

Abstract
Understanding the heterogeneity of dysregulated pathways associated with the development of hepatocellular carcinoma (HCC) may provide prognostic and therapeutic avenues for disease management. As HCC involves a complex process of genetic and epigenetic modifications, we evaluated expression of both polyadenylated transcripts and microRNAs from HCC and liver samples derived from two cohorts of patients undergoing either partial hepatic resection or liver transplantation. Copy number variants were inferred from whole genome low‐pass sequencing data, and a set of 56 cancer‐related genes were screened using an oncology panel assay. HCC was associated with marked transcriptional deregulation of hundreds of protein‐coding genes. In the partially resected livers, diminished transcriptional activity was observed in genes associated with drug catabolism and increased expression in genes related to inflammatory responses and cell proliferation. Moreover, several long noncoding RNAs and microRNAs not previously linked with HCC were found to be deregulated. In liver transplant recipients, down‐regulation of genes involved in energy production and up‐regulation of genes associated with glycolysis were detected. Numerous copy number variants events were observed, with hotspots on chromosomes 1 and 17. Amplifications were more common than deletions and spanned regions containing genes potentially involved in tumorigenesis. Colony stimulating factor 1 receptor (CSF1R), fibroblast growth factor receptor 3 (FGFR3), fms‐like tyrosine kinase 3 (FLT3), nucleolar phosphoprotein B23 (NPM1), platelet‐derived growth factor receptor alpha polypeptide (PDGFRA), phosphatase and tensin homolog (PTEN), G‐protein‐coupled receptors‐like receptor Smoothened (SMO), and tumor protein P53 (TP53) were mutated in all tumors; another 26 cancer‐related genes were mutated with variable penetrance. Conclusion: Our results underscore the marked molecular heterogeneity between HCC tumors and reinforce the notion that precision medicine approaches are needed for management of individual HCC. These data will serve as a resource to generate hypotheses for further research to improve our understanding of HCC biology. (Hepatology Communications 2018; 00:000‐000)
Multiple processes, including genetic and environmental ones, impact the development of hepatocellular carcinoma (HCC), and therefore individual tumors exhibit a large degree of heterogeneity. HCC development occurs in the setting of cirrhosis, and oncogenesis is further promoted by chronic infection with hepatitis B virus (HBV)1 or hepatitis C virus (HCV)2 exposure to aflatoxin B1 from Aspergillus,3 alcohol abuse,4 and nonalcoholic steatohepatitis.5 HCC has a worldwide distribution,6 but its incidence is particularly high in Southeast Asia and Sub‐Saharan Africa because of HBV infection and exposure to aflatoxin B1 from contaminated grains.7 Furthermore, the prevalence of HCC is growing in Western countries as a result of chronic HCV infection.6 Despite advances in HCC diagnosis and treatment, the proportion of surgically resectable tumors remains low and recurrence of HCC after resection is high; even though liver transplantation often provides a favorable outcome, this option is costly and available to a minority of patients.8
Broadly speaking, the natural history of HCC can be divided into molecular, preclinical, and clinical/symptomatic phases.9 The molecular phase refers to tumor biogenesis, which regularly follows preneoplastic stages characterized by chronic hepatitis and cirrhosis, which entails transformation of hepatocytes, biliary epithelial cells, or stem cells.10, 11 Genetic alterations in differentiated cells result in the generation of highly proliferative cells that are recalcitrant to apoptosis, whereas transformation of stem cells leads to aberrant cellular differentiation.9 The initial epigenetic changes in transcription are often triggered by an increased exposure to growth factors and proinflammatory cytokines; thereafter, carcinogenesis is driven by chromosomal rearrangements leading to amplification of proto‐oncogenes and loss of tumor suppressor genes. HBV integration into the genome may also promote hepatocarcinogenesis by insertional mutagenesis,12 and HBV and HCV have the potential to directly modulate host pathways that contribute to hepatocyte transformation.13, 14 Specifically, chronic viral infection may promote re‐expression of telomerase reverse transcriptase to limit telomere shortening and induce chromosomal instability by disrupting mitosis checkpoints.15, 16 In the preclinical phase, the tumor may or may not be detectable by imaging and patients are asymptomatic. The clinical phase is associated with symptoms and usually occurs when the tumor is sized between 4‐8 cm.17
Studying the ontogeny of HCC may provide insight for therapeutic intervention at earlier stages of disease development. A high‐resolution picture of HCC development is currently being forged by systems biology approaches that integrate high‐throughput data on cancer genomics, epigenomics, transcriptomics, proteomics, and metabolomics into computational frameworks.18 Although considerable progress has been achieved, our understanding of the molecular diversity of HCC is still in its infancy. These studies advance with a view to generating improved diagnostic and management strategies and the basis for effective surveillance programs.
Carcinogenesis is associated with aberrant genomic and transcriptional landscapes that extend beyond protein‐coding genes into several classes of structurally and functionally different noncoding RNAs.19 Herein, we report the findings from a multiomic study conducted to investigate the molecular heterogeneity of HCC in livers derived from patients undergoing either hepatic resection (LR) or liver transplantation (LT). Our results provide a series of novel insights into the molecular heterogeneity of HCC that can be further investigated in search of biomarkers or therapeutic drug targets.
Materials and Methods
SAMPLES, NUCLEIC ACIDS, AND LIBRARIES
Explanted liver samples were collected according to protocols reviewed and approved by the Human Research Ethics Board of the University of Alberta. Three explanted livers were dissected immediately after surgery with the help of a pathologist, and tumorous and nontumorous tissues were snap frozen less than 1 hour after explanting. Twelve resected HCC liver samples and corresponding matched nontumor liver tissue were acquired from the tumor bank at the Cross Cancer Institute, University of Alberta. Resected samples were banked frozen specimens of the so‐called gold quality, which were snap frozen in a maximal period of 30 minutes after resection.
Total RNA was extracted with TRIzol Reagent (Invitrogen) as per the manufacturer's recommendation, and the same prep was used for RNA sequencing (RNAseq) and small RNAs library construction. Genomic DNA was extracted using the DNeasy Blood & Tissue kit (QIAGEN) following the manufacturer's protocol. RNA integrity and concentration were determined using an Agilent Bioanalyzer and a Bioanalyzer chip from the RNA 6000 nano kit. DNA was quantified fluorometrically in a Qubit instrument and a double‐stranded DNA HS assay kit.
For construction of RNAseq libraries, the Illumina TruSeq RNA Sample Preparation Kit V2 was used per the manufacturer's instructions. Libraries from explanted livers were sequenced in a HiSeq 2500 instrument, using a paired‐end 300‐cycles protocol at an average depth of ∼40 million paired end reads per sample. Samples from resected livers were sequenced in a MiSeq instrument using a paired‐end 150‐cycles protocol at an average depth of ∼2 million paired‐end reads per sample. Small RNAs libraries (including miRNAs) were built with the TruSeq Small RNA Library Prep Kit (Illumina) per manufacturer's instructions. Libraries were sequenced using a MiSeq instrument and 50 cycles single‐end kits, at an average depth of 1 million reads per sample. Whole genome libraries for evaluation of copy number variants (CNVs) were constructed using the Illumina Nextera XT library prep kit, according to the manufacturer's instructions. Libraries were sequenced in a MiSeq instrument using a paired‐end 500‐cycles kit at an average coverage of 0.15×. For detection of mutations in oncogenes, the Accel‐Amplicon 56G Oncology Panel v2 (Swift Biosciences) was used per provided protocols. Libraries were sequenced in a MiSeq instrument using a paired‐end 500‐cycles kit at an average depth of 500,000 paired‐end reads per sample. All samples were sequenced following a workflow that included demultiplexing and adapter trimming.
BIOINFORMATICS
Low‐quality sequences (Q < 25) were trimmed off with the Fastq‐Mcf software, and only paired‐end reads with a length of at least 75% of the original length were kept for further processing.
RNAseq Libraries
Sequences were aligned to the GRCh38.81 assembly of the human genome using the TopHat2 aligner. Reads that aligned to each gene sequence were counted with HTSeq. Differential expression analysis was conducted with the DESeq2 R package, and genes that were deregulated 2‐fold or more and had a false discovery rate (FDR) <0.05 were considered truly differentially expressed.
microRNA Libraries
Libraries adapters were removed using in‐house Perl scripts. Trimmed sequences were aligned to version 21 of the miRbase mature sequences with the Bowtie aligner. Aligned sequences were parsed with in‐house Perl scripts, and differential expression analysis was conducted as above.
CNVs
Sequences were aligned to the GRCh38.81 assembly of the human genome using Bowtie2. Sam files were then used to infer CNVs using the copy number estimation by a mixture of PoissonS (CN‐MOPS) R package, using paired nontumor samples as references.
Detection of Mutations in Oncogenes
Mutations were detected using the Genome Analyzer Toolkit (GATK) pipeline. In brief, sequences were aligned to the g1k_v37.fasta human genome sequence provided by GATK (https://software.broadinstitute.org/gatk/) with the software BWA. Reads were tagged, sorted, and indexed with the Picard tools (http://broadinstitute.github.io/picard). Base qualities were recalibrated and indels were realigned with GATK. Variants were called with the HaplotypeCaller of GATK and finally filtered (Fisher strand > 30; QualByDepth < 2.0) with the VariantFiltration program of GATK. All plots were created using the R programing language or Circos.
Data Availability
Sequences of all libraries generated in this study are available for research purposes. However, to ensure confidentiality to patients involved in the study, approval of requests by the University of Alberta ethics committee is required.
Results
RNAseq PROFILE OF HCC
RNAseq was performed on HCC or surrounding liver tissue from a total of 15 patients that underwent surgery. Of these, 12 patients had LR; half of the LR patients had an established diagnosis of cirrhosis and a third suffered from chronic viral hepatitis (see http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full for details). HCC and noncancer liver samples were also collected from 3 patients with a diagnosis of alcoholic hepatitis undergoing LT; 2 of these patients also suffered from chronic HCV infection (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). To explore the reproducibility of our own findings, we reanalyzed RNAseq data of 50 paired tumor‐control LR samples from The Cancer Genome Atlas (TCGA) database20 (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full); these samples are referred to as the TCGA cohort.
RNAseq PROFILE OF SAMPLES FROM LR
The nontumor samples from the patients who had LR were observed to cluster relatively close to each other by principal component analysis (PCA), whereas the tumor samples were far more dispersed (Fig. 1A). Two tumors (2 and 4) showed displacement from the rest, which appeared to form two separate clusters with tumors 3, 5, 10, and 11 aggregating together, while the remainder formed another less‐compact cluster (Fig. 1A). Using the combination of an FDR <0.05 and a fold change >2 as thresholds for statistical significance, 726 genes were found to be up‐regulated and 1,066 genes down‐regulated in the HCC samples (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full; Fig. 1B). The heat map representation of abundance of 200 deregulated genes with the largest variance showed the heterogeneity in expression across tumors (Fig. 1C). In concordance with the separation observed in the PCA plot, groups of tumors formed separate clades in the dendrogram, where tumors 3, 5, 10, and 11 aggregated to the left and tumors 2 and 4 aggregated to the right of the other tumors.
Figure 1.
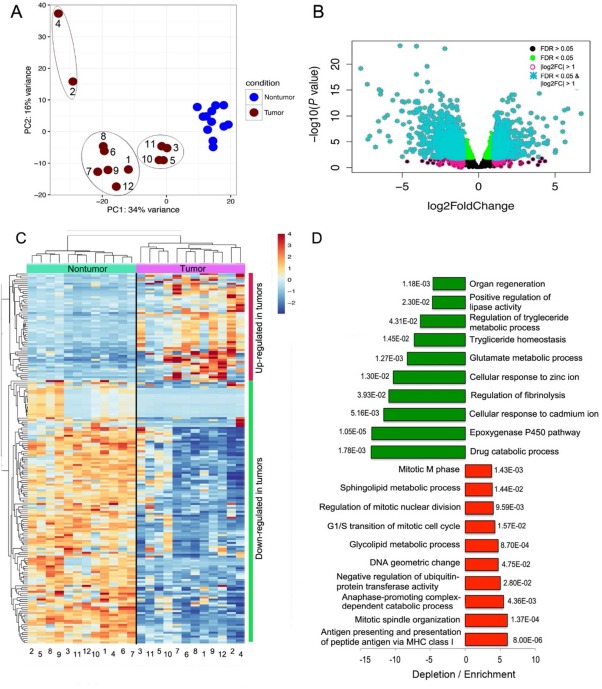
Transcriptional deregulation in HCC resected tumors hints at down‐regulation of drug catabolic processes and activation of the immune system. (A) PCA separated tumor and nontumor samples. While nontumor samples formed a tight cluster, the tumor samples dispersed along the two axes (components), forming apparent subclusters. (B) Classification of differentially expressed genes according to FDR and log2 fold change. Deregulated genes with an FDR <0.05 (green dots) and log2 fold change > 1 were considered truly differentially expressed (turquoise asterisks). (C) Hierarchical clustering of the 200 deregulated genes that exhibited the largest variance also separated tumor and nontumor samples into separate branches of a dendrogram, with larger distances in the tumor branches. Tumors were divided into the same subclusters illustrated in the PCA plot. (D) GO analyses were conducted in the Gene Ontology Consortium web portal (http://www.geneontology.org/). The 10 most enriched or most depleted terms are presented along with the corresponding P values. In general, the magnitude of depletion is larger than the magnitude of enrichment. The most diminished terms are related to drug catabolic processes and the epoxygenase P450 pathway; the top enriched terms are related to antigen presentation, DNA synthesis, and mitosis. Abbreviation: MHC, major histocompatibility complex.
The magnitude of differences in expression between HCC versus surrounding liver tissue was greater for down‐regulated versus up‐regulated genes (Fig. 1B). For example, 136 genes were down‐regulated 10‐fold or more compared to 50 up‐regulated genes (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). The same trend was also observed in the TCGA cohort (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). Among the most down‐regulated genes were those encoding members of the type‐C lectin domain family 4 (CLEC4M* and CLEC4G*), and the most up‐regulated genes included an aldehyde dehydrogenase (ALDH3A1) and a glutamate transporter (SLC7A11*) (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full; genes marked with asterisks were also found deregulated in the TCGA cohort in http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full; see Simon et al.21). Genes responsible for oxidative phosphorylation were markedly diminished in HCC, including subunits of the adenosine triphosphate (ATP) synthase complex (mitochondrially encoded [MT]‐ATP6, MT‐ATP8), cytochrome C oxidase complex (MT‐CO1, MT‐CO2, MT‐CO3), the nicotinamide adenine dinucleotide dehydrogenase complex (MT‐ND1, MT‐ND2, MT‐ND3, MT‐ND4, MT‐ND4L, MT‐ND5, MT‐ND6), ubiquinol cytochrome C reductase (MT‐CYB), the 12S and 16S mitochondrial ribosomal RNAs (MT‐RNR1, MT‐RNR2), a transfer RNA (MT transfer RNA proline [MT‐TP]), and many metallothioneins (Fig. 2; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full).
Figure 2.
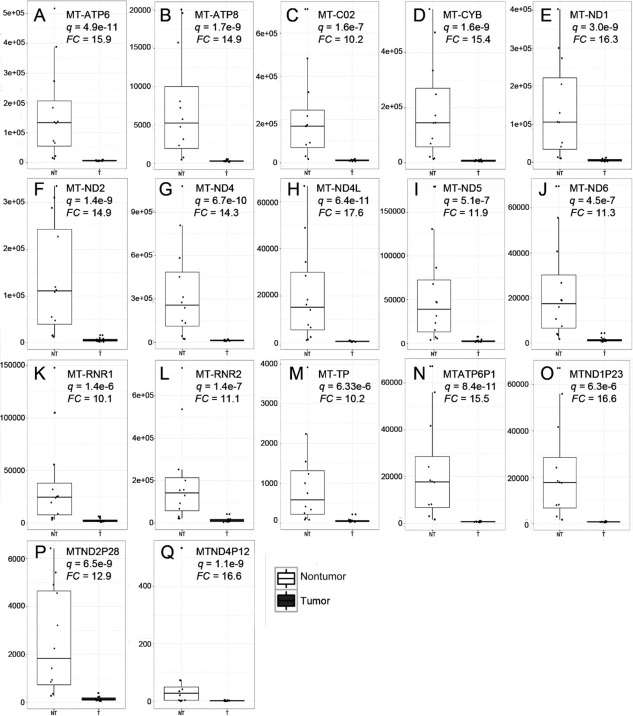
HCC is associated with down‐regulation of enzymes mediating oxidative phosphorylation. Genes that were found down‐regulated 10‐fold or more are shown: subunits of the ATP synthase (A, B), a subunit of the cytochrome C oxidase (C), a subunit of the respiratory chain protein ubiquinol cytochrome C reductase (D), subunits of the NADH dehydrogenase (E‐J), mitochondrial ribosomal RNAs (K, L), a mitochondrial tRNA (M) and pseudogenes for the mitochondrially encoded NADH ubiquinone oxidoreductase core subunits (N‐Q). The importance of pseudogenes on disease is currently under intense debate. These results correspond only to the LR samples and not to the LT or TCGA cohorts. Abbreviations: FC, fold change; CYB, ubiquinol cytochrome C reductase; NADH, nicotinamide adenine dinucleotide; RNR, 16S mitochondrial ribosomal RNA; tRNA, transfer RNA; TP, transfer RNA proline; NT, nontumor; T, tumor. Dots represent values of individual samples. Upper and lower borders of boxes represent upper limits of the third and first quartiles, respectively. Line in the middle of boxes represent the median. Whiskers represent scores outside the middle 50% of values.
In the analyses of long noncoding RNAs (lncRNAs),22 several were found substantially down‐regulated, with the exception of cytoskeleton regulator RNA (LINC00152 or CYTOR), which was found up‐regulated 3.6‐fold (Fig. 3). Especially interesting are LINC01093 and LINC01595, which were found 30‐ and 10‐fold down‐regulated. However, most lncRNAs were down‐regulated in some but not all tumors.
Figure 3.
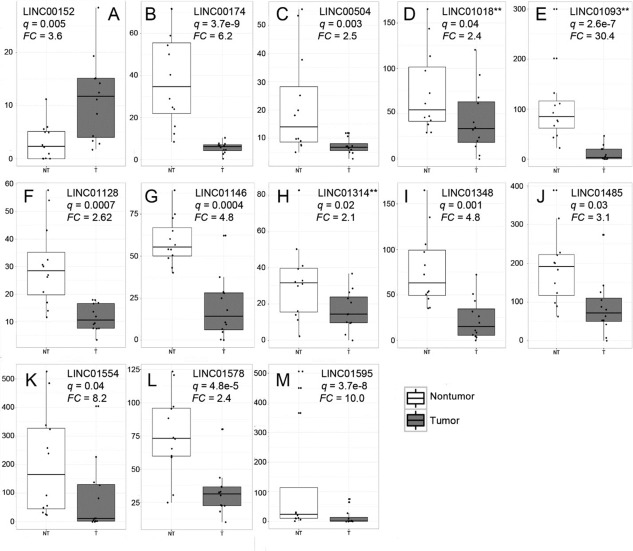
Many lncRNAs were found deregulated in resected HCC samples. (A) LINC00152 was the only lncRNA found up‐regulated in tumor tissue. All other lncRNAs found deregulated were down‐regulated in tumor samples (B‐M). **lncRNAs that were found deregulated in the resected and explanted HCC cohorts. Abbreviations: FC, fold change; NT, nontumor, T, tumor. Dots represent values of individual samples. Upper and lower borders of boxes represent upper limits of the third and first quartiles, respectively. Line in the middle of boxes represent the median. Whiskers represent scores outside the middle 50% of values.
In gene ontology (GO) analyses, the main biological process deregulated was “Drug catabolic processes” with a 13.5‐fold decrease (P = 1.8e–3; Fig. 1D), which was found down‐regulated in the TCGA cohort as well (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). This observation is in agreement with the large number of markedly down‐regulated genes belonging to the cytochrome P450 (CYP) superfamily of enzymes (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full) and to a lesser extent glutathione transferase genes (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). Notably, down‐regulation of drug catabolic processes is associated with cancer drug resistance or low responsiveness. Another GO term significantly down‐regulated was the “Epoxygenase P450 pathway” (Fig. 1D), also mediated by CYP enzymes (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). In addition to xenobiotics, CYP2C and CYP2J enzymes metabolize arachidonic acid to metabolically active epoxyeicosatrienoic acids, which are transient signaling molecules regulating immune activation and inflammation. Accordingly, we found that CYP2AC8 and CYP2C9 were down‐regulated in cancer tissue 7.1‐fold and 5.2‐fold, respectively (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). The up‐regulated pathways included those related to mitosis, lipid processing, and antigen presentation, among others (Fig. 1D).
RNAseq PROFILE OF HCC FROM LT RECIPIENTS
To gain insights into the transcriptional deregulation associated with the later stages of liver disease, we analyzed HCC and nontumor liver tissue from 3 LT recipients. The transcriptional profiles in nontumor tissues were more similar among each other than they were to their cancer counterparts (Fig. 4A), further highlighting transcriptional heterogeneity in HCC samples. In agreement with results from LR samples, a greater number of differentially expressed genes were down‐regulated (574 down‐regulated versus 343 up‐regulated), and the magnitude in fold change was larger for down‐regulated genes (Fig. 4B; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). The RNA expression profile in tumors was heterogeneous across the 200 most variable deregulated genes (Fig. 4C). The repertoire of deregulated genes was substantially different in LT and LR; indeed, only 69 genes were congruently deregulated in both HCC tumor sample groups with the same polarity (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). In GO analysis, the most decreased gene expression term was “Short‐chain fatty acid catabolism”; short‐chain fatty acids, especially butyrate, are associated with cellular homeostasis.23 Other GO terms associated with the production of energy through cellular respiration, including the tricarboxylic acid cycle and consequently oxidative phosphorylation, and terms related to the production of amino acids were also found depleted (Fig. 4D). Some terms were moderately up‐regulated, including “Cellular response to decreased oxygen levels” and “Response to hypoxia.”
Figure 4.
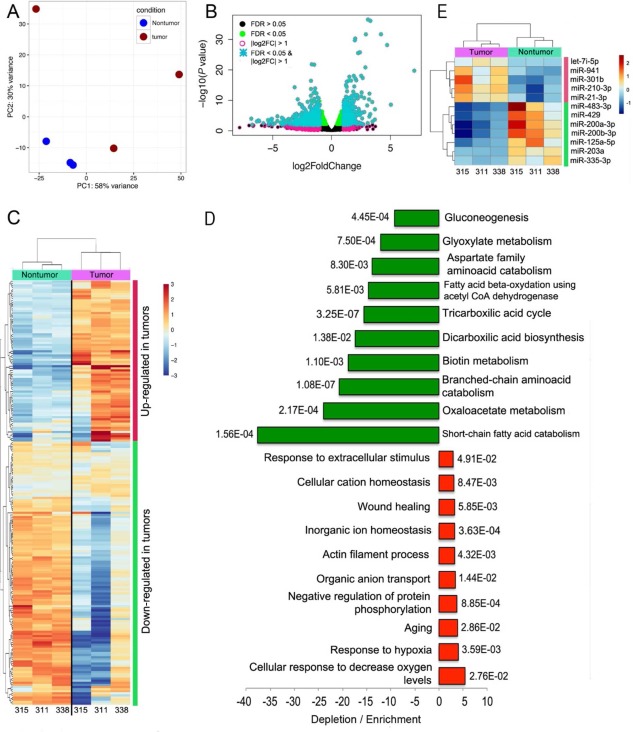
Transcriptional deregulation in HCC from LT recipients suggests general reduction of energy‐producing processes. (A) PCA separated nontumor from tumor samples that adopted a more dispersed distribution along the two principal components. One of the tumors (315) was in close proximity to nontumor samples. (B) Classification of truly differentially expressed genes was performed using an FDR <0.05 (green dots) and log2 fold change > 1 (turquoise asterisks). (C) Hierarchical clustering using the 200 deregulated genes that exhibited the largest variance also separated tumor and nontumor samples into separate branches of a dendrogram, with larger distances in the tumor branches. (D) Gene ontology analyses show the 10 most enriched or depleted terms with the corresponding P values. The magnitude is larger for depletion than for enrichment. (E) Few miRNAs were found deregulated in tumor samples, most of which have been characterized in the context of HCC. Dots represent values of individual samples. Upper and lower borders of boxes represent upper limits of the third and first quartiles, respectively. Line in the middle of boxes represent the median. Whiskers represent scores outside the middle 50% of values. Abbreviations: CoA, coenzyme A; let‐7, lethal‐7 family of miRNAs.
microRNAseq PROFILE IN LR AND LT SAMPLES
We explored the microRNA (miRNA) profile in tumor and nontumor tissues from the patient samples from this study and the TCGA cohort. Using the same thresholds (FDR, <0.05; fold change, >2), we found 30 and 44 miRNAs significantly up‐regulated or down‐regulated, respectively, in our LR samples (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full; Fig. 5B). PCA showed tightly clustered nontumor samples with a dispersed distribution of tumor samples into three discernible clusters (Fig. 5A). Notably, samples 7 and 8 were distinct from the other tumor samples, which may be attributable to the up‐regulation of the “19C miRNA cluster”24 (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). The top 96 deregulated miRNAs segregated into discrete blocks of overexpressed or underexpressed miRNAs upon hierarchical clustering (Fig. 5C), where samples 2 and 4 diverged from the rest of the tumor samples in the cladogram and formed a branch closer to the nontumor samples.
Figure 5.
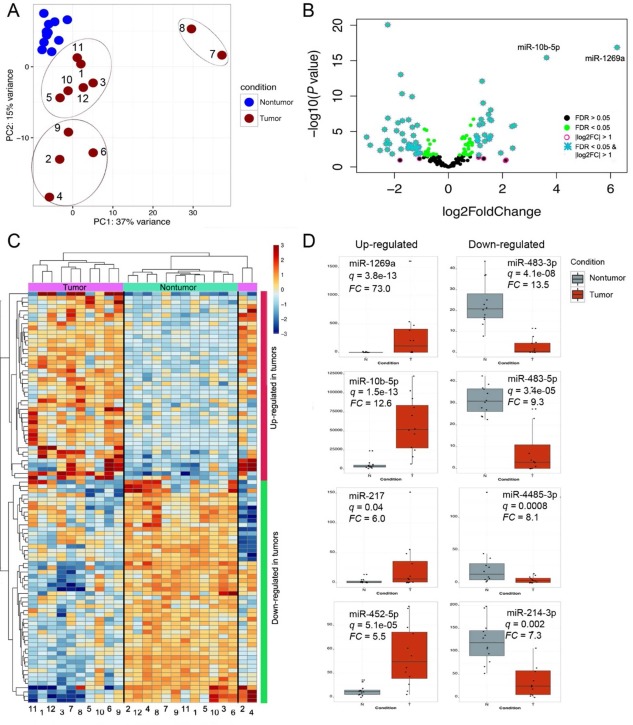
miRNA profiling reveals deregulated miRNAs that have not been previously characterized in HCC. (A) PCA of the miRNA expression data separated nontumor samples from tumor samples. Tumors 7 and 8 were considerably different from the rest of tumors in the two other sub‐clusters. (B) Classification of truly differentially expressed miRNAs was performed with an FDR <0.05 (green dots) and log2 fold change > 1 (turquoise asterisks). miR‐10b‐5p and miR‐1269a were substantially more deregulated than the rest of the miRNAs and for this reason are explicitly shown here. (C) Hierarchical clustering of 96 deregulated miRNAs exhibiting the largest variance separated most tumor from nontumor samples, except for tumors 2 and 4, which formed a branch apart and closer to nontumor samples. (D) The top four up‐regulated or down‐regulated miRNAs are shown. Abbreviation: FC, fold change.
The most up‐regulated miRNAs were miR‐1269a (73‐fold), miR‐10b‐5p (12.6‐fold), miR‐217 (6‐fold), and miR‐452‐5p (5.5‐fold). The most down‐regulated miRNAs were the two isoforms of miR‐483 (3p [13.5‐fold] and 5p [9.3‐fold]), miR‐4485‐3p (8.1‐fold), and miR‐214‐3p (7.3‐fold). Notably, all the miRNAs were also found deregulated with the same polarity in the TCGA cohort except for miR‐483‐3p and miR‐4485‐3p (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). In the LT samples, only five and seven up‐regulated and down‐regulated miRNAs were found, probably due to the small number of samples analyzed (Fig. 4E).
CNVs
CNVs include amplification or deletions of chromosomal fragments and are often associated with a gain of oncogenes and loss of tumor suppressor genes in tumor samples. CNVs were analyzed in each of the HCC samples regarding their respective noncancer liver tissue (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). In general, amplifications were observed more often than deletions (Fig. 6A; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). In the LR samples, CNVs were not distributed homogeneously within the HCC genome but were found at higher frequency on chromosomes 1, 8, 17, and Y; some chromosomes displayed few variants, such as 2, 3, 4, 13, 14, and 18, and the remainder exhibited an intermediate phenotype (Fig. 6B).
Figure 6.
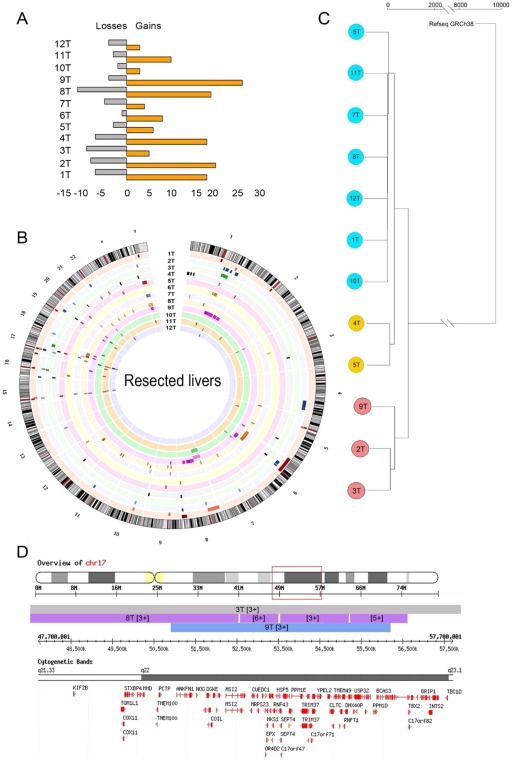
CNV profile is highly variable between samples and is generally associated with amplification of oncogenes. (A) Number of CNV events (losses and gains) in each of the liver resection HCC samples. (B) Ideogram depicting the location and size of the CNV events in each liver resection HCC sample (copy number shown in http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full). Each sample is represented in a distinct color. (C) A dendrogram was derived from the CNV profiles using hierarchical clustering with complete linkage and the GRCh38.81 human genome assembly as root. For each tumor sample, its nontumor counterpart was used as reference. (D) A representative region on chromosome 17 amplified in tumor samples 3, 8, and 9 is shown encoding genes associated with cell proliferation and metastasis. Genes TOM1L1, MSI2, MRPS23, RNF43, and INTS2 were found up‐regulated in the RNAseq libraries. (Also see http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full showing CNV on chromosomes 1 and 17; cytogenetic bands and genes included in the corresponding transect are shown for the sake of clarity). Abbreviations: chr, chromosome; INTS2, integrator complex subunit 2; MRPS23, mitochondrial ribosomal protein S23; MS12 minisatellites detected by probe MMS12; RNF43, ring finger protein 43; T, tumor sample; TOM1L1, target of myb1‐like 1 membrane trafficking protein.
Hierarchical clustering of CNVs delineated three subgroups of resected tumors (Fig. 6C), reflecting the spatial distribution of CNVs observed in the ideogram (Fig. 6B). CNVs that were present in several cancer samples were further characterized to investigate the hypothesis that they were selected due to conferring advantage for cancer progression. For example, CNV events on chromosome 17 between q21.33 and q23.1 for tumor samples 3T, 8T, and 9T show amplifications from one additional copy in samples 3 and 9 to four additional copies in a subfragment amplified in tumor 8T (Fig. 6D). In comparison with the RNAseq data, some of the genes contained in this region were found up‐regulated, including target of myb1‐like 1 membrane trafficking protein (TOM1L1 [2.35‐fold]), minisatellites detected by probe MMS12 (MSI2 [2.32‐fold]), mitochondrial ribosomal protein S23 (MRPS23 [2.36‐fold]), ring finger protein 43 (RNF43 [3.26‐fold]), and integrator complex subunit 2 (INTS2 [2.71‐fold]). Similarly, regions repeatedly amplified on chromosome 1 and chromosome 17 expressing oncogenes were also found (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full).
DIVERSITY OF MUTATIONS IN COMMON CANCER‐RELATED GENES
We used the Accel‐Amplicon 56G Oncology Panel v2 to screen for mutations in genes previously reported in cancer. Invariably, all tumors in our resected or explanted cohorts harbored mutations in the proto‐oncogenes colony stimulating factor 1 receptor (CSF1R) and the nucleolar phosphoprotein B23 (NPM1); the G‐protein‐coupled receptors, such as receptor Smoothened (SMO); the platelet‐derived growth factor receptor alpha polypeptide (PDGFRA); the fms‐like tyrosine kinase 3 (FLT3); the fibroblast growth factor receptor 3 (FGFR3); as well as in the tumor suppressors phosphatase and tensin homologue (PTEN) and the cell cycle control protein tumor protein P53 (TP53). We found 26 other genes that were mutated at variable frequency (Table 1; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full).
Table 1.
Cancer‐Related Genes Mutated in Each of the Analyzed Samples and Number of Mutations Detected per Gene
| Gene | 1T | 2T | 3T | 4T | 5T | 6T | 7T | 8T | 9T | 10T | 11T | 12T | 311T | 315T | 338T |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ABL1 | 1 | ||||||||||||||
| AKT1 | |||||||||||||||
| ALK | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| APC | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||
| ATM | 1 | 1 | |||||||||||||
| BRAF | |||||||||||||||
| CDH1 | |||||||||||||||
| CDKN2A | |||||||||||||||
| CSF1R | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| CTNNB1 | 1 | 1 | 1 | 2 | 1 | 2 | |||||||||
| DDR2 | |||||||||||||||
| DNMT3A | 2 | 2 | 2 | 2 | 2 | ||||||||||
| EGFR | 1 | 1 | 3 | 2 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 4 | 1 | ||
| ERBB2 | |||||||||||||||
| ERBB4 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||
| EZH2 | |||||||||||||||
| FBXW7 | 1 | ||||||||||||||
| FGFRl | 1 | 3 | 2 | ||||||||||||
| FGFR2 | |||||||||||||||
| FGFR3 | 3 | 2 | 1 | 3 | 3 | 2 | 3 | 4 | 2 | 2 | 2 | 2 | 1 | 2 | 4 |
| FLT3 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 |
| FOXL2 | |||||||||||||||
| GNA11 | 1 | 1 | 1 | 1 | |||||||||||
| GNAQ | |||||||||||||||
| GNAS | 1 | ||||||||||||||
| HNF1A | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||
| HRAS | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||
| 1DH1 | 1 | ||||||||||||||
| 1DH2 | 1 | 2 | |||||||||||||
| JAK2 | |||||||||||||||
| JAK3 | 1 | 1 | 1 | ||||||||||||
| KDR | 2 | 2 | 4 | 4 | 4 | 4 | 2 | 1 | 1 | 1 | 4 | 1 | 1 | ||
| K1T | 2 | 2 | 2 | 2 | 1 | 2 | 1 | ||||||||
| KRAS | 1 | ||||||||||||||
| MAP2K1 | |||||||||||||||
| MET | 2 | ||||||||||||||
| MLH1 | |||||||||||||||
| MPL | 1 | ||||||||||||||
| MSH6 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||
| NOTCH1 | |||||||||||||||
| NPM1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| NRAS | |||||||||||||||
| PDGFRA | 6 | 4 | 4 | 3 | 5 | 1 | 1 | 4 | 4 | 5 | 2 | 3 | 4 | 4 | 5 |
| P1K3CA | 2 | 1 | |||||||||||||
| PTEN | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 |
| PTPN11 | 1 | ||||||||||||||
| RB1 | |||||||||||||||
| RET | 2 | 4 | 2 | 2 | 3 | 1 | 3 | 1 | 1 | 3 | 1 | ||||
| SKT11 | |||||||||||||||
| SMAD4 | 1 | 1 | 1 | ||||||||||||
| SMARCBl | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||
| SMO | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 5 | 3 | 1 | 1 | 1 | 2 | 1 | 1 |
| SRC | |||||||||||||||
| TP53 | 4 | 4 | 3 | 4 | 5 | 5 | 3 | 3 | 3 | 4 | 3 | 4 | 5 | 4 | 4 |
| TSC1 | 1 | ||||||||||||||
| VHL |
Discussion
Next‐generation sequencing technologies and bioinformatics approaches have contributed dramatically to describe the molecular heterogeneity of HCC, but our understanding of the processes contributing to hepatocarcinogenesis is far from complete. The molecular diversity observed among HCC samples likely reflects the impact of different factors that contribute to hepatocarcinogenesis,6 including the documented intrinsic mutability caused by integrated HBV.25 Herein, we contribute an additional data set derived from cohorts of HCC tumors from resected livers and LT recipients and reanalyzed transcriptome data from 50 HCC tumors and paired controls from the TCGA project.20
In transcriptome studies, multiple genes were found deregulated in HCC, likely with different biological implications in LR and LT samples. In LR samples, the emergence of drug resistance, induction of inflammation, and increased cell proliferation were observed; these are processes all associated with tumor growth.26 In LT, a marked depletion of processes related to energy production was observed as well as moderate enrichment of functions related to hypoxia tolerance, which together point toward the well‐characterized strong reliance of cancer cells on glycolysis for energy production.27 It is also conceivable that the increased human leukocyte antigen class I antigen presentation in HCC may well represent the presence of infiltrating macrophages responding to the hypoxia inducible factor‐α in the hypoxic microenvironment.28 LR and LT represent snapshots of different stages of HCC, and the differences observed in the molecular profile of such samples likely reflect temporal divergences in cancer cell physiology. For instance, initial stages of tumorigenesis are associated with highly proliferative cellular phenotypes (tumor growth). Later, when tumor cells have proliferated excessively, a hypoxic microenvironment promotes a shift from oxidative phosphorylation toward glycolysis. Other metabolic processes are also expected to slow in older tumor cells due to the progressive perturbation of cell metabolism.
Individual genes warrant further investigation. For instance, the C‐type lectin CLEC4G (or LSECtin), found to be 156‐fold down‐regulated in our study and 538‐fold in the TCGA cohort, has been reported to favor metastasis of colon carcinoma to the liver, likely by promoting adhesion of cancer cells in the liver.29 In addition, LSECtin antagonizes activated T cells in the liver to maintain a tolerogenic environment under normal physiologic conditions.30 Thus, down‐regulation of LSECtin in HCC tumors may contribute to inflammation and diminished adhesion that favors both tumorigenesis and metastasis.
We also observed up‐regulation of lncRNAs, such as LINC00152, which has been implicated in the progression of gastric cancer and HCC.31, 32 Similarly, down‐regulated LINC01018 may also contribute to hepatocellular carcinogenesis as epigenetic silencing of this lncRNA has been shown to promote HCC proliferation.33 Diminished LINC01093 has been associated with HCC progression, and this was the most deregulated lncRNA in our study, with a decreased fold change of 30.4.34, 35 Thus, studying the mechanisms whereby LINC01093 antagonizes HCC might be a promising avenue for the management of HCC. The other lncRNAs reported in Fig. 3 have not been implicated in HCC, and their potential contribution to disease remains to be elucidated.
MiRNAs are another important class of regulatory noncoding RNAs involved in liver diseases.36 The most up‐regulated miRNA in this study was miR‐1269a (73‐fold), which has been linked with relapse and metastasis of colorectal cancer,37 relapse of squamous cell esophageal carcinoma,38 and proliferation of HCC by down‐regulation of forkhead box O1 (FOXO1).39 One interesting down‐regulated miRNA in our study was miR‐4485‐3p, which was found deregulated in a gastric cancer cell line40 and shown to block angiogenesis in Kaposi's sarcoma.41 Given its therapeutic potential as an antagonist of angiogenesis, further research is warranted. The number of deregulated miRNAs in LT samples was small (n = 9); this is likely a consequence of the limited number of samples in this case (n = 3). Of those, five miRNAs were also found deregulated in LR samples. The four miRNAs that were not found deregulated in LR samples might represent miRNAs for which deregulation is associated with later stages of HCC.
CNVs were abundant in our HCC samples, especially those on chromosome 17, which suggests high susceptibility to structural variations in such chromosomes. Some regions of chromosome 17 found amplified contained genes that were up‐regulated in our RNAseq data. These include MRPS23, associated with metastatic phenotypes of uterine cervical cancer42 and breast cancer43; MSI2, which promotes metastasis of non‐small cell lung cancer through regulation of tumor growth factor β44; and TOM1L1, which promotes breast cancer cell invasion.45 Furthermore, 34 of 56 genes that were screened were mutated to different degree. In similar studies, including a larger number of samples, several genes have been reported frequently lacerated in HCC. These include TP53, catenin beta 1 (CTNNB1), Janus kinase 1 (JAK1), axin 1 (AXIN1), and FGF3.46, 47, 48 The tumor suppressors TP53 and PTEN and six other genes were found mutated in all our samples. CTNNB1 was found mutated in 6/15 of our tumors, while the rest were not found mutated or were not tested here.
Most of the phase III trials of molecular‐targeted therapies for HCC have been disappointing,49 and currently the only first‐line molecular‐targeted therapy approved for HCC is sorafenib,50 with benefit limited to 3 months survival. Therefore, the current goal of HCC research is to identify targetable pathways amenable to intervention, but this is easier said than done. For example, our clustering techniques applied to RNAseq, miRNAseq, and CNV profiling led to substantially different subsets of tumor samples, underscoring the intrinsic limitation of stratification of tumors based on single layers of information. Another shortfall of this study was the limited number of samples and lack of clinical data, with the resulting inability to link phenotypic with genotypic data. One possible solution for this problem could be the incorporation of multiple omics data sets together with clinical metadata and disease outcome information; machine‐learning approaches could then be used to model the combined contribution of several layers of information with disease outcome. Performance of this computational approach will improve as data sets from different laboratories across the world continue to describe the heterogeneity of HCC. However, current algorithms for managing HCC de‐emphasize tissue sampling unless there is diagnostic uncertainty. This may well change if precision medicine with the multiomics approaches can identify aberrant pathways amenable for targeted therapy. Accordingly, it is hoped that the information generated in this study will promote hypotheses testing to increase our comprehension of liver cancer biology.
Author names in bold designate shared co‐first authorship.
Abbreviations
- CLEC4
type‐C lectin domain family 4
- CNV
copy number variant
- CYP
cytochrome P450
- FDR
false discovery rate
- GATK
Genome Analyzer Toolkit
- GO
gene ontology
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- LINC00152 (or CYTOR)
cytoskeleton regulator RNA
- lncRNA
long noncoding RNA
- LR
hepatic resection
- LSECtin
type‐C lectin domain family 4
- LT
liver transplantation
- miRNA #miR
microRNA
- MT
mitochondrially encoded
- MT‐ATP
adenosine triphosphate synthase complex
- MT‐CO
cytochrome C oxidase complex
- MT‐ND
nicotinamide adenine dinucleotide dehydrogenase complex
- PCA
principal component analysis
- RNAseq
RNA sequencing
- TCGA
The Cancer Genome Atlas
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full.
Supporting Information
Potential conflict of interest: Dr. Mason consults, is on the speakers' bureau, and received grants from Intercept. He holds intellectual property rights with RevTree.
Supported by funding from Alberta Innovates Technology Futures‐Innovates Centres of Research Excellence (to G.K.S.W.; RES00110334), a Senior Scholar Award from Alberta Heritage Foundation for Medical Research (to A.L.M.; award 200800252), and a studentship from the Canadian Liver Foundation (to Z.L.).
Contributor Information
Juan Jovel, Email: jovel@ualberta.ca.
Andrew L. Mason, Email: am16@ualberta.ca
REFERENCES
- 1. Chang K‐M, Liu M. Chronic hepatitis B: immune pathogenesis and emerging immunotherapeutics. Curr Opin Pharmacol 2016;30:93‐105. [DOI] [PubMed] [Google Scholar]
- 2. Lingala S, Ghany MG. Natural history of hepatitis C. Gastroenterol Clin North Am 2015;44:717‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kew MC. Aflatoxins as a cause of hepatocellular carcinoma. J Gastrointestin Liver Dis 2013;22:305‐310. [PubMed] [Google Scholar]
- 4. Abenavoli L, Masarone M, Federico A, Rosato V, Dallio M, Loguercio C, et al. Alcoholic hepatitis: pathogenesis, diagnosis and treatment. Rev Recent Clin Trials 2016;11:159‐166. [DOI] [PubMed] [Google Scholar]
- 5. Charrez B, Qiao L, Hebbard L. Hepatocellular carcinoma and non‐alcoholic steatohepatitis: the state of play. World J Gastroenterol 2016;22:2494‐2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Balogh J, Victor D, Asham EH, Burroughs SG, Boktour M, Saharia A, et al. Hepatocellular carcinoma: a review. J Hepatocell Carcinoma 2016;3:41‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi‐Lakeh M, MacIntyre MF, et al. Global Burden of Disease Cancer Collaboration. The global burden of cancer 2013. JAMA Oncol 2015;1:505‐527. Erratum in: JAMA Oncol 2015;1:690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morise Z, Kawabe N, Tomishige H, Nagata H, Kawase J, Arakawa S, et al. Recent advances in liver resection for hepatocellular carcinoma. Front Surg 2014;1:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trevisani F, Cantarini MC, Wands JR, Bernardi M. Recent advances in the natural history of hepatocellular carcinoma. Carcinogenesis 2008;29:1299‐1305. [DOI] [PubMed] [Google Scholar]
- 10. D'Errico A, Baccarini P, Fiorentino M, Ceccarelli C, Bonazzi C, Ponzetto A, et al. Histogenesis of primary liver carcinomas: strengths and weaknesses of cytokeratin profile and albumin mRNA detection. Hum Pathol 1996;27:599‐604. [DOI] [PubMed] [Google Scholar]
- 11. Yao Z, Mishra L. Cancer stem cells and hepatocellular carcinoma. Cancer Biol Ther 2009;8:1691‐1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rajput P, Shukla SK, Kumar V. The HBx oncoprotein of hepatitis B virus potentiates cell transformation by inducing c‐Myc‐dependent expression of the RNA polymerase I transcription factor UBF. Virol J 2015;12:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gearhart TL, Bouchard MJ. The hepatitis B virus X protein modulates hepatocyte proliferation pathways to stimulate viral replication. J Virol 2010;84:2675‐2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene 2006;25:3834‐3847. [DOI] [PubMed] [Google Scholar]
- 15. Forgues M, Difilippantonio MJ, Linke SP, Ried T, Nagashima K, Feden J, et al. Involvement of Crm1 in hepatitis B virus X protein‐induced aberrant centriole replication and abnormal mitotic spindles. Mol Cell Biol 2003;23:5282‐2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sell S, Leffert HL. Liver cancer stem cells. J Clin Oncol 2008;26:2800‐2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dhanasekaran R, Venkatesh SK, Torbenson MS, Roberts LR. Clinical implications of basic research in hepatocellular carcinoma. J Hepatol 2016;64:736‐745. Erratum in: J Hepatol 2016;64:1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet 2013;45:1127‐1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chakravarthi BV, Nepal S, Varambally S. Genomic and epigenomic alterations in cancer. Am J Pathol 2016;186:1724‐1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tomczak K, Czerwińska P, Wiznerowicz M. The cancer genome atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn) 2015;19:A68‐A77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simon EP, Freije CA, Farber BA, Lalazar G, Darcy DG, Honeyman JN, et al. Transcriptomic characterization of fibrolamellar hepatocellular carcinoma. Proc Natl Acad Sci U S A 2015;112:E5916‐E5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deniz E, Erman B. Long noncoding RNA (lincRNA), a new paradigm in gene expression control. Funct Integr Genomics 2017;17:135‐143. [DOI] [PubMed] [Google Scholar]
- 23. Sivaprakasam S, Prasad PD, Singh N. Benefits of short‐chain fatty acids and their receptors in inflammation and carcinogenesis. Pharmacol Ther 2016;164:144‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Flor I, Bullerdiek J. The dark side of a success story: microRNAs of the C19MC cluster in human tumours. J Pathol 2012;227:270‐274. [DOI] [PubMed] [Google Scholar]
- 25. Zhao L‐H, Liu X, Yan H‐X, Li W‐Y, Zeng X, Yang Y, et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat Commun 2016;7:12992. Erratum in: Nat Commun 2016;7:13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yokota J. Tumor progression and metastasis. Carcinogenesis 2000;21:497‐503. [DOI] [PubMed] [Google Scholar]
- 27. Devic S. Warburg effect ‐ a consequence or the cause of carcinogenesis? J Cancer 2016;7:817‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang J, Zhang Q, Lou Y, Fu Q, Chen Q, Wei T, et al. Hypoxia‐inducible factor‐1α/interleukin‐1β signaling enhances hepatoma epithelial‐mesenchymal transition through macrophages in a hypoxic‐inflammatory microenvironment. Hepatology 2018;67:1872‐1889. [DOI] [PubMed] [Google Scholar]
- 29. Zuo Y, Ren S, Wang M, Liu B, Yang J, Kuai X, et al. Novel roles of liver sinusoidal endothelial cell lectin in colon carcinoma cell adhesion, migration and in‐vivo metastasis to the liver. Gut 2013;62:1169‐1178. [DOI] [PubMed] [Google Scholar]
- 30. Tang L, Yang J, Liu W, Tang X, Chen J, Zhao D, et al. Liver sinusoidal endothelial cell lectin, LSECtin, negatively regulates hepatic T‐cell immune response. Gastroenterology 2009;137:1498‐1508.e1‐e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou J, Zhi X, Wang L, Wang W, Li Z, Tang J, et al. Linc00152 promotes proliferation in gastric cancer through the EGFR‐dependent pathway. J Exp Clin Cancer Res 2015;34:135. Erratum in: J Exp Clin Cancer Res 2016;35:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cao L, Zhang Q, Cheng S, Chen Z, Hua Z, Yang J, et al. Long non‐coding RNAs and genes contributing to the generation of cancer stem cells in hepatocellular carcinoma identified by RNA sequencing analysis. Oncol Rep 2016;36:2619‐2624. [DOI] [PubMed] [Google Scholar]
- 33. Zheng H, Yang S, Yang Y, Yuan S‐X, Wu F‐Q, Wang L‐L, et al. Epigenetically silenced long noncoding‐SRHC promotes proliferation of hepatocellular carcinoma. J Cancer Res Clin Oncol 2015;141:1195‐1203. [DOI] [PubMed] [Google Scholar]
- 34. Waxman S, Wurmbach E. De‐regulation of common housekeeping genes in hepatocellular carcinoma. BMC Genomics 2007;8:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jin B, Wang W, Du G, Huang G‐Z, Han L‐T, Tang Z‐Y, et al. Identifying hub genes and dysregulated pathways in hepatocellular carcinoma. Eur Rev Med Pharmacol Sci 2015;19:592‐601. [PubMed] [Google Scholar]
- 36. Otsuka M, Kishikawa T, Yoshikawa T, Yamagami M, Ohno M, Takata A, et al. MicroRNAs and liver disease. J Hum Genet 2017;62:75‐80. [DOI] [PubMed] [Google Scholar]
- 37. Bu P, Wang L, Chen K‐Y, Rakhilin N, Sun J, Closa A, et al. miR‐1269 promotes metastasis and forms a positive feedback loop with TGF‐β. Nat Commun 2015;6:6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jang H‐J, Lee H‐S, Burt BM, Lee GK, Yoon K‐A, Park Y‐Y, et al. Integrated genomic analysis of recurrence‐associated small non‐coding RNAs in oesophageal cancer. Gut 2017;66:215‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang X‐W, Shen G‐Z, Cao L‐Q, Jiang X‐F, Peng H‐P, Shen G, et al. MicroRNA‐1269 promotes proliferation in human hepatocellular carcinoma via downregulation of FOXO1. BMC Cancer 2014;14:909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu J, Ma L, Wang Z, Wang L, Liu C, Chen R, et al. MicroRNA expression profile of gastric cancer stem cells in the MKN‐45 cancer cell line. Acta Biochim Biophys Sin (Shanghai) 2014;46:92‐99. [DOI] [PubMed] [Google Scholar]
- 41. Chang H‐C, Hsieh T‐H, Lee Y‐W, Tsai C‐F, Tsai Y‐N, Cheng C‐C, et al. c‐Myc and viral cofactor Kaposin B co‐operate to elicit angiogenesis through modulating miRNome traits of endothelial cells. BMC Syst Biol 2016;10(Suppl. 1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lyng H, Brøvig RS, Svendsrud DH, Holm R, Kaalhus O, Knutstad K, et al. Gene expressions and copy numbers associated with metastatic phenotypes of uterine cervical cancer. BMC Genomics 2006;7:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gatza ML, Silva GO, Parker JS, Fan C, Perou CM. An integrated genomics approach identifies drivers of proliferation in luminal‐subtype human breast cancer. Nat Genet 2014;46:1051‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kudinov AE, Deneka A, Nikonova AS, Beck TN, Ahn Y‐H, Liu X, et al. Musashi‐2 (MSI2) supports TGF‐β signaling and inhibits claudins to promote non‐small cell lung cancer (NSCLC) metastasis. Proc Natl Acad Sci U S A 2016;113:6955‐6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chevalier C, Collin G, Descamps S, Touaitahuata H, Simon V, Reymond N, et al. TOM1L1 drives membrane delivery of MT1‐MMP to promote ERBB2‐induced breast cancer cell invasion. Nat Commun 2016;7:10765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL, et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology 2013;58:1693‐1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 2015;47:505‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, et al. Whole‐genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res 2013;23:1422‐1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Llovet JM, Zucman‐Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Prim 2016;2:16018. [DOI] [PubMed] [Google Scholar]
- 50. Heimbach JK, Kulik LM, Finn RS, Sirlin CB, Abecassis MM, Roberts LR, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018;67:358‐80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1197/full.
Supporting Information
Data Availability Statement
Sequences of all libraries generated in this study are available for research purposes. However, to ensure confidentiality to patients involved in the study, approval of requests by the University of Alberta ethics committee is required.