

Abstract
B cells express an antigen‐specific B‐cell receptor (BCR) and may contribute to liver inflammation by recognizing shared antigens in the gut and liver. Herein, we used high‐throughput BCR sequencing of the immunoglobulin heavy chain, specifically the complementarity‐determining region 3 (CDR3), to characterize the B‐cell repertoire of freshly‐frozen paired gut and liver tissue samples from patients with primary sclerosing cholangitis (PSC) and concurrent inflammatory bowel disease (IBD) (PSC‐IBD, n = 10) and paired formalin‐fixed paraffin‐embedded (FFPE) tumor‐adjacent normal colon and liver tissue from patients with colorectal liver metastases (controls, n = 10). We observed significantly greater numbers of B cells (P < 0.01) and unique B‐cell clonotypes (P < 0.05) in gut samples compared to liver samples of patients with PSC‐IBD, whereas BCR sequences in FFPE normal gut and liver samples were nearly absent (14 ± 5 clonotypes; mean ± SD; n = 20). In PSC‐IBD, an average of 8.3% (range, 1.6%‐18.0%) of B‐cell clonotypes were found to overlap paired gut and liver samples following the exclusion of memory clonotypes reported in the blood of healthy controls. Overlapping gut and liver clonotypes showed stronger evidence of antigen‐driven activation compared to non‐overlapping clonotypes, including shorter CDR3 lengths and higher counts of somatic hypermutation (P < 0.0001). Conclusion: A proportion of gut and liver B cells originate from a common clonal origin (i.e., likely to recognize the same antigen) in patients with PSC which suggests B‐cell antigens are shared across the gut–liver axis. (Hepatology Communications 2018; 00:000‐000)
Primary sclerosing cholangitis (PSC) is a chronic inflammatory and fibrosing hepatobiliary disease strongly associated with inflammatory bowel disease (IBD) in a majority of patients.1, 2, 3, 4 No effective medical treatments exist for patients with PSC‐IBD and the pathologic basis of the gut–liver relationship in PSC‐IBD remains unclear. Activated lymphocytes responding to microbial products (antigens, metabolites, toxins) circulating within the enterohepatic axis5, 6, 7, 8 might be disease relevant as patients with PSC‐IBD show a distinct microbiota in the gut compared to patients with ulcerative colitis and healthy controls.9 Supporting this hypothesis, we have detected that a greater number and proportion of gut and liver T cells share a common clonal origin in patients with PSC‐IBD compared to non‐PSC‐IBD controls.10, 11 These findings suggest immune cells migrating from the gut to the liver drive hepatic inflammation and antigens shared between both tissue compartments may trigger the activation of adaptive immunity in PSC‐IBD.
The pathogenic role of B cells in PSC‐IBD is poorly established as a broad spectrum of non‐specific autoantibodies to biliary and colonic epithelial antigens, neutrophil granulocytes (perinuclear anti‐neutrophil cytoplasmic antibodies [pANCA]), and several ubiquitous self‐proteins appear in affected individuals.1, 2, 3, 4, 12 Although disease‐specific autoantibodies have not been identified in sera,12 we have demonstrated that B cells infiltrating liver explants of patients with PSC‐IBD can be isolated and cultured to produce a diverse repertoire of autoantibodies13 whose specificities may point toward relevant self‐antigens or cross‐reactive exogenous targets.14 Thus, a better appreciation of B‐cell reactivities in PSC‐IBD may uncover useful biomarkers similar to patients with primary biliary cholangitis (PBC) where liver‐infiltrating B cells produce disease‐associated anti‐mitochondrial antibodies to pyruvate dehydrogenase complex‐E2.13 A better understanding of B‐cell reactivities in PSC‐IBD may also help define patient subgroups, including specific genetic and disease characteristics of pANCA‐positive versus seronegative patients.15
As PSC‐IBD lacks a validated antibody response and suitable material from patients and appropriate controls is rare, studying the B‐cell specificity in PSC‐IBD by traditional methodologies has been challenging and controversial.16 Thus, we sought to characterize the specificity of gut and liver B cells in patients with PSC‐IBD, using high‐throughput B‐cell receptor (BCR) sequencing. We aimed to determine if gut‐ and liver‐infiltrating B cells in patients with PSC‐IBD carry the same BCRs (i.e., are clonally related and able to recognize the same antigen) and whether B cells of common clonal origin show features of antigen recognition.
Materials and Methods
STUDY POPULATIONS
Paired, snap‐frozen, inflamed colonic biopsies from the ascending colon (1‐5 mg) and explanted liver tissue (50‐60 mg) from a Norwegian cohort of patients with PCS‐IBD (n = 10) were used in this study (see Table 1 for detailed clinical characteristics of the PSC‐IBD study cohort at the time of sample collection). Genomic DNA isolated from the same tissue samples was previously used to study shared T‐cell clonality in the gut and liver of patients with PSC‐IBD.11 PSC was diagnosed on the basis of accepted criteria with typical findings of bile duct irregularities on cholangiography.17 IBD diagnosis and classification was based on commonly accepted clinical, endoscopic, and histopathologic criteria.18 Nine patients were diagnosed with ulcerative colitis, and 1 patient had Crohn's disease. Colonic biopsies were collected during routine colonoscopy, and active colonic inflammation was graded according to the Mayo ulcerative colitis endoscopic subscoring system.19 The median time from colonoscopy to liver transplantation was 3.5 years (range, 0.6‐7.3 years; Supporting Table S1). Paired formalin‐fixed, paraffin‐embedded (FFPE), tumor‐adjacent normal gut and liver tissue was collected at Queen Elizabeth Hospital Birmingham (Birmingham, United Kingdom) from patients with colon cancer who developed liver metastases (n = 10; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1200/full).
Table 1.
Clinical Details of the PSC‐IBD Study Cohort
| Characteristics | PSC‐IBD (n = 10) | |
|---|---|---|
| At Colonoscopy | At Liver Transplantation | |
| Age, years | 44 ± 11 | 47 ± 10 |
| Male sex, % | 60 | 60 |
| Bilirubin, μmol/L | 25 ± 15 | 16 ± 10 |
| INR | 1.1 ± 0.1 | 1.1 ± 0.1 |
| ALT, U/L | 190 ± 156 | 86 ± 79 |
| ALP, U/L | 318 ± 259 | 231 ± 126 |
| Albumin, g/L | 39 ± 6 | 40 ± 6 |
| Platelets, 109/L | 298 ± 101 | 293 ± 125 |
Values are mean ± SD.
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; INR, international normalized ratio.
ETHICS
This study was performed in accordance with the Declaration of Helsinki and was approved by local research ethics committees (Oslo University Hospital Rikshospitalet [2012‐286] and Human Biomaterials Research Centre University of Birmingham [15/NW/0079]). Written informed consent was obtained from all patients.
HIGH‐THROUGHPUT SEQUENCING AND ANALYSIS OF THE BCR IMMUNOGLOBULIN HEAVY CHAIN
Genomic DNA was extracted from affected colonic biopsies and explanted whole liver tissue, using QIAGEN AllPrep DNA/RNA micro and mini kits (Valencia, CA) according to the manufacturer's protocol. Genomic DNA from paired, FFPE, tumor‐adjacent normal gut and liver tissue was extracted using QIAGEN QIAamp DNA FFPE tissue kits according to the manufacturer's protocol.
BCR sequencing of the immunoglobulin heavy chain (IgH) complementarity determining region 3 (CDR3) was performed by Adaptive Biotechnologies (Seattle, WA) using genomic DNA samples extracted as described above. Unique B‐cell clonotypes were defined as productive BCR nucleotide rearrangements containing in‐frame and distinct CDR3 amino acid sequences lacking a stop codon and containing a conserved cysteine residue at the 3′ variable gene and terminal phenylalanine residue at the 5′ joining gene segments.20, 21, 22 B‐cell clonality was analyzed using the Adaptive Biotechnologies immunoSEQ platform (http://www.adaptive-biotech.com/immunoseq/analyzer). B‐cell clonality was calculated as 1 – (Shannon's Entropy)/log2(number of nucleotide clonotypes). Clonal distribution of private B‐cell clonotypes was calculated by clustering BCR template sequences detected at a frequency of 1, 2 to 10, or greater than 10. Clonotype overlap was calculated as (number of shared BCR templates per amino acid clonotype)/(total number of BCR templates in the total liver and gut sample) × 100, following the exclusion of memory BCR sequences from the blood of healthy controls23; this is publicly accessible through Adaptive Biotechnologies immuneACCESS web portal (https://clients.adaptivebiotech.com/immuneaccess).
STATISTICAL ANALYSES
All values are presented as mean ± SD unless otherwise stated. Nonparametric paired and unpaired t tests were used for comparative analysis between paired and unpaired gut and liver clonotypes. In addition, we used one‐way analysis of variance for multiple unpaired group analysis of CDR3 lengths and somatic hypermutation (SHM) counts for nonparametric data (D'Agostino and Pearson omnibus normality test). GraphPad Prism 6.0 (La Jolla, CA) was used for statistical analyses and figure graphs. P < 0.05 was regarded as statistically significant.
Results
B‐CELL CLONALITY IN THE GUT IS GREATER THAN THE LIVER IN PATIENTS WITH PSC‐IBD
To assess the B‐cell clonality in the gut and liver of patients with PSC‐IBD (Table 1; Supporting Table S1), we first determined the approximate number of B cells within each gut or liver sample by calculating the sum of genomic DNA templates for productive BCR nucleotide rearrangements. Gut samples contained a significantly greater number of B cells (gut, 13,937 ± 4,710 per μg of sample DNA; liver, 5,949 ± 5,476 per μg of sample DNA; P < 0.01) (Fig. 1A) and a greater number of unique B‐cell clonotypes (gut, 7,083 ± 3,637 per μg of sample DNA; liver, 3,348 ± 2,117 per μg of sample DNA; P < 0.05) (Fig. 1B) compared to liver samples. B‐cell clonotypes were defined as productive BCR nucleotide rearrangements with distinct in‐frame CDR3 amino acid sequences lacking a stop codon and containing a conserved cysteine residue at the 3′ variable gene and terminal phenylalanine residue at the 5′ joining gene segments.20, 21, 22
Figure 1.
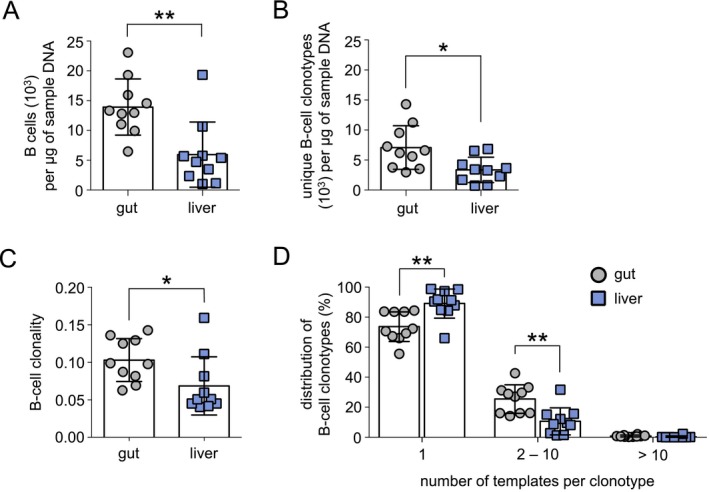
B‐cell clonality in the gut is greater than in the liver in patients with PSC‐IBD. High‐throughput BCR sequencing was completed on paired gut and liver samples from patients with PSC‐IBD (n = 10). (A) B cell/μg of tissue DNA as measured by the total number of templates for a specific BCR rearrangement/μg of sample DNA. B‐cell clonotypes were defined as a productive nucleotide BCR rearrangement with a distinct IgH CDR3. (B) Number of unique B‐cell clonotypes/μg of tissue DNA. (C) Clonality of the gut and liver B‐cell compartment as defined by the relative expansion of each B‐cell clonotype and calculated as 1 − (Shannon's Entropy)/log2 × (number of productive nucleotide clonotypes). A clonality of 1 represents a monoclonal sample, whereas a clonality of 0 represents a completely polyclonal sample. (D) Clonal distribution of each B‐cell clonotype as defined by the number of BCR templates in the sample. B‐cell clonotypes detected only once per sample most likely represent naive B cells, whereas B‐cell clonotypes found at 2‐10 templates have expanded. Clonotypes found at greater than 10 templates per sample represent highly‐expanded B cells. Horizontal lines indicate the mean, and error bars indicate SD. Statistical significance was determined using non‐parametric paired t tests, *P < 0.05, **P < 0.01.
To evaluate the relative expansion of each B‐cell clonotype, we next compared the clonality of the total clonotypes in gut and liver samples from patients with PSC‐IBD whereby the relative abundance of each clonotype was determined as 1 – (Shannon's Entropy)/log2 × (number of productive nucleotide clonotypes). A clonality of 0 represents a polyclonal sample, and clonality of 1 reflects a monoclonal sample. Using this index for relative abundance, the clonality of gut samples was calculated as marginally higher than liver samples from patients with PSC‐IBD (gut, 0.103 ± 0.003; liver, 0.068 ± 0.004; P < 0.01) (Fig. 1C). Both B‐cell repertories appeared substantially more clonal than sorted and sequenced CD19+CD27+ memory B cells from the peripheral blood of healthy controls.23 Together, these findings indicate that B cells in the gut undergo greater clonal expansion compared to the liver. In agreement with these results, we detected a significantly higher proportion of expanded B cells with 2 to 10 templates per clonotype compared to the liver (gut, 25.5% ± 3.0% liver, 10.6% ± 2.9% P < 0.01) and a lower percentage of B cells detected at a DNA template of 1 (gut, 73.6% ± 3.1% liver, 89.1% ± 3.1% P < 0.01) (Fig. 1C). The proportion of highly expanded B cells with more than 10 templates per clonotype was rare in both the gut and liver of PSC‐IBD samples, and the differences in mean were statistically insignificant (gut, 0.8 ± 0.2% liver, 0.3 ± 0.2% P = 0.06).
BCR SEQUENCES ARE RARE IN TUMOR‐ADJACENT NORMAL FFPE GUT AND LIVER TISSUE
Next we compared B‐cell clonality of paired gut and liver samples from patients with PSC‐IBD to tumor‐adjacent normal gut and liver FFPE tissue from patients with colon cancer who developed colorectal metastases in the liver. We detected on average 49,479 ± 6,479 B‐cell clonotypes per gut and liver PSC‐IBD sample (n = 20) and a near absence of B‐cell clonotypes in FFPE samples (14 ± 5, n = 20) (Supporting Table S3) that prevented any further comparison of B‐cell clonality among PSC‐IBD and control samples. Very few B cells infiltrating the gut and liver were also detected by CD20 immunohistochemistry in control tissue (data not shown), indicating that tissue‐resident B cells infiltrating normal gut and liver tissue are relatively rare compared to gut and liver tissue of patients with PSC‐IBD.
PRIVATE B‐CELL CLONOTYPES OVERLAP THE GUT AND LIVER IN PATIENTS WITH PSC‐IBD
As B‐cell clonotypes were nearly undetectable in control samples ( S3), we sought to determine if B cells infiltrating the gut and liver recognized the same or similar antigens in PSC‐IBD by comparing the number of overlapping B‐cell clonotypes in paired (n = 10) and unpaired (n = 90) gut and liver patient samples (Fig. 2A). To better restrict our analysis to clonotypes of potential relevance to PSC‐IBD, we excluded all memory B‐cell clonotypes reported in a BCR repository of over 30 million BCR sequences from healthy controls23 as these expanded clonotypes are likely directed toward common infectious pathogens or generated during vaccination than relevant antigens involved in the pathogenesis of PSC‐IBD.24, 25, 26, 27 Using this approach, we detected a substantial number of BCR sequences that overlapped between paired gut and liver samples (private clonotypes: range, 51‐729) (Fig. 2A,C; Supporting Table S4) and a complete absence of overlapping clonotypes shared among unpaired samples (public clonotypes, P < 0.0001) (Fig. 2B,C). As expected, a greater number of private clonotypes were detected in paired gut and liver samples, a finding that is similar to studies of peripheral blood that show private B‐cell populations are more common than public clonotypes across patients.23 These results are also in line with our previous T‐cell receptor (TCR) sequencing data of gut and liver samples from the same patients11 showing that private T‐cell clonotypes greatly outnumber public T‐cell clonotypes (private clonotypes, 1,532 ± 772; public clonotypes, 111 ± 77; P < 0.0001) (Supporting Fig. S1).
Figure 2.
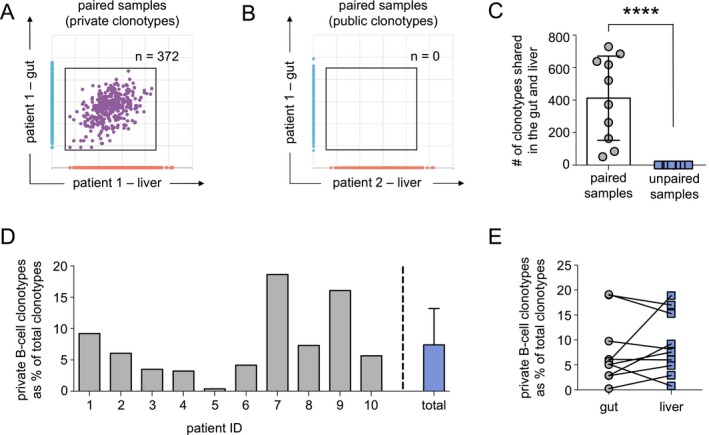
Private B‐cell clonotypes overlapping the gut and liver are detected in patients with PSC‐IBD. (A) Representative scatter plot indicates the number of B‐cell clonotypes shared in the gut and liver sample from the same patient (paired). Clonotypes restricted to the gut of patient 1 are shown in blue (y axis), clonotypes in the liver of the same patient are shown in orange (x axis). Shared clonotypes (private clonotypes) detected in both the gut and liver (gut–liver clonotypes) are gated and shown in purple (circles). (B) Representative scatter plot shows the absence of clonotypes shared in a gut and liver sample from different patients (unpaired sample; public gut–liver clonotypes). Gut clonotypes exclusive to patient 1 are shown in blue (y axis), and clonotypes restricted to the liver of patient 2 are shown in orange (x axis). Gate highlights the absence of public gut–liver clonotypes. (C) Number of private (paired samples) and public gut–liver (unpaired samples) clonotypes detected in each patient (n = 10). (D) Proportion (%) of private gut–liver clonotypes found within total clonotypes of each patient (gray bars) and average percentage for all PSC‐IBD samples (n = 10, blue bar). (E) Overall proportion (%) of the total gut (gray circles), and liver (blue squares) clonotypes proportion represented by private gut–liver clonotypes. Horizontal lines and bars indicate the mean, and error bars indicate SD. Statistical significance was determined using non‐parametric unpaired t tests; ****P < 0.0001.
Although public gut–liver clonotypes were absent among unpaired gut and liver samples, private B‐cell clonotypes in paired samples constituted 8.3% (range, 1.6%‐18.0%) (Fig. 2D) of the total gut and liver B‐cell clonotypes, whereas private T‐cell clonotypes represented on average 32.0% ± 11.7% of the total T‐cell clonotypes, a higher frequency then previously reported11 as clonotypes detected in matched blood samples at a frequency of less than 0.01% were not excluded in this study given BCR clonality in blood was not analyzed (Supporting Fig.S1). Private gut–liver B‐cell clonotypes appeared as a higher proportion of the total gut clonotypes in 5/10 samples and represented a greater proportion of the liver compartment in the remaining 5/10 samples (Fig. 2E). Overall, these findings indicate that a substantial proportion of B cells of common clonal origin overlap the gut and liver within patients (private clonotypes); however, B‐cell clonotypes overlapping both the gut and liver across patients (public clonotypes) are absent or extremely rare and may require pre‐enrichment strategies for detection.28
PUBLIC GUT AND PUBLIC LIVER B‐CELL CLONOTYPES ARE DETECTED IN PSC‐IBD
As we failed to detect any public gut–liver clonotypes, we questioned whether public clonotypes restricted to the gut or the liver are found among patients with PSC‐IBD (n = 10). Indeed, we found an average of 15 public gut clonotypes (range, 1‐28) and 34 public liver clonotypes (range, 1‐134) per patient following the exclusion of clonotypes reported in the memory B‐cell database of healthy controls (Fig. 3A,B). Collectively, these clonotypes represented a very limited proportion of the total gut and liver clonotypes (public gut clonotypes, 0.014% ± 0.008% public liver clonotypes, 0.49% ± 1.05%). Of the total public gut clonotypes (n = 70) and public liver clonotypes (n = 166), the majority were only seen in 2 patients (public gut clonotypes, 65; public liver clonotypes, 163) (Fig. 3D); however, four public gut clonotypes and three public liver clonotypes were found in 3/10 patients, and a single clonotype restricted to gut samples was detected in 4/10 patients (Table 2). These findings indicate that public clonotypes restricted to the gut or liver are present across patients and suggest that similar antigens restricted to or exclusively expressed in each compartment may elicit clonal expansion of B cells that recognize common antigenic triggers in PSC‐IBD.
Figure 3.
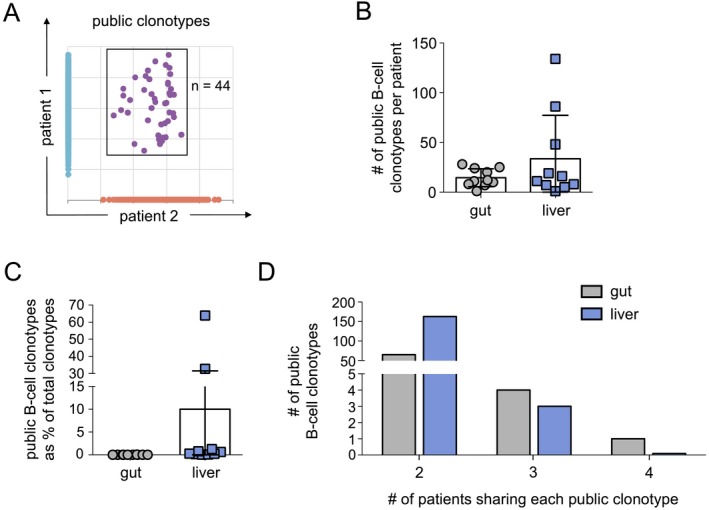
Public gut and public liver B‐cell clonotypes are found in patients with PSC‐IBD. (A) Representative scatter plot indicates the number of shared B‐cell clonotypes between liver samples from different patients. Clonotypes exclusive to patient 1 are shown in blue (y axis), clonotypes restricted to patient 2 are shown in orange (x axis), and shared clonotypes (public clonotypes) detected in both samples are in purple and gated. (B) Number of public gut and public liver B‐cell clonotypes detected in each patient (n = 10). (C) Public B‐cell clonotypes as a proportion (%) of the total gut and liver clonotypes among each patient (n = 10). (D) Number of public gut and public liver B‐cell clonotypes shared among n = 2, n = 3, and n = 4 patients. No public gut or public liver clonotypes were found in more than 4 patients. Bars indicate the mean, and error bars indicate the SD. Differences between gut and liver clonotypes in (B) and (C) are not significant as determined by non‐parametric unpaired t tests; P > 0.05.
Table 2.
Public Gut and Liver B‐Cell Clonotypes Detected in Greater Than 3 Patients With PSC‐IBD
|
Unique CDR3 Amino Acid Sequence |
Tissue |
Number of Patients (n = 10) |
Number of Reads | Relative Abundance (%) |
|---|---|---|---|---|
| CARLEMATILDAFDIW | gut | 4 | 18 | 0.005 |
| CARDLMTTGWFDPW | gut | 3 | 15 | 0.004 |
| CARRGEMATITGAFDIW | gut | 3 | 5 | 0.001 |
| CARTWIHVWTPDFDYW | gut | 3 | 37 | 0.01 |
| CARLQMATILDAFDIW | gut | 3 | 4 | 0.001 |
| CARSGYIYGADAFDIW | liver | 3 | 241 | 0.160 |
| CAVGSSRDSPFYNWFDPW | liver | 3 | 100 | 0.066 |
| CAKDWGLVWVWFDPW | liver | 3 | 35 | 0.023 |
PRIVATE AND PUBLIC B‐CELL CLONOTYPES SHOW EVIDENCE OF ANTIGEN‐DRIVEN EXPANSION AND PREFERENTIAL IMMUNOGLOBULIN HEAVY CHAIN VARIABLE FAMILY USAGE IN PSC‐IBD
B cells repeatedly exposed to the same antigen in the presence of helper T cells and antigen‐presenting cells undergo affinity maturation, a selection process that ultimately results in the creation of a higher affinity BCR variant.29 Several mechanisms contribute to affinity maturation, including the deletion of nucleotides from the CDR3 region and SHM of the nucleotides in the variable gene region.30 To determine if overlapping clonotypes in PSC‐IBD show evidence of affinity maturation, we first established the CDR3 lengths for nonexpanded clonotypes (“naive”) and highly expanded clonotypes (“memory”) by selecting 50 rare clonotypes (BCR template = 1) and the 50 most expanded clonotypes from each patient (n = 10, total n = 500 naive and memory clonotypes). The average CDR3 length for naive and memory clonotypes differed significantly (naive, 54.3 ± 11.8 nt; memory, 51.9 ± 10.8; P < 0.0001) (Fig. 4A), which is consistent with findings that affinity maturation results in shorter CDR3 lengths.23 We next compared the CDR3 lengths of overlapping private and public clonotypes and determined that private gut–liver clonotypes and public gut clonotypes appeared significantly different than naive (private gut–liver, 52.0 ± 10.3 nt; public gut, 42.0 ± 6.6 nt; naive, 54.3 ± 11.8 nt; P < 0.0001) (Fig. 4A); however, the average CDR3 length of public liver clonotypes (54.28 ± 9.9 nt) was not significantly different compared to private clonotypes (P > 0.05). Public gut clonotypes appeared to undergo the strongest affinity maturation because CDR3 length for these clonotypes was significantly shorter (P < 0.0001) than memory clonotypes whereas private gut–liver clonotypes were similar (P > 0.05). Applying the same definitions for naive and memory, we compared SHM counts and confirmed they were significantly lower for naive than memory clonotypes (naive, 1.30 ± 0.6; memory, 6.13 ± 3.2; P < 0.0001), which again was consistent with reported results.23 SH3 counts for private gut–liver clonotypes, public gut clonotypes, and public liver clonotypes were all significantly higher than naive clonotypes (P < 0.0001) but similar to one another (private gut–liver, 5.5 ± 3.2; public gut, 5.4 ± 3.4; public liver, 5.1 ± 2.7; P > 0.05) (Fig. 4B). These data suggest that private gut–liver clonotypes as well as public gut and public liver clonotypes have experienced antigen stimulation relative to unique non‐overlapping B‐cell subsets in patients with PSC‐IBD.
Figure 4.
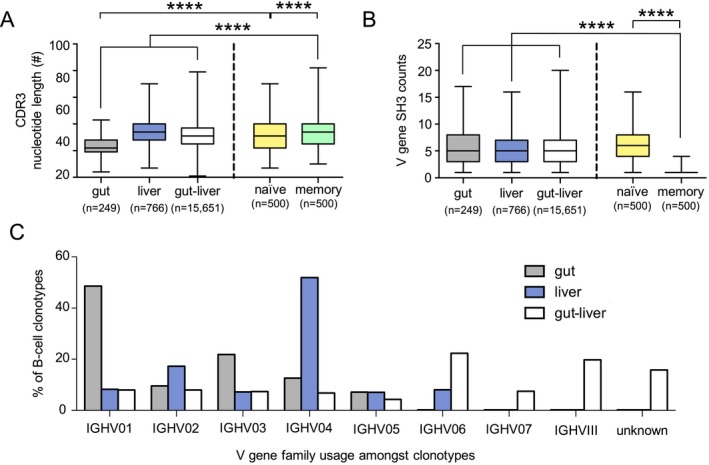
Private and public B‐cell clonotypes in patients with PSC‐IBD show evidence of antigen‐stimulation and differential IGHV family usage. (A) Number of nucleotides per CDR3 in public gut (gray bars; n = 249), public liver (blue bars, n = 766), and private gut–liver (white bars, n = 15,651) B‐cell clonotypes. CDR3 lengths of the 50 least frequent clonotypes from each of the 10 patients (naive, yellow bars; n = 500) and 50 most abundant clonotypes (memory, green bars; n = 500) are shown. Horizontal lines crossing the bars indicate the mean, bars indicate the SD, and error bars represent the minimum and maximum values detected. Statistical significance was calculated using one‐way analysis of variance, ****P = 0.0001. (B) Number of variable gene SHM counts detected in public gut (gray bars, n = 249), public liver (blue bars, n = 766), and private gut–liver (white bars) B‐cell clonotypes. SHM counts of the 50 least frequent clonotypes from each of the 10 patients (naive, yellow bars; n = 500) and 50 most abundant clonotypes (memory, green bars; n = 500) are shown. Statistical significance was calculated using one‐way analysis of variance, P < 0.0001. (C) Relative mean percentage (%) of the IGHV family usage in public gut clonotypes (gray bars, n = 249), public liver clonotypes (blue bars, n = 766), and private gut–liver clonotypes (white bars, n = 15,651) is shown. Abbreviation: IGHV, immunoglobulin heavy chain variable.
Lastly, we evaluated the usage of immunoglobulin heavy chain variable (IGHV) family segments to address if private gut–liver clonotypes, public gut clonotypes, and public liver clonotypes express a stereotypic germline variable family BCR rearrangement pattern (Fig. 4C). The IGHV locus contains over 50 functional genes that are classified into seven families based on nucleotide sequence homology.31 We found IGHV1 family usage in an average of 48.6% of public gut clonotypes, whereas 51.9% of public liver clonotypes used IGHV4. Private gut–liver clonotypes also showed a distinct IGHV family usage as 22.4% of clonotypes used IGHV06, 7.5% used IGHV07, and 18.8% used IGHVIII. Together, these data demonstrate a distinct IGHV usage pattern among private and public clonotypes in PSC‐IBD.
Discussion
This study is the first to report BCR signatures in paired gut and liver tissue samples from patients with PSC‐IBD. We demonstrated that a significantly greater number of B cells and unique B‐cell clonotypes are present in gut compared to liver of patients with PSC‐IBD. We also showed that B cells of common clonal origin are detected among both tissue compartments in PSC‐IBD and that these overlapping clonotypes showed reduced CDR3 lengths and higher counts of SHM indicative of antigen‐driven activation. In addition to clonal T‐cell overlap between the gut and liver,11 our findings support that adaptive immune responses are shared between the gut and liver and may contribute to chronic liver inflammation in patients with PSC‐IBD.
Our data indicate that the gut of patients with PSC‐IBD is populated with a less diverse and more expanded B‐cell repertoire compared to the liver. Contrasting B‐cell clonality between the gut and liver may relate to the progression of disease as gut samples in our study were collected on average 3.5 years before liver transplantation (range, 0.6‐7.3 years) and possibly at a time point when antigenic triggers were more abundant. Future studies using gut and liver specimens collected at closer time points or together at the time of liver transplantation may improve the identification of disease‐relevant B cells, although we detected no consistent trend relating to the time between tissue collection and high or low percentage of private clonotypes observed in our small sample size.
Similar to our previous work on gut and liver T‐cell clonality in PSC‐IBD,11 we found that the productive clonality of the gut and liver B‐cell compartment to range from 0.04 to 0.16, values that are higher than those reported in the blood of patients with systemic lupus erythematosus32 and comparable with those observed in the pancreatic draining lymph nodes of patients with type 1 diabetes.33 These findings suggest that B cells in the gut and liver of patients with PSC‐IBD are more oligoclonal and expanded compared to those in the periphery and support further study of gut and liver tissue as a means of determining potential antigenic triggers in PSC‐IBD.
We also observed that an average of 8.3% (range, 1.6%‐18.0%) of shared B‐cell clonotypes overlap between paired gut and liver samples (private gut–liver clonotypes) from patients with PSC‐IBD. Notably, these overlapping clonotypes showed reduced CDR3 lengths and higher counts of SHM indicative of antigen‐driven activation. As both our TCR and BCR sequencing data were limited to the CDR3 region, we were unable to define potential associations between TCRs and BCRs in the same paired gut and liver samples but such relationships may exist as pathogenic T cells and B cells in PSC‐IBD may recognize different epitopes of the same protein or molecular complex. Further studies combining sequencing analysis of the entire TCR and BCR with in silico modeling may enable such comparisons.
Unexpectedly, we failed to detect any public sharing of the overlapping clonotypes among paired gut and liver samples from patients with PSC‐IBD (i.e., overlapping gut–liver clonotypes were truly private and only found within the same patient). The lack of public gut–liver clonotypes suggests that a diverse range of antigens in each patient may trigger the clonal expansion of B cells, which may possibly explain the absence of a PSC‐specific antibody response.13, 15 Our inability to detect public gut–liver B‐cell clonotypes may also reflect a limitation of our data set as our BCR sequencing was restricted to the CDR3 region of the IgH. Although the IgH CDR3 region confers the greatest contribution to BCR specificity,34 the CDR1 and CDR2 regions also participate in antigen recognition.35 It should be noted that BCR sequencing does not confirm antigen‐specificity but simply infers that B cells of a common clonal origin have presumably expanded in response to similar antigen triggers.
In addition to detecting a substantial proportion of private B‐cell clonotypes in paired gut and liver samples, we discovered the presence of public B‐cell clonotypes restricted to the gut or liver and public T‐cell clonotypes overlapping the gut and liver in patients with PSC‐IBD. The significance of these public B‐cell and T‐cell clonotypes is unknown as public clonotypes represented <1% of the total clonotypes in each compartment but suggests that triggering antigens may be shared among patient subsets. It should also be noted that the number of private T‐cell clonotypes exceeded the number of private B‐cell clonotypes detected in samples from the same patients. This finding suggests that T‐cell expansion is stronger than B‐cell expansion in PSC‐IBD, which may explain the apparent lack of disease‐specific antibody signature in patients with PSC‐IBD. This finding could also imply that T‐cell responses are less specific in PSC‐IBD and that private B‐cell clonotypes recognize distinct antigens in each patient as B‐cell clonotypes detected in the liver and periphery of patients with PBC and alcoholic liver disease also appear patient‐specific.36 Although appealing from the perspective of antigen discovery, public and private clonotypes may also reflect methodologic errors or unreported clonotypes generated in response to common infections; thus, studies involving the use of a greater number of patients and controls is necessary.
A major limitation of our study was our inability to address whether shared clonality between the gut and liver of patients with PSC‐IBD is disease‐specific as differences in percentage of private B‐cells did not correlate with any patient characteristics, including scores for Model for End‐Stage Liver Disease, degree of macroscopic or microscopic gut inflammation detected at colonoscopy, bilirubin, international normalize ratio, alkaline phosphatase, alanine aminotransferase, albumin, or platelet counts as well as human leukocyte antigen risk alleles. This finding suggests private clonotype percentages do not correlate with disease status, although the variation in private clonotypes among patients may reflect sampling variability because limited access to patient material restricted BCR sequencing analysis to small amounts of gut and liver tissue. The use of larger tissue samples combined with selective BCR sequencing of plasmablasts and plasma cells may provide a more accurate representation of relevant B‐cell clonotypes that may correlate with specific patient features.
As extensive sharing of clonotypes among tissues within the gastrointestinal tract has been reported,37 our results may not be unique to PSC‐IBD, and the expansion of private clonotypes across the gut–liver axis may be a common phenomenon in many disease settings. Paired gut and liver samples from IBD patients without PSC would serve as the ideal control to address this question; however, these patients do not undergo routine liver biopsy or liver transplantation and thus liver samples are not typically available. Sampling of matched gut tissue from other liver disorders, such as PBC, alcoholic liver disease, or nonalcoholic steatohepatitis, is extremely rare in clinical practice as is the sampling of matched tissue from patients that undergo colectomy for IBD without clinical features of liver dysfunction. Paired gut and liver tissue is theoretically available from PSC patients without IBD; however, the majority of such patients will develop IBD in the future.38 BCR sequencing of gut and liver B cells in animal models of PSC and IBD may bring greater clarity to this important question, but given that a proportion of the clonotypes overlap the gut and liver, our data strongly support the prospective and longitudinal collection of gut tissue as a proxy for studying early immune responses in the liver of patients with PSC‐IBD. Unlike liver tissue, collection of gut samples represents a feasible approach for obtaining both patient and appropriate ‘control’ samples because patients with PSC‐IBD and IBD without PSC (controls) undergo routine colonoscopies to monitor disease progression.
In addition to the lack of appropriate disease controls, several technological and methodologic constraints should be acknowledged in this study. Very few BCR sequences were retrieved using genomic DNA from control liver and gut tissue (normal tissue adjacent to tumor), and these low numbers restricted informative comparisons to PSC‐IBD samples. Whether low numbers of B cells are present in the normal setting or due to genomic DNA degradation resulting from formalin‐fixation or other technological error is unknown, but our results clearly illustrate the difficulty in collecting paired gut and liver control tissue to verify the disease relevance of our findings. The use of genomic DNA from whole tissue instead of purified B cells was another limitation of our study as deep sequencing still resulted in poor retrieval of BCR sequences. In hindsight, the use of purified B cells from fresh tissue samples allowing for single cell analysis of the light chain as well as the heavy chain would have enabled more sophisticated receptor modeling analyses, metadata comparisons between TCR and BCR clonotypes and clonal lineage analyses, which together could better inform the nature and origin of relevant antigenic triggers. Our challenges underscore that bulk analysis of immune‐cell specificities is unlikely to yield meaningful conclusions and that future studies should begin with well‐characterized, purified cell populations (i.e., memory B cells) analyzed preferably at a single‐cell level from multiple tissue compartments collected as closely in time as possible.
Overall, despite technological and methodologic obstacles, our findings support the concept that lymphocytes of similar antigen specificities circulate within the gut–liver axis. These findings justify intensified efforts to apply immunophenotyping in parallel with single‐cell sorting and other ‘omics’ approaches to determine if B cells recognizing specific antigenic triggers drive pathogenicity in patients with PSC‐IBD.
Author names in bold designate shared co‐first authorship.
Abbreviations
- BCR
B‐cell receptor
- CDR
complementarity‐determining region
- FFPE
formalin‐fixed, paraffin‐embedded
- IBD
inflammatory bowel disease
- IgH
immunoglobulin heavy chain
- IGHV
immunoglobulin heavy chain variable
- PBC
primary biliary cholangitis
- PSC
primary sclerosing cholangitis
- SHM
somatic hypermutation
- TCR
T‐cell receptor
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1200/full.
Supporting Information
Acknowledgment
We thank the patients who participated in this research study and Ashnila Janmohamed for reviewing clinical information.
Potential conflict of interest: Dr. Jørgensen has consulted and advised for Celltrion, Intercept Pharmaceuticals, and Tillotts Pharma. Dr. Hirschfield has consulted and advised for Dignity Sciences, GlaxoSmithKline, Intercept Pharmaceuticals, NGM Bio, Novartis, and Shire and is on the speakers' bureau for Falk Pharma. The other authors have nothing to report.
Support provided by the Norwegian PSC Research Center to B.K.C., E.K.K.H., and T.H.K., by the Southeastern Norway Regional Health Authority (Grant No. 2015024) to T.H.K., by the European Union Seventh Framework Programme (FP7‐PEOPLE‐2013‐COFUND grant agreement #609020 Scientia Fellows to B.K.C.), and the National Institute for Health Research Birmingham Biomedical Research Centre to B.K.C., G.M.H., and E.L.
This article presents independent research, and the views expressed are those of the author(s) and not necessarily those of the National Health Service, the National Institute for Health Research, or the Department of Health.
Contributor Information
Brian K. Chung, Email: b.k.chung@medisin.uio.no
Gideon M. Hirschfield, Email: g.hirschfield@bham.ac.uk
References
- 1. Saha K, Case R, Wong PK. A simple method of concentrating monoclonal antibodies from culture supernatant by ultrafiltration. J Immunol Methods 1992;151:307‐308. [DOI] [PubMed] [Google Scholar]
- 2. Karlsen TH, Franke A, Melum E, Kaser A, Hov JR, Balschun T, et al. Genome‐wide association analysis in primary sclerosing cholangitis. Gastroenterology 2010;138:1102‐1111. [DOI] [PubMed] [Google Scholar]
- 3. Loftus EV. PSC‐IBD: a unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut 2005;54:91‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu JZ, Hov JR, Folseraas T, Ellinghaus E, Rushbrook SM, Doncheva NT, et al.; UK‐PSCSC Consortium; International PSC Study Group; International IBD Genetics Consortium . Dense genotyping of immune‐related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet 2013;45:670‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Farci P, Diaz G, Chen Z, Govindarajan S, Tice A, Agulto L, et al. B cell gene signature with massive intrahepatic production of antibodies to hepatitis B core antigen in hepatitis B virus‐associated acute liver failure. Proc Natl Acad Sci U S A 2010;107:8766‐8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cabibi D, Tarantino G, Barbaria F, Campione M, Craxì A, Di Marco V. Intrahepatic IgG/IgM plasma cells ratio helps in classifying autoimmune liver diseases. Dig Liver Dis 2010;42:585‐592. [DOI] [PubMed] [Google Scholar]
- 7. Grant AJ, Lalor PF, Salmi M, Jalkanen S, Adams DH. Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet 2002;359:150‐157. [DOI] [PubMed] [Google Scholar]
- 8. Moreira RK, Revetta F, Koehler E, Washington MK. Diagnostic utility of IgG and IgM immunohistochemistry in autoimmune liver disease. World J Gastroenterol 2010;16:453‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kummen M, Holm K, Anmarkrud JA, Nygård S, Vesterhus M, Høivik ML, et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017;66:611‐619. [DOI] [PubMed] [Google Scholar]
- 10. Liaskou E, Klemsdal Henriksen EK, Holm K, Kaveh F, Hamm D, Fear J, et al. High‐throughput T‐cell receptor sequencing across chronic liver diseases reveals distinct disease‐associated repertoires. Hepatology 2016;63:1608‐1619. [DOI] [PubMed] [Google Scholar]
- 11. Henriksen EK, Jørgensen KK, Kaveh F, Holm K, Hamm D, Olweus J, et al. Gut and liver T‐cells of common clonal origin in primary sclerosing cholangitis‐inflammatory bowel disease. J Hepatol 2017;66:116‐122. [DOI] [PubMed] [Google Scholar]
- 12. Hov JR, Boberg KM, Karlsen TH. Autoantibodies in primary sclerosing cholangitis. World J Gastroenterol 2008;14:3781‐3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chung BK, Guevel BT, Reynolds GM, Gupta Udatha DB, Henriksen EK, Stamataki Z, et al. Phenotyping and auto‐antibody production by liver‐infiltrating B cells in primary sclerosing cholangitis and primary biliary cholangitis. J Autoimmun 2017;77:45‐54. [DOI] [PubMed] [Google Scholar]
- 14. Kain R, Exner M, Brandes R, Ziebermayr R, Cunningham D, Alderson CA, et al. Molecular mimicry in pauci‐immune focal necrotizing glomerulonephritis. Nat Med 2008;14:1088‐1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hov JR, Boberg KM, Taraldsrud E, Vesterhus M, Boyadzhieva M, Solberg IC, et al. Antineutrophil antibodies define clinical and genetic subgroups in primary sclerosing cholangitis. Liver Int 2017;37:458‐465. [DOI] [PubMed] [Google Scholar]
- 16. Op De Beeck K, Van den Bergh K, Vermeire S, Decock S, Derua R, Waelkens E, et al. Immune reactivity to beta‐tubulin isotype 5 and vesicular integral‐membrane protein 36 in patients with autoimmune gastrointestinal disorders. Gut 2011;60:1601‐1602. [DOI] [PubMed] [Google Scholar]
- 17. Karlsen TH, Folseraas T, Thorburn D, Vesterhus M. Primary sclerosing cholangitis ‐ a comprehensive review. J Hepatol 2017:1298‐1323. [DOI] [PubMed] [Google Scholar]
- 18. Lennard‐Jones JE. Classification of inflammatory bowel disease. Scand J Gastroenterol Suppl 1989;170:2‐6. [DOI] [PubMed] [Google Scholar]
- 19. Jørgensen KK, Grzyb K, Lundin KE, Clausen OP, Aamodt G, Schrumpf E, et al. Inflammatory bowel disease in patients with primary sclerosing cholangitis: Clinical characterization in liver transplanted and nontransplanted patients. Inflamm Bowel Dis 2012;18:536‐545. [DOI] [PubMed] [Google Scholar]
- 20. Suwannalai P, Scherer HU, van der Woude D, Ioan‐Facsinay A, Jol‐van der Zijde CM, van Tol MJ, et al. Anti‐citrullinated protein antibodies have a low avidity compared with antibodies against recall antigens. Ann Rheum Dis 2011;70:373‐379. [DOI] [PubMed] [Google Scholar]
- 21. Yousfi Monod M, Giudicelli V, Chaume D, Lefranc MP. IMGT/JunctionAnalysis: the first tool for the analysis of the immunoglobulin and T cell receptor complex V‐J and V‐D‐J JUNCTIONs. Bioinformatics 2004;20(Suppl. 1):i379‐i385. [DOI] [PubMed] [Google Scholar]
- 22. Huang H, Benoist C, Mathis D. Rituximab specifically depletes short‐lived autoreactive plasma cells in a mouse model of inflammatory arthritis. Proc Natl Acad Sci U S A 2010;107:4658‐4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. DeWitt WS, Lindau P, Snyder TM, Sherwood AM, Vignali M, Carlson CS, et al. A public database of memory and naive B‐cell receptor sequences. PLoS ONE 2016;11:e0160853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scott MG, Tarrand JJ, Crimmins DL, McCourt DW, Siegel NR, Smith CE, et al. Clonal characterization of the human IgG antibody repertoire to Haemophilus influenzae type b polysaccharide. II. IgG antibodies contain VH genes from a single VH family and VL genes from at least four VL families. J Immunol 1989;143:293‐298. [PubMed] [Google Scholar]
- 25. Silverman GJ, Lucas AH. Variable region diversity in human circulating antibodies specific for the capsular polysaccharide of Haemophilus influenzae type b. Preferential usage of two types of VH3 heavy chains. J Clin Invest 1991;88:911‐920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wrammert J, Koutsonanos D, Li G‐M, Edupuganti S, Sui J, Morrissey M, et al. Broadly cross‐reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J Exp Med 2011;208:181‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jackson KJ, Liu Y, Roskin KM, Glanville J, Hoh RA, Seo K, et al. Human responses to influenza vaccination show seroconversion signatures and convergent antibody rearrangements. Cell Host Microbe 2014;16:105‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lindop R, Arentz G, Chataway TK, Thurgood LA, Jackson MW, Reed JH, et al. Molecular signature of a public clonotypic autoantibody in primary Sjögren's syndrome: A “forbidden” clone in systemic autoimmunity. Arthritis Rheum 2011;63:3477‐3486. [DOI] [PubMed] [Google Scholar]
- 29. Sage PT, Sharpe AH. T follicular regulatory cells. Immunol Rev 2016;271:246‐259. [DOI] [PubMed] [Google Scholar]
- 30. Teng G, Papavasiliou FN. Immunoglobulin somatic hypermutation. Annu Rev Genet 2007;41:107‐120. [DOI] [PubMed] [Google Scholar]
- 31. Matsuda F, Ishii K, Bourvagnet P, Kuma KI, Hayashida H, Miyata T, et al. The complete nucleotide sequence of the human immunoglobulin heavy chain variable region locus. J Exp Med 1998;188:2151‐2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu S, Hou XL, Sui WG, Lu QJ, Hu YL, Dai Y. Direct measurement of B‐cell receptor repertoire's composition and variation in systemic lupus erythematosus. Genes Immun 2017;18:22‐27. [DOI] [PubMed] [Google Scholar]
- 33. Seay HR, Yusko E, Rothweiler SJ, Zhang L, Posgai AL, Campbell‐Thompson M, et al. Tissue distribution and clonal diversity of the T and B cell repertoire in type 1 diabetes. JCI Insight 2016;1:e88242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu JL, Davis MM. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity 2000;13:37‐45. [DOI] [PubMed] [Google Scholar]
- 35. Ohno S, Mori N, Matsunaga T. Antigen‐binding specificities of antibodies are primarily determined by seven residues of VH. Proc Natl Acad Sci U S A 1985;82:2945‐2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tan YG, Wang XF, Zhang M, Yan HP, Lin DD, Wang YQ, et al. Clonal characteristics of paired infiltrating and circulating B lymphocyte repertoire in patients with primary biliary cholangitis. Liver Int 2018;38:542‐552. [DOI] [PubMed] [Google Scholar]
- 37. Meng W, Zhang B, Schwartz GW, Rosenfeld AM, Ren D, Thome JJ, et al. An atlas of B‐cell clonal distribution in the human body. Nat Biotechnol 2017;35:879‐884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Riley TR, Schoen RE, Lee RG, Rakela J. A case series of transplant recipients who despite immunosuppression developed inflammatory bowel disease. Am J Gastroenterol 1997;92:279‐282. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1200/full.
Supporting Information