

ABSTRACT
SOX9 controls cell lineage fate and differentiation in major biological processes. It is known as a potent transcriptional activator of differentiation-specific genes, but its earliest targets and its contribution to priming chromatin for gene activation remain unknown. Here, we address this knowledge gap using chondrogenesis as a model system. By profiling the whole transcriptome and the whole epigenome of wild-type and Sox9-deficient mouse embryo limb buds, we uncover multiple structural and regulatory genes, including Fam101a, Myh14, Sema3c and Sema3d, as specific markers of precartilaginous condensation, and we provide evidence of their direct transactivation by SOX9. Intriguingly, we find that SOX9 helps remove epigenetic signatures of transcriptional repression and establish active-promoter and active-enhancer marks at precartilage- and cartilage-specific loci, but is not absolutely required to initiate these changes and activate transcription. Altogether, these findings widen our current knowledge of SOX9 targets in early chondrogenesis and call for new studies to identify the pioneer and transactivating factors that act upstream of or along with SOX9 to prompt chromatin remodeling and specific gene activation at the onset of chondrogenesis and other processes.
KEY WORDS: Chondrogenesis, Epigenetic regulation, Pioneer transcription factor, Precartilaginous condensation, SOX9
Summary: SOX9 directly upregulates genes specifically expressed in precartilaginous condensations and well-known early-cartilage markers, but only partially contributes to the initiation of chromatin remodeling at precartilage and early-cartilage loci.
INTRODUCTION
Chondrogenesis is a unique and essential process in development, in adult homeostasis, and in the repair of the vertebrate skeleton (Berendsen and Olsen, 2015; Kozhemyakina et al., 2015). It generates embryonic cartilage primordia that prefigure most adult skeletal structures. These primordia give rise to cartilage growth plates, i.e. fast-growing templates and signaling centers for developing bones, in addition to life-long cartilage structures that support airways, the auditory system and synovial joints. Cartilage structures also impact the development and function of such skeleton-associated structures as the vascular network, tendons, ligaments and muscles (Berendsen and Olsen, 2015; Eshkar-Oren et al., 2009; Huang et al., 2015; Pryce et al., 2009; Subramanian and Schilling, 2015). A deep understanding of the molecular mechanisms that underlie chondrogenesis is therefore necessary to fully understand the foundations of skeletogenesis, and to decipher the causes and improve treatments for cartilage malformation (chondrodysplasias) and adult-onset (namely osteoarthritis) diseases (Martel-Pelletier et al., 2016; Warman et al., 2011).
The first hallmark of chondrogenesis is precartilaginous condensation (PC). PC starts when multipotent skeletogenic cells commit to chondrogenesis, strengthen their cytoskeleton, adopt a round shape and establish gap junctions (Edwards et al., 2010; Kim et al., 2009; Ray and Chapman, 2015). Prechondrocytes initiate the avascular and aneural nature of cartilage by excluding other cell types from PCs, and then undergo overt chondrocyte differentiation. Although many structural and regulatory genes have already been described that control chondrocyte differentiation (Kozhemyakina et al., 2015; Liu et al., 2017), the genes involved in committing progenitor cells to chondrogenesis and in generating PC remain ill defined. These genes likely include regulators of epigenetic and transcriptional events.
The SOX9 transcription factor has pivotal roles in cell-fate determination and differentiation in multiple processes (Jo et al., 2014; Lefebvre and Dvir-Ginzberg, 2017; Symon and Harley, 2017). It is already expressed in multipotent skeletogenic cells; it continues to be expressed during chondrocyte lineage progression, except in terminally differentiating growth plate chondrocytes; and it has been proposed to drive chondrocyte specification and differentiation (Akiyama et al., 2002; Ng et al., 1997; Wright et al., 1995). At the molecular level, SOX9 is known to activate many chondrocyte differentiation markers through direct binding to enhancers and promoters (Liu and Lefebvre, 2015; Ohba et al., 2015). Sox9 was first shown to be essential at the onset of chondrogenesis, when mouse chimeras were created using Sox9-null and wild-type cells (Bi et al., 1999). At embryonic day (E) 11.5, these chimeras exhibited intermingled mutant and wild-type cells in limb bud mesenchyme. By E12.5, however, wild-type cells had formed PCs, whereas mutant cells had apparently been expelled to the outskirts of the PC (Barna and Niswander, 2007; Bi et al., 1999). In a subsequent study, Sox9 inactivation in early limb-bud mesenchyme (Sox9fl/flPrx1Cre) was found to preclude PC formation and cause widespread cell apoptosis (Akiyama et al., 2002). To date, the molecular role of SOX9 in commitment to chondrogenesis and in PC remains largely unknown.
Pioneer transcription factors elicit cell-fate changes by: (1) accessing naive (silent and unmarked) chromatin; (2) recruiting factors capable of freeing chromatin from nucleosomal packing; (3) inducing epigenetic changes conducive to transcriptional activation or repression; and (4) assembling transcriptional complexes (Iwafuchi-Doi and Zaret, 2016; Zaret et al., 2016). The chondrogenic pioneer factors remain unknown, but SOX9 is a strong candidate, given its requirement for chondrogenesis. It was put forward as a pioneer in hair follicle stem cells because markers of these cells became active upon forced expression of SOX9 in epidermal cells (Adam et al., 2015). However, it remains unknown whether SOX9 changed the chromatin landscape of these genes. High-throughput sequencing assays of immunoprecipitated chromatin (ChIP-seq) have shown that SOX9 binds in differentiated chondrocytes to genomic regions that carry the characteristic histone 3 lysine 27 acetylation (H3K27ac) modification of active enhancers, and lack histone 3 lysine 27 trimethylation (H3K27me3) transrepression marks (Liu and Lefebvre, 2015; Ohba et al., 2015). However, no study has yet shown whether SOX9 itself elicits this active-transcription state.
Using whole-transcriptome and whole-epigenome sequencing approaches for mouse embryo limb buds, we identify genes specifically expressed and likely involved in PC. We show that SOX9 directly upregulates these genes and well-known early-cartilage markers, and that its actions in chondrogenic cells have little impact on neighboring cell types. We also provide evidence that SOX9 only partially contributes to inducing epigenetic modifications at precartilage and early-cartilage loci. Beyond expanding knowledge of the molecular regulation of early events in chondrogenesis, our findings also call for further studies to identify the pioneer factors that act upstream of SOX9 or along with SOX9 to commit progenitor cells to the chondrocyte lineage and initiate chondrogenesis.
RESULTS
SOX9 strengthens the actin cytoskeleton of prechondrocytes
We characterized the contributions of Sox9 at the onset of chondrogenesis using mouse embryos at E11.5 and E12.5. For E11.5, we generated Sox9−/− and Sox9fl/− or Sox9+/− littermates using Cre-recombinase transgenes inactivating Sox9 in the mother and father germ lines (Akiyama et al., 2004). As reported (Akiyama et al., 2002), these embryos had limb buds morphologically similar to those of wild-type embryos, with no PCs yet (Fig. 1A). For E12.5, we conditionally inactivated Sox9 in early limb-bud mesenchyme (Sox9fl/flPrx1CreER) because Sox9−/− embryos die around E12.0 (Akiyama et al., 2004). E12.5 Sox9fl/fl embryos had readily recognizable digital condensations, whereas, as reported for Sox9fl/flPrx1Cre embryos (Akiyama et al., 2002), Sox9fl/flPrx1CreER littermates did not (Fig. 1B). To test whether Sox9-null presumptive chondrocytes consolidated their actin cytoskeleton, we stained E12.5 limb-bud sections with phalloidin-iFluor-488, a reagent that binds specifically to filamentous actin (F-actin). In control paws, F-actin densely lined contacts between Sox9-expressing prechondrocytes, but was sparse in distal digital mesenchyme and Sox9-negative interdigital regions (Fig. 1Ca-e). It was equally sparse throughout the limb buds of mutant embryos (Fig. 1Cf-j). We concluded that condensing prechondrocytes require SOX9 at least in part to reinforce and re-organize their cytoskeleton.
Fig. 1.

Requirement for SOX9 in precartilaginous condensation. (A,B) H&E staining of hindlimb bud sections from E11.5 Sox9fl/− and Sox9−/− (A), and E12.5 Sox9fl/fl and Sox9fl/flPrx1CreER embryo littermates (B). Arrow, digit 3 PC. (C) Visualization of the actin cytoskeleton in E12.5 Sox9flfl and Sox9flflPrx1CreER mouse limb buds. Sections were stained for SOX9 (red), F-actin (green) and DNA (blue). (Ca,f) Low-magnification images of limb buds. (Cb-d,g-i) High-magnification images within actual (Sox9fl/fl) and failed (Sox9fl/flPrx1CreER) PCs. (Ce,Cj) Images within actual and presumptive interdigital mesenchyme. (Cb-e,g-j) Top panels, merged images for SOX9, F-actin and DNA; bottom panels, F-actin.
SOX9 may affect early limb bud gene expression cell- and non-cell-autonomously
We performed RNA-seq assays on E11.5 and E12.5 control and mutant embryo paws in order to identify changes in gene expression due to Sox9 inactivation. Sox9fl/− embryos were a good proxy for wild-type littermate controls because their phenotype at E11.5 and later on was mild compared with that of Sox9 homozygous mutants (Bi et al., 2001; Akiyama et al., 2002). Together, the four experimental groups expressed 12,880 genes. Of all genes, 22% (2838) had their RNA level differing by ≥1.5-fold in at least one group compared with others (Table S1).
Mutants upregulated seven genes at E11.5 and 23 at E12.5 (Table S2A; Fig. 2A,B). Two of the genes, Gm11681 and Cdh22, were upregulated at both stages. Gm11681 lies immediately downstream of Sox9. It encodes a long non-coding RNA with no ortholog in other genomes and no known function. Cdh22, encoding cadherin 22, is expressed in the zone of polarizing activity at E11.5, and in interdigital mesenchyme at E12.5 (Kitajima et al., 1999). The genes that were upregulated only in E11.5 mutant limbs were not known to be involved in specific developmental pathways (Table S2B). Genes that were upregulated only in E12.5 mutants included Grem1, which encodes a BMP signaling antagonist (gremlin; Table S2C). Grem1 is expressed in subectodermal limb bud mesenchyme at E12.5 (Zúñiga et al., 1999). Most of the other genes that were upregulated in E12.5 mutants were erythroid markers, possibly reflecting a change in the ratio of chondrogenic versus non-chondrogenic cells in mutants or a non-cell-autonomous influence of SOX9 on non-chondrogenic cells. In summary, all of the genes that were upregulated in Sox9-mutant limb buds were expressed outside of the Sox9 domain and are unlikely to be relevant to PC.
Fig. 2.

Transcriptome changes in Sox9-deficient limb buds. (A) Scatter plots comparing RNA levels in E11.5 Sox9fl/− and Sox9−/− (left) and E12.5 Sox9fl/fl and Sox9fl/flPrx1CreER (right) embryo limb buds. Each dot shows the average NRPKM value for three to four biological replicates for one RNA species. Gray dots within the zone delineated with green lines correspond to RNAs whose levels changed ≤1.5-fold in mutants. Gray dots outside of the green lines correspond to RNAs whose levels showed a non-significant >1.5-fold change. Red and blue dots correspond to genes significantly upregulated and downregulated ≥1.5-fold, respectively, in mutant samples. Gene numbers are indicated. (B) Venn diagram showing the numbers (white) and percentages of genes whose expression changed significantly upon Sox9 inactivation. (C) Scatter plots comparing RNA levels in E11.5 Sox9fl/− and E12.5 Sox9fl/fl control (left) and E11.5 Sox9−/− and E12.5 Sox9fl/flPrx1CreER-mutant (right) limb buds. (D) Venn diagram showing the numbers and percentages of genes whose expression changed significantly between E11.5 and E12.5. C, control; M, mutant.
Only 23 genes were downregulated in mutant limb buds at E11.5, compared with 72 at E12.5. Key chondrocyte markers, namely Col2a1 (collagen 2), and Sox5 and Sox6 (transcription factors promoting chondrocyte differentiation), were among the 17 genes that were downregulated at both stages (Table S2D). This gene group also included genes that may strengthen the actin cytoskeleton or affect cell-cell interaction during PC: Fam101a (filamin-interacting protein), Sema3d (semaphorin 3d) and Enpp2 (ectonucleotide pyrophosphatase/phosphodiesterase 2). The genes that were downregulated only at E11.5 included Dhrs3, which encodes a dehydrogenase/reductase that attenuates retinoic acid signaling during early-embryo patterning (Kam et al., 2013); other genes had no known roles that would suggest their involvement at the onset of chondrogenesis (Table S2E). The genes that were downregulated only at E12.5 included more major chondrocyte-differentiation markers, such as Matn1 (matrilin 1), Acan (aggrecan) and Papss2 (3′-phosphoadenosine 5′-phosphosulfate synthase) (Table S2F). They also included genes with possible roles in PC, such as Ablim3 (actin-binding LIM protein 3), Nog (noggin), Sema3c (semaphoring 3c) and Myh14 (non-muscle myosin heavy chain 14). This manual analysis thus identified many chondrocyte markers among the genes that were downregulated in Sox9-mutant limb buds, and also identified multiple genes that are possibly involved in PC. Accordingly, ingenuity pathway analysis (IPA) listed chondrocyte differentiation and cartilage development as the most affected biological processes at E12.5 (Table S3).
The analysis of temporal changes in gene expression revealed that 661 genes were upregulated (≥1.5-fold) in control paws between E11.5 and E12.5, versus 396 in mutants (Fig. 2C,D and Table S4A-C). The overlap was 340 genes. At the same time, 108 genes had their expression significantly decreased in both control and mutant paws, with an overlap of 57 genes (Fig. 2C,D and Table S4D-F). This analysis thus revealed many more genes affected in mutants than the analysis of differences between controls and mutants at either E11.5 or E12.5. The difference in outcome mainly reflected the fact that many genes were more strongly upregulated in controls than in mutants between E11.5 and E12.5. IPA indicated that the 340 genes upregulated in both controls and mutants between E11.5 and E12.5 are involved in many biological processes (Table S5A). The most relevant to limb morphogenesis were skeletal myogenesis, skeletogenesis and vascular network development. The processes that changed only in controls were related to angiogenesis, but most listed genes control many processes, including chondrogenesis, rather than only angiogenesis (Table S5B). For example, they included Thbs1 and Thbs2, which encode pericellular matrix proteins (thrombospondins 1 and 2) and are highly expressed in chondrogenic and other areas, and Ctgf (connective tissue growth factor), which encodes a matricellular protein important in chondrogenesis and other processes (Ivkovic et al., 2003; Kyriakides et al., 1998). The top biological processes identified for genes upregulated in only mutant groups included cell death and survival, possibly forewarning the massive cell death occurring in E13.5 Sox9-deficient paws (Akiyama et al., 2002) (Table S5C). The genes that were downregulated between E11.5 and E12.5 in both controls and mutants, and in controls only, contribute to biological processes involved in early limb development, such as genes for HOX transcription factors and morphogens (Table S5D,E). The genes that were downregulated in mutants only, are involved in various biological processes, but most are highly expressed in cartilage (Table S5F).
Taken together, these analyses of transcriptomic changes occurring in E11.5 and E12.5 control and Sox9-deficient limb buds revealed, as expected, many known chondrocyte markers, but also uncovered numerous genes with known or potential roles in PC, and suggested that SOX9 may have both cell-autonomous (direct) roles in PC and non-cell-autonomous (indirect) roles in concurrent processes.
PC failure has little impact on non-chondrogenic processes in early limb buds
Cartilage formation occurs concomitantly with the development of other structures, and drastically changes the morphology of limb buds. Our transcriptome analyses of whole limb buds thus gave us the opportunity to address the important issue of whether cartilage failure had an impact on non-chondrogenic processes in Sox9-mutant limb buds.
Growth and patterning of early limb buds are governed by crosstalk between multiple morphogen-induced pathways (Delgado and Torres, 2017). As mentioned earlier, RNA-seq data indicated elevated expression of Grem1 in E12.5 mutant paws. Comparison of RNA levels measured in RNA-seq and qRT-PCR validation assays revealed that this effect was due to a failure in downregulation of the gene in mutants between E11.5 and E12.5 (Fig. 3A,B). Other genes for limb bud morphogens, such as Bmp2 and Shh, and their receptors and target genes were similarly expressed in E11.5 and E12.5 control limbs, or were upregulated or downregulated between the two stages; however, these changes were unaffected by Sox9 inactivation. We thus concluded that Sox9 expression in chondrogenic cells only discretely affects early limb-bud growth and patterning pathways.
Fig. 3.

Impact of SOX9 on osteochondroprogenitor and non-chondrogenic cell markers in limb buds. Bar graphs show the RNA levels determined using RNA-seq (A,C,E-G) or qRT-PCR (B,D) for various genes in limb buds from control embryos (blue bars) and Sox9 mutant littermates (red bars) at E11.5 (light shades) or E12.5 (dark shades). Each panel features gene markers for specific processes or cell types, as indicated. Each bar is the mean±s.d. for three or four biological replicates. Dotted lines for RNA-seq assays indicate the threshold value for significant expression (NRPKM=3). ΔP<0.05 for differences between controls and mutants; *P<0.05 for differences between E11.5 and E12.5 (Student's t-tests).
We next examined the expression levels of genes for transcription factors that specify limb-bud mesenchyme and downstream lineages in order to determine whether SOX9 affects the fate of these cells. The RNA levels for Prrx1, an early limb bud mesenchyme marker, and for several osteochondroprogenitor and tenogenic markers, such as Twist1, Sox8, Sp7, Erg, Mkx and Scx, were not significantly changed in E11.5 and E12.5 Sox9-deficient limb buds compared with controls (Fig. 3C,D). In contrast, Foxc1 and Foxc2, which mark osteochondroprogenitors and remain expressed in early chondrocytes, were downregulated in Sox9 mutants (Kuratani et al., 1994; Loebel et al., 2012; Hernández-Hernández et al., 2017; Iwamoto et al., 2007). Hence, Sox9 inactivation only mildly affected expression of key markers of limb-bud mesenchyme and downstream lineages, and thus did not prompt major cell fate changes by E12.5.
Myogenic cells arise from progenitors that are distinct from the limb osteochondro- and tenogenic progenitors, but that migrate into the limb-bud mesenchyme concomitantly with the onset of chondrogenesis. Expression of Myf5 and Myog, which encode transcription factors that are crucial for skeletal myoblast specification and differentiation, was not significantly altered in mutant paws (Fig. 3E). Likewise, expression of genes coding for endothelial-cell markers, such as Pecam and Sox18 (Marcelo et al., 2013), and expression of the genes for vascular endothelial growth factors, were not altered either (Fig. 3F,G). The latter findings surprised us because it has previously been proposed that SOX9 promotes vascularization around PCs by increasing Vegfa expression in prechondrocytes (Eshkar-Oren et al., 2009). It must be noted that our findings dismiss a crucial role for SOX9 in vascular cell development, but they do not exclude a role in the distribution of blood vessels around PCs.
Taken together, these data indicated that PC failure in Sox9-deficient limb buds had no major impact on cell-lineage fate determination outside chondrogenesis until at least E12.5. We therefore focused the rest of our study on the roles of SOX9 in nascent chondrocytes.
SOX9 robustly increases the expression of chondrocyte-marker genes
Early chondrocyte-differentiation markers, such as Col2a1, Wwp2 and Sox5, were already highly expressed in E11.5 control limb buds and their RNA levels increased by less than twofold by E12.5 (Fig. 4A). These levels were reduced by less than twofold in Sox9 mutants at both stages. Late chondrocyte-differentiation markers, such as Col9a3, Sox6 and Acan, were expressed weakly or insignificantly in E11.5 control paws, and were upregulated by more than twofold by E12.5 (Fig. 4B). These genes were not significantly expressed in mutant paws at E11.5, and the few of them that became expressed by E12.5 exhibited RNA levels more than twofold lower than in controls. This led us to draw the unexpected conclusion that SOX9 is dispensable in the initiation of early-chondrocyte-differentiation marker expression. Using well calibrated qRT-PCR assays (Fig. S1), we confirmed that Col2a1, Sox5 and Sox6 were significantly expressed in E11.5 and E12.5 mutant paws (Fig. 4C). The actual reduction in RNA level in mutants was larger for Col2a1 when determined by qRT-PCR (fivefold at E11.5 and sevenfold at E12.5) than by RNA-seq, but it was similar for the other genes. RNA in situ hybridization (ISH) assays showed, as expected, that Col2a1 was expressed robustly and along with Sox9 in chondrogenic cells in E11.5 and E12.5 control limb buds (Fig. 4D). It was also expressed in control ectoderm, as has previously been reported (Nakamura et al., 2006), and similarly expressed in the ectoderm of mutant paws, but was weakly detectable in mutant mesenchyme. We concluded that chondrocyte-differentiation markers may rely on SOX9 for activation in chondrogenic cells, but the earliest markers do not require SOX9 to become active.
Fig. 4.

Impact of SOX9 on expression of chondrocyte differentiation markers. (A,B) Bar graphs showing the RNA levels measured using RNA-seq for early (A) and late (B) chondrocyte markers in the limb buds of E11.5 Sox9fl/− and Sox9−/− embryos (light blue and light red, respectively) and E12.5 Sox9fl/fl and Sox9fl/flPrx1CreER embryos (dark blue and dark red, respectively). Each bar is the mean±s.d. for three or four biological replicates. Dotted lines indicate the threshold value for significant expression (NRPKM=3). ∆P<0.05 for differences between controls and mutants; *P<0.05 for differences between E11.5 and E12.5 (Student’s t-tests). (C) Relative RNA levels measured by qRT-PCR for major chondrocyte differentiation markers. Data are normalized to values obtained in E11.5 controls. The differences between mutants and controls are indicated in percentages. ΔP<0.05 for differences between controls and mutants; *P<0.05 for differences between E11.5 and E12.5 (Student's t-tests). (D) RNA ISH of control and mutant embryo limb sections with Col2a1 and Sox9 RNA probes. In controls, Sox9 and Col2a1 RNAs produced strong signals in E11.5 chondrogenic mesenchyme (arrows) and E12.5 PCs. Col2a1 RNA is detected in the ectoderm (arrowheads) at both stages, and in E11.5 mutant mesenchyme (arrows). Asterisks indicate patchy signals seen for Sox9 and Col2a1 RNAs in E12.5 mutant mesenchyme, identifying cells in which Sox9 escaped inactivation. Scale bar: 50 µm.
SOX9 upregulates precartilage-marker genes
Our transcriptome profiling experiments gave us a unique opportunity to increase the current knowledge of genes expressed during PC and dependent upon SOX9. Tnc (tenascin C) and Nog have previously been shown to mark PCs (Hall and Miyake, 2000; Mackie et al., 1987; Pizette and Niswander, 2000) and were thus unsurprisingly found to be upregulated between E11.5 and E12.5 in a SOX9-dependent manner (Fig. 5A). It has previously been proposed that Cdh2 (N-cadherin) promotes PC (Oberlender and Tuan, 1994), but it was equally expressed in controls and mutants at both stages, indicating that PC does not involve a change in expression of this gene.
Fig. 5.

Impact of SOX9 on PC marker expression. (A) RNA levels measured using RNA-seq for PC markers in E11.5 Sox9fl/− and Sox9−/− and E12.5 Sox9fl/fl and Sox9fl/flPrx1CreER embryo limb buds. Values are expressed as mean±s.d. for three to four biological replicates. The dotted line indicates the threshold value for significant expression (NRPKM=3). ΔP<0.05 for differences between controls and mutants; *P<0.05 for differences between E11.5 and E12.5 (Student's t-tests). (B) Relative RNA levels measured by qRT-PCR for PC markers. Data are normalized to values obtained in E11.5 controls. ΔP<0.05 for differences between controls and mutants; *P<0.05 for differences between E11.5 and E12.5 (Student's t-tests). (C) RNA ISH of E11.5 Sox9fl/− and Sox9−/− and E12.5 Sox9fl/fl and Sox9fl/flPrx1CreER embryo limb bud sections with Sema3c and Sema3d probes. (D) Effect of Sema3c and Sema3d silencing on chondrogenesis. Limb bud mesenchymal cells were transfected with siRNAs during micromass formation, as indicated. Micromasses were stained with Alcian Blue 5 days later. Left, representative pictures. Right, quantification of staining extent. The percentage of stained area is shown as mean±s.d. for triplicates. (E) Effect of exogenous SEMA3C on chondrogenesis. One day after plating, limb bud micromasses were treated with SEMA3C and BMP7 as indicated. PNA and Alcian Blue staining were performed 2 and 4 days later, respectively. Left, representative pictures. Right, quantification of staining. For PNA, mean fluorescence intensities were calculated for two or three replicates in two independent experiments. For Alcian Blue, the percentage of stained area was calculated using two or three replicates in two independent experiments. *P<0.05 for differences between experimental groups, as indicated in the graphs (one-way ANOVA and Bonferroni post-hoc tests).
Several genes that have not previously been linked to PC caught our attention because they were more strongly upregulated in controls than in mutants between E11.5 and E12.5, and encode proteins that interact with the actin cytoskeleton. Among them, Ablim3 encodes an actin-binding LIM protein 3 (Matsuda et al., 2010); Enpp2 encodes autotaxin, which is important for cartilage formation and actin stress fiber formation (Nishioka et al., 2016); Fam101a encodes a filamin-interacting protein, is highly expressed in developing cartilage, and promotes actin dynamics and cartilage formation in redundancy with Fam101b (Mizuhashi et al., 2014); and Myh14 encodes the non-muscle myosin heavy chain 14, an actin-cytoskeleton-interacting protein. Sema3c and Sema3d also caught our attention because they encode short-range morphogens that have previously been shown to control cell-cell adhesion and repulsion in processes such as collective endothelial cell migration and cardiogenic mesenchyme formation (Hamm et al., 2016). qRT-PCR assays for Fam101a, Myh14, Sema3c and Sema3d, and RNA ISH assays for Sema3c and Sema3d consolidated our RNA-seq findings (Fig. 5B,C), and led us to conclude that these genes may promote PC downstream of SOX9.
We used a standard assay for limb bud cell micromass in vitro (Underhill et al., 2014) to test whether Sema3c and/or Sema3d might influence chondrogenesis. Knockdown of Sema3c either alone or in combination with Sema3d significantly reduced the ability of the cells to form cartilaginous nodules (Fig. 5D and Fig. S2). Exogenous addition of SEMA3C protein increased cell condensation and slowed down overt chondrogenesis, whether the cells were stimulated by the addition of BMP7 or not (Fig. 5E). These gain-of-function effects were mild, however, possibly because of endogenous expression of semaphorins. These functional data in vitro thus further suggest that Sema3c and/or Sema3d might have key roles in the early steps of chondrogenesis.
SOX9 directly transactivates precartilage-marker genes
SOX9 upregulates many chondrocyte-specific genes by cooperatively binding with SOX5 and/or SOX6 on enhancers associated with these genes (Liu and Lefebvre, 2015). To test whether SOX9 similarly controls precartilage-markers, we generated ChIP-seq profiles for H3K27ac (active enhancer mark) in E12.5 limb buds and compared them with SOX9 ChIP-seq profiles (Garside et al., 2015). We found that SOX9 associated with four enhancers located upstream of Fam101a and with the Myh14 promoter (Fig. 6A). It was also bound to several enhancers within Sema3c and Sema3d. For each of these genes, we selected the region most efficiently bound by SOX9 (highest ChIP-seq peak) and tested it in a reporter assay in the non-chondrocytic HEK-293 T cell line. As expected, the enhancer-less reporter had minimal activity, even upon forced expression of SOX9 and/or SOX6 (Fig. 6B). The Fam101a region had no effect in the absence of exogenously expressed SOX protein, whereas the Myh14, Sema3c and Sema3d regions, and an Acan enhancer region used as a positive control, augmented the promoter activity up to 6.6-fold. In each case, SOX6 alone was inactive, but SOX9 alone, or combined with SOX6, significantly upregulated the reporters. Together with RNA-seq findings, these ChIP-seq and reporter data concurred that SOX9 directly transactivates precartilage-marker genes.
Fig. 6.
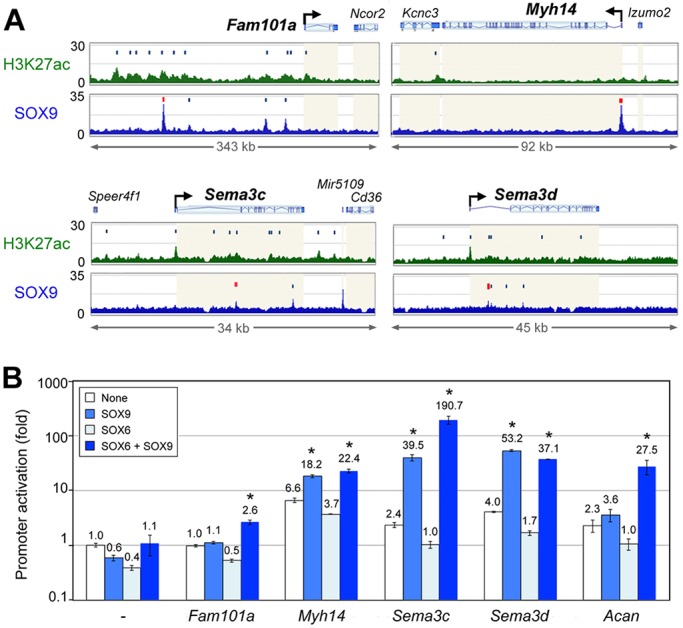
Direct activation of PC markers by SOX9. (A) SOX9 and H3K27ac ChIP-seq profiles in PC-marker loci in E12.5 wild-type mouse limb buds. Dark blue vertical bars, peak summits; red vertical bar, SOX9-bound enhancer functionally tested in B; light beige highlight, gene bodies. (B) Activation of reporters driven by precartilage-marker enhancers in HEK-293T cells forced to express no protein (none), SOX9 and/or SOX6. Data are mean±s.d. of technical triplicates for a representative experiment. *P<0.05 (Student's t-test).
SOX9 promotes epigenetic changes at precartilage- and cartilage-marker genes
In addition to transactivating precartilage- or cartilage-specific genes, SOX9 may direct chondrogenesis through pioneer functions. To test this possibility, we characterized epigenetic profiles in Sox9-control and Sox9-mutant limb buds through ChIP-seq assays. We first carried out global analyses by averaging epigenetic profiles for the entire genome or for 623 genes listed by IPA as chondrocyte-differentiation and cartilage-development markers (Table S7). Both analyses revealed lower average signals for active promoters (H3K4me3) and active transcription (H3K36me3) in E11.5 Sox9 mutant limb buds compared with controls (Fig. 7A and Fig. S3). These differences were accompanied by larger average signals for transrepression (H3K27me3), but unchanged signals for poised or active enhancers (H3K4me1) and active enhancers (H3K27ac). By E12.5, Sox9 mutants no longer showed reduced marks for active promoters and transcription. They had partially caught up with removing transrepression marks, and were now displaying weaker active-enhancer marks. We concluded that SOX9 helps to remove transrepression marks and to add active-enhancer marks in early limb bud cells.
Fig. 7.

Impact of SOX9 on global and Col2a1 epigenetic profiles in mouse limb buds. (A) Average profiles of histone modifications obtained in ChIP-seq assays of E11.5 Sox9+/+ and Sox9−/− (left) and E12.5 Sox9fl/fl and Sox9fl/flPrx1CreER (right) embryo limb buds for 623 cartilage-related genes (Table S7). The characteristic bimodal shape of the H3K4me1 profile is more apparent at E12.5 than at E11.5, likely because peaks are smaller. (B) Histone modification profiles at the Col2a1 and neighboring gene loci in E11.5 Sox9+/+ and Sox9−/− and E12.5 Sox9fl/fl and Sox9fl/flPrx1CreER embryo limb buds. H3K27ac peaks are labeled P1 to P5, and their summits are identified with vertical bars. (C) Comparison of H3K27ac (green) and SOX9 (blue) peak profiles at the Col2a1 locus for E9.5 to E14.5 limbs and for newborn mouse rib cartilage. The P1 to P5 peaks for H3K27ac are indicated. Their summits, and the summits of additional peaks, are marked with vertical bars. Light beige highlight indicates gene bodies.
We next examined epigenetic modifications at individual loci, starting with Col2a1 and its ubiquitously expressed 5′ neighbor Senp1 (sentrin-specific protease-1). Promoter and active-transcription marks indicated that both genes were expressed in control and Sox9-null limb buds at E11.5 and E12.5 (Fig. 7B). These marks were weaker in mutants than in controls for Col2a1, but not for Senp1. Transrepression marks were absent within and around Senp1, but present at the Col2a1 promoter, where they were stronger in mutants than controls at both developmental stages. No significant difference was detected between controls and mutants for H3K4me1 signatures at either locus. Although H3K27ac marks were identical at the Senp1 locus in control and mutant paws, differences were observed at the Col2a1 locus. Five major active-enhancer peaks (designated as P1 to P5) were detected within and upstream of the Col2a1 gene body in control paws at E11.5 and E12.5. P1 and P2 (located upstream of the gene) were not detected in E11.5 mutants and were weaker in E12.5 mutants than in controls. P3 (located close to P2) and P4 (located at the level of the promoter and first intron) were detected in mutants, but were smaller than in controls at both stages. Contrasting with P1 to P4, P5 (encompassing exons 16 to 19) was unaffected in mutants. This peak intrigued us because, unlike others, it was not detected in newborn mouse cartilage (Ohba et al., 2015) and RCS chondrocytic cells (Liu and Lefebvre, 2015). We therefore compared the H3K27ac profiles available for various developmental stages (Fig. 7C). P5 was the most prominent peak in the Col2a1 locus in E9.5 limb buds, whereas P4 and P3 were small. The relative heights of these peaks changed over time. P5 became small by E14.5, and P4 became prominent, followed by P3 and P1. By birth, P5 was no longer protruding from an H3K27ac peak that had a low intensity but covered the entire Col2a1 gene, whereas P4, P3 and P1 remained prominent, and additional small peaks became detectable. Interestingly, SOX9 ChIP-seq assays performed at E12.5 detected P1 to P4, but not P5, and assays performed at birth showed the same SOX9 peaks and additional ones, but not P5. Taken together, these data supported a model whereby SOX9 binds to multiple enhancers in the Col2a1 locus, contributes to removing transrepression marks at the promoter, and helps to increase active-promoter and active-enhancer marks, but does not bind to and does not epigenetically modify the P5 enhancer located in the middle of the gene. We speculate that this enhancer drives Col2a1 expression in limb bud ectoderm, whereas the others are primarily active in the chondrocyte lineage.
The loci for other precartilage- and cartilage-marker genes showed epigenetic changes consistent with RNA-seq data and with global and Col2a1 epigenetic changes, although with variations among genes. For example, the Fam101a and Sema3d loci showed an increase in active-promoter and active-enhancer marks, and a decrease in transrepression marks between E11.5 and E12.5 (Fig. 8A). According to the stronger SOX9 dependency of Fam101a, the active-promoter mark was more affected by Sox9 inactivation in Fam101a than in Sema3d. Wwp2 and Mir140, which resides within the 3′ end of Wwp2, are both highly expressed in developing cartilage and required for proper chondrogenesis (Miyaki et al., 2010). Despite its high expression from E11.5 and modest SOX9 dependency, Wwp2 showed hardly any change in active-promoter and transrepression marks in E11.5 and E12.5 control and Sox9-mutant limb buds (Fig. 8A). In contrast, Mir140 had an active-promoter mark that strongly increased between E11.5 and E12.5, and was tightly dependent upon Sox9 expression at both stages. Many enhancers were located between the two gene promoters, were occupied by SOX9 and showed SOX9-dependent increases in H3K27ac marks. Other cartilage markers, such as Col9a3 and Acan, had stronger transrepression marks in Sox9-deficient limb buds than in controls at E11.5, and these marks decreased by E12.5 independently of SOX9. Concomitantly, and in agreement with the stronger upregulation of Col9a3 than Acan between E11.5 and E12.5, Col9a3 showed increases in active-promoter and active-enhancer marks that were largely SOX9 dependent, whereas the Acan locus maintained low active-promoter and active-enhancer marks.
Fig. 8.

Impact of SOX9 on the epigenetic profiles of various loci in limb buds. (A) Histone modification profiles for PC and early-cartilage gene loci in E11.5 Sox9+/+ and Sox9−/− and E12.5 Sox9fl/fl and Sox9fl/flPrx1CreER embryo limb buds. (B) Histone modification profiles for various genes in E11.5 Sox9+/+ and Sox9−/− and E12.5 Sox9fl/fl and Sox9fl/flPrx1CreER embryo limb buds. In both panels the summits of H3K27ac and SOX9 peaks are marked with vertical bars. Light beige highlight indicates gene bodies.
We then analyzed ENCODE data to determine whether the epigenetic modifications observed at pre- and cartilage loci in Sox9-mutant limb buds were also seen in tissues that do not normally express Sox9, such as embryonic and adult liver (RPKM values for Sox9 were 0.2 and 0.9 in E14.5 and 8-week-old mouse liver, respectively, and 30.6 for E14.5 limbs; ENCODE BioProject PRJNA66167). No significant marks of active promoter (H3K4me3), enhancer (H3K27ac) and transcription (H3K36me3) were detected at the Col2a1 and Acan loci in these tissues (Fig. S4). However, poised enhancer (H3K4me1) and repression (H3K27me3) marks were present within or upstream of the gene bodies in adult liver (data were not available for fetal liver). These findings thus support the notion that precartilage- and cartilage-specific genes are in a poised or repressed state in non-chondrogenic tissues, and that the epigenetic marks of active transcription observed in Sox9-deficient limb buds may genuinely reflect Sox9-independent initiation of the chondrogenic program.
To determine whether SOX9 solely modifies the epigenetic status of precartilage and early-cartilage genes, we analyzed markers of late-stage growth-plate chondrocytes, such as Ihh (Indian hedgehog) and Runx2, and genes active in limb bud cells that do not express Sox9, such as α-globin genes and the tenocyte markers Scx, Mkx, Tnmd and Kera (Fig. 8B). Interestingly, although active-promoter and active-enhancer marks were not affected by Sox9 inactivation, transrepression marks were stronger at most genes in E11.5 Sox9 mutants than in controls, as seen for pre- and early-cartilage genes.
In conclusion, SOX9 is dispensable in the initiation of important epigenetic modifications at the onset of chondrogenesis. It nevertheless helps remove transrepression marks in a non-specific manner, which is likely to facilitate its identification of specific targets. Upon binding to its targets, SOX9 then enhances active-transcription signatures and engages in transactivation.
DISCUSSION
This study increased our understanding of mechanisms whereby skeletogenic cells initiate chondrogenesis and are directed by SOX9 in this process. It uncovered several genes that are controlled by SOX9 and that are likely to be crucial for PC by strengthening the actin cytoskeleton and by promoting homotypic cell-cell adhesion and heterotypic cell-cell repulsion. Furthermore, this study disconcerted our expectations by revealing that SOX9 helps but is not required for pioneer actions eliciting removal of epigenetic transrepression marks and adding active-promoter and active-enhancer marks at precartilage- and cartilage-specific loci. SOX9 is thus a crucial chondrogenic transcription factor, but not a master pioneer factor in nascent chondrogenesis.
PC initiates major morphological changes in embryos and it is SOX9 dependent. This study proposed several candidate effectors of this process, including Fam101a and other actin-cytoskeleton regulators. FAM101A and FAM101B interact with filamins to facilitate actin bundling (Gay et al., 2011). Actin bundles become particularly abundant during PC and allow cells to generate force and resist mechanical deformation. Fam101a/Fam101b-null mice exhibit vertebral fusions and delayed growth, and primary chondrocytes that are derived from them have fewer actin bundles (Mizuhashi et al., 2014). Although inactivation of Fam101a, Fam101b and any other PC-candidate gene was not reported to affect PC in vivo, the failure of PC in the absence of SOX9 may occur because multiple genes are simultaneously downregulated. Similarly, the single inactivation of any cartilage-matrix component gene does not preclude chondrogenesis, but the combined downregulation of all genes in Sox9 mutants does.
Cartilage is devoid of vascular, neural and any other non-chondrocytic cells. This exclusive cellular composition is initiated during PC through as yet elusive mechanisms. Its SOX9 dependency was first suggested when Sox9+/+/Sox9−/− mouse chimeras were found to expel Sox9−/− cells from exclusively Sox9+/+ PCs (Bi et al., 1999). This intriguing observation implied the existence of a cell-autonomous mechanism of homotypic adhesion of Sox9+/+ chondrogenic cells along with a mechanism of heterotypic cell exclusion driven by Sox9-expressing cells. It was later proposed that the exclusion of endothelial cells from PCs was orchestrated by VEGF produced by prechondrocytes in a SOX9-dependent manner (Eshkar-Oren et al., 2009). In the present study, however, the VEGF genes and endothelial-cell markers were expressed at normal levels in Sox9-deficient limb buds. SOX9 is thus unlikely to control VEGF expression in prechondrocytes and must use another mechanism to prevent PC vascularization. Interestingly, our findings that Sema3c and Sema3d are highly expressed in PCs and significantly downregulated in the absence of Sox9 offer a new molecular basis for the homotypic adhesion of prechondrocytes and exclusion of other cell types. Previous in vitro studies showed that SEMA3D promotes homotypic adhesion of mesenchymal cells and exclusion of endothelial cells from cardiogenic tissue (Hamm et al., 2016), and similar roles were shown for SEMA3C, SEMA3D and other SEMA3 proteins in various processes (Gaur et al., 2009; Liu and Halloran, 2005; Mammoto et al., 2011). Our gain- and loss-of-function experiments in vitro provide additional evidence that SEMA3C and/or SEMA3D may facilitate PC. In vivo, the knockdown of sema3d in zebrafish embryos was found to cause dramatic reductions and malformations of lower jaw cartilage, suggesting that the fish gene has important roles in jaw chondrogenesis and that these roles are not compensated for by other genes (Berndt and Halloran, 2006). Skeletal defects were not reported for Sema3c-null and Sema3d-null mice (Degenhardt et al., 2013; Feiner et al., 2001). As the two genes encode similar proteins and are co-expressed in mouse PCs, it is possible that they act redundantly in mammals and thus, unlike single-null mutants, double-null mutants would be skeletally compromised.
In addition to endothelial cell markers, genes encoding important limb-bud patterning factors and key specification and differentiation factors for other cell types also showed normal expression in Sox9-deficient limb buds. These cell types included limb-bud progenitor cells, osteochondroprogenitors, tenocytes and myoblasts. Thus, although it is accepted that the formation of the musculoskeletal system involves spatial and temporal coordination between all tissues, our data indicated that crosstalk between chondrocytic and other cell types does not initiate or does not require SOX9 before E12.5. Supporting this conclusion, previous studies have shown that tenogenesis is necessary for bone ridge formation and that mechanical force generated by muscle is necessary for joint formation, but that bone ridge and joint defects were not detected in mutant mice before E13.5 (Blitz et al., 2009; Kahn et al., 2009). Also supporting our conclusion, massive cell death, likely involving chondrogenic and other cells, has previously been detected in Sox9-deficient limb buds at E13.5, but not earlier (Akiyama et al., 2002).
RNA-seq and ChIP-seq assays recently widened the known spectrum of genes transactivated by SOX9 in differentiated chondrocytes from dozens to thousands (Garside et al., 2015; Liu and Lefebvre, 2015; Ohba et al., 2015). We further broadened this spectrum by identifying SOX9 targets in prechondrocytes and we also started to address the issue of pioneer actions of SOX9 in the chondrocyte lineage. Forced expression of SOX9 has previously been shown to be sufficient to activate genes characteristic of differentiated chondrocytes or hair follicle stem cells in cells not committed to these lineages, but molecular evidence of pioneer functions for SOX9 has not yet been put forward (Adam et al., 2015; Bell et al., 1997; Ikeda et al., 2004; Kishi et al., 2011). The fact that forcedly expressed SOX9 activated chondrocyte markers in only few cell types suggests that other factors have chondrogenic pioneer functions upstream of SOX9, or along with SOX9. Consistent with this idea, we showed that SOX9 helps to broadly remove transrepression marks and consolidate histone marks of active transcription at precartilage- and cartilage-specific loci, but is dispensable to initiate these events.
Pioneer transcription factors act at multiple levels. They first bind to closed chromatin. In agreement with this capability, SOX9 has previously been shown to bind and displace nucleosomes on a Col2a1 chromatin template in vitro (Coustry et al., 2010; Furumatsu et al., 2005). Once bound to closed chromatin, pioneer factors recruit histone-modifying enzymes. SOX9 binds the genome of differentiated chondrocytes to enhancers that harbor H3K27ac marks and are occupied by p300 (Ohba et al., 2015). This protein, and its close relative CBP, are both histone acetyltransferases and transcriptional co-activators, and have been shown to physically interact with SOX9 and help SOX9 transactivate a Col2a1 reporter in vitro (Furumatsu et al., 2005; Tsuda et al., 2003). SOX9 thus likely recruits CBP/p300 in vivo to establish or secure active-enhancer signatures in precartilage or cartilage-specific genes. The removal of H3K27me3 repressor marks typically involves histone demethylases. The ARID5B transcriptional co-activator physically associates with SOX9 and recruits the histone demethylase PHF2 to SOX9 target genes in chondrocytes in vitro (Hata et al., 2013), and the histone demethylase KDM4B has been proposed to promote chondrogenesis downstream of TGFβ (Lee et al., 2016; Yapp et al., 2016). Many histone demethylases are broadly expressed, and we did not find evidence that SOX9 upregulates any of their genes in limb buds. More studies are needed to definitively determine which histone demethylases work with SOX9 to prime precartilage or cartilage-specific genes for activation. In addition, studies involving ATAC-seq or DNase-seq assays are needed to definitively test whether SOX9 has chondrogenic pioneer factor activities and to what extent these activities are necessary and sufficient to modify chromatin accessibility at chondrocyte-specific loci.
Beyond suggesting genuine chondrogenic pioneer functions for SOX9, our findings also suggested that SOX9 may not be the first factor to access and remodel chromatin at chondrogenic loci, but may intervene secondarily to secure and reinforce active-chromatin states. Concerted actions have been shown in pluripotent embryonic stem cells for the four master regulators, SOX2, POU5F1 (also known as OCT4), KLF4 and MYC (Soufi et al., 2015), and several transcription factor types have been shown to serve as pioneers in other cells. Best known are Forkhead transcription factors, with FOXA and its GATA partners having served as pioneer paradigms in hepatogenesis (Golson and Kaestner, 2016; Iwafuchi-Doi and Zaret, 2016). Two Forkhead genes, Foxc1 and Foxc2, are expressed in skeletogenic mesenchyme and are necessary for proper chondrogenesis (Kume et al., 1998; Motojima et al., 2016; Zhao et al., 2015). We showed here that they are expressed in Sox9-deficient limb buds, albeit at a lower level than in controls. Their proteins could thus initiate chondrogenesis before, together with or downstream of SOX9. In the GATA family, Gata6 is expressed in mouse embryo PCs, but its roles have yet to be assessed (Alexandrovich et al., 2008). Multiple pioneer factors are, therefore, likely to control chondrogenesis.
By showing that SOX9 may make key pioneer contributions in early chondrogenesis, our study suggests that SOX9 could also have similar actions in other processes, and more SOX proteins could have pioneer actions than is currently known (Hou et al., 2017). Beyond its roles in pluripotent stem cells, SOX2 initiates an essential SOX code in neurogenesis. Crucial in neural stem cells, it is succeeded by SOX4 and SOX11 (SOXC group proteins) in nascent neuronal cells, and by SOX8, SOX9 and SOX10 (SOXE group proteins) in overt neurogenesis (Bergsland et al., 2011; Reiprich and Wegner, 2015). A similar code might operate in chondrogenesis, as Sox4 and Sox11 are expressed and are instrumental in skeletogenic mesenchyme (Bhattaram et al., 2014). SOX8 and SOX10 act in redundancy with SOX9 in several lineages, including Sertoli cells and neural crest cells (Barrionuevo and Scherer, 2010; Reiprich and Wegner, 2015). Sox8 is also co-expressed with Sox9 in skeletogenic cells (Sock et al., 2001), and its modest but significant expression is unchanged in Sox9-deficient limb buds. SOX8 and SOX9 may thus share functions in prechondrocytes, with SOX8 being able to epigenetically modify and initiate the expression of precartilage or cartilage genes in the absence of SOX9. This hypothesis would reconcile evidence from previous studies that SOX9 has pioneer functions in vitro and in vivo, and from the present study that SOX9 is dispensable for initial chondrogenic actions. Taken together, this study further illuminates our understanding of chondrogenesis and provides leads for new investigations towards fully deciphering the pioneer mechanisms that initiate chondrogenesis and cell-fate determination in various developmental, physiological and disease processes.
MATERIALS AND METHODS
Mice
Mice were used as approved by the Cleveland Clinic Institutional Animal Care and Use Committee. All were on a mixed genetic background (CD1/129/C57BL6). Sox9 mutant and control littermates were generated using breeders carrying Sox9 wild-type and conditional-null alleles (Kist et al., 2002), and PrmCre (O'Gorman et al., 1997), Zp3Cre (Lewandoski et al., 1997) or Prx1CreER transgenes (Kawanami et al., 2009). PrmCre and Zp3Cre are expressed in the male and female germ lines, respectively. They were used to generate Sox9−/− embryos (Akiyama et al., 2004). Prx1CreER was used to inactivate Sox9 in limb bud mesenchyme before the onset of skeletogenesis. Pregnant females carrying Sox9fl/flPrx1CreER embryos were injected intraperitoneally with 0.5 mg tamoxifen/10 g of body weight at E9.5. Genotyping was carried out using previously described PCR strategies (Kist et al., 2002; Kawanami et al., 2009).
RNA-seq assay
Mouse embryo limb buds were harvested in ice-cold PBS, incubated in RNAlater (Thermo Fisher Scientific) at 4°C overnight and stored at −20°C. Total RNA was extracted and purified using the RNeasy Mini Kit (Qiagen) after limb-bud homogenization in the kit RLT lysis buffer using disposable pellet pestles (Fisherbrand). RNA quality and quantity were assessed using a 2100 Bioanalyzer (Agilent Technologies). Only samples with an RNA integrity number (RIN) >9 were processed for RNA-seq assays. Libraries were generated from 250 ng RNA using the TruSeq Stranded Total RNA Sample Prep Kit (Illumina) and sequenced with an Illumina HiSeq 2500 System (Genomics Core Facility, University of Chicago).
RNA-seq data were analyzed using the Strand NGS pipeline, as described in Liu and Lefebvre (2015). In brief, single-end reads were aligned to the mouse genome (mm10), and unmapped and duplicate reads filtered out. RNA levels were expressed as numbers of reads per kilobase of exons per million of total reads (NRPKM). Differential RNA levels among samples were determined using one-way ANOVA followed by post-hoc Tukey HSD test. Asymptotic analysis was used for P-value computation, followed by the Benjamin-Hochberg procedure for multiple-testing correction. For a gene to be included in downstream analyses, its RNA level average had to be ≥3 NRPKM in at least one sample type, and ≥1.5-fold different in at least one sample type compared with others, with P≤0.05. Gene ontology analysis was performed using IPA (Qiagen).
qRT-PCR assay
Total RNA was prepared using TRIzol (Life Technologies) and the RNeasy Mini Kit. cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). qPCR was performed using the StepOne Plus Real Time PCR system (Thermo Fisher Scientific) and SYBR Green PCR Master Mix (Thermo Fisher Scientific). Primers and PrimeTime qPCR Primer Assays (Integrated DNA Technologies) are listed in Table S6A. PCR consisted of one cycle at 95°C for 10 s followed by 40 cycles at 95°C for 5 s and 60°C for 30 s. Relative mRNA levels were calculated using the ΔΔCt method, with Actb or Hprt levels as references.
ChIP-seq assay
Embryo limb buds were collected in ice-cold PBS and fixed in DMEM containing 1% formaldehyde and 10% fetal calf serum for 15 min at room temperature. After stopping crosslinking with 0.125 M glycine for 5 min, limb buds were washed three times in ice-cold PBS, homogenized with a pestle in buffer 1 [50 mM HEPES, pH 7.5, 140 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 10% glycerol, 0.5% IGEPAL CA-630 (Sigma-Aldrich), 0.25% Triton X-100 and 1× Protease Inhibitor Cocktail (PIC; Roche)], and left on ice for 10 min. After centrifugation, cell pellets were resuspended in buffer 2 [10 mM Tris-HCl, pH 8, 200 mM NaCl, 1 mM EDTA, 0.5 mM ethylene glycol tetraacetic acid (EGTA) and PIC] and incubated at room temperature for 10 min, on a shaker, to extract chromatin. Following centrifugation, chromatin pellets were stored at −80°C. Chromatin from 20-40 limbs was pooled according to genotype in buffer 3 (10 mM Tris-HCl, pH 8, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 5% sarkosyl and PIC) and processed as described in Liu and Lefebvre (2015). Briefly, chromatin was sheared into 100-500 bp fragments using a Bioruptor sonication system (Diagenode). Supernatants were recovered by centrifugation at 6000 g for 5 min. Then, 2 µg of H3K27ac (Abcam, ab4729), H3K4me1 (Abcam, ab8895), or H3K4me3 (Abcam, ab8580) rabbit polyclonal antibodies, 5 µg of H3K27me3 mouse monoclonal antibody (Abcam, ab6002), or 5 µg of H3K36me3 rabbit polyclonal antibodies (Abcam, ab9050) were coupled with 20 µl Dynabeads (Life Technologies) and incubated with chromatin fragments in 1 ml of buffer 3 at 4°C overnight. All antibodies have been validated in numerous previous studies, including Liu and Lefebvre (2015). Magnetic beads were washed with RIPA buffer (1% IGEPAL, 1% sodium deoxycholate, 1 mM EDTA, 50 mM HEPES, pH 7.5, and 0.5 M LiCl) and immunoprecipitated chromatin fragments recovered in elution buffer (50 mM Tris-HCl, pH 8, 10 mM EDTA, and 1% SDS). After reverse-crosslinking at 65°C overnight, DNA was purified by phenol/chloroform/isoamyl alcohol extraction and ethanol precipitation. Libraries were generated using TruSeq ChIP library preparation kits (Illumina) and single-end reads were obtained using a HiSeq 2500 platform (Illumina).
ChIP-seq data FASTQ files were aligned to the mouse genome (mm10) using the Strand NGS pipeline. Unmapped and duplicate reads were filtered out. Peak calling for SOX9 and histone modifications was carried out using MACS software (version 1.4.5) with default setting (effective genome size, 1.87e+09; band width, 300; model fold, 10, 30; P-value cutoff, 1.00e-05; and range for calculating regional lambda, 1 to 10 kb). Corresponding input libraries were used as controls. Peaks with a false discovery rate <1% were retained for analysis. Strand NGS software was used for peak visualization and region comparison, and the PAVIS tools to assign SOX9 peaks and modified-histone regions to the nearest genes, as has previously been described (Huang et al., 2013; Liu and Lefebvre, 2015). ChIP-seq data were downloaded from the GEO repository for SOX9 in E12.5 limb buds (GSE73225; Garside et al., 2015); for H3K27ac in E9.5 and E10.5 limb buds (GSE45456; Andrey et al., 2013); for H3K27ac in E14.5 limb buds (GSE31039; ENCODE/LICR); for SOX9 and H3K27ac in newborn ribs (GSE69108; Ohba et al., 2015); and liver samples (GSE31038, ENCODE Project Consortium, 2012).
Histology, immunostaining and RNA ISH
Mouse embryos were fixed in 4% paraformaldehyde overnight at 4°C. Paraffin embedding, sectioning at 7 µm and section staining with Hematoxylin and Eosin (H&E) were performed following standard protocols. To make frozen sections, fixed embryos were impregnated sequentially in 10%, 15% and 20% sucrose, and were left to sink in a 1:1 mix of 20% sucrose and OCT overnight at 4°C. Limb buds were embedded in a 1:3 mix of 20% sucrose and OCT. Cryosections were made at 10 µm in thickness.
For immunostaining, frozen sections were washed in PBS for 5 min and in PBT (PBS, 1% BSA and 0.1% Triton X-100) for 5 min thrice. They were blocked in PBT containing 10% donkey serum for 1 h at 4°C and then incubated overnight at 4°C in a humidified chamber with SOX9 antibody (Millipore, AB5535, 1:1000; validated in Yao et al., 2015 and Liu and Lefebvre, 2015). Following three 10 min washes in PBT, sections were incubated with Alexa Fluor 549-conjugated donkey anti-rabbit secondary antibody (Thermo Fisher Scientific, A21207, 2 µg/ml) and CytoPainter Phalloidin-iFluor 488 Reagent (Abcam, ab176753, 1:1000) for 2 h at room temperature. Signals were visualized and captured by confocal microscopy. Images were processed using Adobe Photoshop CS6 software.
RNA ISH was carried out using anti-sense RNA probes synthesized with digoxigenin (DIG) RNA labeling mix (Roche) from linearized plasmid templates (Table S6B). Mouse embryo sections were deparaffinized, rehydrated, washed in PBS containing 0.1% diethylpyrocarbonate (DEPC-PBS), post-fixed in 4% formaldehyde for 20 min and treated with 20 µg/ml of proteinase K in DEPC-PBS for 7.5 min. After an additional post-fixation in 4% formaldehyde for 20 min and two washes in DEPC-PBS, sections were acetylated with 1.5% triethanolamine in 0.03 N HCl and 0.25% acetic anhydride to reduce electrostatic binding of probes. They were then washed, dehydrated and air-dried. Probes were diluted at 0.25 ng/kb/µl in hybridization buffer (10 mM Tris-HCl, pH 7.5, 600 mM NaCl, 1 mM EDTA, 0.25% SDS, 0.2% dextran sulfate, 0.02% Ficoll 400, 0.02% polyvinylpyrrolidone, 0.02% bovine serum albumin, 200 µg/ml yeast tRNA and 50% formamide) and added onto each slide for hybridization at 60°C overnight. After post-hybridization washes and treatment with 20 µg/ml RNAse A and 1 µg/ml RNAse T1 (Thermo Fisher Scientific, 12091-021 and AM2283, respectively) in NTE buffer (10 mM Tris-HCl, pH 8, 0.5 M NaCl, and 5 mM EDTA) for 30 min at 37°C, sections were incubated in 1% blocking reagent in MAB buffer (0.1 M maleic acid, pH 7.5, and 0.15 M NaCl) for 1 h. Alkaline phosphatase-conjugated digoxygenin antibody (Sigma-Aldrich, 11093274910, 1:5000) was added for 16 h at 4°C. After washes in MAB and alkaline phosphatase buffer (0.1 M Tris-HCl, pH 9.5, 0.1 M NaCl, 50 mM MgCl2, 0.1% Tween 20, and 2 mM levamisole), slides were incubated with BM purple solution (Roche) at 4°C in the dark until specific or background signal appeared (24 to 96 h). The reaction was stopped with 10 mM EDTA in PBS. Slides were mounted with DAKO aqueous medium. Images were captured under microscopy with a digital camera and processed using Adobe Photoshop CS6 software.
Micromass culture
Mesenchymal cells were isolated from E11.5 embryo limb buds as described by Underhill et al. (2014). Briefly, limb buds were digested with dispase to obtain single-cell suspensions. Cells were re-suspended in medium at 2×107/ml and plated as 10 µl micromasses. After 2 h, DMEM/F12 medium with 10% FBS, 50 µg/ml ascorbic acid, 1 mM sodium pyruvate and 1 mM L-cysteine was gently added. For loss-of-function experiments, each micromass was transfected with 10 pmol control siRNA or specific siRNA for Sema3c or Sema3d (Thermo Fisher Scientific) and 1.5 µl RNAiMAX (Thermo Fisher Scientific). For gain-of-function experiments, recombinant mouse SEMA3C and human BMP7 (R&D Systems) were added to the medium at 500 ng/ml and 100 ng/ml, respectively, at culture days one and three. Staining with 10 µg/ml rhodamine-labeled peanut agglutinin (PNA, Vector Laboratories) was performed after fixation with 4% PFA. Staining with 1% Alcian blue 8GX (Fisher Scientific) was performed following fixation with 4% formalin and a wash with 0.2 M HCl. Staining intensities were quantified using NIH ImageJ software.
Reporter assay
Selected SOX9-bound genomic regions were amplified by PCR using mouse DNA and specific primers (Table S6C). PCR products were cloned in a reporter plasmid upstream a minimal Col2a1 promoter driving the firefly luciferase gene (Lefebvre et al., 1996) and sequence-verified. Thirty thousand HEK-293T cells (recently amplified from an ATCC CRL-3216 vial) were cultured in each well of 24-well plates overnight and then transfected with a mixture including DMEM, FuGENE6 (Promega), 150 ng reporter, 33.3 ng pGFP plasmid, 25 ng pSV2bgal plasmid, 50 ng SOX9 expression plasmid, 50 ng SOX6 expression plasmid and empty plasmid (up to 400 ng DNA). After 40-42 h, cells were collected in Tropix lysis buffer (100 mM potassium phosphate, pH 7.8, 1 mM DTT, 0.2% Triton X-100). Reporter activities were measured using a Dual-Light luciferase and β-galactosidase reporter gene assay system (Thermo Fisher Scientific). Promoter activation folds were calculated by dividing the activities of reporters containing a promoter and enhancer region by the activity of the related promoter-only reporter. They were normalized for transfection efficiency and calculated as mean±s.d. for technical triplicates. The statistical significance of fold changes among samples was determined using the two-tailed Student's t-test (P<0.05).
Supplementary Material
Acknowledgements
We thank Pallavi Bhattaram and other members of the Lefebvre laboratory for precious advice throughout the study. Imaging of fluorescent signals on tissue sections was carried out using a Leica SP5 confocal/multi-photon microscope purchased with partial funding from the National Institutes of Health to the Imaging Core of the Lerner Research Institute (SIG grant 1S10RR026820-01).
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: C.-F.L., M.A., A.H., V.L.; Methodology: C.-F.L., M.A., A.H., V.L.; Software: C.-F.L., M.A., A.H., V.L.; Validation: C.-F.L., M.A., A.H., V.L.; Formal analysis: C.-F.L., M.A., A.H., V.L.; Investigation: C.-F.L., M.A., A.H., V.L.; Resources: V.L.; Data curation: V.L.; Writing - original draft: C.-F.L., M.A., A.H., V.L.; Writing - review & editing: C.-F.L., M.A., A.H., V.L.; Visualization: C.-F.L., M.A., A.H., V.L.; Supervision: V.L.; Project administration: V.L.; Funding acquisition: V.L.
Funding
This work was funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (AR46249 and AR68308 to V.L.). Deposited in PMC for release after 12 months.
Data availability
All raw sequencing data can be found in the GEO database (GSE114522).
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.164459.supplemental
References
- Adam R. C., Yang H., Rockowitz S., Larsen S. B., Nikolova M., Oristian D. S., Polak L., Kadaja M., Asare A., Zheng D. et al. (2015). Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 521, 366-370. 10.1038/nature14289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H., Chaboissier M. C., Martin J. F., Schedl A. and de Crombrugghe B. (2002). The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813-2828. 10.1101/gad.1017802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H., Chaboissier M.-C., Behringer R. R., Rowitch D. H., Schedl A., Epstein J. A. and de Crombrugghe B. (2004). Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc. Natl. Acad. Sci. USA 101, 6502-6507. 10.1073/pnas.0401711101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrovich A., Qureishi A., Coudert A. E., Zhang L., Grigoriadis A. E., Shah A. M., Brewer A. C. and Pizzey J. A. (2008). A role for GATA-6 in vertebrate chondrogenesis. Dev. Biol. 314, 457-470. 10.1016/j.ydbio.2007.12.001 [DOI] [PubMed] [Google Scholar]
- Andrey G., Montavon T., Mascrez B., Gonzalez F., Noordermeer D., Leleu M., Trono D., Spitz F. and Duboule D. (2013). A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science 340, 1234167 10.1126/science.1234167 [DOI] [PubMed] [Google Scholar]
- Barna M. and Niswander L. (2007). Visualization of cartilage formation: insight into cellular properties of skeletal progenitors and chondrodysplasia syndromes. Dev. Cell 12, 931-941. 10.1016/j.devcel.2007.04.016 [DOI] [PubMed] [Google Scholar]
- Barrionuevo F. and Scherer G. (2010). SOX E genes: SOX9 and SOX8 in mammalian testis development. Int. J. Biochem. Cell Biol. 42, 433-436. 10.1016/j.biocel.2009.07.015 [DOI] [PubMed] [Google Scholar]
- Bell D. M., Leung K. K., Wheatley S. C., Ng L. J., Zhou S., Ling K. W., Sham M. H., Koopman P., Tam P. P. and Cheah K. S. (1997). SOX9 directly regulates the type-II collagen gene. Nat. Genet. 16, 174-178. 10.1038/ng0697-174 [DOI] [PubMed] [Google Scholar]
- Berendsen A. D. and Olsen B. R. (2015). Bone development. Bone 80, 14-18. 10.1016/j.bone.2015.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsland M., Ramskold D., Zaouter C., Klum S., Sandberg R. and Muhr J. (2011). Sequentially acting Sox transcription factors in neural lineage development. Genes Dev. 25, 2453-2464. 10.1101/gad.176008.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt J. D. and Halloran M. C. (2006). Semaphorin 3d promotes cell proliferation and neural crest cell development downstream of TCF in the zebrafish hindbrain. Development 133, 3983-3992. 10.1242/dev.02583 [DOI] [PubMed] [Google Scholar]
- Bhattaram P., Penzo-Méndez A., Kato K., Bandyopadhyay K., Gadi A., Taketo M. M. and Lefebvre V. (2014). SOXC proteins amplify canonical WNT signaling to secure nonchondrocytic fates in skeletogenesis. J. Cell Biol. 207, 657-671. 10.1083/jcb.201405098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W., Deng J. M., Zhang Z., Behringer R. R. and de Crombrugghe B. (1999). Sox9 is required for cartilage formation. Nat. Genet. 22, 85-89. 10.1038/8792 [DOI] [PubMed] [Google Scholar]
- Bi W., Huang W., Whitworth D. J., Deng J. M., Zhang Z., Behringer R. R. and de Crombrugghe B. (2001). Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc. Natl. Acad. Sci. USA 98, 6698-6703. 10.1073/pnas.111092198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitz E., Viukov S., Sharir A., Shwartz Y., Galloway J. L., Pryce B. A., Johnson R. L., Tabin C. J., Schweitzer R. and Zelzer E. (2009). Bone ridge patterning during musculoskeletal assembly is mediated through SCX regulation of Bmp4 at the tendon-skeleton junction. Dev. Cell 17, 861-873. 10.1016/j.devcel.2009.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coustry F., Oh C.-D., Hattori T., Maity S. N., de Crombrugghe B. and Yasuda H. (2010). The dimerization domain of SOX9 is required for transcription activation of a chondrocyte-specific chromatin DNA template. Nucleic Acids Res. 38, 6018-6028. 10.1093/nar/gkq417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt K., Singh M. K., Aghajanian H., Massera D., Wang Q., Li J., Li L., Choi C., Yzaguirre A. D., Francey L. J. et al. (2013). Semaphorin 3d signaling defects are associated with anomalous pulmonary venous connections. Nat. Med. 19, 760-765. 10.1038/nm.3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado I. and Torres M. (2017). Coordination of limb development by crosstalk among axial patterning pathways. Dev. Biol. 429, 382-386. 10.1016/j.ydbio.2017.03.006 [DOI] [PubMed] [Google Scholar]
- Edwards G. O., Coakley W. T., Ralphs J. R. and Archer C. W. (2010). Modelling condensation and the initiation of chondrogenesis in chick wing bud mesenchymal cells levitated in an ultrasound trap. Eur. Cell Mater. 19, 1-12. 10.22203/eCM.v019a01 [DOI] [PubMed] [Google Scholar]
- Eshkar-Oren I., Viukov S. V., Salameh S., Krief S., Oh C.-D., Akiyama H., Gerber H.-P., Ferrara N. and Zelzer E. (2009). The forming limb skeleton serves as a signaling center for limb vasculature patterning via regulation of Vegf. Development 136, 1263-1272. 10.1242/dev.034199 [DOI] [PubMed] [Google Scholar]
- Feiner L., Webber A. L., Brown C. B., Lu M. M., Jia L., Feinstein P., Mombaerts P., Epstein J. A. and Raper J. A. (2001). Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development 128, 3061-3070. [DOI] [PubMed] [Google Scholar]
- Furumatsu T., Tsuda M., Yoshida K., Taniguchi N., Ito T., Hashimoto M., Ito T. and Asahara H. (2005). Sox9 and p300 cooperatively regulate chromatin-mediated transcription. J. Biol. Chem. 280, 35203-35208. 10.1074/jbc.M502409200 [DOI] [PubMed] [Google Scholar]
- Garside V. C., Cullum R., Alder O., Lu D. Y., Vander Werff R., Bilenky M., Zhao Y., Jones S. J., Marra M. A., Underhill T. M. et al. (2015). SOX9 modulates the expression of key transcription factors required for heart valve development. Development 142, 4340-4350. 10.1242/dev.125252 [DOI] [PubMed] [Google Scholar]
- Gaur P., Bielenberg D. R., Samuel S., Bose D., Zhou Y., Gray M. J., Dallas N. A., Fan F., Xia L., Lu J. et al. (2009). Role of class 3 semaphorins and their receptors in tumor growth and angiogenesis. Clin. Cancer Res. 15, 6763-6770. 10.1158/1078-0432.CCR-09-1810 [DOI] [PubMed] [Google Scholar]
- Gay O., Gilquin B., Nakamura F., Jenkins Z. A., McCartney R., Krakow D., Deshiere A., Assard N., Hartwig J. H., Robertson S. P. et al. (2011). RefilinB (FAM101B) targets filamin A to organize perinuclear actin networks and regulates nuclear shape. Proc. Natl. Acad. Sci. USA 108, 11464-11469. 10.1073/pnas.1104211108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golson M. L. and Kaestner K. H. (2016). Fox transcription factors: from development to disease. Development 143, 4558-4570. 10.1242/dev.112672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall B. K. and Miyake T. (2000). All for one and one for all: condensations and the initiation of skeletal development. BioEssays 22, 138-147. [DOI] [PubMed] [Google Scholar]
- Hamm M. J., Kirchmaier B. C. and Herzog W. (2016). Sema3d controls collective endothelial cell migration by distinct mechanisms via Nrp1 and PlxnD1. J. Cell Biol. 215, 415-430. 10.1083/jcb.201603100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata K., Takashima R., Amano K., Ono K., Nakanishi M., Yoshida M., Wakabayashi M., Matsuda A., Maeda Y., Suzuki Y. et al. (2013). Arid5b facilitates chondrogenesis by recruiting the histone demethylase Phf2 to Sox9-regulated genes. Nat. Commun. 4, 2850 10.1038/ncomms3850 [DOI] [PubMed] [Google Scholar]
- Hernández-Hernández J. M., Garcia-Gonzalez E. G., Brun C. E. and Rudnicki M. A. (2017). The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 72, 10-18. 10.1016/j.semcdb.2017.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L., Srivastava Y. and Jauch R. (2017). Molecular basis for the genome engagement by Sox proteins. Semin. Cell Dev. Biol. 63, 2-12. 10.1016/j.semcdb.2016.08.005 [DOI] [PubMed] [Google Scholar]
- Huang W., Loganantharaj R., Schroeder B., Fargo D. and Li L. (2013). PAVIS: a tool for peak annotation and visualization. Bioinformatics 29, 3097-3099. 10.1093/bioinformatics/btt520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A. H., Riordan T. J., Pryce B., Weibel J. L., Watson S. S., Long F., Lefebvre V., Harfe B. D., Stadler H. S., Akiyama H. et al. (2015). Musculoskeletal integration at the wrist underlies the modular development of limb tendons. Development 142, 2431-2441. 10.1242/dev.122374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T., Kamekura S., Mabuchi A., Kou I., Seki S., Takato T., Nakamura K., Kawaguchi H., Ikegawa S. and Chung U.-I. (2004). The combination of SOX5, SOX6, and SOX9 (the SOX trio) provides signals sufficient for induction of permanent cartilage. Arthritis. Rheum. 50, 3561-3573. 10.1002/art.20611 [DOI] [PubMed] [Google Scholar]
- Ivkovic S., Yoon B. S., Popoff S. N., Safadi F. F., Libuda D. E., Stephenson R. C., Daluiski A. and Lyons K. M. (2003). Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development 130, 2779-2791. 10.1242/dev.00505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi-Doi M. and Zaret K. S. (2016). Cell fate control by pioneer transcription factors. Development 143, 1833-1837. 10.1242/dev.133900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M., Tamamura Y., Koyama E., Komori T., Takeshita N., Williams J. A., Nakamura T., Enomoto-Iwamoto M. and Pacifici M. (2007). Transcription factor ERG and joint and articular cartilage formation during mouse limb and spine skeletogenesis. Dev. Biol. 305, 40-51. 10.1016/j.ydbio.2007.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo A., Denduluri S., Zhang B., Wang Z., Yin L., Yan Z., Kang R., Shi L. L., Mok J., Lee M. J. et al. (2014). The versatile functions of Sox9 in development, stem cells, and human diseases. Genes Dis. 1, 149-161. 10.1016/j.gendis.2014.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn J., Shwartz Y., Blitz E., Krief S., Sharir A., Breitel D. A., Rattenbach R., Relaix F., Maire P., Rountree R. B. et al. (2009). Muscle contraction is necessary to maintain joint progenitor cell fate. Dev. Cell 16, 734-743. 10.1016/j.devcel.2009.04.013 [DOI] [PubMed] [Google Scholar]
- Kam R. K., Shi W., Chan S. O., Chen Y., Xu G., Lau C. B.-S., Fung K. P., Chan W. Y. and Zhao H. (2013). Dhrs3 protein attenuates retinoic acid signaling and is required for early embryonic patterning. J. Biol. Chem. 288, 31477-31487. 10.1074/jbc.M113.514984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawanami A., Matsushita T., Chan Y. Y. and Murakami S. (2009). Mice expressing GFP and CreER in osteochondro progenitor cells in the periosteum. Biochem. Biophys. Res. Commun. 386, 477-482. 10.1016/j.bbrc.2009.06.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. K., Kim S.-J., Kang S.-S. and Jin E.-J. (2009). Curcumin inhibits cellular condensation and alters microfilament organization during chondrogenic differentiation of limb bud mesenchymal cells. Exp. Mol. Med. 41, 656-664. 10.3858/emm.2009.41.9.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi S., Abe H., Akiyama H., Tominaga T., Murakami T., Mima A., Nagai K., Kishi F., Matsuura M., Matsubara T. et al. (2011). SOX9 protein induces a chondrogenic phenotype of mesangial cells and contributes to advanced diabetic nephropathy. J. Biol. Chem. 286, 32162-32169. 10.1074/jbc.M111.244541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kist R., Schrewe H., Balling R. and Scherer G. (2002). Conditional inactivation of Sox9: a mouse model for campomelic dysplasia. Genesis 32, 121-123. 10.1002/gene.10050 [DOI] [PubMed] [Google Scholar]
- Kitajima K., Koshimizu U. and Nakamura T. (1999). Expression of a novel type of classic cadherin, PB-cadherin in developing brain and limb buds. Dev. Dyn. 215, 206-214. [DOI] [PubMed] [Google Scholar]
- Kozhemyakina E., Lassar A. B. and Zelzer E. (2015). A pathway to bone: signaling molecules and transcription factors involved in chondrocyte development and maturation. Development 142, 817-831. 10.1242/dev.105536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume T., Deng K.-Y., Winfrey V., Gould D. B., Walter M. A. and Hogan B. L. M. (1998). The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell 93, 985-996. 10.1016/S0092-8674(00)81204-0 [DOI] [PubMed] [Google Scholar]
- Kuratani S., Martin J. F., Wawersik S., Lilly B., Eichele G. and Olson E. N. (1994). The expression pattern of the chick homeobox gene gMHox suggests a role in patterning of the limbs and face and in compartmentalization of somites. Dev. Biol. 161, 357-369. 10.1006/dbio.1994.1037 [DOI] [PubMed] [Google Scholar]
- Kyriakides T. R., Zhu Y.-H., Yang Z. and Bornstein P. (1998). The distribution of the matricellular protein thrombospondin 2 in tissues of embryonic and adult mice. J. Histochem. Cytochem. 46, 1007-1015. 10.1177/002215549804600904 [DOI] [PubMed] [Google Scholar]
- Lee H.-L., Yu B., Deng P., Wang C.-Y. and Hong C. (2016). Transforming growth factor-beta-induced KDM4B promotes chondrogenic differentiation of human mesenchymal stem cells. Stem Cells 34, 711-719. 10.1002/stem.2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre V. and Dvir-Ginzberg M. (2017). SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect. Tissue Res., 58, 2-14. 10.1080/03008207.2016.1183667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre V., Zhou G., Mukhopadhyay K., Smith C. N., Zhang Z., Eberspaecher H., Zhou X., Sinha S., Maity S. N. and de Crombrugghe B. (1996). An 18-base-pair sequence in the mouse proalpha1(Mizuhashi et al.) collagen gene is sufficient for expression in cartilage and binds nuclear proteins that are selectively expressed in chondrocytes. Mol. Cell. Biol. 16, 4512-4523. 10.1128/MCB.16.8.4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandoski M., Wassarman K. M. and Martin G. R. (1997). Zp3-cre, a transgenic mouse line for the activation or inactivation of loxP-flanked target genes specifically in the female germ line. Curr. Biol. 7, 148-151. 10.1016/S0960-9822(06)00059-5 [DOI] [PubMed] [Google Scholar]
- Liu Y. and Halloran M. C. (2005). Central and peripheral axon branches from one neuron are guided differentially by Semaphorin3D and transient axonal glycoprotein-1. J. Neurosci. 25, 10556-10563. 10.1523/JNEUROSCI.2710-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.-F. and Lefebvre V. (2015). The transcription factors SOX9 and SOX5/SOX6 cooperate genome-wide through super-enhancers to drive chondrogenesis. Nucleic Acids Res. 43, 8183-8203. 10.1093/nar/gkv688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.-F., Samsa W. E., Zhou G. and Lefebvre V. (2017). Transcriptional control of chondrocyte specification and differentiation. Semin. Cell Dev. Biol. 62, 34-49. 10.1016/j.semcdb.2016.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loebel D. A. F., Hor A. C. C., Bildsoe H., Jones V., Chen Y.-T., Behringer R. R. and Tam P. P. L. (2012). Regionalized Twist1 activity in the forelimb bud drives the morphogenesis of the proximal and preaxial skeleton. Dev. Biol. 362, 132-140. 10.1016/j.ydbio.2011.11.020 [DOI] [PubMed] [Google Scholar]
- Mackie E. J., Thesleff I. and Chiquet-Ehrismann R. (1987). Tenascin is associated with chondrogenic and osteogenic differentiation in vivo and promotes chondrogenesis in vitro. J. Cell Biol. 105, 2569-2579. 10.1083/jcb.105.6.2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammoto T., Mammoto A., Torisawa Y.-S., Tat T., Gibbs A., Derda R., Mannix R., de Bruijn M., Yung C. W., Huh D. et al. (2011). Mechanochemical control of mesenchymal condensation and embryonic tooth organ formation. Dev. Cell 21, 758-769. 10.1016/j.devcel.2011.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelo K. L., Goldie L. C. and Hirschi K. K. (2013). Regulation of endothelial cell differentiation and specification. Circ. Res. 112, 1272-1287. 10.1161/CIRCRESAHA.113.300506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel-Pelletier J., Barr A. J., Cicuttini F. M., Conaghan P. G., Cooper C., Goldring M. B., Goldring S. R., Jones G., Teichtahl A. J. and Pelletier J.-P. (2016). Osteoarthritis. Nat. Rev. Dis. Primers 2, 16072 10.1038/nrdp.2016.72 [DOI] [PubMed] [Google Scholar]
- Matsuda M., Yamashita J. K., Tsukita S. and Furuse M. (2010). abLIM3 is a novel component of adherens junctions with actin-binding activity. Eur. J. Cell. Biol. 89, 807-816. 10.1016/j.ejcb.2010.07.009 [DOI] [PubMed] [Google Scholar]
- Miyaki S., Sato T., Inoue A., Otsuki S., Ito Y., Yokoyama S., Kato Y., Takemoto F., Nakasa T., Yamashita S. et al. (2010). MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 24, 1173-1185. 10.1101/gad.1915510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuhashi K., Kanamoto T., Moriishi T., Muranishi Y., Miyazaki T., Terada K., Omori Y., Ito M., Komori T. and Furukawa T. (2014). Filamin-interacting proteins, Cfm1 and Cfm2, are essential for the formation of cartilaginous skeletal elements. Hum. Mol. Genet. 23, 2953-2967. 10.1093/hmg/ddu007 [DOI] [PubMed] [Google Scholar]
- Motojima M., Tanimoto S., Ohtsuka M., Matsusaka T., Kume T. and Abe K. (2016). Characterization of kidney and skeleton phenotypes of mice double heterozygous for Foxc1 and Foxc2. Cells Tissues Organs 201, 380-389. 10.1159/000445027 [DOI] [PubMed] [Google Scholar]
- Nakamura E., Nguyen M.-T. and Mackem S. (2006). Kinetics of tamoxifen-regulated Cre activity in mice using a cartilage-specific CreER(T) to assay temporal activity windows along the proximodistal limb skeleton. Dev. Dyn. 235, 2603-2612. 10.1002/dvdy.20892 [DOI] [PubMed] [Google Scholar]
- Ng L.-J., Wheatley S., Muscat G. E. O., Conway-Campbell J., Bowles J., Wright E., Bell D. M., Tam P. P. L., Cheah K. S. and Koopman P. (1997). SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev. Biol. 183, 108-121. 10.1006/dbio.1996.8487 [DOI] [PubMed] [Google Scholar]
- Nishioka T., Arima N., Kano K., Hama K., Itai E., Yukiura H., Kise R., Inoue A., Kim S.-H., Solnica-Krezel L. et al. (2016). ATX-LPA1 axis contributes to proliferation of chondrocytes by regulating fibronectin assembly leading to proper cartilage formation. Sci. Rep. 6, 23433 10.1038/srep23433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlender S. A. and Tuan R. S. (1994). Expression and functional involvement of N-cadherin in embryonic limb chondrogenesis. Development 120, 177-187. [DOI] [PubMed] [Google Scholar]
- O'Gorman S., Dagenais N. A., Qian M. and Marchuk Y. (1997). Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc. Natl. Acad. Sci. USA 94, 14602-14607. 10.1073/pnas.94.26.14602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba S., He X., Hojo H. and McMahon A. P. (2015). Distinct transcriptional programs underlie Sox9 regulation of the mammalian chondrocyte. Cell Rep. 12, 229-243. 10.1016/j.celrep.2015.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizette S. and Niswander L. (2000). BMPs are required at two steps of limb chondrogenesis: formation of prechondrogenic condensations and their differentiation into chondrocytes. Dev. Biol. 219, 237-249. 10.1006/dbio.2000.9610 [DOI] [PubMed] [Google Scholar]
- Pryce B. A., Watson S. S., Murchison N. D., Staverosky J. A., Dunker N. and Schweitzer R. (2009). Recruitment and maintenance of tendon progenitors by TGFbeta signaling are essential for tendon formation. Development 136, 1351-1361. 10.1242/dev.027342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray P. and Chapman S. C. (2015). Cytoskeletal reorganization drives mesenchymal condensation and regulates downstream molecular signaling. PLoS ONE 10, e0134702 10.1371/journal.pone.0134702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiprich S. and Wegner M. (2015). From CNS stem cells to neurons and glia: sox for everyone. Cell Tissue Res. 359, 111-124. 10.1007/s00441-014-1909-6 [DOI] [PubMed] [Google Scholar]
- Sock E., Schmidt K., Hermanns-Borgmeyer I., Bosl M. R. and Wegner M. (2001). Idiopathic weight reduction in mice deficient in the high-mobility-group transcription factor Sox8. Mol. Cell. Biol. 21, 6951-6959. 10.1128/MCB.21.20.6951-6959.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A., Garcia M. F., Jaroszewicz A., Osman N., Pellegrini M. and Zaret K. S. (2015). Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555-568. 10.1016/j.cell.2015.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A. and Schilling T. F. (2015). Tendon development and musculoskeletal assembly: emerging roles for the extracellular matrix. Development 142, 4191-4204. 10.1242/dev.114777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symon A. and Harley V. (2017). SOX9: a genomic view of tissue specific expression and action. Int. J. Biochem. Cell Biol. 87, 18-22. 10.1016/j.biocel.2017.03.005 [DOI] [PubMed] [Google Scholar]
- Tsuda M., Takahashi S., Takahashi Y. and Asahara H. (2003). Transcriptional co-activators CREB-binding protein and p300 regulate chondrocyte-specific gene expression via association with Sox9. J. Biol. Chem. 278, 27224-27229. 10.1074/jbc.M303471200 [DOI] [PubMed] [Google Scholar]
- Underhill T. M., Dranse H. J. and Hoffman L. M. (2014). Analysis of chondrogenesis using micromass cultures of limb mesenchyme. Methods Mol. Biol. 1130, 251-265. 10.1007/978-1-62703-989-5_19 [DOI] [PubMed] [Google Scholar]
- Warman M. L., Cormier-Daire V., Hall C., Krakow D., Lachman R., LeMerrer M., Mortier G., Mundlos S., Nishimura G., Rimoin D. L. et al. (2011). Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. 155, 943-968. 10.1002/ajmg.a.33909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright E., Hargrave M. R., Christiansen J., Cooper L., Kun J., Evans T., Gangadharan U., Greenfield A. and Koopman P. (1995). The Sry-related gene Sox9 is expressed during chondrogenesis in mouse embryos. Nat. Genet. 9, 15-20. 10.1038/ng0195-15 [DOI] [PubMed] [Google Scholar]
- Yao B., Wang Q., Liu C.-F., Bhattaram P., Li W., Mead T. J., Crish J. F. and Lefebvre V. (2015). The SOX9 upstream region prone to chromosomal aberrations causing campomelic dysplasia contains multiple cartilage enhancers. Nucleic Acids Res. 43, 5394-5408. 10.1093/nar/gkv426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yapp C., Carr A. J., Price A., Oppermann U. and Snelling S. J. B. (2016). H3K27me3 demethylases regulate in vitro chondrogenesis and chondrocyte activity in osteoarthritis. Arthritis Res. Ther. 18, 158 10.1186/s13075-016-1053-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret K. S., Lerner J. and Iwafuchi-Doi M. (2016). Chromatin scanning by dynamic binding of pioneer factors. Mol. Cell 62, 665-667. 10.1016/j.molcel.2016.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Zhou W., Yao Z., Wan Y., Cao J., Zhang L., Zhao J., Li H., Zhou R., Li B. et al. (2015). Foxp1/2/4 regulate endochondral ossification as a suppresser complex. Dev. Biol. 398, 242-254. 10.1016/j.ydbio.2014.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zúñiga A., Haramis A.-P. G., McMahon A. P. and Zeller R. (1999). Signal relay by BMP antagonism controls the SHH/FGF4 feedback loop in vertebrate limb buds. Nature 401, 598-602. 10.1038/44157 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.