 (1%) | – | – | – | – |
| Weight loss | – | – | 1 (4%) | 1 (1%) | – | – | – | – |
|
| ||||||||
| Nervous system | ||||||||
|
| ||||||||
| Hearing loss | 2 (9%) | – | – | 2 (3%) | – | – | – | – |
| Neuropathy: motor | – | – | 1 (4%) | 1 (1%) | – | – | – | – |
| Seizure | – | – | 1 (4%) | 1 (1%) | – | – | – | – |
| Urinary retention (neurogenic bladder) | – | – | 1 (4%) | 1 (1%) | – | – | – | – |
|
| ||||||||
| Metabolic | ||||||||
|
| ||||||||
| Potassium, serum-low (hypokalemia) | – | 1 (3%) | – | 1 (1%) | – | - | 2 (8%) | 2 (3%) |
| Calcium, serum-low (hypocalcemia) | – | – | – | – | – | – | 1 (4%) | 1 (1%) |
| Glucose, serum-low (hypoglycemia) | – | – | – | – | 1 (4%) | – | – | 1 (1%) |
|
| ||||||||
| Cutaneous | ||||||||
|
| ||||||||
| Rash/desquamation | 2 (9%) | 1 (3%) | 1 (4%) | 4 (5%) | – | – | – | – |
| Rash: dermatitis associated with radiation | – | 1 (3%) | – | 1 (1%) | – | – | – | – |
|
| ||||||||
| Pulmonary | ||||||||
|
| ||||||||
| Aspiration | – | 1 (3%) | 0.0% | 1 (1%) | – | – | – | – |
| Hypoxia | – | 1 (3%) | 0.0% | 1 (1%) | – | – | – | – |
| Pulmonary/Upper Respiratory - Other | – | – | 3.8% | 1 (1%) | – | – | – | – |
|
| ||||||||
| Musculoskeletal | ||||||||
|
| ||||||||
| Pain, Extremity-limb | 1 (4%) | 1 (3%) | – | 2 (3%) | – | – | – | – |
| Pain, Abdomen NOS | – | 1 (3%) | – | 1 (1%) | – | – | – | – |
Data are number of patients (%). Grade 1 and 2 events were not recorded as specified in the protocol. Also not recorded were: Grade 3 elevation in ALT or AST which occurred within 7 days after methotrexate infusion; grade 3 and 4 hematologic toxicities that occurred from the beginning of induction therapy through the end of consolidation therapy for low and high risk patients; grade 3 and 4 hematologic toxicities that occurred from the beginning of induction therapy through the end of induction for the intermediate risk patients; grade 3 or 4 electrolyte abnormalities that occurred from the beginning of induction chemotherapy to the end of maintenance therapy were not recorded unless these resulted in hospitalization. Events were classified according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0. Attribution to therapy was based on investigators’ judgment. No deaths (grade 5) events occurred during the study. LR = Low risk. IR = Intermediate risk. HR= High risk. WBC = white blood cell. NOS = not otherwise specified. AST = aspartateaminotransferase. ALT = alanineaminotransferase.
Discussion
Despite a low-toxicity profile and a statistically significant difference in the outcome of the risk groups, the SJYC07 clinical trial was not a therapeutic success. The 5-year event-free survival of 31% for the entire medulloblastoma cohort did not improve upon previously reported event-free survival rates; the 5-year event-free survival of 25% for intermediate-risk patients failed to suggest a benefit for patients who received combined chemotherapy and focal radiotherapy; the frequent occurrence of progression/relapse during maintenance did little to suggest remission could be maintained on oral chemotherapy; and the 5-year event-free survival of 55% for patients who risk-stratified into a low-risk category fell short of the 5-year progression-free survival reported for similar cohorts receiving intraventricular or high-dose regimens.(10, 13)
A higher event-free survival for M0, R0, medulloblastoma patients < 3 years old with DN/MBEN histology was observed in a risk adapted model over patients with elevated risk characteristics. However, the increase in intensity afforded by different consolidation regimens (i.e. focal RT for intermediate-risk, and topotecan and cyclophosphamide for high risk) failed to improve the event-free survival over that which has been historically reported. In fact the event-free survival results from all three trial arms are very similar when clinically and morphologically matched to those reported in an international meta-analysis of early childhood medulloblastoma treated with a variety of treatment regimens.(8) Moreover when compared to Children’s Oncology Group (COG) P9934 study which used a similar approach of induction chemotherapy followed by focal RT the results are very similar for non-desmoplastic medulloblastoma (P9934 4-year event-free survival 23±12% vs SJYC07 5-year event-free survival of 25±12).(7) Of interest, is the low local relapse rate and, similarly, high distant relapse rate of the intermediate risk patients which hints at effective local control but absent distant control from focal RT.
Reports on the pharmacokinetic findings of the different drugs used, such as high-dose IV methotrexate, and the neurocognitive outcomes of young children with brain tumors treated on this trial will be forthcoming. These pharmacokinetic results might give insight into differences in outcomes between patients who receive intraventricular methotrexate and those who do not. Furthermore neurocognitive results may help understand the effects of focal RT on tumors such as ependymoma or choroid plexus carcinoma that may still continue to benefit from this intermediate-risk approach.
Notwithstanding the therapeutic shortcomings in medulloblastoma, a great deal of knowledge was gained from this study; specifically, an improved understanding of how progression-free survival associated with molecular patterns derived from the invaluable tissue contributions of the patients.
The statistically significant difference observed in progression-free survival according to consensus molecular subgroup is of paramount importance. While the SJYC07 risk stratification reached statistical significance, re-stratification of the cohort by subgroup demonstrated a similar effect, suggesting that risk-stratification by molecular sub-classification is a viable and robust alternative to stratification on clinical risk factors. Nineteen SHH patients were risk-stratified to the intermediate- (n=7) and high-risk groups (n=12) and, despite the change in therapy, progression-free survival was not different from the low-risk arm. Furthermore, removal of SHH patients from the intermediate- and high-risk arms revealed that the progression-free survival of G3 and G4 was very low (5-year progression-free survival of 8±8% and 13±12%, respectively) and reinforced that this reduced-intensity approach should not be continued for non-SHH medulloblastoma. In fact, given that most patients progressed after the intensive chemotherapy portion of therapy and the salvage rate for patients who received craniospinal irradiation was encouraging, our data suggest that delayed, or perhaps reduced-dose, craniospinal irradiation strategies warrant exploration for this population. Equally, or arguably more importantly, novel strategies that employ precision-based approaches warrant further consideration such that neuro-cognitive quality of life is not sacrificed for better survival outcomes.
Still, while the lower than expected progression-free survival of low-risk DN/MBEN group (n=23; 5-year 55%, 95% CI 33-77%) and the 5-year 51% (95% CI 35-68%) observed for the entire SHH cohort (n=42) should not be interpreted as progress, these figures facilitated the observation of a significant clinical distinction between iSHH-I and iSHH-II subtypes. Over 75% of patients with iSHH-II tumors survived without craniospinal irradiation, myeloablative chemotherapy, or any intra-ventricular therapy in comparison to only 25% of iSHH-I patients. Equally impressive was that iSHH-II patients who did not receive focal radiation therapy (n=14) had a 5-year progression-free survival of >90% as compared to just 25% for iSHH-I (n=20). iSHH-I was significantly enriched for SUFU aberrations (including 5/6 pathogenic SUFU germline variants), supporting the recently published finding of inferior outcomes in iSHH medulloblastomas harboring germline SUFU mutations.(26) iSHH-I was also significantly enriched for chromosome 2 gain over iSHH-II, consistent with what was recently described for the SHH-beta group defined by Cavalli et al.(21)
The small sample size and the confounding between molecular, morphologic, and clinical characteristics did not allow for definitive conclusions regarding whether molecular classification outperforms morphological- or clinical-based classification. However multiple aspects pertinent to the importance of molecular diagnosis and sub-classification were suggested in this study. As expected from prior analyses of retrospective cohorts and our molecular cohort, early childhood medulloblastoma divided into 3 consensus subgroups (SHH, G3, and G4),(20–22) but importantly, age at diagnosis had a defining role in subgroup prevalence. In the molecular cohort we observed greater than 70% of children <3 years-old were SHH, while in patients ≥3, this dropped to 30%. Conversely, non-SHH medulloblastoma represented only about one-third of children <3, but more than two-thirds between 3- and 6-years. This age variability suggests that subgroup prevalence is linked to development and that age cutoffs in clinical trials will radically alter the study population by unwittingly selecting certain subgroups over others. In the SJYC07 trial select 3-5 year-olds (M0, non-LCA, non-MYC or -MYCN amplified) were included in the intermediate-risk group with the hope that focal radiation would benefit this population. The risk classification scheme of the trial and the natural age distribution of subgroups combined to fill this intermediate-risk group with a majority of non-SHH patients and there was no significant difference in outcome. However, the potential of confounding by age remained and the indiscriminate use of age to define cohorts on clinical trials should be re-examined.
Two ETMRs, both noted by pathology to be uncharacteristic for medulloblastoma, but which also tested negative for the C19MC amplicon common to ETMR, were diagnosed and registered as medulloblastoma on the trial. While this enrollment had little effect on the overall outcome of the study, it supports the notion that methylation-based classification can assist in accurate characterization of challenging cases and reduce the number of confounding diagnoses enrolled on a clinical trial [Capper et al Nature. In press].
With regard to histology, 39 (93%) of the 42 SHH medulloblastomas from the SJYC07 cohort were DN/MBEN and 3 (7%) were CMB, thus generating highly similar survival outcomes for SHH vs. DN/MBEN cohorts (compare figure 2D and 5A). In contrast, recent publications report 50-60% of SHH in young children as DN/MBEN and 30-40% as CMB.(21, 22, 27) Therefore, while it is agreed that DN/MBEN histology remains inextricably linked to the SHH subgroup, there appears to be a lack of uniformity across cohorts as to what constitutes a SHH tumor with classic histology. It has long been recognized that inter-tumoral variability can lead to discrepant calls even among experienced pathologists,(28, 29) and since many trials use DN/MBEN histology for risk-stratification, then discrepancies in histologic diagnosis may account for widely discrepant clinical trial outcomes. Hence, in order to capture and assess uniform cohorts on clinical trials, molecular classification should be placed ahead of morphologic classification (but not replace it) as is now recommended in the 2016 World Health Organization classification of tumors of the CNS.(30)
While this study reports on a large molecularly assembled cohort of early childhood medulloblastoma and utilizes a prospectively acquired clinical cohort to report outcomes, we note the limitations regarding both sample power and bias towards single trial therapy. Given its rarity, medulloblastoma in early childhood is a challenging disease to study and the molecular analyses performed here on 190 medulloblastoma would be strengthened by the addition of more samples. Moreover, our outcome findings can only be interpreted relative to the therapy received. In retrospect the inclusion of R+ and/or LCA histology might have introduced unfavorable bias to the intermediate-risk group, however given that only 3 (9%) of 32 patients had these features the effect was nominal. We therefore strongly recommend validation in other clinical trial cohorts such that different therapies are evaluated relative to the observed molecular patterns. Since the most striking clinical finding from this study is that iSHH-II patients benefit from reduced-intensity therapy, it is important to compare this result to other reduced-intensity trials (i.e. COG clinical trial, ACNS1221(31)). Furthermore, because higher progression-free survival in DN/MBEN patients has been reported on HITT studies and with myeloablative regimens, it is necessary to assess the prevalence of iSHH-I vs iSHH-II in these cohorts in order to understand if more intensive therapies are more efficacious than what was observed here. Also, the outcome of non-SHH medulloblastoma (G3/G4) treated with intra-ventricular or myeloablative regimens needs to be assessed and analyzed for benefit.
In conclusion, we have reported on a prospective clinical trial cohort, described a large molecularly defined cohort of early childhood medulloblastoma, and added clinical context by exploring associations with survival. As such, we have identified a new subset of medulloblastoma (iSHH-II) that can be treated with reduced-intensity chemotherapy. We have described extremely poor progression-free survival for non-SHH tumors (G3/G4) and identified a subset of SHH (iSHH-I) that require more intensive and/or novel therapies. We propose that these results be replicated and validated on other recently completed pediatric brain tumor trials that have enrolled early childhood medulloblastomas so that the implications of these findings can be interpreted in a broader context. The findings described here will heavily influence risk-stratification approaches and treatment selection on the next generation of early childhood medulloblastoma trials.
Supplementary Material
Research in Context.
Evidence before this study
We searched PubMed to identify articles published between January 1, 2005 and December 31, 2017, using the search terms “medulloblastoma”, “infant OR young children OR subgroups”. Several clinical and biology studies were identified and reviewed. Overall, early childhood medulloblastoma was determined to be very challenging to study and treat for a number of reasons: (1) The incidence is rare and therefore most studies are characterized by small sample sizes. (2) The definition has been inconsistent and the term “infant” has been used to describe any child less than 3-years-old on some trials or as old as 6 on others. (3) The treatment strategies have been heterogeneous and changed from a delayed craniospinal irradiation approach in the 1980s and early 1990s to an approach that promoted craniospinal irradiation avoidance in the late 1990s and early 2000s. (4) Biologic heterogeneity, although recognized in different histologic variants, has yet to be adequately defined due to absent technology.
Nevertheless, three treatment strategies emerged as frontrunners for the treatment of this disease: (1) an approach that utilized conventional chemotherapy with high-dose myeloablative chemotherapy, (2) an approach that utilized conventional chemotherapy with both systemic and intra-ventricular methotrexate and (3) an approach that combined conventional chemotherapy with focal radiation therapy in lieu of craniospinal irradiation. Each strategy contained potential risks and benefits and each had its advocates, but all were confounded by the underlying complexities inherent to early childhood medulloblastoma as discussed above.
Added value of this study
The risk-adapted approach of this study, which utilized components of all three treatment strategies on a clinical trial population of children with medulloblastoma aged 0-5 years, provides a wide-ranging account of early childhood medulloblastoma across multiple modalities. Our results show a tiered survival pattern across all three risk groups with the low-risk group displaying improved survival over intermediate- and high-risk groups. Event-free survival of these risk groups, however, did not improve on historical outcomes causing early closure of the trial.
Most importantly, this study identifies biological heterogeneity to account for the outcome variability seen between, as well as within, medulloblastoma subgroups. Using DNA methylation profiling and next-generation sequencing (NGS), we look beyond tumor morphology and into the molecular landscape. We show SHH subgroup to be a favorable predictor of outcome and Group 3 and Group 4 disease to have extremely poor progression-free survival associated with this treatment approach. Furthermore, we identify a subtype within SHH that has a very good progression-free survival on what would be currently considered “reduced-intensity” therapy.
Implications of all the available evidence
This study lays the framework for the study of early childhood medulloblastoma. Distinctions uncovered by molecular analysis associate with progression-free survival and advance our understanding of the disease. These results will shape future medulloblastoma trials for young children into molecularly driven risk-adapted models that will ultimately transform practice.
Acknowledgments
Dr Indelicato reported non-financial support from Ion Beam Applications, outside the submitted work. Dr Ellison reported a patent Methods and compositions for typing subgroups of medulloblastoma 13/818,213 issued to St. Jude Children’s Research Hospital. Dr. Jones has a patent DNA methylation-based method for classifying tumor species pending. Dr. Waszak reports grants from European Molecular Biology Organization (EMBO), grants from Swiss National Science Foundation (SNSF), during the conduct of the study.
Footnotes
Author Contributions
GWR and PAN co-led the study and provided project supervision. GWR, VAR, and PAN prepared figures and wrote the manuscript. VAR, IB, SMW, KSS, JOK, DTWJ, MK, SMP, VAR, and PAN did bioinformatics analysis. GWR, DCB, AB, PGF, SP, JRC, TH, DJI, FB, PK, NDS, ZP, TEM, CFS, BAO, JOK, DTWJ, TS, PL, MK, AK, SMP, RJG, RPS, DWE, AG, and PAN gathered samples and patient data and provided clinical interpretation. BAO and DWE provided central pathology review. CAB and AO-T performed statistical analysis of clinical and bioinformatics data. RPS, RJG, CFS, TEM, AO-T, and AG designed and wrote the clinical trial. AG was the principal investigator of the clinical trial. All authors contributed and reviewed the final manuscript.
Competing financial interests
All other authors declare no competing financial interests
References
- 1.Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, et al. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro-oncology. 2017;19(suppl_5):v1–v88. doi: 10.1093/neuonc/nox158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. The Lancet Oncology. 2006;7(10):813–20. doi: 10.1016/S1470-2045(06)70867-1. [DOI] [PubMed] [Google Scholar]
- 3.Packer RJ, Gajjar A, Vezina G, Rorke-Adams L, Burger PC, Robertson PL, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24(25):4202–8. doi: 10.1200/JCO.2006.06.4980. [DOI] [PubMed] [Google Scholar]
- 4.Edelstein K, Spiegler BJ, Fung S, Panzarella T, Mabbott DJ, Jewitt N, et al. Early aging in adult survivors of childhood medulloblastoma: long-term neurocognitive, functional, and physical outcomes. Neuro-oncology. 2011;13(5):536–45. doi: 10.1093/neuonc/nor015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dhall G, Grodman H, Ji L, Sands S, Gardner S, Dunkel IJ, et al. Outcome of children less than three years old at diagnosis with non-metastatic medulloblastoma treated with chemotherapy on the “Head Start” I and II protocols. Pediatric blood & cancer. 2008;50(6):1169–75. doi: 10.1002/pbc.21525. [DOI] [PubMed] [Google Scholar]
- 6.Geyer JR, Sposto R, Jennings M, Boyett JM, Axtell RA, Breiger D, et al. Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children’s Cancer Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23(30):7621–31. doi: 10.1200/JCO.2005.09.095. [DOI] [PubMed] [Google Scholar]
- 7.Ashley DM, Merchant TE, Strother D, Zhou T, Duffner P, Burger PC, et al. Induction chemotherapy and conformal radiation therapy for very young children with nonmetastatic medulloblastoma: Children’s Oncology Group study P9934. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30(26):3181–6. doi: 10.1200/JCO.2010.34.4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rutkowski S, von Hoff K, Emser A, Zwiener I, Pietsch T, Figarella-Branger D, et al. Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28(33):4961–8. doi: 10.1200/JCO.2010.30.2299. [DOI] [PubMed] [Google Scholar]
- 9.Pompe RS, von Bueren AO, Mynarek M, von Hoff K, Friedrich C, Kwiecien R, et al. Intraventricular methotrexate as part of primary therapy for children with infant and/or metastatic medulloblastoma: Feasibility, acute toxicity and evidence for efficacy. Eur J Cancer. 2015;51(17):2634–42. doi: 10.1016/j.ejca.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 10.Cohen BH, Geyer JR, Miller DC, Curran JG, Zhou T, Holmes E, et al. Pilot Study of Intensive Chemotherapy With Peripheral Hematopoietic Cell Support for Children Less Than 3 Years of Age With Malignant Brain Tumors, the CCG-99703 Phase I/II Study. A Report From the Children’s Oncology Group. Pediatr Neurol. 2015;53(1):31–46. doi: 10.1016/j.pediatrneurol.2015.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rutkowski S, Bode U, Deinlein F, Ottensmeier H, Warmuth-Metz M, Soerensen N, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. The New England journal of medicine. 2005;352(10):978–86. doi: 10.1056/NEJMoa042176. [DOI] [PubMed] [Google Scholar]
- 12.Saha A, Salley CG, Saigal P, Rolnitzky L, Goldberg J, Scott S, et al. Late effects in survivors of childhood CNS tumors treated on Head Start I and II protocols. Pediatric blood & cancer. 2014;61(9):1644–52. doi: 10.1002/pbc.25064. quiz 53-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Bueren AO, von Hoff K, Pietsch T, Gerber NU, Warmuth-Metz M, Deinlein F, et al. Treatment of young children with localized medulloblastoma by chemotherapy alone: results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro-oncology. 2011;13(6):669–79. doi: 10.1093/neuonc/nor025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fouladi M, Gururangan S, Moghrabi A, Phillips P, Gronewold L, Wallace D, et al. Carboplatin-based primary chemotherapy for infants and young children with CNS tumors. Cancer. 2009;115(14):3243–53. doi: 10.1002/cncr.24362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stewart CF, Iacono LC, Chintagumpala M, Kellie SJ, Ashley D, Zamboni WC, et al. Results of a phase II upfront window of pharmacokinetically guided topotecan in high-risk medulloblastoma and supratentorial primitive neuroectodermal tumor. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2004;22(16):3357–65. doi: 10.1200/JCO.2004.10.103. [DOI] [PubMed] [Google Scholar]
- 16.Bowers DC, Aquino VM, Leavey PJ, Bash RO, Journeycake JM, Tomlinson G, et al. Phase I study of oral cyclophosphamide and oral topotecan for children with recurrent or refractory solid tumors. Pediatric blood & cancer. 2004;42(1):93–8. doi: 10.1002/pbc.10456. [DOI] [PubMed] [Google Scholar]
- 17.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer cell. 2007;11(1):69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 18.Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24(12):1924–31. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 19.Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta neuropathologica. 2012;123(4):465–72. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547(7663):311–7. doi: 10.1038/nature22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer cell. 2017;31(6):737–54 e6. doi: 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwalbe EC, Lindsey JC, Nakjang S, Crosier S, Smith AJ, Hicks D, et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. The Lancet Oncology. 2017 doi: 10.1016/S1470-2045(17)30243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hovestadt V, Remke M, Kool M, Pietsch T, Northcott PA, Fischer R, et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta neuropathologica. 2013;125(6):913–6. doi: 10.1007/s00401-013-1126-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gajjar AJ, Robinson GW. Medulloblastoma-translating discoveries from the bench to the bedside. Nature reviews Clinical oncology. 2014;11(12):714–22. doi: 10.1038/nrclinonc.2014.181. [DOI] [PubMed] [Google Scholar]
- 25.Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta neuropathologica. 2012;123(4):473–84. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guerrini-Rousseau L, Dufour C, Varlet P, Masliah-Planchon J, Bourdeaut F, Guillaud-Bataille M, et al. Germline SUFU mutation carriers and medulloblastoma: clinical characteristics, cancer risk and prognosis. Neuro-oncology. 2017 doi: 10.1093/neuonc/nox228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lafay-Cousin L, Smith A, Chi SN, Wells E, Madden J, Margol A, et al. Clinical, Pathological, and Molecular Characterization of Infant Medulloblastomas Treated with Sequential High-Dose Chemotherapy. Pediatric blood & cancer. 2016;63(9):1527–34. doi: 10.1002/pbc.26042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McManamy CS, Pears J, Weston CL, Hanzely Z, Ironside JW, Taylor RE, et al. Nodule formation and desmoplasia in medulloblastomas-defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol. 2007;17(2):151–64. doi: 10.1111/j.1750-3639.2007.00058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellison DW, Kocak M, Figarella-Branger D, Felice G, Catherine G, Pietsch T, et al. Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. J Negat Results Biomed. 2011;10:7. doi: 10.1186/1477-5751-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta neuropathologica. 2016;131(6):803–20. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 31.Lafay-Cousin L, Bouffet E, Onar A, Billups C, Hawkins C, Eberhart C, et al. ACNS1221: A phase II study for the treatment of non metastatic desmoplastic medulloblastoma in children less than 4 years of age - A report from the Children Oncology Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2017;35(15_suppl):10505. doi: 10.1200/JCO.19.00845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.