

Abstract
Vitamin B1 is an essential compound in all organisms acting as a cofactor in key metabolic reactions. It is formed by the condensation of two independently biosynthesized molecules referred to as the pyrimidine and thiazole moieties. In bacteria and plants, the biosynthesis of the pyrimidine moiety, 4-amino-5-hydroxymethyl-2-methylpyrimidine phosphate (HMP-P), requires a single enzyme, THIC (HMP-P synthase). The enzyme uses an iron–sulfur cluster as well as a 5’-deoxyadenosyl radical as cofactors to rearrange the 5-amino-imidazole ribonucleotide (AIR) substrate to the pyrimidine ring. So far, the only structure reported is the one from the bacteria Caulobacter crescentus. In an attempt to structurally characterize an eukaryotic HMP-P synthase, we have determined the high-resolution crystal structure of THIC from Arabidopsis thaliana at 1.6 Å. The structure is highly similar to its bacterial counterpart although several loop regions show significant differences with potential implications for the enzymatic properties. Furthermore, we have found a metal ion with octahedral coordination at the same location as a zinc ion in the bacterial enzyme. Our high-resolution atomic model shows a metal ion with multiple coordinated water molecules in the close vicinity of the substrate binding sites and is an important step toward the full characterization of the chemical rearrangement occurring during HMP-P biosynthesis.
Keywords: Thiamin biosynthesis, HMP-P synthase, X-ray structure, SAM radical dependent enzyme, Metal binding site
1. Introduction
Vitamin B1 in its form as thiamin pyrophosphate (TPP) is an essential cofactor for key cellular metabolic enzymes in all forms of life. It is involved in acetyl-CoA biosynthesis, the tricarboxylic acid cycle, the pentose phosphate pathway, isoprenoid biosynthesis through the non-mevalonate pathway, as well as the Calvin-Benson cycle in plants (Jurgenson et al., 2009). Recent studies also suggest a role of this vitamin as a stress protectant in plants (Baxter et al., 2007; Bettendorff and Wins, 2009; Tunc-Ozdemir et al., 2009). De novo thiamin biosynthesis occurs only in bacteria, fungi, and plants. Therefore, auxotrophic organisms such as animals are dependent on its dietary uptake. The biosynthesis of thiamin is regulated by end product feedback inhibition in bacteria and plants, mediated through riboswitches (Winkler et al., 2002). This regulatory mechanism involves the binding of TPP to pre-mRNA elements present in certain genes involved in thiamin biosynthesis or transport (Winkler et al., 2002). In yeast, thiamin biosynthesis is regulated by a series of transcription factors instead of riboswitch control (Tang et al., 1994; Zurlinden and Schweingr-uber, 1997).
The general mechanism of thiamin biosynthesis involves three phases. The first phase leads to the independent formation of thia-zole and pyrimidine moieties. In the next phase, these heterocycles are coupled together to form thiamin monophosphate. The final phase results in the formation of the cofactor form of vitamin B1, TPP. The process of thiazole heterocycle formation is relatively well understood both chemically and structurally in prokaryotes and eukaryotes (Chatterjee et al., 2006, 2007; Jurgenson et al., 2006; Kriek et al., 2007). The formation of the pyrimidine moiety, 4-ami-no-5-hydroxymethyl-2-methylpyrimidine phosphate (HMP-P), is catalyzed by the THIC enzyme in bacteria and in plants or by the THI5 proteins in yeast (Wightman and Meacock, 2003; Lawhorn et al., 2004; Raschke et al., 2007; Ishida et al. 2008; Lai et al., 2012; Coquille et al., 2012). Isotopic labeling and reconstitution studies have shown that THIC uses 5-amino-imidazole ribonucleotide (AIR) as a substrate (Zeidler et al., 2003; Lawhorn et al., 2004). Moreover, biochemical and spectroscopic analyses have demonstrated that THIC is a [4Fe–4S] cluster protein belonging to the S-adenosyl methionine (SAM) radical superfamily (Raschke et al., 2007; Chatterjee et al., 2008; Martinez-Gomez and Downs, 2008; Martinez-Gomez et al., 2009; Dowling et al., 2012). THIC is proposed to utilize the reductive cleavage of SAM to generate a transient 5’-deoxyadenosyl radical (dAdo·), which in turn generates a substrate radical species as observed for various other SAM radical-dependent reactions (Frey, 2001; Wang and Frey, 2007). The conversion of AIR to HMP-P is a complex intramolecular rearrangement, involving the breakage, and re-forming of multiple bonds as well as the generation of several radical species during the catalytic cycle (Chatterjee et al., 2010). However, the exact reaction mechanism is not completely elucidated. EPR spectroscopic studies indicate that the dAdo· radical generates a protein-bound radical, which then reacts with the substrate (Martinez-Gomez et al., 2009). Although, NMR analysis seems to demonstrate that the dAdo· radical reacts directly with the substrate (Chatterjee et al., 2010). While the X-ray crystal structure of the bacterial THIC has assisted in elucidating certain aspects of the mechanism of the reaction (Chatterjee et al., 2008), further structural data are needed to gain insight into the architecture of an eukaryotic homolog.
In this paper, we report the high resolution crystal structure (1.6 Å) of THIC from the model plant Arabidopsis thaliana. We compare it with the structure of its homolog from the bacterium Caulobacter crescentus (Chatterjee et al., 2008). In particular, two large loops masking several residues important for the aerobic activity are absent in the eukaryotic protein. Although the [4Fe–4S] cluster C-terminal binding domain is not visible, our high-resolution crystal structure substantiates interesting features; notably the presence of a large electron density peak near the AIR and SAM binding sites, in which we built a cobalt ion. The high resolution diffraction data shows numerous coordinated water molecules in the close proximity of the site where the dAdo· radical is proposed to be formed. These coordinated water molecules may mimic the position of protein atoms upon iron–sulfur cluster binding to the missing C-terminal fragment.
2. Material and methods
2.1. Purification and enzymatic activity of A. thaliana THIC
The protein used in our experiments is a N-terminally truncated version of A. thaliana THIC, lacking the first 71 amino acids (ΔΝ71–AtTHIC; Raschke et al., 2007). This version of the protein was previously reported as a soluble protein, whereas the full-length one is not (Raschke et al., 2007). The deleted region is likely to incorporate a chloroplastic targeting peptide and is predicted as mostly disordered by the program Phyre (Raschke et al., 2007; Kelley and Sternberg, 2009). Therefore, we first wanted to confirm that this version of the protein is active. For this part of the study, the construct designed to express ΔN71–AtTHIC fused to a C-terminal hexa-histidine affinity tag was introduced into the Escherichia coli BL21 (DE3) strain. Cells were incubated at 37 °C with shaking at 180 rpm until the OD600 reached 0.5 and then placed for 1.5 h at 4 °C. The medium was supplemented with 0.47 mM ferrous ammonium sulfate and 1.1 mM cysteine per 1.5 L culture. Expression was induced by addition of 0.1 mM IPTG, followed by growth for 15 h at 15 °C with shaking at 90 rpm. Prior to harvesting, the cultures were placed at 4 °C for 3 h. The harvested cell pellet was resuspended and lyzed in 100 mM Tris – HCl, pH 7.5, containing 2 mM dithio-threitol (DTT), 0.2 mg/mL lysozyme and 100 U of benzonase in an anaerobic chamber (from COY Laboratory Products). This mixture was then cooled in an ice-bath for 2 h. After sonication and centrifugation at 25,000g, the enzyme was purified from the extract using standard Ni2+–NTA chromatography (Qiagen) in 100 mM Tris–HCl, pH 7.5, containing 300 mM sodium chloride, 2 mM DTT and either 20 mM or 250 mM imidazole for washing and elution steps, respectively. The purified protein was exchanged into 100 mM potassium phosphate, pH 7.5, containing 30% glycerol and 2 mM DTT using an Econo-Pac® 10DG Desalting column. As shown in Fig. 1A, the anaerobically purified DN71-AtTHIC presents a characteristic absorption profile with a maximum at 410 nm and a shoulder at 320 nm. The strong decrease observed for both features after reduction with sodium dithionite confirms the presence of the Fe–S cluster bound to the protein.
Fig.1.
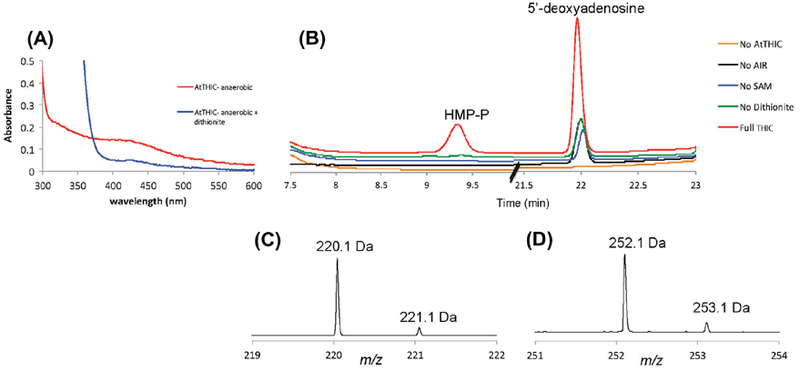
ΔN71-AtTHIC activity measurement. (A) The red trace corresponds to anaerobically purified ΔN71–AtTHIC showing a UV–visible spectrum with a maximum at 410 mm and a shoulder at 320 nm, characteristic of a [4Fe–4S] cluster. Upon addition of dithionite, reduction of the cluster leads to the UV–visible spectrum shown in the blue trace with a characteristic absorption maximum at 425 nm. (B) HPLC profiles for the formation of HMP-P and 5ʹ-deoxyadenosine as measured by the absorbance at 254 nm. The red trace clearly indicates that the HMP-P is formed (elution time 9.4 min) only in assays where ΔN71–AtTHIC, AIR, SAM, and dithionite are present (All). No HMP-P formation is detected in controls where eitherAIR, SAM, ΔN71–AtTHIC or dithionite is absent. High amounts of 5ʹ-deoxyadenosine formation are seen in assays where ΔN71–AtTHIC, AIR, SAM, and dithionite are present (All). Loweramounts of 5ʹ-deoxyadenosine (elution time 22 min) are seen in assays where eitherAIR, SAM ordithionite is absent and no 5ʹ-deoxyadenosine is detected in assays where ΔN71–AtTHIC is absent. This suggests that a small amount of 5ʹ-deoxyadenosine co-purifies with ΔΝ71–AtTHIC. (C and D) ESI-MS (positive mode) of the reaction products eluting at 9.4 min (C, HMP-P) and 22 min (D, 5ʹ-deoxyadenosine).
2.2. Crystallization and structure determination
For crystallization studies, the ΔN71–AtTHIC protein was expressed aerobically in the E. coli BL21 (DE3) pRARE strain (Raschke et al., 2007). The cells were incubated at 30 °C overnight in autoinduction medium (1χ N–Z-amine AS containing 0.5% glycerol, 0.05% glucose, 0.2% α-lactose, 25 mM ammonium sulfate, 50 mM potassium dihydrogen phosphate, 50 mM disodium hydrogen phosphate, and 1 mM magnesium sulfate). After harvesting by centrifugation at 5000g, the cell pellets were resuspended and lyzed in 50 mM sodium phosphate, pH 7.5, containing 300 mM sodium chloride, 10 mM imidazole, and 5 mM β-mercaptoethanol. Cell debris was removed by centrifugation at 25,000g. The ΔΝ71–AtTHIC protein present in the supernatant was purified by Ni2+–NTA chromatography (Qiagen) followed by a gel-filtration step using a preparative Superdex 200 column (GE Healthcare) preequilibrated in crystallization buffer (20 mM Hepes, pH 7.5, containing 150 mM NaCl, 1 mM β-mercaptoethanol, and 10% (v/v) glycerol). The ΔN71–AtTHIC protein was concentrated to 15 mg/ml and used immediately for crystallization. Crystals of ΔΝ71–AtTHIC were obtained by a proteolysis assay coupled with the sitting-drop vapor diffusion technique at 18 °C in 96-well crystallization plates (Swiscii). Specifically, the protease, a-chymotrypsin, was added to the protein sample at a ratio of 1:500 w/w immediately prior to setting up the crystallization trials. The crystallization drops were then prepared by mixing 0.4 μl of this protease/protein solution with an equal volume of the crystallization solutions. High diffracting ΔN71–AtTHIC needles appeared after three days in the Hampton Research Crystal screen 2: 0.01 M cobalt (II) chloride hexahydrate, 0.1 M sodium acetate trihydrate pH 4.6, 1 M 1,6-hexanediol (condition #11). The crystals were directly flash frozen in liquid nitrogen. Diffraction data were collected at the Swiss Light-Source beamline PXIII at 100 K (SLS, Villigen-Paul Scherrer Institute). The diffraction spots were integrated and scaled with the XDS package (Kabsch, 2010). The statistics for the ΔN71–AtTHIC data set are reported in Table 1.
Table 1.
Crystallographic data collection and refinement statistics.
| Δ71–AtTHIC crystal | |
|---|---|
| Data collection | |
| Wavelength (Å) | 0.97795 |
| Space group | P3221 |
| Unit cell dimensions (Å) | |
| a, b, c (Å) | 107.0, 107.0, 88.3 |
| a, b, c (°) | 90.0, 90.0, 120.0 |
| Resolution (Å) | 41.05–1.6 (1.64–1.60) |
| No. of unique reflections | 77’101 (5’668) |
| Redundancy | 10(9.95) |
| Rmeas1 | 14.6 (127.1) |
| I/σ (I) | 13.58 (2.35) |
| Completeness (%) | 100 (100) |
| Matthews coefficient/Solvent content | 1 mol/AU 2.75/55.23 |
| Refinement | |
| Rwork (%)2 | 15.19 |
| Rfree (%)3 | 17.6 |
| No. of atoms | |
| Protein | 3821 |
| Cobalt | 1 |
| Hexane-1,6-diol | 24 |
| Water | 701 |
| Mean B-factors (Å2) | |
| Protein | 14.90 |
| Cobalt | 12.25 |
| Hexane-1,6-diol | 32.27 |
| Solvent | 30.16 |
| RMSD from ideal geometry | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.068 |
| Residues in favored region of the Ramachandran plot (%) | 98.7 |
| Residues in allowed region of the Ramachandran plot (%) | 1.3 |
Values in parentheses are for the highest-resolution shell.
With Ih the intensity of reflection h, and nh the multiplicity.
With Fobs and Fcalc the observed and calculated structure factors respectively and h the reflections indices.
Rfree: Cross-validation of Rwork.
The structure of the A. thaliana THIC protein was determined by molecular replacement using the program Phaser from the CCP4 package (McCoy et al., 2007). We used the coordinates of the C. crescentus ThiC protein (PDB ID 3EPM; Chatterjee et al., 2008) as a reference. The asymmetric unit contains 1 molecule. To build our protein model, we first removed model bias by rounds of simulated annealing performed with the program Phenix (Adams et al., 2010), followed by the calculation of difference Fourier maps. We rebuilt the ΔN71–AtTHIC model in the graphic program COOT (Emsley and Cowtan, 2004). The model was finally refined using the same programs by iterative rounds of energy minimization, B-factor, and anisotropic refinements followed by calculation of composite omit and difference Fourier maps. Numerous water molecules and ions were added in the late stage of the refinement. Our model shows good stereochemistry as indicated by the program PROCHECK with no residues in the disallowed regions of the Ramachandran plot (Laskowski et al., 1993). The final model statistics are shown in Table 1. Although the ΔN71-AtTHIC crystal diffracted to high resolution (~1.6 Å), residues 72–84 and 557–644 are not visible in the electron density map. These two protein segments were probably cleaved by the α-chymotrypsin protease during the crystallization; although we cannot exclude that they were simply disordered in the crystal.
3. Results and discussion
3.1. Activity measurement of the DN71-AtTHIC
The activity of ΔN71-AtTHIC as isolated was assessed under anaerobic conditions at room temperature with reaction mixtures containing all or various combinations of ΔN71–AtTHIC, AIR, SAM, and an excess of dithionite (Fig. 1B; Chatterjee et al., 2008). At the end of the reaction, the enzyme was removed by ultrafiltration using a 10kDa cut-off filter and the products were analyzed by HPLC coupled to mass spectrometry (Fig. 1C,D; Chatterjee et al., 2008). Activity was observed only when all the reaction components were present, confirming that despite the absence of the first 71 amino acids, the truncated AtTHIC is active as it converts AIR into HMP-P and SAM into 5’-deoxyadenosine (Fig. 1B–D). However, if we assume that ΔN71–AtTHIC is fully reconstituted with a [4Fe–4S] cluster, the ratio of HMP-P/THIC is 0.7/1. The observation of only a single turnover is likely due to improper reconstitution of the (4Fe–4S) cluster and requires further investigation.
3.2. Monomer structure
The THIC monomer consists of three domains namely the N-terminal domain (residues 85–223), the central domain (residues 224–519), and the C-terminal domain (residues 520–556) as shown in Fig. 2A. As mentioned previously, the last 87 residues of AtTHIC, which includes the binding site for the iron–sulfur cluster, were not visible in our electron density map and as such were not built in our final model. The N-terminal domain of AtTHIC forms a blanket-like structure (Fig. 2A,B). The presence of such a domain is unique to THIC enzymes from aerobic species (Dougherty and Downs, 2006). This domain plays a structural role in two ways: (i) it seals off the N-terminal face of the central (β/α)8 TIM barrel domain, where the putative active site is found (Fig. 2A,B) and (ii) it extends over the C-terminal domain such that the C-terminus has a defined orientation near/above the active site (Fig. 2D). The central domain houses the (β/α)8 TIM barrel fold (Fig. 2A,C). The cavity at the C-terminal face of the barrel delineates the part of the active site responsible for the association with the substrate AIR near a conserved cysteine residue, cysteine 484 in A. thaliana THIC (Fig. 2C; Chatterjee et al., 2008). Recently, the SAM molecule was proposed to be stabilized near residue glutamate 489 from the central domain (Dowling et al., 2012). The structure and sequence of the central domain are highly conserved between prokaryotic and eukaryotic anaerobic and aerobic species (Fig. 3A and Fig. S2; Chatterjee et al., 2008). The visible part of the C-terminal domain is composed of three antiparallel a-helices forming a sheet-like structure sandwiched between a loop of the aerobic specific N-terminal domain (Dougherty and Downs, 2006) and the TIM barrel of the symmetry-related molecule (Fig. 2D). Notably, the three helices of the C-terminal domain are stabilized by a network of hydrogen bonds between residues serine 121 and glutamate 124 from the N-terminal domain and asparate 540 and arginine 537 from the C-terminal domain (Fig. 2D). This domain is structurally very similar to ThiC from C. crescentus (Fig. 3B) and is very well conserved at the sequence level (Fig. 3A). The rest of the C-terminal region is missing in our atomic structure (residues 557–644) and includes the conserved CX2CX4C motif predicted to bind the [4Fe–4S] cluster in THIC enzymes (Dougherty and Downs, 2006; Raschke et al., 2007).
Fig.2.
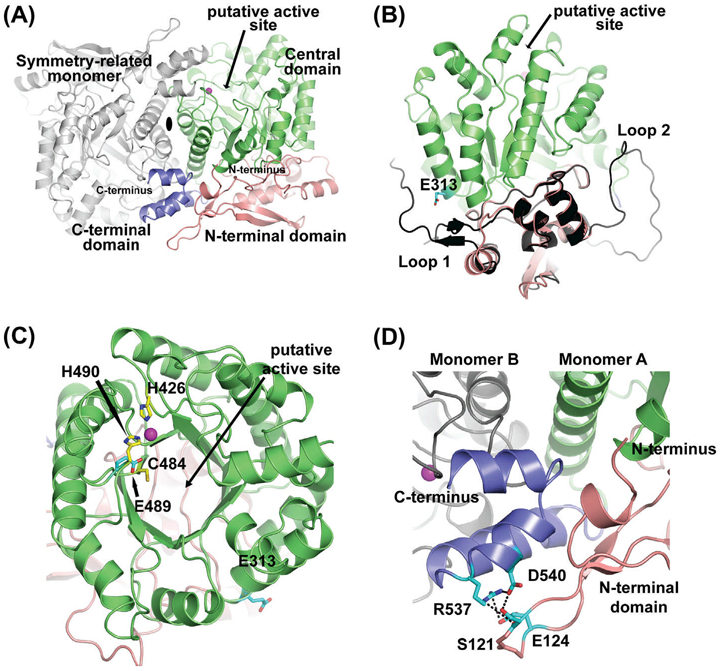
Crystal structure of Arabidopsis THIC. (A) The THIC dimer is shown using a cartoon representation. The dimer is formed by two crystallographically-related molecules around a 2-fold axis (shown as a black ellipse). The N-terminal, central, and C-terminal domains are labeled and colored in salmon, green, and dark blue, respectively. The active site is indicated with an arrow and the position of the metal ion as magenta-colored spheres. The gray-colored molecule is the symmetry-related Arabidopsis THIC monomer. (B) Side view of the Arabidopsis THIC monomer superimposed onto the C. crescentus N-terminal domain structure (colored in black). The two loops present in C. crescentus are absent in the A. thaliana atomic model (labeled as loop 1 and loop 2). Residue E313 is shown as sticks colored according to atom types (carbon cyan, and oxygen red). (C) Top view of the green-colored central domain displaying a typical (β/α)8 TIM barrel fold. The two histidines that partially coordinate the metal ion (H426, H490), one cysteine (C484) involved in the catalytic cycle, the glutamate (E489) implicated in the SAM molecule binding as well as E313 are shown as sticks colored according to atom types (carbon yellow or cyan, oxygen red, and nitrogen blue). (D) Close-up view of the blue-colored C-terminal domain, sandwiched between the N-terminal domain of monomer A colored in salmon and the central domain loops of monomer B in gray. Residues S121, E124, R537, and D540 are shown as sticks and colored to atom types (carbon in cyan, oxygen and nitrogen as before).
Fig.3.
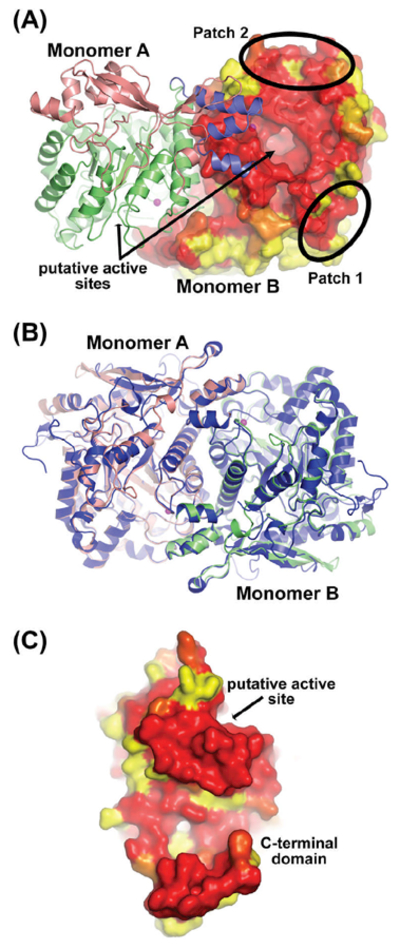
Conservation and similarities of the A. thaliana THIC structure compared to its bacterial counterpart. (A) Mixed cartoon and surface representation of the A. thaliana THIC dimer. Monomer A is colored as in Fig.2A and monomer B is shown in molecular surface mode colored according to sequence conservation (yellow for no conservation, red for full conservation). The sequence alignment used to calculate the conservation is provided in the supplementary information (Fig. S2). The two black ellipses indicate conserved sequence patches that are referred to in the text. (B) Superimposition of the dimeric THIC atomic models from C. crescentus (in dark blue) and A. thaliana (in salmon and green). (C) The ΔN71–AtTHIC is shown from the interface side to appreciate the high sequence conservation. The THIC monomer is shown in molecular surface mode colored as in panel A.
3.3. DN71–AtTHIC is dimeric
The crystal structure of A. thaliana ΔN71–AtTHIC contains a single molecule in the asymmetric unit. This monomer forms a crystallographic dimer with a symmetry-related molecule (Fig. 2A). Size exclusion chromatography of the purified protein shows two peaks, a major peak with an elution volume corresponding to an apparent molecular mass of ~130 kDa by extrapolation from the elution volume of protein molecular mass standards and a minor peak corresponding to higher molecular mass complexes (Fig. S1). Electrospray ionization mass spectrometry revealed a molecular mass of 65,451 Da for ΔDN71–AtTHIC. Therefore, this data indicates that ΔN71–AtTHIC is predominantly dimeric in solution, as observed for the bacterial C. crescentus ThiC (Chatterjee et al., 2008). Superimposition of the ΔN71–AtTHIC structure with that of C. crescentus ThiC demonstrates a striking similarity between the monomer folds (Fig. 3B). Furthermore, the ΔΝ71–AtTHIC crystallographic dimer perfectly superimposes with the C. crescentus dimer indicating that the interface used for dimer formation is the same in both cases (Fig. 3B). The observed interface between the two ΔN71–AtTHIC monomers buries a surface of 2475 Å2 (~13.2% of solvent accessible area per monomer) as calculated using the PISA server (Krissinel and Henrick, 2007). Like its bacterial homolog, the dimer interface of the eukaryotic AtTHIC is strongly conserved at the amino acid sequence level (Fig. 3C; Deprez et al., 2005). The main interaction surfaces involve several helices of the two (b/a)8 TIM barrels and the three-helix sheet of the two C-terminal domains, which are joined in an antiparallel arrangement (Fig. 3B). As for its prokaryotic counterpart, the Arabidopsis THIC dimer presents a domain swapped arrangement with the C-terminal domain of one monomer covering the active site of the second monomer (Fig. 3A; the C-terminal domain of monomer A covers the active site of monomer B). This kind of domain swapped organization is also found in the adenosylcobalamin (AdoCbl)-dependent enzyme ornithine 4,5-aminomutase (Wolthers et al., 2010). The AdoCbl radical enzyme superfamily and the SAM radical superfamily (to which AtTHIC belongs) are thought to be evolutionary related (Frey, 2001; Berkovitch et al., 2004). In fact, both of these superfamilies utilize a 5’-deoxyadenosyl radical for their enzymatic activity and some members share structural features, notably a (β/α)8 TIM barrel fold (Dowling et al., 2012). The dimerization is therefore functionally important as the C-terminus of one monomer is likely to contribute to the active site of the neighboring monomer with the two molecules being arranged in a head to tail orientation.
3.4. Differences between the prokaryotic and the eukaryotic enzymes
The sequence conservation within the THIC protein family is relatively high, particularly within the following regions (Fig. 3A and Fig. S2): (i) the central domain formed by the (β/α)8 TIM barrel, which contains the putative active site and makes part of the dimer interface; (ii) two large patches located on the external side of the Arabidopsis THIC TIM barrel (Fig. 3A). The latter conserved patches appear to be too far away from the proposed active site location to be involved in the reaction mechanism. However, one can speculate that the strong conservation across evolution is indicative of a functionally relevant region in the protein. These regions could for example be used to stabilize the C-terminal protein fragment that is not visible in our crystal structure. Moreover, upon comparison with the C. crescentus structure, the Arabidopsis THIC protein has lost two long loops within the N-terminal domain, one of which covers conserved patch 1 in the bacterial counterpart (compare Figs. 2B,3A). Several residues found within patch 1 have been studied in more detail due to their specific conservation in aerobic species (Dougherty and Downs, 2006). Notably, the absence of the extended loop 1 in our structure exposes glutamate 313 from the central domain to the solvent (compare Fig. 2B,C). It has been reported previously that mutation of the equivalent residue in S. enterica (E324K) makes the S. enterica enzyme specifically inactive under aerobic conditions (Dougherty and Downs, 2006). This residue is therefore affecting enzyme activity, although it is located more than 25 A from the active site residue, cysteine 484 (Fig. 2C; Dougherty and Downs, 2006). Another residue differentially conserved in aerobic species, arginine 537, is involved in a network of hydrogen bonds with residues serine 121, glutamate 124, and aspartate 540 (Fig. 2D; Dougherty and Downs, 2006). Mutation of the equivalent residue in S. enterica (R548H) led to reduced growth in thiamin free medium (Dougherty and Downs, 2006). We predict that mutating this residue would destabilize the C-terminal three-helix sheet, possibly affecting the positioning of the [4Fe-4S] cluster near the active site, and as such would modify the overall activity of the enzyme.
3.5. Metal ion binding site
Each monomer in the structure of A. thaliana THIC contains a large peak of electron density near the assumed active site. Our 2Fo–Fc Fourier electron density map calculated at 1.6 Å clearly indicates that the ion has an octahedral coordination sphere, using two strictly conserved histidine residues (H426 and H490) and four water molecules (Figs. 2C and 4A). Furthermore, the anomalous Fourier electron density map also shows a strong peak at the same location. Notably, the bacterial ThiC structure had a zinc ion bound between the two equivalent histidine residues (Chatterjee et al., 2008). In an attempt to identify the bound ion, we quantified various elements in our AtTHIC protein preparation, prior to its crystallization, by mass spectrometry (Supplementary Table S1). This experiment indicates that our preparation of AtTHIC has several metal ions present in sub-stoichiometric amounts, although none of these ions were part of the buffer used during the last purification step (Supplementary Table S1). The presence of multiple metal ions indicates that AtTHIC has a metal binding site (albeit weak). As our crystallization buffer contains 10 mM of cobalt chloride, a metal ion which has anomalous signal at the X-ray wavelength used for data collection, we decided to position a cobalt ion in our electron density peak (Fig. 4A). The high diffraction properties of our crystals made possible the precise refinement of the coordinated water molecules around the placed cobalt ion (coordinating distances between 2.13 and 2.85 A). It appears that AtTHIC has a low affinity metal ion-binding site although we do not know its exact role especially as the iron–sulfur cluster and a large protein region are missing from our crystal structure. As the protein contributes only two ligands in our structure (H426 and H490), we would like to emphasize that the missing C-terminal fragment (residues 557–644) may contribute additional protein ligands and as such replace some of the coordinated water molecules. Notably, the residue equivalent to histidine 426 in S. enterica was shown to be sensitive to the formation of the 5’-deoxyadenine radical generated during HMP-P biosynthesis, which indicates that it is in close proximity (Martinez-Gomez et al., 2009). Simultaneously, the glutamate 489 residue has been recently proposed to be essential for positioning the SAM molecule and forms a hydrogen bond with one of the water molecules coordinating the associated metal ion (Fig. 4A,B; Chatterjee et al., 2008; Dowling et al., 2012). Based on the homology model of the dAdo-bound ThiC protein published recently, we cannot help but speculate that the presence of a metal ion may, together with the iron–sulfur cluster, modulate the reactivity of the SAM molecule (Fig. 4B; Dowling et al., 2012). It is unlikely that both the bacterial and the eukaryotic ThiC enzymes have kept a metal ion-binding site if it was not used during the reaction mechanism. Interestingly, the arginine 386 has two distinct conformations visible in the electron density maps, which indicates that its side chain is highly flexible and could stabilize reaction intermediates and/or the phosphate part of the substrate AIR, as observed in the C. crescentus structure (Fig. 4B).
Fig.4.
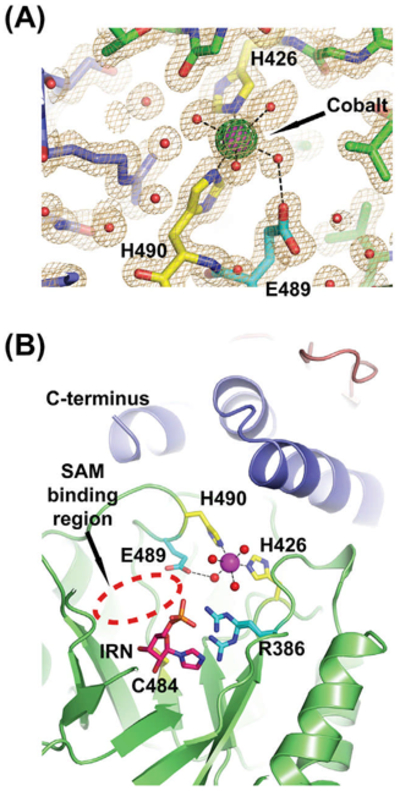
The A. thaliana THIC protein coordinates a metal ion. (A) Close-up view of the fitted cobalt ion with the Fourier electron density map contoured at 1.5σ (light orange) and the anomalous Fourier map contoured at 5σ (green). The ion has six ligands in a octahedral geometry. The ligands are the two NE atoms from histidines 426 and 490 (shown as yellow sticks) and four water molecules (shown as red spheres). Metal ion coordination is indicated by black dashed lines. (B) Top view of the predicted A. thaliana THIC active site cavity. The central domain of one monomer is shown in green together with domains of the second monomer (colored in dark blue for the C-terminal domain and salmon for the N-terminal). The protein is shown using a cartoon representation. The various residues discussed in the text are shown as sticks (R386, H426, C484, E489, H490). R386 has two conformations. The imidazole ribotide (IRN) molecule bound to the C. crescentus protein (PDB code 3EPN) is also shown to delineate the substrate binding area after superimposition of the two protein models. The SAM binding region is indicated by a red dashed ellipse. Atoms are colored according to their types (carbon yellow, cyan or magenta; oxygen, red; sulfur, gold; phosphate, orange; nitrogen, blue, and cobalt purple). The C-terminus indicated the end of the visible part of ΔN71-AtTHIC.
4. Conclusions
Overall, the determined structure of THIC from A. thaliana is the first available model for an eukaryotic member of this essential enzyme family. The 1.6 Å resolution of A. thaliana THIC compares very well with the previous 2.8 Å resolution of C. crescentus ThiC with the exceptions of two loops that are missing in the eukaryotic protein. Our structure provides a better picture of the metal binding site located near the putative active site, a metal binding site that was also observed in the bacterial ThiC structure. We suggest that the metal ion is potentially relevant for the reaction mechanism by providing activated water molecules, susceptible to relay the dAdo” radical. A structure of the protein with an iron–sulfur cluster bound remains to be determined to fully apprehend the overall enzymatic reaction.
4.1. Accession number
The atomic coordinates of Arabidopsis THIC have been deposited in the Protein Data Bank with accession code PDB 4N7Q.
Supplementary Material
Acknowledgments
We wish to thank Dr. Markus Kaufmann (University of Geneva) for performing preliminary gel filtration experiments, the SLS (Swiss Light Source) for beam time allocation and the staff of beamline X06DA (PXIII) at SLS for help with data collection. We gratefully acknowledge financial support from the E. Boninchi, the E&L Schmidheiny and the Marc Birkigt Foundations. Furthermore, we specifically thank the University of Geneva and the Swiss National Science Foundation (SNSF) R’equip grant no.316030–128787 for the development of the crystallographic platform. The work was supported by the SNSF (PP00A-119186 and SINERGIA project CRSI33_127506 to T.B.F., 31003A-124909 and 31003A-140924 to S.T.) and by the University of Geneva.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jsb.2013.10.005.
References
- Adams PD et al. , 2010. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta. Crystallogr. D Biol. Crystallogr 66 (Pt 2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter CJ et al. , 2007. The metabolic response of heterotrophic Arabidopsis cells to oxidative stress. Plant Physiol 143 (1), 312–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovitch F, Nicolet Y, Wan JT,Jarrett JT, Drennan CL, 2004. Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science 303 (5654), 76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettendorff L, Wins P, 2009. Thiamin diphosphate in biological chemistry: new aspects of thiamin metabolism, especially triphosphate derivatives acting other than as cofactors. FEBSJ 276 (11), 2917–2925. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Hazra AB, Abdelwahed S, Hilmey DG, Begley TP, 2010. A ‘‘radical dance’’ in thiamin biosynthesis: mechanistic analysis of the bacterial hydroxymethylpyrimidine phosphate synthase. Angew. Chem. Int. Ed. Engl 49 (46), 8653–8656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Jurgenson CT, Schroeder FC, Ealick SE, Begley TP, 2006. Thiamin biosynthesis in eukaryotes: characterization of the enzyme-bound product of thiazole synthase from Saccharomyces cerevisiae and its implications in thiazole biosynthesis. J. Am. Chem. Soc 128 (22), 7158–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Jurgenson CT, Schroeder FC, Ealick SE, Begley TP, 2007. Biosynthesis of thiamin thiazole in eukaryotes: conversion of NAD to an advanced intermediate. J. Am. Chem. Soc 129 (10), 2914–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Li Y, Zhang Y, Grove TL, Lee M, Krebs C, Booker SJ, Begley TP, Ealick SE, 2008. Reconstitution of ThiC in thiamine pyrimidine biosynthesis expands the radical SAM superfamily. Nat. Chem. Biol 4 (12), 758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquille S, Roux C, Fitzpatrick TB, Thore S, 2012. The last piece in the vitamin B1 biosynthesis puzzle: structural and functional insight into yeast 4-amino-5-hydroxymethyl-2-methylpyrimidine phosphate (HMP-P) synthase. J. Biol. Chem 287 (50), 42333–42343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deprez C, Lloubes R, Gavioli M, Marion D, Guerlesquin F, Blanchard L, 2005. Solution structure of the E. coli TolA C-terminal domain reveals conformational changes upon binding to the phage g3p N-terminal domain.J. Mol. Biol 346 (4), 1047–1057. [DOI] [PubMed] [Google Scholar]
- Dougherty MJ, Downs DM, 2006. A connection between iron and sulfur cluster metabolism and the biosynthesis of 4-amino-5-hydroxymethyl-2-methylpyrimidine pyrophosphate in Salmonella enterica. Microbiology 152 (Pt 8), 2345–2353. [DOI] [PubMed] [Google Scholar]
- Dowling DP, Vey JL, Croft AK, Drennan CL, 2012. Structural diversity in the AdoMet radical enzyme superfamily. Biochim. Biophys. Acta 11 (1824), 1178–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K, 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60 (Pt 12 Pt 1), 2126–2132. [DOI] [PubMed] [Google Scholar]
- Frey PA, 2001. Radical mechanisms of enzymatic catalysis. Annu. Rev. Biochem 70, 121–148. [DOI] [PubMed] [Google Scholar]
- Ishida S, Tazuya-Murayama K, Kijima Y, Yamada K, 2008. The direct precursor of the pyrimidine moiety of thiamin is not urocanic acid but histidine in Saccharomyces cerevisiae. J. Nutr. Sci. Vitaminol. (Tokyo) 54 (1), 7–10. [DOI] [PubMed] [Google Scholar]
- Jurgenson CT, Begley TP, Ealick SE, 2009. The structural and biochemical foundations of thiamin biosynthesis. Annu. Rev. Biochem 78, 569–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgenson CT, Chatterjee A, Begley TP, Ealick SE, 2006. Structural insights into the function of the thiamin biosynthetic enzyme Thi4 from Saccharomyces cerevisiae. Biochemistry 45 (37), 11061–11070. [DOI] [PubMed] [Google Scholar]
- Kabsch W, 2010. Xds. Acta Crystallogr. D Biol. Crystallogr 66 (Pt 2), 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ, 2009. Protein structure prediction on the web: a case study using the phyre server. Nat. Protoc 4 (3), 363–371. [DOI] [PubMed] [Google Scholar]
- Kriek M, Martins F, Leonardi R, Fairhurst SA, Lowe DJ, Roach PL, 2007. Thiazole synthase from Escherichia coli: an investigation of the substrates and purified proteins required for activity in vitro. J. Biol. Chem 282 (24), 17413–17423. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K, 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372 (3), 774–797. [DOI] [PubMed] [Google Scholar]
- Lai RY et al. , 2012. Thiamin pyrimidine biosynthesis in Candida albicans: a remarkable reaction between histidine and pyridoxal phosphate. J. Am. Chem. Soc 134 (22), 9157–9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Moss DS, Thornton JM, 1993. Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol 231 (4), 1049–1067. [DOI] [PubMed] [Google Scholar]
- Lawhorn BG, Mehl RA, Begley TP, 2004. Biosynthesis of the thiamin pyrimidine: the reconstitution of a remarkable rearrangement reaction. Org. Biomol. Chem 2 (17), 2538–2546. [DOI] [PubMed] [Google Scholar]
- Martinez-Gomez NC, Downs DM, 2008. ThiC is an [Fe–S] cluster protein that requires AdoMet to generate the 4-amino-5-hydroxymethyl-2-methylpyrimidine moiety in thiamin synthesis. Biochemistry 47 (35), 9054–9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Gomez NC, Poyner RR, Mansoorabadi SO, Reed GH, Downs DM, 2009. Reaction of AdoMet with ThiC generates a backbone free radical. Biochemistry 48 (2), 217–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, 2007. Phaser crystallographic software. J. Appl. Crystallogr 40 (Pt 4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschke M, Burkle L, Muller N, Nunes-Nesi A, Fernie AR, Arigoni D, Amrhein N, Fitzpatrick TB, 2007. Vitamin B1 biosynthesis in plants requires the essential iron sulfur cluster protein, THIC. Proc. Natl. Acad. Sci. U.S.A 104 (49), 19637–19642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang CS, Bueno A, Russell P, 1994. Ntf1+ encodes a 6-cysteine zinc finger-containing transcription factor that regulates the nmt1 promoter in fission yeast. J. Biol. Chem 269 (16), 11921–11926. [PubMed] [Google Scholar]
- Tunc-Ozdemir M, Miller G, Song L, Kim J, Sodek A, Koussevitzky S, Misra AN, Mittler R, Shintani D, 2009. Thiamin confers enhanced tolerance to oxidative stress in Arabidopsis. Plant Physiol 151 (1), 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SC, Frey PA, 2007. S-adenosylmethionine as an oxidant: the radical SAM superfamily. Trends Biochem. Sci 32 (3), 101–110. [DOI] [PubMed] [Google Scholar]
- Wightman R, Meacock PA, 2003. The THI5 gene family of Saccharomyces cerevisiae: distribution of homologues among the hemiascomycetes and functional redundancy in the aerobic biosynthesis of thiamin from pyridoxine. Microbiology 149 (Pt 6), 1447–1460. [DOI] [PubMed] [Google Scholar]
- Winkler W, Nahvi A, Breaker RR, 2002. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 419 (6910), 952–956. [DOI] [PubMed] [Google Scholar]
- Wolthers KR, Levy C, Scrutton NS, Leys D, 2010. Large-scale domain dynamics and adenosylcobalamin reorientation orchestrate radical catalysis in ornithine 4,5-aminomutase. J. Biol. Chem 285 (18), 13942–13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler J, Sayer BG, Spenser ID, 2003. Biosynthesis of vitamin B1 in yeast. Derivation of the pyrimidine unit from pyridoxine and histidine. Intermediacy of urocanic acid. J. Am. Chem. Soc 125 (43), 13094–13105. [DOI] [PubMed] [Google Scholar]
- Zurlinden A, Schweingruber ME, 1997. Identification of a DNA element in the fission yeast Schizosaccharomyces pombe nmt1 (thi3) promoter involved in thiamine-regulated gene expression. J. Bacteriol 179 (18), 5956–5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.