

Abstract
Thiamin is synthesized by most prokaryotes and by eukaryotes such as yeast and plants. In all cases, the thiazole and pyrimidine moieties are synthesized in separate branches of the pathway and coupled to form thiamin phosphate. A final phosphorylation gives thiamin pyrophosphate, the active form of the cofactor. Over the past decade or so, biochemical and structural studies have elucidated most of the details of the thiamin biosynthetic pathway in bacteria. Formation of the thiazole requires six gene products, and formation of the pyrimidine requires two. In contrast, details of the thiamin biosynthetic pathway in yeast are only just beginning to emerge. Only one gene product is required for the biosynthesis of the thiazole and one for the biosynthesis of the pyrimidine. Thiamin can also be transported into the cell and can be salvaged through several routes. In addition, two thiamin degrading enzymes have been characterized, one of which is linked to a novel salvage pathway.
Keywords: degradation, salvage, transport, vitamin B1
INTRODUCTION
When Christiaan Eijkman received the 1929 Nobel Prize in Medicine (see “The Eijkman Nobel Prize” sidebar) “for his discovery of the antineuritic vitamin” (1) dubbed thiamin through the work of Casimir Funk (2), it seemed unlikely that nearly 80 years later thiamin would still be the subject of active research, producing exciting advances in chemistry and biology. We now know that the elaborate process of thiamin biosynthesis utilizes many previously unprecedented biochemical mechanisms. Even the regulation of thiamin was shown to use a sophisticated method of transcriptional control through the use of the thiamin riboswitch (3, 4).
In all organisms, the thiazole and pyrimidine moieties of thiamin monophosphate (ThMP) are generated in separate branches of the pathway and then joined by a coupling enzyme. ThMP is converted to the active form of the cofactor thiamin diphosphate (ThDP) by a specific kinase. The best-studied thiamin biosynthetic pathways are those of Escherichia coli and Bacillus subtilis, which utilize very similar pathways, yet differ in some notable ways. The enzymes involved in the thiamin biosynthesis pathways for prokaryotes are illustrated in Figure 1. Table 1 gives the gene and enzyme names. Although at one time the gene names varied between E. coli and B. subtilis, the currently accepted standards, which are the same for both species, are used here.
Figure 1.
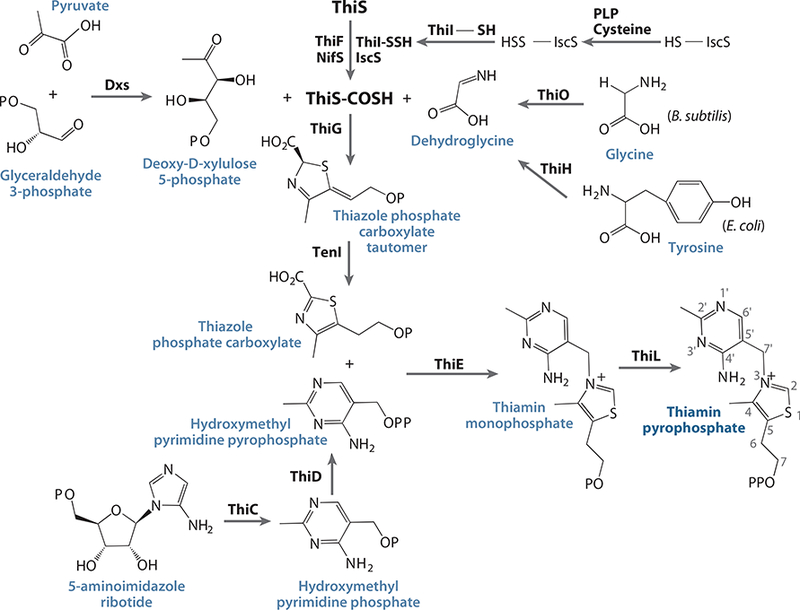
Complete de novo thiamin biosynthetic pathway in bacteria.
Table 1.
The bacterial thiamin biosynthetic enzymes
| Gene product | Enzyme function | Protein Data Bank identifier and organism for representative structure |
|---|---|---|
| ThiC | Hydroxymethyl pyrimidine synthase | 3EPM, Caulobacter crescentus |
| ThiE | Thiamin phosphate synthase | 1G69, Bacillus subtilis |
| ThiF | Adenyltransferase | 1ZUD, Escherichia coli ThiS-ThiF complex |
| ThiS | Sulfur carrier protein | 1ZUD, E. coli ThiS-ThiF complex 1TYG, B. subtilis ThiS-ThiG complex |
| ThiG | Thiazole synthase | 1TYG, B. subtilis ThiS-ThiG complex |
| ThiO | Glycine oxidase | 1NG3, B. subtilis |
| ThiH | Thiazole synthase | None |
| ThiI | Sulfur transferase | 2C5S, Bacillus anthracis |
| NifS | Sulfur donor | 1EG5, Thermatoga maritima |
| ThiM | Thiazole kinase | 1ESQ, B. subtilis |
| ThiN | Thiamin pyrophosphokinase | None |
| ThiD | Hydroxymethyl pyrimidine (phosphate) kinase | 1JXI, Salmonella typhimurium |
| ThiL | Thiamin phosphate kinase | 3C9T, Aquifex aeolicus |
| ThiK | Thiamin kinase | None |
| Dxs | Deoxy-D-xylulose 5-phosphate synthase | 2O1S, E. coli |
| TbpA | Thiamin binding protein | 2QRY, E. coli |
Collaboration between chemists, enzymologists, and structural biologists has produced a clear understanding of how prokaryotes produce this essential cofactor. The X-ray crystal structures and biochemical functions of nearly every enzyme in this pathway have been determined. Studies on the formation of the thiazole, the pyrimidine, and thiamin itself, along with a variety of required kinases, are an important contribution to the vast array of biochemical knowledge in the field of vitamin biosynthesis. In this review, we highlight the enzymological and structural studies that have elucidated thiamin biosynthesis in prokaryotes. Mechanisms and protein structures, where possible, are described for each step. Thiamin salvage, transport, and regulation of thiamin biosynthetic genes are also discussed. The final section highlights a divergent thiazole biosynthetic pathway in Saccharomyces cerevisiae. The conclusion presents areas of further thiamin biochemical research in both bacteria and higher organisms.
THIAZOLE BIOSYNTHESIS IN PROKARYOTES
The thiazole moiety (4-methyl-5-β- hydroxyethylthiazole or THZ) is made through three distinct steps (Figure 1). First, glyceraldehyde 3-phosphate and pyruvate are coupled together by 1-deoxy-D-xylulose 5-phosphate synthase (Dxs) to give 1-deoxy-D-xylulose5-phosphate (DXP). Next, the sulfur carrier protein ThiS undergoes an adenylylation by ThiF, followed by a sulfur transfer step using ThiI (E. coli) and IscS (NifS) to yield a thiocarboxy at its C terminus. It is this sulfur atom that is incorporated into the THZ ring of thiamin. Finally, glycine (by ThiO in B. subtilis) or tyrosine (by ThiH in E. coli) is converted to dehydroglycine. The thiocarboxy C terminus of ThiS, along with DXP and dehydroglycine, are all coupled together by thiazole synthase, ThiG, to give thiazole phosphate carboxylate tautomer. The enzyme TenI (B. subtilis) then aromatizes the thiazole tautomer to the thiazole phosphate carboxylate. The key enzymes of THZ formation are discussed in the following sections.
Deoxy-D-Xylulose 5-Phosphate Synthase
The first step in thiazole biosynthesis utilizes Dxs to produce DXP from glyceraldehyde 3-phosphate and pyruvate. Paradoxically, despite being required for the biosynthesis of thiamin, Dxs itself requires ThDP for activity. How bacteria evolved to use a biosynthetic enzyme that requires the very metabolite that it makes is unknown. However, Dxs is also used in the production of pyridoxol (7) and isopentenyl pyrophosphate (PP) (8–11), so it is possible that the function of DXP synthase may have evolved during a time when bacteria produced thiamin through a more ancient pathway.
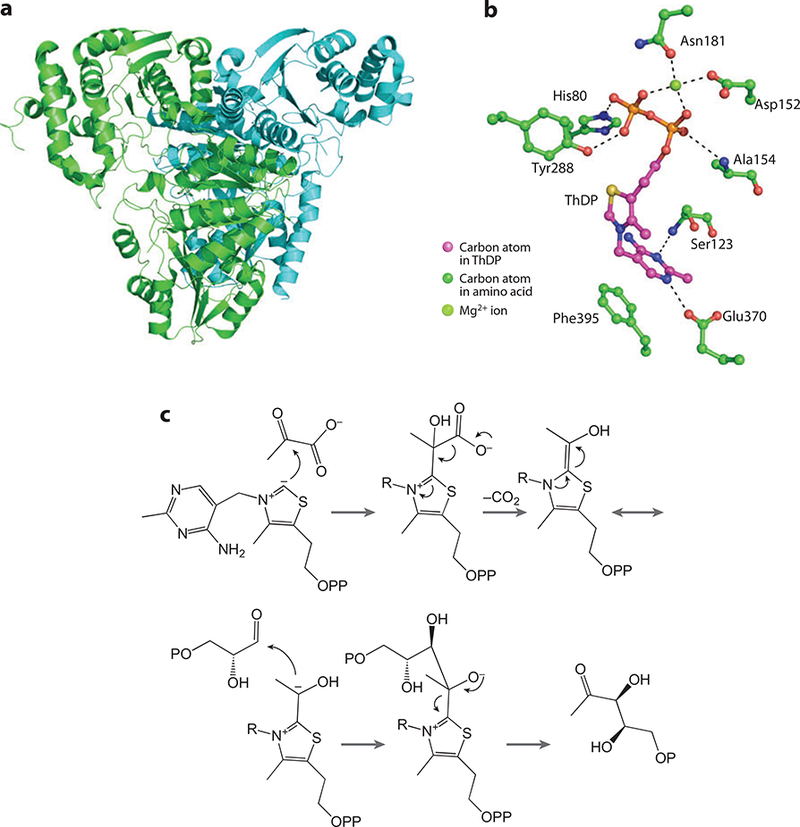
Dxs exists as a dimer, with the majority of the 3900-Å2 protomer interface consisting of hydrophobic residues. The crystal structures of Dxs from E. coli and Deinococcus radiodurans show that the monomer contains three distinct domains (Figure 2a) (12). Domains I (residues1–319), II (residues 320–495), and III (residues 496–629) contain five-, six- and five-stranded β-sheets, respectively. All β-sheets are parallel with the exception of domain III, which has the first β-strand antiparallel to the other four. Dxs is most structurally similar to transketolase (13), pyruvate dehydrogenase E1 subunit (14), and 2-oxoisovalerate dehydrogenase (15). Though these enzymes catalyze similar reactions and use ThDP as a cofactor, the arrangement of domains is different from that seen in Dxs. Dxs is the only enzyme for which the active site is contained within a single monomer—between domains I and II—and not between two monomers as with the other examples.
Figure 2.
(a) The 1-deoxy-D-xylulose 5-phosphate synthase (Dxs) crystal structure. (b) Active-site residues. (c) The reaction mechanism for Dxs.
The active site of Dxs is shown in Figure 2b. The key interactions involved in ThDP binding include hydrogen bonds to the side chain oxygen atom of Glu370 and the amide nitrogen atom of Ser123 of N1 and N3 of the pyrimidine ring, respectively. The thiazole ring does not interact directly with the protein. The PP moiety forms several interactions with the protein. His80 and Tyr288 donate hydrogen bonds to the β-phosphate, and the amide nitrogen of Ala154 donates a hydrogen bond to the α-phosphate. A magnesium ion binds to both phosphate groups as well as to Asn181 and Asp152. The final ligands include the carbonyl oxygen atom from Met185 and, most likely, a water molecule.
The mechanism is depicted in Figure 2c and is typical of ThDP-utilizing enzymes. ThDP exists as an ylide, with the C2 carbon atom from the thiazole moiety acting as the nucleophile that attacks the C2 carbonyl carbon of pyruvate. Loss of CO2 gives the eneamine intermediate, which exists in resonance with the corresponding zwitterion. This acts as the nucleophile attacking the aldehyde group of glyceraldehyde 3-phosphate. Release of the product restores the thiamin ylide.
Sulfur Carrier Protein
The sulfur atom in the THZ ring originates from the thiocarboxylated sulfur carrier protein ThiS. The idea that a protein could be used as a sulfur carrier in the biosynthesis of a cofactor was first postulated in 1993 by Pitterle & Rajagopalan (16) while studying genes involved in the production of molybdopterin. Since then, the identification, function, and, in some instances, the structure of other sulfur carrier proteins have been solved. We now know that sulfur carrier proteins have a ubiquitinlike β-grasp fold and a diglycyl C terminus and are posttranslationally modified to have a thiocarboxy C terminus. They are used not only in the biosynthesis of thiamin, but also in molybdopterin (17), cysteine (18), and thioquinolobactin (19). The structure of ThiS has been solved by NMR (20), and X-ray crystal structures of ThiS in complex with the thiamin biosynthetic enzymes ThiF (21) and ThiG (22) have also been solved. ThiS consists of a five-stranded mixed β-sheet with an α-helix, which crosses over between strands β2 and β3, and a 310-helix between strands β4 and β5 (Figure 3a). Despite being structurally similar, there is little sequence similarity between ThiS and ubiquitin. In light of the structure of ThiS, it has been suggested that ubiquitin derives its origin from a prokaryotic ancestor because it also undergoes an adenylylation step followed by AMP displacement by a sulfur nucleophile (20).
Figure 3.
(a) Crystal structure of the ThiF-ThiS complex. (b) Modeled structure of ATP in the ThiF active site.
ThiS is part of the E. coli thiCEFSGH operon, which contains most of the genes necessary to make thiamin (23). ThiS was found to be posttranslationally modified with a thiocarboxy C terminus in thil+ E. coli strains, but it was not modified in thiI− strains (24). The protein ThiF was bound to ThiS tightly enough to be copurified. The purified ThiFThiS complex produced PP when incubated with ATP, and further analysis by electrospray ionization Fourier transform mass spectrometry (ESI/FTMS) showed a mass increase of 329 Da, consistent with the addition of AMP and loss of PP from ATP (24). ESI/FTMS analysis of ThiS-ThiF and thiI+ E. coli strains showed that a mass increase of 16 Da was localized to the final residue on the C terminus of ThiS (24). The mass increase corresponds to a sulfur atom delivered by ThiI (NifS in B. subtilis), displacing the C-terminal oxygen atom. This generates ThiS-COS−, which is the source of the sulfur atom in the thiazole ring.
Adenylyltransferase
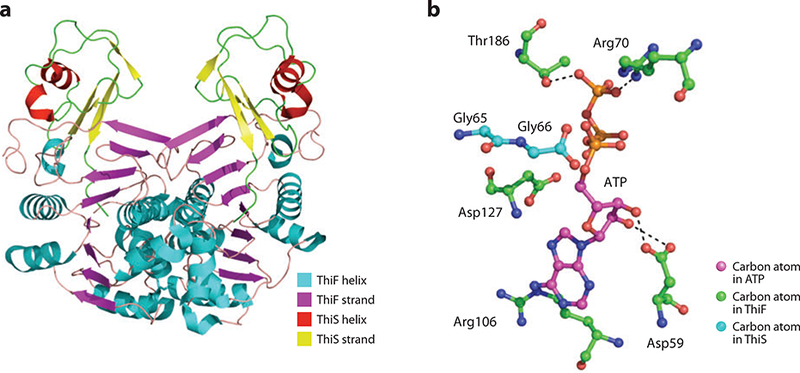
The sequence and structure of ThiF are similar to those of the molybdopterin biosynthetic enzyme MoeB, the ubiquitin-conjugating enzyme E1 and the ubiquitin-related modifier 1-conjugating enzyme Uba4; each is an adenylyltransferase. This observation led to the conclusion that the ThiF mechanism may be similar to that of the ubiquitin-conjugating system that cross-links ubiquitin through a conserved cysteine residue. Crystal structures of ThiF alone (25) and the ThiF-ThiS complex (21) show how the structure of ThiF changes upon ThiS binding. A crossover loop, encompassing residues 181–185, is not visible in the complex structure but is ordered in the uncomplexed ThiF structure. This loop contains a conserved cysteine residue (Cys184) required for catalysis. It was found that the α-phosphate of a modeled AMP molecule is about 20 Å away from the thiol moiety of Cys184. However, this residue lies on a flexible loop and may still reach the substrate from that distance.
The ThiF-ThiS complex exists as a dimer of ThiF with one ThiS molecule bound to each ThiF protomer. The ThiS molecules do not interact with each other, which results in a cleft, approximately 20 Å wide, in the overall structure (Figure 3a). The protein-protein interface between ThiF and ThiS is 60% hydrophobic, with strands β3 and β4, Leu58, and the last seven residues of the C terminus from ThiS contributing the most interactions. The hydrophilic interactions include 14 hydrogen bonds, seven bridging waters, and a salt bridge. ThiF binds to ThiS using strands β5- β8, residues preceding helices α4 and α9, and helix α10.
The structure of the ThiF-ATP complex was used to generate a model of ATP bound to the active site of the ThiS-ThiF complex (Figure 3b). Asp127 is positioned near the α-phosphate and is predicted to bind a magnesium ion that would activate the α-phosphate for a nucleophilic attack required for adenylyltransferase activity. The C terminus of ThiS is also near the α-phosphate, facilitating transfer of AMP. The 2’ and 3’ oxygen atoms of the ribose ring donate hydrogen bonds to the carboxylate moiety of Asp59. The adenine ring is held by Arg106 through a cation-π interaction. The γ-phosphate accepts hydrogen bonds from the γ-oxygen atom of Thr186 and the guanidinium moiety of Arg70.
Sulfur Transfer
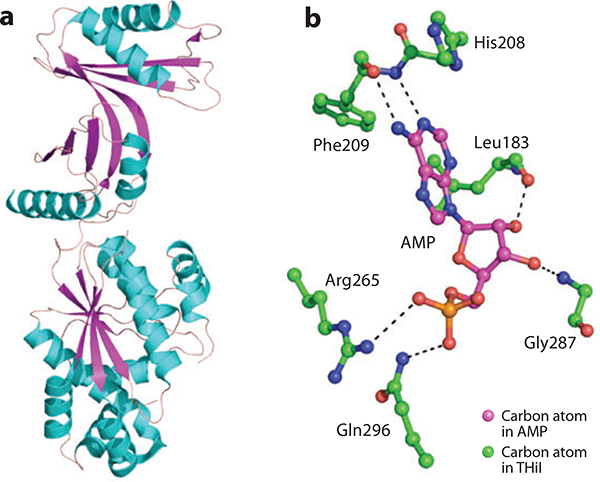
The structures of ThiI from Bacillus anthracis (26) and Pyrococcus horikoshii (27) have been determined and show that ThiI contains three separate domains (Figure 4a). The N-terminal domain is ferridoxin like, the connecting domain is an RNA-binding THUMP domain and the C-terminal domain is a PP-binding domain. The N-terminal domain consists of a four-stranded antiparallel β-sheet and two α-helices. This region is structurally similar to a wide range of proteins that adopt a ferridoxin-like fold. The final β-strand of the ferridoxin-like domain becomes the first β-strand of the following THUMP domain. The THUMP domain consists of five β-strands and two α-helices. The final domain contains a PP-binding loop and is structurally similar to the “N-type” ATP pyrophosphatases that catalyze an adenylylation step. ThiI is required both for sulfur transfer to ThiS in E. coli and for modifying uridine to 4-thiouridine in some prokaryotic tRNAs (28). IscS is also required for the biosynthesis of iron-sulfur clusters and may be responsible for sulfur incorporation in molybdopterin (29).
Figure 4.
(a) Crystal structure of ThiI. (b) Active-site residues that interact with bound AMP.
Figure 4b shows the protein-AMP-binding interactions that are seen in the PP-loop domain. The carbonyl oxygen atom and amide nitrogen atom of Phe209 form hydrogen bonds with N7 and N5 of the adenine ring, respectively. The carbonyl oxygen atom of Leu183 and the amide nitrogen atom of Gly287 form hydrogen bonds with the 2´ and 3´ oxygen atoms of the ribose ring, respectively. The phosphate moiety of AMP accepts hydrogen bonds from the side chains of Arg265 and Gln296.
Glycine Oxidase
The main difference between the B. subtilis pathway and the E. coli pathway occurs at the generation of dehydroglycine, which provides the final atoms for the formation of the thiazole. In B. subtilis, the reaction is catalyzed by ThiO, a flavoenzyme that uses glycine as a substrate to generate the glycine imine (30). The ThiO monomer has two separate domains (Figure 5a). One domain belongs to the glutathione reductase type 2 family and is responsible for binding flavin adenine dinucleotide (FAD), and the other domain binds substrate. ThiO is a tetramer with 222-point symmetry. Each ThiO protomer interacts with each of the other protomers in the tetramer. Openings in the quaternary structure allow the substrate to enter the active site of the substrate-binding domain.
Figure 5.
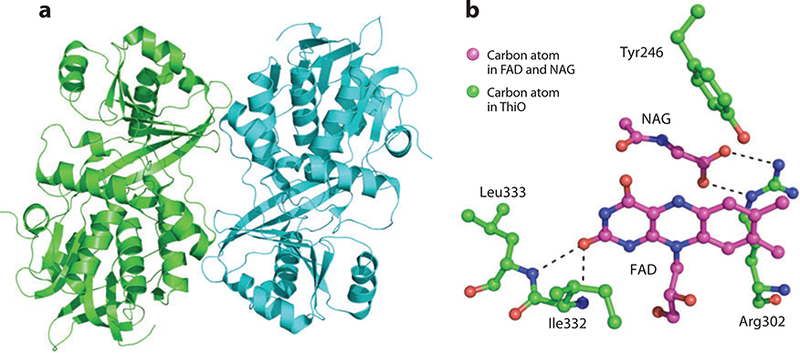
(a) X-ray structure of glycine oxidase, ThiO. (b) Active-site residues that interact with N-acetylglycine (NAG) and the isoalloxizine ring of flavin adenine dinucleotide (FAD).
The active site of ThiO contains an FAD cofactor. Figure 5b depicts the active site of ThiO with the glycine analog N-acetylglycine (NAG) bound. The carboxylate moiety of NAG binds to the guanidinium moiety of Arg302, which positions the substrate for oxidization by the flavin ring. The O7 oxygen atom of the flavin ring accepts hydrogen bonds from the amide nitrogen atoms of Ile332 and Leu333. The ring also forms π-stacking interactions with Tyr246.
ThiH is the E. coli enzyme that generates dehydroglycine. Its substrate is tyrosine, and there is no sequence similarity to ThiO. The structure of ThiH has not yet been determined. In E. coli, ThiH copurifies with ThiG and has been shown to contain an iron-sulfur cluster (31). Sequence alignments show strong similarity to biotin synthase, a radical S-adenosylmethionine (SAM) enzyme (32). In addition, in vitro reconstitution of the E. coli thiazole biosynthethic pathway requires the ThiH-ThiG complex, ThiS-thiocarboxylate, tyrosine, SAM, and NADPH and occurs only under anaerobic conditions (33–36). In contrast, ThiO from the B. subtilis pathway is capable of making THZ in vitro using only ThiG along with sulfide, oxygen, and glycine (37).
The difference in the pathways between ThiO and ThiH lies in the fact that B. subtilis is an obligate aerobe, whereas E. coli can grow under aerobic and anaerobic conditions. Direct oxidation of glycine in an anaerobic environment is not likely to take place and requires a different catalytic strategy involving the use of an adenosyl radical intermediate. Kriek et al. (36) have shown that the conversion of tyrosine to dehydroalanine is initiated through the generation of a tyrosine radical that ultimately leads to dehydroglycine and p-cresol.
Thiazole Synthase
All the components required for THZ formation—ThiS-thiocarboxylate, DXP, and dehydroglycine—are assembled by thiazole synthase ThiG. ThiG shows some substrate tolerance as it is also capable of using 1,4- dideoxy-D-xylulose-5-phosphate as a substrate (38).
The crystal structure of the ThiG-ThiS complex reveals an octamer with 222-point symmetry (Figure 6a). ThiG has a (βα)8 fold, and each protomer binds to two separate ThiG protomers in addition to one ThiS molecule. One ThiG protomer interacts with helices α 7 and α8 of the twofold related helices in the other. The other ThiG-ThiG interaction involves the β6/α6 loop of one ThiG protomer that interacts with the C-terminal helix α8. Two areas on ThiG make up the majority of the protein-protein interactions with ThiS. The C-terminal tail of ThiS passes through a clamp loop consisting of 14 residues. Hydrophobic residues on this loop bind to the hydrophobic region between the β -sheet and α-helix of ThiS after the C-terminal tail of ThiS has passed through it. These residues are structurally similar to hydrophobic residues in the enzymes MoaD (17) and NEDD8 (39), which have been responsible for interacting with their respective binding partners. The second area is mostly hydrophobic and involves strands β3, β4, and β5 of ThiS and β1, β2, α1, α2, α3, and a loop region connecting α3 to β4. Although hydrophobic contacts make up the majority of protein-protein interactions in the ThiGThiS structure (62%), three salt bridges and 11 hydrogen bonds contribute as well.
Figure 6.
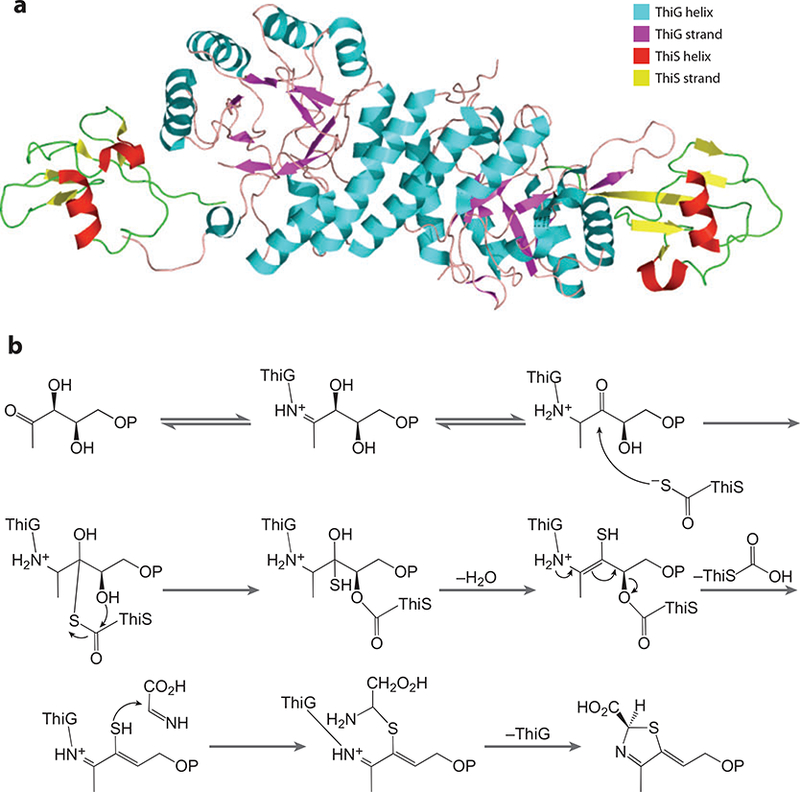
(a) X-ray structure of the ThiG-ThiS complex. (b) The reaction mechanism of ThiG.
As described above, ThiF also binds ThiS but exhibits a different (Rossmann-like) fold. The residues in ThiS that bind both ThiF and ThiG are similar and are mostly located in the C terminus. In both cases, approximately 70% of all the protein-protein interactions with ThiS are hydrophobic. The fact that ThiS is capable of binding to proteins with such dissimilar folds, and differing electrostatic surfaces, supports the proposal that ThiS could be a predecessor to ubiquitin—a protein able to bind to many different targets in the degradation signaling pathway via the proteasome.
The reaction catalyzed by ThiG is outlined in Figure 6b. The final product is not thiazole phosphate (THZ-P), as has long been believed, but rather a carboxy thiazole phosphate tautomer, which must aromatize to form the final product. This step occurs very slowly and is catalyzed by the aromatase TenI in B. subtilis (A. Hazra, A. Chatterjee, & T. Begley, unpublished data).
Aromatization of the Thiazole Ring
The final protein involved in thiamin biosynthesis, TenI, has a known structure, but initially its catalytic activity was unclear (40). It is now known that TenI facilitates the aromatization of the thiazole tautomer product of ThiG (A. Hazra, A. Chatterjee, & T. Begley, unpublished data). Because the structure of TenI (Figure 7a) is similar to thiamin phosphate synthase (ThiE) (41), it was believed that TenI might catalyze the same reaction. However, the active site of TenI contains a leucine residue in the place of a glycine residue in ThiE, which prohibits proper binding of the substrates necessary to generate ThMP.
Figure 7.
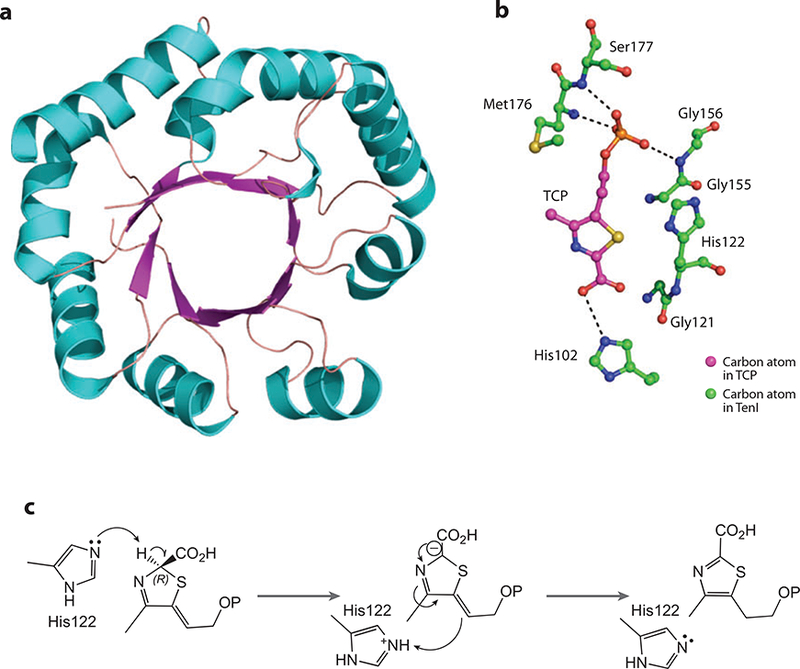
(a) X-ray structure of TenI. (b) Active-site residues interacting with thiazole carboxylate phosphate (TCP). (c) The proposed reaction mechanism of TenI
The structure of TenI complexed with carboxy thiazole phosphate (TCP) has been determined (Y. Han, Y. Zhang, & S. Ealick, unpublished data). The phosphate moiety accepts hydrogen bonds from the amide nitrogen atoms of Gly156, Met176, and Ser177 (Figure 7b). The carboxylate binds to the ε-nitrogen atom of His102. The proposed aromatization mechanism involves His122 acting as a base to remove a hydrogen from C2 and replacing it on C6 of TCP (Figure 7c).
HYDROXYMETHYL PYRIMIDINE BIOSYNTHESIS
The 4-amino-5-hydroxymethyl-2-methyl- pyrimidine phosphate (HMP-P) ring is generated through a complicated rearrangement reaction catalyzed by ThiC, using 5-aminoimidazole ribotide (AIR) as the substrate. ThiD then phosphorylates HMP-P to give HMP-PP.
Hydroxymethyl Pyrimidine Phosphate Synthase
In contrast to THZ-P, biosynthesis of the HMP-P moiety of thiamin in prokaryotes requires only one enzyme; however, it involves one of the most complicated rearrangement reactions in primary metabolism. Figure 1 shows that AIR is converted to HMP in a single enzyme-catalyzed reaction. Labeling studies have identified the origin of all of the HMP-P atoms (Figure 8c) (42–46). AIR is also an intermediate on the purine biosynthetic pathway.
Figure 8.
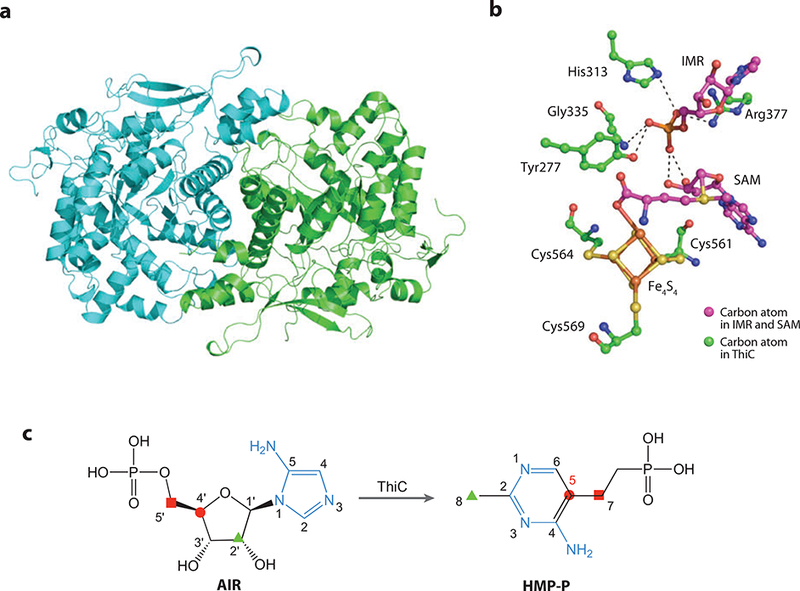
(a) X-ray structure of 4-amino-5-hydroxymethyl-2-methylpyrimidine phosphate synthase, ThiC. (b) Active-site model showing residues interacting with desamino AIR (IMR), S-adenosylmethionine (SAM), and the Fe4S4 cluster. (c) The origin of the atoms of HMP-P derived from isotopic labeling studies.
The crystal structure of ThiC from Caulobacter crescentus has been determined (47) and consists of an N-terminal domain, a core (βα)8 barrel domain, and a C-terminal domain, containing three absolutely conserved cysteine residues (Figure 8a). The N-terminal domain has a three-stranded antiparallel β-sheet and four α-helices that fold over the core domain. Sequence alignments show that the N-terminal domain is much smaller in anaerobes and cyanobacteria compared to aerobic organisms. Part of the C-terminal domain is disordered in the crystal structure. Three helices in this domain occur after the final helix of the (βα)8 domain and make up a significant portion of the protein-protein interface in the ThiC dimer. Though the characteristic iron sulfur cluster motif CX2CX4C is present in the ThiC sequence, the cluster is not present when ThiC is overexpressed in E. coli and must be reconstituted in vitro.
The active site of ThiC, modeled with the substrate analog desamino AIR (IMR) along with the cofactor SAM and a [4Fe-4S] cluster, is shown in Figure 8b. SAM and the [4Fe-4S] cluster were modeled into the active site using other radical SAM enzyme structures as a guide, and the IMR is derived from X-ray crystal data. Hydrogen bonds to the phosphate group of IMR come from the side chains of Tyr277, His313, and Arg377 as well as the amide nitrogen atom of Gly335. The iron atoms of the iron sulfur cluster bind to the thiol side chains of Cys561, Cys564, Cys569, and the carboxylate moiety of SAM.
ThiC is required for HMP biosynthesis in plants as well. Sequence comparisons between Arabidopsis thaliana and the prokaryotic HMP synthase enzymes from E. coli, B. subtilis, Salmonella typhimurium, and Azotobacter vinelandii show significant similarity (48). It has been shown that ThiC in plants and algae also requires an iron sulfur cluster, as do their prokaryotic counterparts (49).
4-Amino-5-hydroxymethyl-2- methylpyrimidine Phosphate Kinase
The crystal structure of ThiD shows that it is a dimer with each monomer adopting an αβα sandwich fold consisting of eight α-helices, two 310-helices, and ten β-strands (Figure 9a). The dimeric interaction between ThiD protomers is relatively flat and consists of the first 70 C-terminal residues and a C-terminal loop, spanning residues 249–266. ThiD has a fold similar to that of a family of ribokinases, as does ThiK (below). ThiD is able to utilize both HMP and HMP-P as substrates in the same active site (50) and catalyzes both phosphorylation reactions. The pyrimidine ring binds similarly whether the substrate is HMP or HMP-P, but the phosphate group of the latter is able to move into different phosphate-binding pockets through rotation about the C5-C7 bond, thus allowing it to be both a product and a substrate. On the basis of the HMP-P three-dimensional structure, it is appears that HMP binds first, followed by ATP. After the first phosphorylation of HMP, ADP is released, and another molecule of ATP is bound in order to generate HMP-PP.
Figure 9.
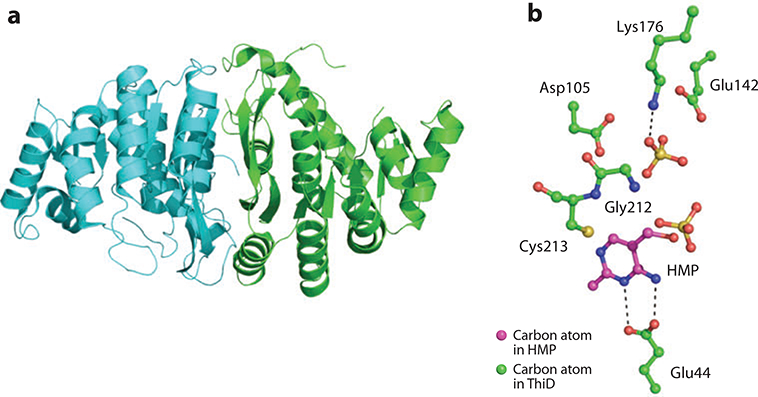
(a) X-ray structure of ThiD. (b) Active-site residues interacting with HMP.
The active site of ThiD is depicted in Figure 9b. The N3 and N5 nitrogen atoms of the pyrimidine ring donate hydrogen bonds to the carboxylate side chain of Glu44. A sulfate ion, which mimics phosphate, is situated close to the hydroxyl moiety of the HMP for the phosphate addition reaction in the α position. The second sulfate-binding site (also mimicking phosphate) is positioned through a hydrogen bond to the amino group of Lys176; this marks the position of the second phosphorylation reaction in the β position. Despite the prevalence of pyrophosphorylated metabolites, the only other known example of a dual kinase activity is thymidine kinase from the human herpes virus 8 (51).
THIAMIN PYROPHOSPHATE BIOSYNTHESIS
ThMP is formed through the coupling reaction of THZ-P and HMP-PP using ThiE. Thiamin phosphate kinase (ThiL) adds the final phosphate group to ThMP to give ThDP, the active form of the cofactor. ThDP can also be formed in one step from thiamin using thiamin pyrophosphokinase. In bacteria, this enzyme is called ThiN. In higher organisms, the thiamin pyrophosphokinase is THI80.
Thiamin Phosphate Synthase
The high-resolution crystal structure of ThiE revealed a (βα)8-barrel fold, where helices α2–8 and α10 surround the central β-strandlined barrel (Figure 10a). Helix α1 spans the N-terminal barrel entrance, and α9 is inserted between β8 and α10. ThiE exists as a dimer where helix α3 from each protomer aligns parallel to each other along the dimer interface, which also contains four salt bridges and three bridging water molecules.
Figure 10.
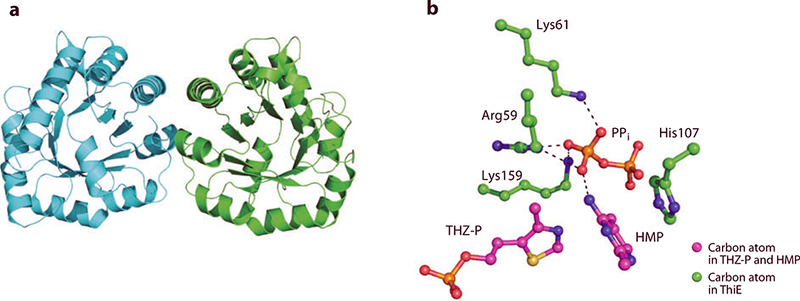
(a) X-ray structure of thiamin phosphate synthase, ThiE. (b) Active-site residues interacting with thiazole phosphate (THZ-P), HMP, and pyrophosphate (PPi).
Figure 10b depicts the active site of the S130A mutant of ThiE. This structure shows separate pyrimidine, THZ-P, and pyrophosphate moieties, suggesting that a carbocation has formed. The pyrimidine forms a hydrogen bond with an oxygen atom of the pyrophosphate group through the N4´ nitrogen atom. The oxygen atom and nitrogen atom of the amide moiety of Gln57 form hydrogen bonds with the N4´ and N3 nitrogen atoms of the pyrimidine ring, respectively. The pyrophosphate group also accepts hydrogen bonds from the side chains of Arg59, Lys61, and Lys159. The phosphate moiety of THZ-P forms hydrogen bonds with the amide nitrogen atoms of Gly168, Ser209, and Ile208, as well as with the hydroxyl moieties of Thr156 and Thr158.
The proposed mechanism of ThMP formation begins with the loss of the PP group from C5 of HMP-PP to generate a carbocation that is stabilized through the delocalized n system of the pyrimidine ring. Following a proton transfer from the N4´ nitrogen atom of the pyrimidine ring to the PP leaving group, a pyrimidine imine methide intermediate is formed, which then undergoes nucleophilic attack from the nitrogen atom of the THZ-P to form ThMP (52). The importance of a serine residue at position 130 (41) is demonstrated by 8000-fold reduction of activity in the alanine mutant (52).
Investigation of cryptic enzymes in E. coli led to the discovery that the previously unannotated gene yjbQ is a thiamin phosphate synthase homolog (53). Cryptic genes capable of duplicating contemporary thiamin biosynthetic enzymes are likely to exist because little intracellular thiamin is required for growth. This increases the likelihood that another enzyme within the proteome may be capable of catalyzing the same reaction. Poor catalytic efficiency of the cryptic enzyme may still be sufficient for cellular growth. The yjbQ gene was transformed into the MC1061ΔthiE strain of E. coli and showed thiamin auxotrophy complementation. Additionally, transformation of the thiE disruption strain with yjbQ genes from Pyrococcus, Sulfolobus, and Thermotoga also exhibited complementation, thereby showing that the thiamin phosphate synthase catalytic activity is a property of the YjbQ protein family. Comparisons between YjbQ and ThiE show that there is no sequence (and likely no structural) similarity, suggesting that these two enzymes come from different pathways.
Thiamin Phosphate Kinase
The final step in cofactor formation is the phosphorylation of ThMP to form ThDP. This reaction is carried out by ThiL in an ATP-dependent manner (54). ThiL contains two domains (Figure 11a). Domain 1 is a half-barrel with four very long β-strands. A ThiL dimer is formed when these β-strands from adjacent protomers join to form an eight-stranded β-barrel at the dimer interface. The second domain is an α/β domain composed of a six-stranded antiparallel β-sheet and a bundle of helices. The dimer interface is mostly hydrophobic with 10 hydrogen bonds and a single disulfide bridge that connects two Cys34 residues: one from each protomer. The structure of ThiL complexed with substrates, products, and analogs suggests that the enzyme utilizes a direct in line phosphate transfer to generate ThDP.
Figure 11.
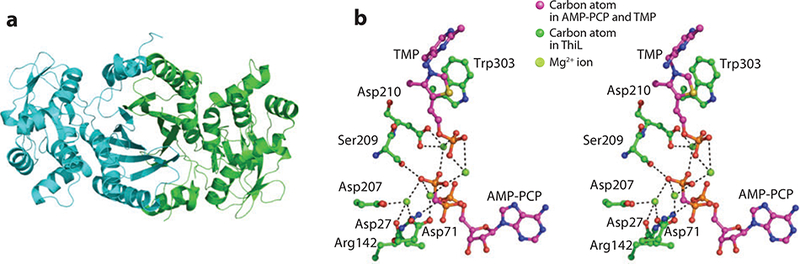
(a) X-ray structure of ThiL. (b) Stereoview of the active site of ThiL with bound thiamin monophosphate (TMP) and ATP analog AMP-PCP
The active site of ThiL is depicted in Figure 11b which shows four of the five Mg2+ ions observed to coordinate the phosphate moieties of ThMP and the AMP analog AMP-PCP. Two of the Mg2+ ions coordinate both the phosphate moiety of ThMP and the γ-phosphate of AMP-PCP, and Ser209 donates a hydrogen bond from the side chain hydroxyl oxygen atom. One Mg2+ ion bridges the two molecules through their phosphate moieties and is coordinated by Asp210. The other two Mg2+ ions interact with the γ-phosphate of AMP-PCP as well as Asp27, Asp71, and Asp207. The α-phosphate of AMP-PCP also accepts a hydrogen bond from Arg142.
A phosphorylated enzyme intermediate was ruled out because no amino acid residue is suitably positioned for phosphorylation. Comparisons with ThDP-binding proteins showed that the pyrimidine moiety is rotated approximately 100° when compared to ThDP in enzymes that require it as a cofactor. This prevents activation of the C2 carbon on the thiazole ring by N4 of the pyrimidine ring to generate the thiamin ylide in the active site of ThiL. ThDP can also be produced by thiamin pyrophosphokinase from B. subtilis (55), which is able to carry out the pyrophosphorylation of thiamin. Most higher organisms also contain this enzyme (56).
THIAMIN DEGRADATION
Thiaminases degrade thiamin into separate thiazole and pyrimidine moieties. Two classes of thiaminases have been identified: thiaminase I (57–61) and thiamine II (62). TenA was shown to be the thiaminase II in B. subtilis (40). Thiaminase I has been associated with early mortality syndrome in predatory fish of the Laurentian Great Lakes and the New York Finger Lakes (63) as well as in Atlantic salmon in the Baltic Sea (64). Fish produce eggs with low levels of thiamin, which leads to a variety of illnesses and finally death between the time the fish are hatched and their first feeding. It is believed that the thiamin deficiency results from the egg-carrying female feeding on the nonnative forage fish alewife (Alosa pseudoharengus) that produces high levels of thiaminase I. The importance of thiaminase I is also indicated in the sidebar “Thiaminase I— The Culprit Behind a Disastrous Nineteenth Century Australian Expedition.”
Catalytically, the difference between thiaminase I and II is seen in the substrates used to cleave the C-N bond connecting the two heterocyclic rings of thiamin. Thiaminase I can use aniline, cysteine, dithiothreitol, pyridine, quinoline, and veratrylamine as substrates (57, 67), and thiaminase II can only use water (Figure 12c) (40). Thiaminase I is also able to accept thiamin analogs, which vary extensively in the thiazole ring moiety but not in the pyrimidine moiety (68). The two classes of thiaminases have been shown to have dissimilar sequences and structures. It is also known that thiaminase II (TenA) promotes the production of degradative enzymes—dubbed Deg proteins in B. subtilis (69). These proteins are expressed in bacteria during the transition between a growth phase and a stationary phase. The protein TenI has been shown to have the opposite effect on the production of Deg proteins (70).
Figure 12.
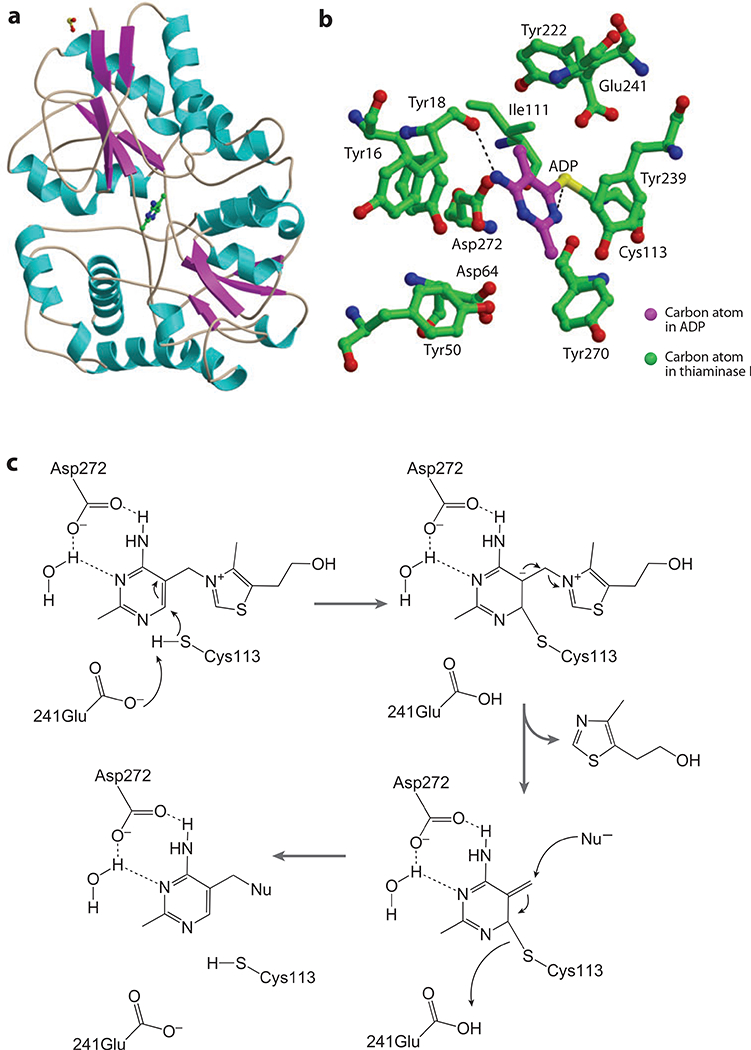
(a) Thiaminase I crystal structure. (b) Active-site residues with the inhibitor 4-amino-2,5-dimethylpyrimidine (ADP). (c) The reaction mechanism for thiaminase I.
Thiaminase I
The structure of thiaminase I from Bacillus thiaminolyticus (Figure 12a) shows an overall fold similar to that of group II periplasmicbinding proteins, such as the maltose-binding protein (71) and the spermidine putrescinebinding protein potD (72, 73). The periplasmic binding proteins consist of two domains, each with an α/β fold. The two domains form a deep cleft and are connected by three crossover segments. This led to the proposal that thiaminase I may have evolved from an ancient periplasmic binding protein responsible for the uptake of thiamin (62). The physiological function of thiaminase I is not yet understood.
The active site of thiaminase I is located in the cleft between the two domains (Figure 12b). Six tyrosine residues and four acidic residues line the cleft. The active site was identified using the mechanism-based inhibitor 4-amino-6-chloro-2,5-dimethylpyrimidine, which bonds irreversibly to the active-site cysteine residue (Cys113). The structural arrangement suggests the mechanism outlined in Figure 12c. Thiamin is positioned in the active site by two hydrogen bonds between the pyrimidine moiety and Asp272, one of which occurs through an intervening water molecule. Glu241 then activates Cys113 for attack at C6 of the pyrimidine to form a zwitterionic intermediate. Nucleophilic attack and protonation by Glu241 result in cleavage of the bond between the thiazole and pyrimidine and release of products.
Thiaminase II
TenA was shown to be thiaminase II, which uses water exclusively as the nucleophile to cleave the C-N bond connecting the pyrimidine and thiazole moieties (40). The crystal structure of TenA shows a bundle of 11 helices that surround a deep acidic pocket (Figure 13a). The walls of the pocket are made of helices α4, α5, α9, and α10 with a total volume of approximately 700 Å3. The biological unit is a tetramer, with 222-point symmetry. Each monomer in the quaternary structure interacts with two other monomers. One interaction involves helices α 7 and all, and the other involves helices α4 and α5.
Figure 13.
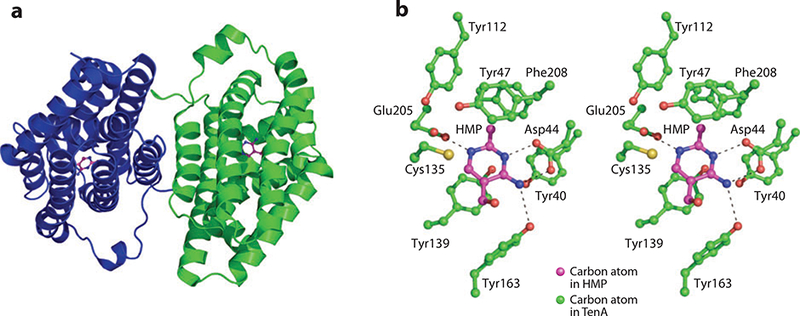
(a) X-ray structure of TenA. (b) Stereoview of the active site of TenA with 4-amino-5-hydroxymethyl-2-methylpyrimidme (HMP) bound.
The active site of TenA is shown in Figure 13b. Several residues, Tyr47, Tyr112, Tyr163, and Phe208, contribute to the π- stacking environment around the HMP ligand. The carboxylate side chain of Glu205 forms a hydrogen bond with the N1 nitrogen atom of the pyrimidine ring. The carboxylate side chain of Asp44 and hydroxyl group of Tyr163 form hydrogen bonds with N3 and N4´, respectively. The catalytic residue Cys135 is positioned near the C2 atom of the pyridine ring and is required for the cleavage of thiamin into its respective heterocycles (Figure 12c).
The biological function of TenA appears to be the salvage of base-degraded thiamin (Figure 14) (74). Some of the inconsistencies, suggesting that TenA’s physiological role is not thiamin degradation, included the observation that it is found in the thiamin biosynthetic operon. Also, TenA has activity specifically with thiamin and not ThMP or ThDP. Because the products of de novo thiamin biosynthesis are the singly and doubly phosphorylated forms of thiamin, TenA would not degrade thiamin produced in the cell at all. TenA is found in Bacillus halodurans, a bacterium that lives in soil pH > 10, which led to the suggestion that TenA might be involved in the salvage of degraded products of thiamin. Additionally, TenA in B. halodurans is clustered near genes associated with a deformylase YlmB and an ABC transporter complex ThiXYZ, also involved in salvage (75).
Figure 14.
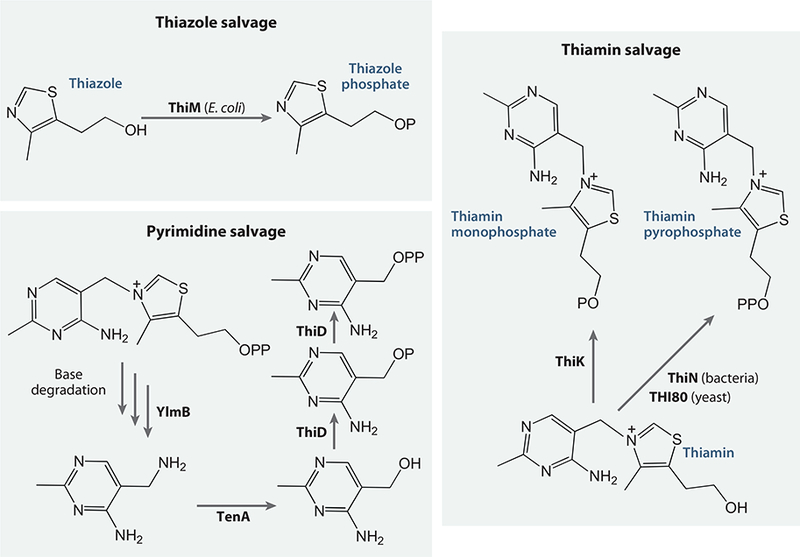
Salvage pathways for thiazole, thiamin, and pyrimidine.
The mechanism of thiamin salvage involves uptake of N-formyl-4-amino-5-aminomethyl-2-methylpyrimidine, generated by base degradation of thiamin, followed by deformylation by YlmB, to give aminopyrimidine and TenA-catalyzed hydrolysis to generate HMP (Figure 14). TenA was shown to have 100 times greater activity against aminopyrimidine than with thiamin, further demonstrating that thiamin is not the natural substrate for this enzyme (76). The putative transporter ThiY was also shown to bind aminopyrimidine, which is then delivered to the ThiXZ transport channel.
THIAMIN SALVAGE
As an alternative to the de novo pathway described above, thiamin, or components of thiamin, can be salvaged (Figure 14). In bacteria, the thiazole alcohol can be converted to THZ-P by thiazole kinase (ThiM). In addition, HMP can be converted to HMP-P by HMPP kinase (ThiD). ThiD is also the enzyme that converts HMP-P to HMP-PP in the de novo pathway. In bacteria, thiamin can be converted to ThMP by thiamin kinase (ThiK) or to ThDP by thiamin pyrophosphokinase (ThiN). The corresponding enzyme in higher organisms is THI80.
Thiazole Kinase
ThiM from B. subtilis (77) and S. typhimurium (78) has been biochemically characterized, and the structure of the B. subtilis ThiM has been determined (Figure 15a) (79). ThiM is a trimer with each protomer containing nine β-strands and 12 α-helices. ThiM from B. subtilis is structurally homologous to ribokinase (80), adenosine kinase (81), and other related kinases. Ribokinase and adenosine kinase each have additional β-sheets that act as a flap over the active site. In the trimeric structure of ThiM, this flap is replaced by residues from an adjacent protomer.
Figure 15.
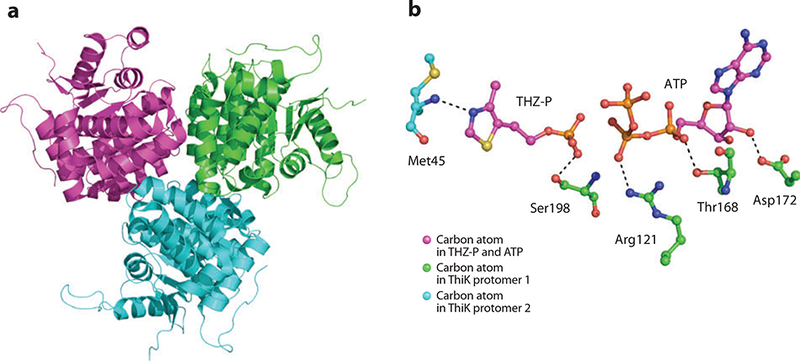
(a) X-ray structure of thiazole kinase (ThiM). (b) Active site of ThiM with thiazole phosphate (THZ-P) and ATP bound.
The active site of ThiM is shown in Figure 15b. The active site contains the product THZ-P and the substrate ATP and is biochemically inert. The γ-phosphate of ATP is seen in close proximity to the phosphate of THZ-P, although it is in a bent conformation to accommodate the presence of the THZ-P. In the absence of the phosphate moiety on thiazole, this phosphate is able to adopt an extended conformation that is suitable for phosphate transfer to the hydroxyl group of the thiazole substrate. The thiazole ring is held in place through a hydrogen bond to the amide nitrogen atom of Met45. The phosphate moiety of THZ-P binds to the side chain of Ser198. Arg121 binds to the β-phosphate of ATP, and Thr168 donates a hydrogen bond to the α-phosphate through the hydroxyl moiety of its side chain. The ribose ring is held in place through a hydrogen bond between the 2´ position of the ribose ring and the carboxylate moiety of Asp172.
Thiamin Pyrophosphokinase
Structures have been published for thiamin pyrophosphokinase (THI80) from yeast and mouse (82, 83). The two enzymes share 26% sequence identity and have similar threedimensional structures. The main difference is that the yeast enzyme has 20 additional Nterminal residues, seven additional C-terminal residues, a 38-residue insertion, and several small insertions. The overall structure of thiamin pyrophosphokinase is a homodimer (Figure 16a). Each monomer consists of an α/β domain and a β-sandwich domain. Both domains contribute to dimer formation. Each of the two active sites is located in a cleft between the N-terminal domain of one protomer and the C-terminal domain of the other. Iterative BLAST searches suggest that the bacterial thiamin pyrophosphate kinase (ThiN) and THI80 share at least some structural homology.
Figure 16.
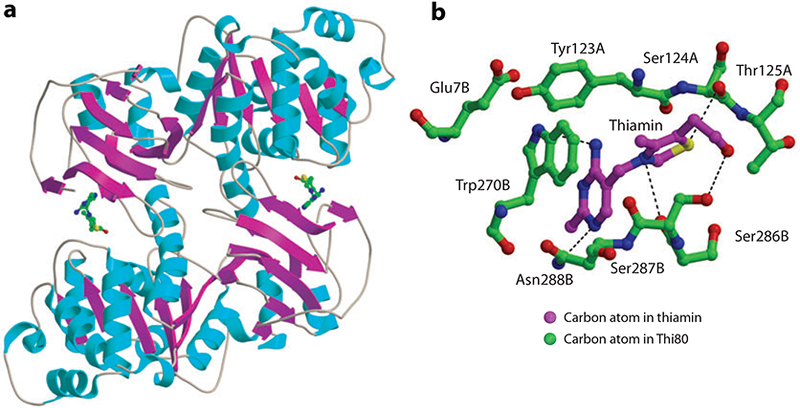
(a) Thiamin pyrophosphokinase (THI80) crystal structure. (b) Active-site residues shown with their chain identifications.
Both the yeast and mouse enzymes contain a bound thiamin at the active site (Figure 16b). The binding site (yeast numbering) is formed mainly by residues 123–125 of one protomer and residues 7, 270, and 286–288 of the other. Primary interactions are formed by Glu7 of one protomer and Tyr123 of the other protomer. Hydrogen bonds are formed with Gln122 and Ser286, and Trp270 forms π-stacking interactions with the pyrimidine. Thiamin is observed in the unusual F conformation, and ThDP is usually in the V conformation when utilized as a cofactor. The ATP-binding site has not yet been characterized.
THIAMIN TRANSPORT
Bacteria are able to use exogenous thiamin and components of thiamin to supplement their own de novo biosynthesis through transmembrane transporters. Thiamin, ThMP, ThDP (84), HMP (85), and THZ (86) can all be taken up into the cell from nutrient media. Uptake of these compounds is poorly understood, but the thiamin-regulated operon tbpAthiPQ in E. coli and S. typhimurium (84) encodes an ABC transporter and includes a periplasmic thiaminbinding protein (TbpA), a transmembrane thiamin channel (ThiP), and an ATPase responsible for active transport into the cell (ThiQ) (84).
Thiamin-Binding Protein
TbpA from E. coli is a dimer with one ThMPbinding site per protomer (Figure 17a) (87). The TbpA protomer has two domains connected by a flexible linker. The TpbA-binding site is in a cleft created between these two domains and is typical of periplasmic binding proteins. TbpA is a monomer in solution, suggesting that the dimer observed in the crystal structure may be an artifact. It is also possible that the dimer forms after ThMP binds, resulting in higher binding affinities in the periplasm, which might increase transport efficiency.
Figure 17.
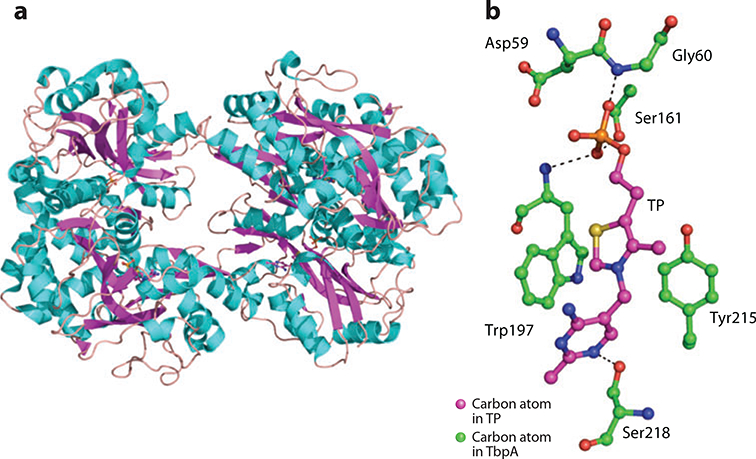
(a) X-ray structure of the thiamin-binding protein TbpA. (b) Thiamin-binding residues are shown with bound thiamin phosphate (TP).
The ThMP-binding site of TbpA is shown in Figure 17b. The phosphate moiety accepts hydrogen bonds from the amide nitrogen atoms of Gly60 and Trp197. The hydroxyl side chain of Ser161 contributes a third hydrogen bond to the phosphate. The pyrimidine moiety accepts a hydrogen bond from the side chain of Ser218 at theN1 position and forms π-stacking interactions with Trp197. The thiazole moiety is sandwiched between the aromatic rings of the Trp197 and Tyr215.
TpbA is similar in structure to thiaminase I and possesses limited sequence similarity, suggesting that both proteins share a common ancestor. Organisms contain thiaminase I because it degrades thiamin. The sequence identity between TpbA and thiaminase I is only 15%, and the residues required to carry out the degradation reaction are not conserved.
YkoF Protein
B. subtilis uses an additional ABC transporter found in the ykoCDEF operon, which encodes for two transmembrane components (YkoC and YkoE), an ATPase (YkoD), and a thiamin/HMP-binding protein (YkoF) for which a crystal structure is available (88). YkoF has a ferridoxin-like fold and two thiamin (N-and C-terminal) binding domains per protomer that have vastly unequal binding affinities (Figure 18a). The high-affinity binding site is 10 μM, whereas the low-affinity binding site is 250 μM (88). Also, there are no direct interactions with the thiazole moiety of ThDP. Because the binding affinity of TpbA is significantly higher at 3.8 nM, it is possible that YkoF may have a biological function other than ThDP uptake.
Figure 18.
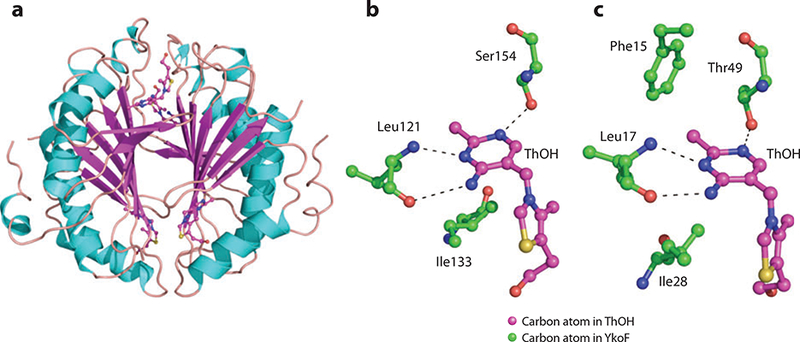
(a) X-ray structure of the YkoF dimer. (b) High-affinity binding site for thiamin alcohol (ThOH). (c) Low-affinity binding site for thiamin alcohol.
Figure 18a shows the structure of the YkoF dimer. Each monomer contains an eight-stranded antiparallel β-sheet with four α-helices stacked against one face. The monomeric structure has approximate twofold symmetry owing to the internal tandem repeat of the ferridoxin-like fold. Superimposing the 74 structurally similar Cα carbon atoms from each resulting domain shows a root mean square deviation of 1.7 Å. The dimer is arranged so that each monomer is positioned head to tail, with the hydrophobic core of each β-sheet making the majority of the protein-protein interface. The resulting dimer is spherical in shape with each monomer as one hemisphere of the globular structure.
Figure 18b, c show the high-affinity and low-affinity binding sites for thiamin, respectively. Both sites are similar, with the same number of hydrogen bonds to the pyrimidine ring. The high-affinity binding site forms hydrogen bonds between the amide nitrogen atom and the carbonyl oxygen atom with N3 and N4´ of the pyrimidine ring, respectively. Another hydrogen bond is accepted by N1 of the ring by the hydroxyl side chain of Ser154. Ile133 forms a hydrophobic region near the thiazole ring. In the low-affinity binding site, the same hydrogen bonds are seen but in this case are contributed by Leu17 and the hydroxyl side chain of Thr49. Phe15 may be involved in π-stacking, and Ile28 provides a similar hydrophobic environment near the thiazole ring as does Ile133 in the high-affinity binding site.
Eukaryotic thiamin uptake primarily utilizes proteins within the ACT transporter family. The recently discovered thiamin-binding protein Thi9 (89) was identified in Schizosaccharomyces pombe and is found within the APC superfamily of proteins; these proteins bind substrates such as choline, basic amino acids, polyamines, and γ-aminobutyric acid (90). Another protein, Bsu1, was also shown to be a pyridoxine-proton symporter, and Bsu1 is similar to exporting proteins from the multidrug resistance family (91). It is likely that Bsu1 is also able to bind HMP. Both Thi9 and Bsu1 are regulated by intracellular concentrations of thiamin.
THIAMIN REGULATION
The regulation of gene expression of most thiamin biosynthetic proteins is controlled at the transcriptional level. The E. coli operons thiCEFSGH, thiMD, and tbpAthiPQ are all regulated by the presence of ThDP through interactions with the THI-box riboswitch on mRNA encoding for each operon. Also, B. subtilis has a thiamin-regulated tenA-tenI-thiOSGFD operon (92) with a Rho-independent transcriptional terminator site upstream from the thiamin-regulated genes (75). In B. subtilis, the genes thiC and ywbI-ThiME are not downregulated by thiamin but rather are partially repressed by thiazole (77, 93). The genes thiI, thiL, dxs, and iscS are the only thiamin biosynthetic enzymes not regulated by a riboswitch and are not clustered with other thiamin biosynthetic genes. In the case of thiI, dxs, and iscS, this could be because these genes are required for other pathways.
The riboswitch is now known as a common regulatory element in gene expression and is present in biosynthetic genes for amino acids, nucleotides, and other vitamins (94) as well as in many different classes of bacteria (75). Some organisms use riboswitches to regulate over 2% of their genome (95–101), thus making the riboswitch a good drug target for pathogenic bacteria (102). The THI-box remains the only example of a riboswitch seen in eukaryotes, and it inhibits translation of thiamin biosynthetic genes by interfering with intron splicing (4, 48, 103).
The THI-box consists of a 5´ untranslated region that forms a thiamin-binding site (104). In gram-positive bacteria, binding of thiamin induces the formation of a Rho-independent transcriptional terminator. In gram-negative bacteria, binding of thiamin masks the Shine-Dalgarno sequence, which is required for the initiation of translation (105). The thiaminbinding domain of the riboswitch is 1000-fold more specific for ThDP than for ThMP, with KD values of 0.1 μΜ and 100 μΜ, respectively (96), thereby ensuring that only the active form of the cofactor inhibits translation.
THI-Box Riboswitch
The fact that ThDP binds to an mRNA molecule presents an interesting problem in that both molecules carry negative charges on their phosphate groups. The recent crystal structure of a THI-box RNA complexed with ThDP sheds light on how the binding occurs (Figure 19a) (3, 4, 106). ThDP binds in a linear conformation as seen in solution. In contrast, ThDP adopts a V conformation in enzymes that utilize it as a cofactor (107). The pyrimidine moiety of ThDP stacks between the conserved purine bases G42 and A43 of the THI-box (Figure 19b). Electrostatic interactions between two magnesium-binding domains, which are found within the binding pocket of the riboswitch, coordinate the phosphate groups of ThDP. Residue G60 interacts with one Mg2+ directly, and U59, A61, and C77 interact with the same Mg2+ ion through a water molecule. The second Mg2+ is coordinated to A75, C77, and G78 via a water molecule as well. Mn2+, Ca2+, and Ba2+ have also been shown to bind the pyrophosphate group (106). The presence of the pyrophosphate group is required for divalent cation coordination as shown by the THI-box complex structure with pyrithiamine, a thiamin analog lacking a pyrophosphate group, which showed the absence of divalent cations (106). Two additional metal-binding sites contribute to the overall structure of the THI-box/ThDP complex. One interacts with U39 and G40 to help position the J2–3 bulge for pyrimidine binding.
Figure 19.
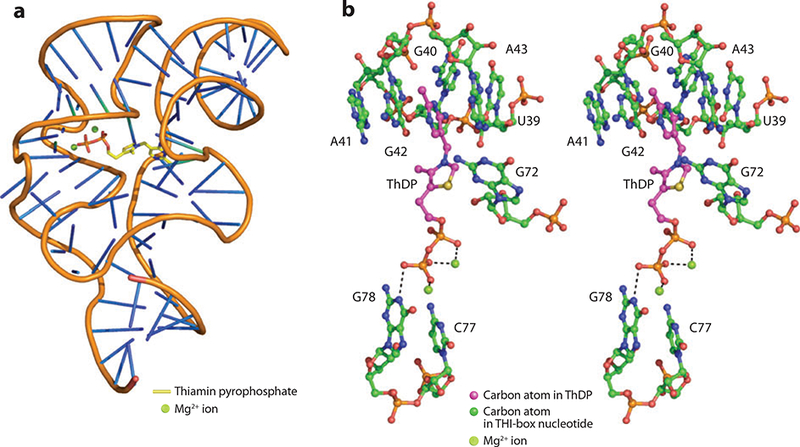
(a) X-ray structure of the THI-box. (b) Stereoview of the ThDP-binding site of the THI-box.
This metal-binding site is able to accommodate Mg2+, Mn2+, and Ba2+. The final metalbinding site connects helices P3 and P5 upon ThDP binding and is only able to bind Mg2+ (106). Figure 19b illustrates the THI-box, with two Mg2+ ions coordinating the pyrophosphate group (3).
EUKARYOTIC THIAMIN BIOSYNTHESIS
Though considerable progress has been made in discovering the biosynthesis, uptake, salvage, and regulation of thiamin in prokaryotes, comparatively little is still known about how eukaryotes make thiamin. As is the case for bacteria, the thiazole and pyrimidine moieties are generated through separate pathways, coupled by a single enzyme and phosphorylated by a variety of kinases (Figure 20). The thiazole ring is made from glycine, cysteine, and a five-carbon sugar, using a single enzyme, Thi4 (108–111).
Figure 20.
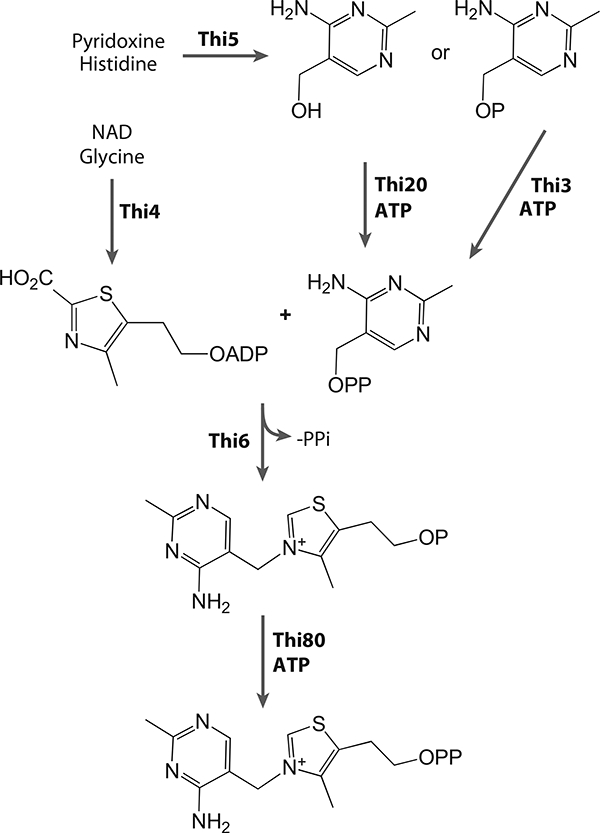
Thiamin biosynthetic pathway in yeast.
Thi5 generates the HMP moiety using pyridoxine and histidine (112, 113). HMP is subsequently pyrophosphorylated by Thi20 (114). The thiazole and pyrimidine are then coupled by the bifunctional enzyme Thi6 (115), which also contains a hydroxyethylthiazole kinase activity (116). Thi20 is also a thiaminase II and has N-and C-terminal sequence homology with B. subtilis ThiD and TenA, respectively.
Hydroxymethylpyrimidine Kinase/Thiamin Phosphate Synthase
The thiamin biosynthetic enzyme Thi3 in plants was implicated in the biosynthesis of the hydroxymethylpyrimidine moiety over 15 years ago (117, 118), but no functional annotation was ascribed. The purification of THI3 from Zea mays and biochemical characterization have revealed that it is a dual-function enzyme (119). The N-terminal domain catalyzes the phosphorylation of HMP-P, and the C-terminal domain generates thiamin phosphate from HMP-PP and THZ-P. Structural predictions, using the Swiss Institute of Bioinformatics modeling server SWISSMODEL, revealed that the N-terminal domain could be similar to the prokaryotic HMP-P kinase enzyme ThiD (50). The predicted residues responsible for HMP-P kinase activity included Gln98 and Met134, which correspond in S. typhimurium to the active-site residues Glu44 and Met80, respectively. Q98L and M134K mutations resulted in loss of HMP-P kinase activity, whereas ThiE activity remained. The C-terminal ThiE domain was predicted to be similar to the ThiE/TenI class of proteins that have a (βα)8-barrel fold. The active-site serine required for catalysis in ThiE from B. subtilis (Ser130) (41, 52) was predicted to be Ser444 in THI3. Mutation of Ser444 to alanine resulted in complete loss of ThiE activity and retention of HMP-P kinase activity. Kinetic analysis also revealed that the ThiE activity is uncompetitively inhibited by the presence of HMP-PP and ATP.
Thiazole Synthase
Recent discoveries in yeast thiamin biosynthesis have focused on the formation of the thiazole ring. The enzyme Thi4 in yeast—Thi1 in A. thaliana—is a homooctamer with 35 kDa protomers (30 kDa for THI1) (Figure 21a). Orthologs of THI4 in Fusarium oxysporum, F. solani (STI35), and Aspergillus oryzae (THIA) are transcriptionally regulated by ThDP binding to a THI-box riboswitch (103). Thi4 is a dual function enzyme involved with mitochondrial DNA repair (120) in addition to thiazole biosynthesis. The crystal structure of Thi4 (121) shows a proline in the cis conformation (Pro121) that follows a loop region responsible for donating hydrogen bonds to the α- and β-phosphate moieties of adenylated thiazole carboxylate (ADT), an unexpected bound product. It is possible that isomerization of this proline may lead to product release. Interestingly, in N. crassa, a cis-trans prolyl isomerase interacts with Thi4 (122), further supporting this speculation.
Figure 21.
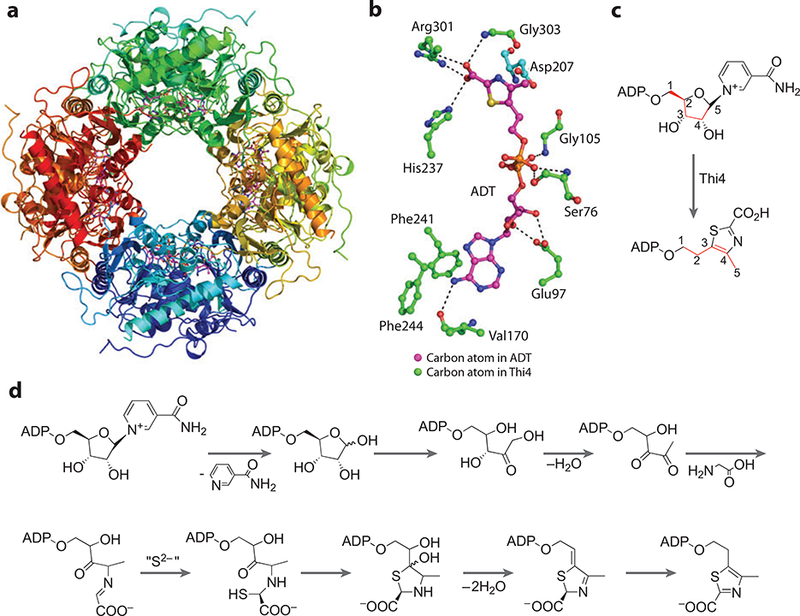
(a) X-ray structure of the Thi4 octamer. (b) Active-site residues that bind adenylated thiazole (ADT). (c) Overall Thi4 reaction with carbon atoms from NAD common to ADT colored red. (d) The mechanism of Thi4.
Figure 21a shows the biological unit of Thi4. The homooctameric structure is arranged as a tetramer of dimers. The dimeric interactions are primarily through helix α4 using both hydrogen bonding and hydrophobic interactions. Four such dimers are arranged into an octamer through interactions with helix α1. The doughnut-shaped octamer is approximately 96 Å across and 65 Å high. The inner ring contains solvent and is approximately 30 Å across. The substrate-binding sites are found on the inner side of the ring.
Figure 21b shows key residues that bind to the ADT molecule. The adenine ring is held through π-stacking interactions with Phe241 and Phe244. The carbonyl oxygen atom of Val170 accepts a hydrogen bond from N7´ of the adenine ring. The 2´ and 3´ hydroxyl groups of the ribose sugar each donate one hydrogen bond to the carboxylate moiety of Glu97. The α-phosphate of ADT accepts a hydrogen bond from the backbone nitrogen atom of Gly105. Ser76 donates two hydrogen bonds to the β-phosphate moiety through its backbone nitrogen atom and the hydroxyl side chain. The carboxylate of the thiazole ring accepts hydrogen bonds from His237, Arg301, and Gly303. Asp207 from the adjacent monomer is shown just above the thiazole ring and is required for ADT formation.
The structures of Thi4 and Thi1 were analyzed for structural homologs using the DALI server (123) and shown to be most similar to several flavoenzymes from the glutathione reductase type II family (31). This was unexpected because typical NAD-binding proteins have a Rossmann fold (124). Surprisingly, one of the top structural homologs is the prokaryotic thiazole biosynthetic enzyme glycine oxidase (ThiO) from B. subtilis. The FAD-binding domain of ThiO overlays with the NAD-binding domain of THI4 with a root mean square difference of 2.4 Å. The second domain showed no closely related structural homologs. In the superposition, the ADP moiety of ADT overlays well with the FAD in ThiO; however, the thiazole carboxylate and isoalloxizine rings, respectively, deviate past the β-carbon. The similarity between Thi4 and flavoenzymes suggests that an ancient predecessor to Thi4 was capable of binding FAD and evolved the ability to bind and catalyze NAD without a rearrangement of topology.
The crystal structures of Thi1 and Thi4 contributed to the identification of the substrates, which for many years remained elusive. Crystal structures of Thi4 (121) and Thi1 (129) showed clear density for ADT in the active site. The unusual metabolite, bound to Thi4 overexpressed in E. coli, was confirmed to be ADT by mass spectrometry and NMR experiments (130). The adenylated ligand suggested that the substrate for Thi4 was a dinucleotide, but ADT was bound so tightly that it could not be removed from the native protein without denaturation; however, C204S and H200N mutants lacked a bound ligand but were still partially active. Thi4 mutants incubated in the presence of NAD and glycine showed four intermediates. The four intermediates were identified as ADT, ADP-ribose, ADP-ribulose, and an ADP-ribulose-glycine adduct (125). This represented a biochemical characterization of the consumption of one cofactor to generate a precursor for another cofactor, a type of “Cofactor Cannibalism” (see sidebar and Figure 21c) (128). Figure 21d shows a proposed Thi4 mechanism consistent with biochemical assays and the X-ray crystal structure.
CONCLUSIONS AND FUTURE ISSUES
Thiamin is synthesized by most prokaryotes and by eukaryotes such as yeast and plants. In all cases, the thiazole and pyrimidine moieties are synthesized in separate branches of the pathway and coupled to form thiamin phosphate. A final phosphorylation gives thiamin pyrophosphate, the active form of the cofactor. Over the past decade or so, biochemical and structural studies have elucidated most of the details of the thiamin biosynthetic pathway in bacteria (Figure 1). Formation of the thiazole requires six gene products (Figure 6), and formation of the pyrimidine requires two. In contrast, details of the thiamin biosynthetic pathway in yeast are only just beginning to emerge (Figure 20). Only one gene product is required for the biosynthesis of the thiazole (Figure 21) and one for the biosynthesis of the pyrimidine.
Considerable progress has been made in understanding the biosynthesis of thiamin in bacteria, and significant inroads have been made for eukaryotes. However, many important questions still remain. The structure of ThiH is not known, although it is likely to be similar to that of biotin synthase. It is also not known how ThiH and ThiG interact. Although structural examples are available for almost all of the prokaryotic enzymes, in many cases, the hard work of preparing and crystallizing complexes will be necessary to fully elucidate the enzyme mechanisms. The recent discovery of a thiamin salvage pathway that utilizes TenA, and the vast number of TenA orthologs, highlights the possibility that other salvage pathways remain to be discovered. Some organisms lack enzymes associated with the standard pathway, and it is likely that completely new pathways or significant variations of the known pathways remain to be discovered.
The field of eukaryotic thiamin biosynthesis still has many questions to be answered, but the remarkable discovery of the structure, function, substrate, and product of thiazole synthase (Thi4/Thi1) shows that new biochemical methods of primary metabolite biosynthesis can still be unveiled. The role of Thi4 in mitochondrial
DNA repair is not understood. Structures are not known for Thi3, Thi5, Thi6, Thi20, and others. The possibility of drug discovery using thiamin biosynthetic enzymes or the THIbox as a target may provide new leads in the field of antimicrobial drug development. The structure of the THI-box shows that it binds ThDP differently than typical thiamin-binding proteins, leading to the possibility that thiamin analogs could be developed that bind to the thiamin riboswitch while not interfering with human thiamin-binding proteins. Little is known about the structure and mechanism behind exogenous thiamin, HMP and THZ uptake, so biochemical and structural studies into thiamin transport will be needed.
Other forms of thiamin are also being discovered. Thiamin triphosphate (ThTP) activates chloride channels (131, 132) and is in high concentration in neuronal cells (133). ThTP is also the phosphate donor for the phosphorylation of a histidine residue in 43K rapsyn, which is found in postsynaptic membranes and is required for proper synapse function (134). A recently discovered adenyltransferase (135) generates an adenylated thiamin species, adenosine thiamin triphosphate (AThTP), which appears in response to carbon starvation in E. coli (136). AThTP is also present in yeast, animal tissue, and the roots of higher plants, illustrating another metabolite found across a diverse range of organisms. Further research on the function of thiamin, its biosynthetic and transport proteins, and its regulation will continue to reveal the biochemical foundations of this essential cofactor.
THE EIJKMAN NOBEL PRIZE.
Christiaan Eijkman’s pioneering work from 1890 to 1900 showed that a component of rice hulls could reverse the effects of beriberi in animals; however, it was not until 1933 that R.R. Williams purified thiamin. Prior to this discovery, Umetaro Suzuki identified rice hull isolates that contained the active ingredient. This work was first published in Japanese and then republished in German in 1912 (5).
The active ingredient called oryzanine, and later patented under the names aberic acid and orizanin, was shown to be an essential dietary component. Dogs fed on a thiamin-deficient diet of polished rice and boiled meat would succumb to beriberi in weeks but would recover quickly when given small amounts of oryzanine. This discovery was published about the time Casimir Funk was able to obtain crystals of a substance preventing polyneuritis, dubbing it “vitamine”—as it was a vital amine—although it was later shown that Funk most likely crystallized nicotinic acid (6). Eijkman further advanced his work after initially attributing the cause of beriberi to poisoning or microbial effects. He won the Nobel Prize in Physiology or Medicine in 1929, “for his discovery of the antineuritic vitamin” (1), also dubbed thiamin through the work of Funk (2).
THIAMINASE I—THE CULPRIT BEHIND A DISASTROUS NINETEENTH CENTURY AUSTRALIAN EXPEDITION.
An expedition headed by Robert O’Hara Burke to explore the inland territory of eastern Australia was launched on August 20, 1860, from the city of Melbourne to the Gulf of Carpentaria—a one-way trek of approximately 2800 km. Poor leadership combined with attrition and bad weather ultimately led to the exhaustion of supplies and the subsequent need to live off the land. One source of nutrition was flour obtained from the nardoo fern (Marsilea drummondii). Unbeknownst to the ill-fated travelers, improper preparation of the fern’s sporocaps produced baking flour with dangerously high levels of heat-resistant thiaminase I (65). Local Aborigines inactivated the thiaminase activity when preparing the flour by diluting it with water while prohibiting contact with other organic sources. Thiaminase I from the nardoo fern loses significant activity in the absence of cosubstrates such as proline, hydroxyl proline, and imidazole (66). Dilution of the enzyme combined with isolation from its cosubstrates successfully inactivates the thiaminase activity. The disregard of this important step led to the death of Burke along with at least two other men from beriberi due to thiamin deficiency.
COFACTOR CANNIBALISM.
The catabolic pathways of many primary metabolites are well understood and are often utilized as a source of carbon or nitrogen for a growing cell. In the case of some newly discovered enzymes, cofactors are catabolized in order to generate other cofactors. The substrate for thiazole synthase (Thi4) in S. cerevisiae that is required to generate thiamin is the cofactor NAD (Figure 20) (125). In the case of the cofactor adenosylcobalamin, the protein BluB catalyzes an oxidative ring opening of the isoalloxazine moiety of FAD to generate the dimethylbenzamidazole ligand, which coordinates the cobalt ion of vitamin B12 (126,127). Another protein in the yeast thiamin biosynthetic pathway has also been found to use a cofactor as a substrate. The enzyme Thi5, which is responsible for generating the HMP moiety of thiamin, uses as substrates pyridoxine and histidine in a complicated rearrangement. Although radical SAM enzymes sometimes require the sacrifice of SAM, in the examples of Thi4, Thi5, and BluB, the cofactor substrates are needed to contribute to the molecular structure of their respective products (128).
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants DK44083 to T.P.B. and DK067081 to S.E.E. We thank Leslie Kinsland for help in preparing this manuscript. Dr. Jurgenson was affiliated with the Department of Chemistry and Chemical Biology, Cornell University when this article was written.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Eijkman C 1990. [Anti-neuritis vitamin and beriberi. Nobel Prize paper. 1929]. Ned. Tijdschr. Geneeskd 134:1654–57 [PubMed] [Google Scholar]
- 2.Funk C 1912. The preparation from yeast and certain foodstuffs of the substance the deficiency of which in diet occasions polyneuritis in birds. J. Physiol . 45:75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serganov A, Polonskaia A, Phan AT, Breaker RR, Patel DJ. 2006. Structural basis for gene regulation by a thiamine pyrophosphate-sensing riboswitch. Nature 441:1167–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thore S, Leibundgut M, Ban N. 2006. Structure of the eukaryotic thiamine pyrophosphate riboswitch with its regulatory ligand. Science 312:1208–11 [DOI] [PubMed] [Google Scholar]
- 5.Suzuki U, Shamimura T, Odake S. 1912. Oryzanine, a component of rice bran, and its physiological significance. Biochem. Z . 43:89–153 [Google Scholar]
- 6.Freidrich W 1988. Thiamin, vitamin B1, aneurin Vitamins , pp. 341–42. Berlin, Ger.: de Gruyter [Google Scholar]
- 7.Hill RE, Himmeldirk K, Kennedy IA, Pauloski RM, Sayer BG, et al. 1996. The biogenetic anatomy of vitamin B6. A 13C NMR investigation of the biosynthesis of pyridoxol in Escherichia coli . J. Biol. Chem 271:30426–35 [DOI] [PubMed] [Google Scholar]
- 8.Lois LM, Campos N, Putra SR, Danielsen K, RohmerM, BoronatA. 1998. Cloning and characterization of a gene from Escherichia coli encoding a transketolase-like enzyme that catalyzes the synthesis of d-1- deoxyxylulose 5-phosphate, a common precursor for isoprenoid, thiamin, and pyridoxol biosynthesis. Proc. Natl. Acad. Sci. USA 95:2105–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lange BM, Wildung MR, McCaskill D, Croteau R. 1998. A family of transketolases that directs isoprenoid biosynthesis via a mevalonate-independent pathway. Proc. Natl. Acad. Sci. USA 95:2100–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sprenger GA, Schorken U, Wiegert T, Grolle S, de Graaf AA, et al. 1997. Identification of a thiamindependent synthase in Escherichia coli required for the formation of the 1-deoxy-D-xylulose 5-phosphate precursor to isoprenoids, thiamin, and pyridoxol. Proc. Natl. Acad. Sci. USA 94:12857–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lange BM, Rujan T, Martin W, Croteau R. 2000. Isoprenoid biosynthesis: the evolution of two ancient and distinct pathways across genomes. Proc. Natl. Acad. Sci. USA 97:13172–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiang S, Usunow G, Lange G, Busch M, Tong L. 2007. Crystal structure of 1-deoxy-D-xylulose 5- phosphate synthase, a crucial enzyme for isoprenoids biosynthesis. J. Biol. Chem 282:2676–82 [DOI] [PubMed] [Google Scholar]
- 13.Nikkola M, Lindqvist Y, Schneider G. 1994. Refined structure of transketolase from Saccharomyces cerevisiae at 2.0 Å resolution. J. Mol. Biol 238:387–404 [DOI] [PubMed] [Google Scholar]
- 14.Arjunan P, Nemeria N, Brunskill A, Chandrasekhar K, Sax M, et al. 2002. Structure of the pyruvate dehydrogenase multienzyme complex E1 component from Escherichia coli at1.85 Å resolution. Biochemistry 41:5213–21 [DOI] [PubMed] [Google Scholar]
- 15.Aevarsson A, Seger K, Turley S, Sokatch JR, Hol WG. 1999. Crystal structure of 2-oxoisovalerate and dehydrogenase and the architecture of 2-oxo acid dehydrogenase multienzyme complexes. Nat. Struct. Biol 6:785–92 [DOI] [PubMed] [Google Scholar]
- 16.Pitterle DM, Rajagopalan KV. 1993. The biosynthesis of molybdopterin in Escherichia coli. Purification and characterization of the converting factor. J. Biol. Chem 268:13499–505 [PubMed] [Google Scholar]
- 17.Lake MW, Wuebbens MM, Rajagopalan KV, Schindelin H. 2001. Mechanism of ubiquitin activation revealed by the structure of a bacterial MoeB-MoaD complex. Nature 414:325–29 [DOI] [PubMed] [Google Scholar]
- 18.Burns KE, Baumgart S, Dorrestein PC, Zhai H, McLafferty FW, Begley TP. 2005. Reconstitution of a new cysteine biosynthetic pathway in Mycobacterium tuberculosis . J. Am. Chem. Soc 127:11602–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Godert AM, Jin M, McLafferty FW, Begley TP. 2007. Biosynthesis of the thioquinolobactin siderophore: an interesting variation on sulfur transfer. J. Bacteriol 189:2941–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C, Xi J, Begley TP, Nicholson LK. 2001. Solution structure of ThiS and implications for the evolutionary roots of ubiquitin. Nat. Struct. Biol 8:47–51 [DOI] [PubMed] [Google Scholar]
- 21.Lehmann C, Begley TP, Ealick SE. 2006. Structure of the Escherichia coli ThiS-ThiF complex, a key component of the sulfur transfer system in thiamin biosynthesis. Biochemistry 45:11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Settembre EC, Dorrestein PC, Zhai H, Chatterjee A, McLafferty FW, et al. 2004. Thiamin biosynthesis in Bacillus subtilis: structure of the thiazole synthase/sulfur carrier protein complex. Biochemistry 43:11647–57 [DOI] [PubMed] [Google Scholar]
- 23.Vander Horn PB, Backstrom AD, >Stewart V, >Begley TP. 1993. Structural genes fo rthiamine biosynthetic enzymes (thiCEFGH) in Escherichia col K-12. J. Bacteriol 175:982–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor SV, Kelleher NL, Kinsland C, Chiu HJ, Costello CA, et al. 1998. Thiamin biosynthesis in Escherichia coli. Identification of this thiocarboxylate as the immediate sulfur donor in the thiazole formation. J. Biol. Chem 273:16555–60 [DOI] [PubMed] [Google Scholar]
- 25.Duda DM, Walden H, Sfondouris J, Schulman BA. 2005. Structural analysis of Escherichia coli ThiF. J. Mol. Biol 349:774–86 [DOI] [PubMed] [Google Scholar]
- 26.Waterman DG, Ortiz-Lombardia M, Fogg MJ, Koonin EV, Antson AA. 2006. Crystal structure of Bacillus anthracis ThiI, a tRNA-modifying enzyme containing the predicted RNA-binding THUMP domain. J. Mol. Biol 356:97–110 [DOI] [PubMed] [Google Scholar]
- 27.Sugahara M, Murai S, Sugahara M, Kunishima N. 2007. Purification, crystallization and preliminary crystallographic analysis of the putative thiamine-biosynthesis protein PH1313 from Pyrococcus horikoshii OT3. Acta Crystallogr. F 63:56–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lauhon CT, Erwin WM, Ton GN. 2004. Substrate specificity for 4-thiouridine modification in Escherichia coli. J. Biol. Chem 279:23022–29 [DOI] [PubMed] [Google Scholar]
- 29.Kessler D 2006. Enzymatic activation of sulfur for incorporation into biomolecules in prokaryotes. FEMS Microbiol. Rev 30:825–40 [DOI] [PubMed] [Google Scholar]
- 30.Settembre EC, Dorrestein PC, Park JH, Augustine AM, Begley TP, Ealick SE. 2003. Structural and mechanistic studies on ThiO, a glycine oxidase essential for thiamin biosynthesis in Bacillus subtilis . Biochemistry 42:2971–81 [DOI] [PubMed] [Google Scholar]
- 31.Dym O, Eisenberg D. 2001. Sequence-structure analysis of FAD-containing proteins. Protein Sci . 10:1712–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. 2004. Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science 303:76–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leonardi R, Fairhurst SA, Kriek M, Lowe DJ, Roach PL. 2003. Thiamine biosynthesis in Escherichia coli: isolation and initial characterization of the ThiGH complex. FEBS Lett . 539:95–99 [DOI] [PubMed] [Google Scholar]
- 34.Leonardi R, Roach Peter L. 2004. Thiamine biosynthesis in Escherichia coli: in vitro reconstitution of the thiazole synthase activity. J. Biol. Chem 279:17054–62 [DOI] [PubMed] [Google Scholar]
- 35.Kriek M, Martins F, Leonardi R, Fairhurst SA, Lowe DJ, Roach PL. 2007. Thiazole synthase from Escherichia coli: an investigation ofthe substates and purified proteins required for activity in vitro. J. Biol. Chem 282:17413–23 [DOI] [PubMed] [Google Scholar]
- 36.Kriek M, Martins F, Challand MR, Croft A, Roach PL. 2007. Thiamine biosynthesis in Escherichia coli: identification of the intermediate and by-product derived from tyrosine. Angew. Chem. Int. Ed. Engl 46:9223–26 [DOI] [PubMed] [Google Scholar]
- 37.Park JH, Dorrestein PC, Zhai H, Kinsland C, McLafferty FW, Begley TP. 2003. Biosynthesis of the thiazole moiety of thiamin pyrophosphate (vitamin B1). Biochemistry 42:12430–38 [DOI] [PubMed] [Google Scholar]
- 38.Chatterjee A, Han X, McLafferty FW, Begley TP. 2006. Biosynthesis of thiamin thiazole: determination of the regiochemistry of the S/O acyl shift by using 1,4-dideoxy-D-xylulose-5-phosphate. Angew. Chem. Int. Ed. Engl 45:3507–10 [DOI] [PubMed] [Google Scholar]
- 39.Walden H, Podgorski MS, Huang DT, Miller DW, Howard RJ, et al. 2003. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol. Cell 12:1427–37 [DOI] [PubMed] [Google Scholar]
- 40.Toms AV, Haas AL, Park JH, Begley TP, Ealick SE. 2005. Structural characterization of the regulatory proteins TenA and TenI from Bacillus subtilis and identification of TenA as a thiaminase II. Biochemistry 44:2319–29 [DOI] [PubMed] [Google Scholar]
- 41.Chiu HJ, Reddick JJ, Begley TP, Ealick SE. 1999. Crystal structure of thiamin phosphate synthase from Bacillus subtilis at 1.25 Å resolution. Biochemistry 38:6460–70 [DOI] [PubMed] [Google Scholar]
- 42.Estramareix B, David S. 1990. Conversion of 5-aminoimidazole ribotide to the pyrimidine of thiamin in enterobacteria: study of the pathway with specifically labeled samples of riboside. Biochim. Biophys. Acta 1035:154–60 [DOI] [PubMed] [Google Scholar]
- 43.Estramareix B, Therisod M. 1984. Biosynthesis of thiamin: 5-aminoimidazole ribotide as the precursor of all the carbon atoms of the pyrimidine moiety. J. Am. Chem. Soc 106:3857–60 [Google Scholar]
- 44.Yamada K, Kumaoka H. 1982. Biosynthesis of thiamin. Incorporation of a two-carbon fragment derived from ribose of 5-aminoimidazole ribotide into the pyrimidine moiety of thiamin. Biochem. Int 5:771–76 [Google Scholar]
- 45.Estramareix B, David S. 1986. Biosynthesis of thiamine: origin of the methyl carbon atom of the pyrimidine moiety in Salmonella typhimurium . Biochem. Biophys. Res. Commun 134:1136–41 [DOI] [PubMed] [Google Scholar]
- 46.Lawhorn BG, Mehl RA, Begley TP. 2004. Biosynthesis of the thiamin pyrimidine: the reconstitution of a remarkable rearrangement reaction. Org. Biomol. Chem 2:2538–46 [DOI] [PubMed] [Google Scholar]
- 47.Chatterjee A, Li Y, Zhang Y, Grove TL, Lee M, et al. 2008. Reconstitution of ThiC in thiamine pyrimidine biosynthesis expands the radical SAM superfamily. Nat. Chem. Biol 4:758–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Croft MT, Moulin M, Webb ME, Smith AG. 2007. Thiamine biosynthesis in algae is regulated by riboswitches. Proc. Natl. Acad. Sci. USA 104:20770–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raschke M, Burkle L, Muller N, Nunes-Nesi A, Fernie AR, et al. 2007. Vitamin B1 biosynthesis in plants requires the essential iron-sulfur cluster protein, THIC. Proc. Natl. Acad. Sci. USA 104:19637–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng G, Bennett EM, Begley TP, Ealick SE. 2002. Crystal structure of 4-amino-5-hydroxymethyl-2-methylpyrimidine phosphate kinase from Salmonella typhimurium at2.3 Å resolution. Structure 10:225–35 [DOI] [PubMed] [Google Scholar]
- 51.Gustafson EA, Schinazi RF, Fingeroth JD. 2000. Human herpesvirus 8 open reading frame 21isa thymidine and thymidylate kinase of narrow substrate specificity that efficiently phosphorylates zidovudine but not ganciclovir. J. Virol 74:684–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peapus DH, Chiu HJ, Campobasso N, Reddick JJ, Begley TP, Ealick SE. 2001. Structural characterization of the enzyme-substrate, enzyme-intermediate, and enzyme-product complexes of thiamin phosphate synthase. Biochemistry 40:10103–14 [DOI] [PubMed] [Google Scholar]
- 53.Morett E, Saab-Rincon G, Olvera L, Olvera M, Flores H, Grande R. 2008. Sensitive genome-wide screen for low secondary enzymatic activities: the YjbQ family shows thiamin phosphate synthase activity. J. Mol. Biol 376:839–53 [DOI] [PubMed] [Google Scholar]
- 54.McCulloch KM, Kinsland C, Begley TP, Ealick SE. 2008. Structural studies of thiamin monophosphate kinase in complex with substrates and products. Biochemistry 47:3810–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Melnick J, Lis E, Park JH, Kinsland C, Mori H, et al. 2004. Identification of the two missing bacterial genes involved in thiamine salvage: thiamine pyrophosphokinase and thiamine kinase. J. Bacteriol 186:3660–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nosaka K, Kaneko Y, Nishimura H, Iwashima A. 1993. Isolation and characterization of a thiamin pyrophosphokinase gene, THI80, from Saccharomyces cerevisiae . J. Biol. Chem 268:17440–47 [PubMed] [Google Scholar]
- 57.Costello CA, Kelleher NL, Abe M, McLafferty FW, Begley TP. 1996. Mechanistic studies on thiaminase I. Overexpression and identification of the active site nucleophile. J. Biol. Chem 271:3445–52 [DOI] [PubMed] [Google Scholar]
- 58.Nicewonger R, Rammelsberg A, Costello CA, Begley TP. 1995. Mechanistic studies on thiaminase I. 1. The stereochemical course of the reaction. Bioorg. Chem 23:512–18 [Google Scholar]
- 59.Kelleher NL, Costello CA, Begley TP, McLafferty FW. 1995. Thiaminase I (42 kDa) heterogeneity, sequence refinement, and active site location from high-resolution tandem mass spectrometry. J. Am. Soc. Mass Spectrom 6:981–84 [DOI] [PubMed] [Google Scholar]
- 60.Kelleher NL, Nicewonger RB, Begley TP, McLafferty FW. 1997. Identification of modification sites in large biomolecules by stable isotope labeling and tandem high resolution mass spectrometry. The active site nucleophile of thiaminase I. J. Biol. Chem 272:32215–20 [DOI] [PubMed] [Google Scholar]
- 61.Wu M, Papish ET, Begley TP. 2000. Mechanistic studies on thiaminase I. Identification of the product of thiamin degradation in the absence of the nucleophilic cosubstrate. Bioorg. Chem 28:45–48 [Google Scholar]
- 62.Campobasso N, Begun J, Costello CA, Begley TP, Ealick SE. 1998. Crystallization and preliminary X-ray analysis of thiaminase I from Bacillus thiaminolyticus: space group change upon freezing of crystals. Acta Crystallogr. D 54:448–50 [DOI] [PubMed] [Google Scholar]
- 63.Honeyfield DC, Brown SB, Fitzsimons JD, Tillitt DE. 2005. Early mortality syndrome in Great Lakes salmonines. J. Aquat. Anim. Health 17:1–3 [Google Scholar]
- 64.Marquenski SV, Brown SB. 1997. Early mortality syndrome in the Great Lakes In Chemically Induced Alterations in Functional Development and Reproduction of Fishes, ed. Rolland RM, Gilbertson M, Peterson RE, pp. 135–52. Pensacola, FL: SETAC Press [Google Scholar]
- 65.Earl JW, McCleary BV. 1994. Mystery of the poisoned expedition. Nature 368:683–84 [DOI] [PubMed] [Google Scholar]
- 66.McCleary BV, Chick BF. 1977. The purification and properties of a thiaminase I enzyme from nardoo (Marsilea drummondii). Phytochemistry 16:207–13 [Google Scholar]
- 67.Lienhard GE. 1970. Kinetic evidence for a (4-amino-2-methyl-5-pyrimidinyl)methyl-enzyme intermediate in the thiaminase I reaction. Biochemistry 9:3011–20 [DOI] [PubMed] [Google Scholar]
- 68.Bos M, Kozik A. 2000. Some molecular and enzymatic properties of a homogeneous preparation of thiaminase I purified from carp liver. J. Protein Chem 19:75–84 [DOI] [PubMed] [Google Scholar]
- 69.Strauch MA. 1993. Regulation of Bacillus subtilis gene expression during the transition from exponential growth to stationary phase. Prog. Nucleic Acid Res. Mol. Biol 46:121–53 [DOI] [PubMed] [Google Scholar]
- 70.Pang AS, Nathoo S, Wong SL. 1991. Cloning and characterization of a pair of novel genes that regulate production of extracellular enzymes in Bacillus subtilis . J. Bacteriol 173:46–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Spurlino JC, Lu GY, Quiocho FA. 1991. The 2.3 Å resolution structure of the maltose- or maltodextrin-bindingprotein, a primaryreceptorofbacterial active transport and chemotaxis. J. Biol. Chem 266:5202–19 [DOI] [PubMed] [Google Scholar]
- 72.Sugiyama S, Vassylyev DG, Matsushima M, Kashiwagi K, Igarashi K, Morikawa K. 1996. Crystal structure of PotD, the primary receptor of the polyamine transport system in Escherichia coli . J. Biol. Chem 271:9519–25 [DOI] [PubMed] [Google Scholar]
- 73.Sugiyama S, Matsuo Y, Maenaka K, Vassylyev DG, Matsushima M, et al. 1996. The1.8 Å X-ray structure ofthe Escherichia coli PotD protein complexed with spermidine and the mechanism ofpolyamine binding. Protein Sci 5:1984–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jenkins AH, Schyns G, Potot S, Sun G, Begley TP. 2007. A new thiamin salvage pathway. Nat. Chem. Biol 3:492–97 [DOI] [PubMed] [Google Scholar]
- 75.Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. 2002. Comparative genomics of thiamin biosynthesis in procaryotes. New genes and regulatory mechanisms. J. Biol. Chem 277:48949–59 [DOI] [PubMed] [Google Scholar]
- 76.Jenkins AL, Zhang Y, Ealick SE, Begley TP. 2008. Mutagenesis studies on TenA: a thiamin salvage enzyme from Bacillus subtilis . Bioorg. Chem 36:29–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang Y, Taylor SV, Chiu HJ, Begley TP. 1997. Characterization of the Bacillus subtilis thiC operon involved in thiamine biosynthesis. J. Bacteriol 179:3030–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Petersen LA, Downs DM. 1997. Identification and characterization of an operon in Salmonella typhimurium involved in thiamine biosynthesis. J. Bacteriol 179:4894–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Campobasso N, Mathews II, Begley TP, Ealick SE. 2000. Crystal structure of 4-methyl-5-ß-hydroxyethylthiazole kinase from Bacillus subtilis at 1.5 Å resolution. Biochemistry 39:7868–77 [DOI] [PubMed] [Google Scholar]
- 80.Sigrell JA, Cameron AD, Jones TA, Mowbray SL. 1998. Structure of Escherichia coli ribokinase in complex with ribose and dinucleotide determined to 1.8 Å resolution: insights into a new family of kinase structures. Structure 6:183–93 [DOI] [PubMed] [Google Scholar]
- 81.Mathews II, Erion MD, Ealick SE. 1998. Structure of human adenosine kinase at 1.5 Å resolution. Biochemistry 37:15607–20 [DOI] [PubMed] [Google Scholar]
- 82.Baker LJ, Dorocke JA, Harris RA, Timm DE. 2001. The crystal structure of yeast thiamin pyrophosphokinase. Structure 9:539–46 [DOI] [PubMed] [Google Scholar]
- 83.Timm DE, Liu J, Baker LJ, Harris RA. 2001. Crystal structure of thiamin pyrophosphokinase. J. Mol. Biol . 310:195–204 [DOI] [PubMed] [Google Scholar]
- 84.Webb E, Claas K, Downs D. 1998. thiBPQ encodes an ABC transporter required for transport of thiamine and thiamine pyrophosphate in Salmonella typhimurium . J. Biol. Chem 273:8946–50 [DOI] [PubMed] [Google Scholar]
- 85.Bellion E, Lash TD. 1983. Transport of 2-methyl-4-amino-5-hydroxymethylpyrimidine by Salmonella typhimurium. Biochim. Biophys. Acta 735:337–40 [DOI] [PubMed] [Google Scholar]
- 86.Bellion E, Lash TD, McKellar BR. 1983. Transport of thiamine and 4-methyl-5-hydroxyethylthiazole by Salmonella typhimurium. Biochim. Biophys. Acta 735:331–36 [DOI] [PubMed] [Google Scholar]
- 87.Soriano EV, Rajashankar KR, Hanes JW, Bale S, Begley TP, Ealick SE. 2008. Structural similarities between thiamin-binding protein and thiaminase-I suggest a common ancestor. Biochemistry 47:1346–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Devedjiev Y, Surendranath Y, Derewenda U, Gabrys A, Cooper DR, et al. 2004. The structure and ligand binding properties of the B. subtilis YkoF gene product, a member of a novel family of thiamin/HMPbinding proteins. J. Mol. Biol 343:395–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vogl C, Klein CM, Batke AF, Schweingruber ME, Stolz J. 2008. Characterization of Thi9, a novel thiamine (vitamin B1) transporter from Schizosaccharomyces pombe . J. Biol. Chem 283:7379–89 [DOI] [PubMed] [Google Scholar]
- 90.Jack DL, Paulsen IT, Saier MH Jr. 2000. The amino acid/polyamine/organocation (APC) superfamily of transporters specific for amino acids, polyamines and organocations. Microbiology 146:1797–814 [DOI] [PubMed] [Google Scholar]
- 91.Goffeau A, ParkJ, Paulsen IT, Jonniaux J-L, Dinh T, et al. 1997. Multidrug-resistant transport proteins in yeast: complete inventory and phylogenetic characterization of yeast open reading frames within the major facilitator superfamily. Yeast 13:43–54 [DOI] [PubMed] [Google Scholar]
- 92.Lee JM, Zhang S, Saha S, Santa Anna S, Jiang C, Perkins J. 2001. RNA expression analysis using an antisense Bacillus subtilis genome array. J. Bacteriol 183:7371–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang Y, Begley TP. 1997. Cloning, sequencing and regulation of thiA, a thiamin biosynthesis gene from Bacillus subtilis . Gene 198:73–82 [DOI] [PubMed] [Google Scholar]
- 94.Grundy FJ, Henkin TM. 2006. From ribosome to riboswitch: control of gene expression in bacteria by RNA structural rearrangements. Crit. Rev. Biochem. Mol. Biol 41:329–38 [DOI] [PubMed] [Google Scholar]
- 95.Winkler WC, Cohen-Chalamish S, Breaker RR. 2002An mRNA structure that controls gene expression by binding FMN. Proc. Natl. Acad. Sci. USA 99:15908–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Winkler W, Nahvi A, Breaker RR. 2002. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 419:952–56 [DOI] [PubMed] [Google Scholar]
- 97.Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR. 2002. Genetic control by a metabolite binding mRNA. Chem. Biol 9:1043. [DOI] [PubMed] [Google Scholar]
- 98.Mironov AS, Gusarov I, Rafikov R, Lopez LE, Shatalin K, et al. 2002. Sensing small molecules by nascent RNA: a mechanism to control transcription in bacteria. Cell 111:747–56 [DOI] [PubMed] [Google Scholar]
- 99.Winkler WC, Nahvi A, Sudarsan N, Barrick JE, Breaker RR. 2003. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nat. Struct. Biol 10:701–7 [DOI] [PubMed] [Google Scholar]
- 100.Mandal M, Boese B, Barrick JE, Winkler WC, Breaker RR. 2003. Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell 113:577–86 [DOI] [PubMed] [Google Scholar]
- 101.Winkler WC. 2005. Metabolic monitoring by bacterial mRNAs. Arch. Microbiol . 183:151–89 [DOI] [PubMed] [Google Scholar]
- 102.Blount KF, Breaker RR. 2006. Riboswitches as antibacterial drug targets. Nat. Biotechnol 24:1558–64 [DOI] [PubMed] [Google Scholar]
- 103.Sudarsan N, Barrick JE, Breaker RR. 2003. Metabolite-binding RNA domains are present in the genes of eukaryotes. RNA 9:644–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Miranda-Rios J, Navarro M, Soberon M. 2001. A conserved RNA structure (thi box) is involved in regulation of thiamin biosynthetic gene expression in bacteria. Proc. Natl. Acad. Sci. USA 98:9736–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Miranda-Rios J 2007. The THI-box riboswitch, or how RNA binds thiamin pyrophosphate. Structure 15:259–65 [DOI] [PubMed] [Google Scholar]
- 106.Edwards TE, Ferré-D´ Amare´ AR 2006. Crystal structures of the thi-box riboswitch bound to thiamine pyrophosphate analogs reveal adaptive RNA-small molecule recognition. Structure 14:1459–68 [DOI] [PubMed] [Google Scholar]
- 107.Muller YA, Lindqvist Y, Furey W, Schulz GE, Jordan F, Schneider G. 1993. A thiamin diphosphate binding fold revealed by comparison of the crystal structures of transketolase, pyruvate oxidase and pyruvate decarboxylase. Structure 1:95–103 [DOI] [PubMed] [Google Scholar]
- 108.White RL, Spenser ID. 1979. Thiamin biosynthesis in Saccharomyces cerevisiae. Origin of carbon-2 of the thiazole moiety. Biochem. J 179:315–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.White RL, Spenser ID. 1979. Biosynthesis of vitamin B1 in yeast. Origin of the thiazole unit. J. Am. Chem. Soc 101:5102–4 [Google Scholar]
- 110.White RL, Spenser ID. 1982. Thiamin biosynthesis in yeast. Origin of the five-carbon unit of the thiazole moiety. J. Am. Chem. Soc 104:4934–43 [Google Scholar]
- 111.Praekelt UM, Byrne KL, Meacock PA. 1994. Regulation of THI4 (MOL1), a thiamine-biosynthetic gene of Saccharomyces cerevisiae . Yeast 10:481–90 [DOI] [PubMed] [Google Scholar]
- 112.Ishida S, Tazuya-Murayama K, Kijima Y, Yamada K. 2008. The direct precursor of the pyrimidine moiety of thiamin is not urocanic acid but histidine in Saccharomyces cerevisiae . J. Nutr. Sci. Vitaminol 54:7–10 [DOI] [PubMed] [Google Scholar]
- 113.Wightman R, Meacock PA. 2003. The THI5 gene family of Saccharomyces cerevisiae: distribution of homologues among the hemiascomycetes and functional redundancy in the aerobic biosynthesis of thiamin from pyridoxine. Microbiology 149:1447–60 [DOI] [PubMed] [Google Scholar]
- 114.Haas AL, Laun NP, Begley TP. 2005. Thi20, a remarkable enzyme from Saccharomyces cerevisiae with dual thiamin biosynthetic and degradation activities. Bioorg. Chem 33:338–44 [DOI] [PubMed] [Google Scholar]
- 115.Webb ME, Marquet A, Mendel RR, Rebeille F, Smith AG. 2007. Elucidating biosynthetic pathways for vitamins and cofactors. Nat. Prod. Rep 24:988–1008 [DOI] [PubMed] [Google Scholar]
- 116.Nosaka K, Nishimura H, Kawasaki Y, Tsujihara T, Iwashima A. 1994. Isolation and characterization of the THI6 gene encoding a bifunctional thiamin-phosphate pyrophosphorylase/hydroxyethylthiazole kinase from Saccharomyces cerevisiae . J. Biol. Chem 269:30510–16 [PubMed] [Google Scholar]
- 117.Schweingruber AM, Dlugonski J, Edenharter E, Schweingruber ME. 1991. Thiamin in Schizosaccha-romyces pombe: dephosphorylation, intracellular pool, biosynthesis and transport. Curr. Genet 19:249–54 [DOI] [PubMed] [Google Scholar]
- 118.Zurlinden A, Schweingruber ME. 1992. Cloning and regulation of Schizosaccharomyces pombe thi2, a gene involved in thiamin biosynthesis. Gene 117:141–43 [DOI] [PubMed] [Google Scholar]
- 119.Rapala-kozik M, Olczak M, Ostrowska K, Starosta A, Kozik A. 2007. Molecular characterization of the thi3 gene involved in thiamine biosynthesis in Zea mays: cDNA sequence and enzymatic and structural properties of the recombinant bifunctional protein with 4-amino-5-hydroxymethyl-2-methylpyrimidine (phosphate) kinase and thiamine monophosphate synthase activities. Biochem. J 408:149–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Machado CR, Praekelt UM, de Oliveira RC, Barbosa AC, Byrne KL, et al. 1997. Dual role for the yeast THI4 gene in thiamine biosynthesis and DNA damage tolerance. J. Mol. Biol 273:114–21 [DOI] [PubMed] [Google Scholar]
- 121.Jurgenson CT, Chatterjee A, Begley TP, Ealick SE. 2006. Structural insights into the function of the thiamin biosynthetic enzyme Thi4 from Saccharomyces cerevisiae . Biochemistry 45:11061–70 [DOI] [PubMed] [Google Scholar]
- 122.Faou P, Tropschug M. 2003. A novel binding protein for a member of CyP40-type cyclophilins: N. crassa CyPBP37, a growth and thiamine regulated protein homolog to yeast Thi4p. J. Mol. Biol 333:831–44 [DOI] [PubMed] [Google Scholar]
- 123.Holm L, Sander C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol 233:123–38 [DOI] [PubMed] [Google Scholar]
- 124.Rossmann MG, Moras D, Olsen KW. 1974. Chemical and biological evolution of nucleotide-binding protein. Nature 250:194–99 [DOI] [PubMed] [Google Scholar]
- 125.Chatterjee A,Jurgenson CT, Schroeder FC, Ealick SE, Begley TP. 2007. Biosynthesis of thiamin thiazole in eukaryotes: conversion of NAD to an advanced intermediate. J. Am. Chem. Soc 129:2914–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Taga ME, Larsen NA, Howard-Jones AR, Walsh CT, Walker GC. 2007. BluB cannibalizes flavin to form the lower ligand of vitamin B12. Nature 446:449–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Campbell GRO, Taga ME, Mistry K, Lloret J, Anderson PJ, et al. 2006. Sinorhizobium meliloti bluB is necessary for production of 5,6-dimethylbenzimidazole, the lower ligand of B12. Proc. Natl. Acad. Sci. USA 103:4634–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ealick SE, Begley TP. 2007. Biochemistry: molecular cannibalism. Nature 446:387–88 [DOI] [PubMed] [Google Scholar]
- 129.Godoi PH, Galhardo RS, Luche DD,Van Sluys MA,Menck CF, Oliva G. 2006. Structure of the thiazole biosynthetic enzyme THI1 from Arabidopsis thaliana. J. Biol. Chem 281:30957–66 [DOI] [PubMed] [Google Scholar]
- 130.Chatterjee A, Jurgenson CT, Schroeder FC, Ealick SE, Begley TP. 2006. Thiamin biosynthesis in eukaryotes: characterization of the enzyme-bound product of thiazole synthase from Saccharomyces cerevisiae and its implications in thiazole biosynthesis. J. Am. Chem. Soc 128:7158–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bettendorff L, Kolb HA, Schoffeniels E. 1993. Thiamine triphosphate activates an anion channel of large unit conductance in neuroblastoma cells. J. Membr. Biol 136:281–88 [DOI] [PubMed] [Google Scholar]
- 132.Bettendorff L, Peeters M, Wins P, Schoffeniels E. 1993. Metabolism of thiamine triphosphate in rat brain: correlation with chloride permeability. J. Neurochem 60:423–34 [DOI] [PubMed] [Google Scholar]
- 133.Bettendorff L, Peeters M, Jouan C, Wins P, Schoffeniels E. 1991. Determination of thiamin and its phosphate esters in cultured neurons and astrocytes using an ion-pair reversed-phase high-performance liquid chromatographic method. Anal. Biochem 198:52–59 [DOI] [PubMed] [Google Scholar]
- 134.Nghiem HO, Bettendorff L, Changeux JP. 2000. Specific phosphorylation of Torpedo 43K rapsyn by endogenous kinase(s) with thiamine triphosphate as the phosphate donor. FASEB J . 14:543–54 [DOI] [PubMed] [Google Scholar]
- 135.Makarchikov AF, Brans A, Bettendorff L. 2007. Thiamine diphosphate adenylyl transferase from E. coli: functional characterization of the enzyme synthesizing adenosine thiamine triphosphate. BMC Biochem . 8:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bettendorff L, Wirtzfeld B, Makarchikov AF, Mazzucchelli G, Frederich M, et al. 2007. Discovery of a natural thiamine adenine nucleotide. Nat. Chem. Biol 3:211–12 [DOI] [PubMed] [Google Scholar]