

Abstract
Acute pulmonary embolism (APE)-induced inflammation contributes to cardiomyocyte injury and dysfunction in the right ventricle (RV) of the heart. The interactions of cyclophilin A with its ligand extracellular matrix metalloproteinase inducer (EMMPRIN or CD147) may be involved in this inflammatory process. To this end, here we induced APE by intravenous injections of microspheres in Sprague-Dawley rats. We found that after the APE, cyclophilin A and CD147 levels increased synchronously in RV tissue following APE and peaked at 24 h. The cyclophilin A inhibitor cyclosporine A attenuated the APE-induced cyclophilin A levels, and a monoclonal antibody of CD147 (anti-CD147) abrogated the elevation of CD147 in the RV but not the increase of cyclophilin A. Importantly, treatment with cyclosporine A, anti-CD147, or both attenuated APE-induced increases in RV systolic pressure, plasma cardiac troponin-I (cTnI) concentrations, the RV/left ventricle diameter ratio, and the Tei index, measured by echocardiography 24 h after APE induction. These beneficial effects were associated with reduced RV neutrophil infiltration and prevention of matrix metalloproteinase 9 (MMP-9) and MMP-2 activation. These findings suggested that inhibiting the cyclophilin A–CD147 interaction attenuates APE-associated RV cardiomyocyte injury and dysfunction by suppressing inflammation. We further proposed that cyclophilin A and CD147 might participate in APE-induced pathological processes by partly activating the ERK1/2 kinase–nuclear factor-κB pathway. We conclude that the cyclophilin A–CD147 interaction may represent a potential therapeutic target for managing APE.
Keywords: inflammation, Western blot, cardiomyopathy, biotechnology, thrombosis, acute pulmonary embolism, CD147, cyclophilin A, EMMPRIN, right ventricular dysfunction, heart disease
Introduction
Acute pulmonary embolism (APE)2 is a common and potentially life-threatening disease with a 30-day all-cause mortality rate between 9 and 11% (1). The presence of right ventricular (RV) dysfunction is closely related to a significant increase in morbidity and mortality following pulmonary embolism (1, 2). Previous studies indicate that an inflammatory response participates in RV dysfunction following APE. In this regard, APE-associated inflammatory cells inflow to RV tissue (1, 3–5), followed by a rapid release and activation of large amounts of matrix metalloproteinases (MMPs) (4–6). Activated MMPs (especially MMP-2 and MMP-9) could degrade cardiac troponin-I (cTnI), myosin light chain-1, and other sarcomeric and cytoskeletal proteins, thus causing myocardial contractile dysfunction following APE (7–9). Numerous studies have shown that anti-inflammatory compounds or the inhibition of MMPs attenuates cardiomyocyte injury and RV dysfunction in models of APE (3, 5, 6, 10, 11); however, the exact mechanism by which inflammatory cells are recruited is still unclear.
Cyclophilin A (CyPA) is a highly conserved and ubiquitously distributed protein that has peptidylprolyl isomerase activity (12, 13). It makes up 0.1–0.6% of all cytosolic proteins and plays an important role in immunomodulation, cell signaling, transcriptional regulation, and protein folding and trafficking (12, 13). Within the last decade, studies have demonstrated that CyPA could be secreted from the cell in response to inflammatory stimuli, such as hypoxia, infection, and oxidative stress (12–15). By interacting with its cell-surface signaling receptor cluster of differentiation 147 (CD147), also known as extracellular matrix metalloproteinase inducer (EMMPRIN), extracellular CyPA could activate ERK/nuclear factor (NF)-κB pathways, stimulate cytokine release, accelerate leukocyte recruitment, and boost MMP activation in stimulus sites (12, 13, 16–19). The CyPA–CD147 interaction associated with the acute or chronic inflammation pathological process has been found in many diseases (12, 13, 20–22). Crucially, inhibition of this interaction ameliorates myocardial inflammation and remodeling in troponin I–induced myocarditis (23), and it reduces infarct size following myocardial ischemia and reperfusion (24–26).
Given this evidence, we hypothesized that CyPA also participates in the pathological progression of cardiomyocyte injury and RV dysfunction following APE. Therefore, the purpose of this study was to investigate the role of extracellular CyPA and CD147 in RV injury and dysfunction following APE and to examine whether inhibition of CyPA with cyclosporine A (CsA) or inhibition of CD147 with CD147 mAb (anti-CD147) attenuates the RV injury and dysfunction associated with APE.
Results
General observations
There were no significant differences in body weight between any of the experimental groups (data not shown). All animals (42/42 rats) survived in the sham group. However, 15.4% (23/149 rats) of all rats died after induction of APE. As shown in Fig. 1A, the lungs of the rats in the APE groups exhibited red-and-white patches, which were observed visually, and upon histopathological examination, the microsphere that was injected could be found in the pulmonary artery of the APE model.
Figure 1.
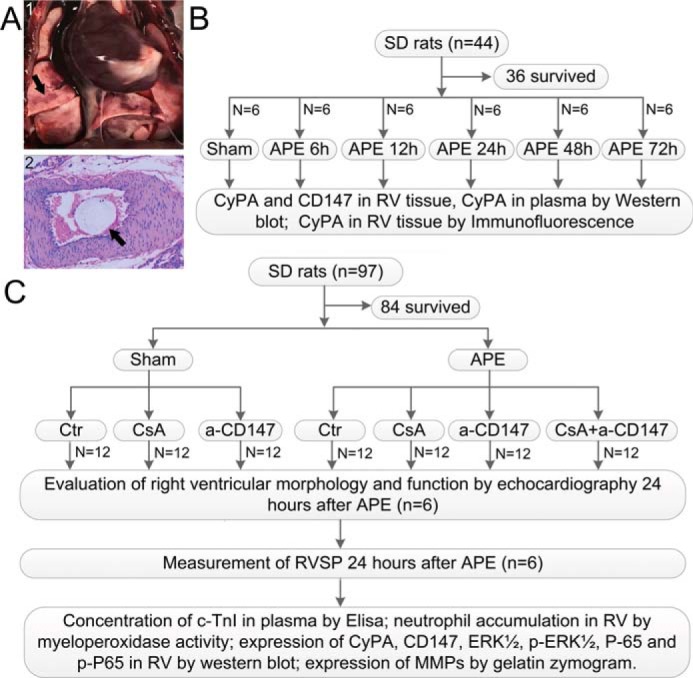
A, gross appearance and histological examination of the lung in APE rat. Panel 1, gross appearance of the embolized lung, and the black arrow indicates red and white patches. Panel 2, histological examination of the embolized pulmonary artery, and the black arrow indicates the microsphere injected. B, first part of the experiment was designed to show the expressions of CyPA in plasma and RV tissue and CD147 in RV tissue by time course after APE and to investigate the suitable time point for further research. C, second part of the experiment was designed to examine the role of CyPA–CD147 in RV dysfunction after APE and indicate the potential mechanisms. CsA was administered 15 min before surgery at a dose of 10 mg/kg by slow i.v. infusion for 5 min, and a-CD147 was administered immediately after the APE model's establishment at a dose of 3 mg/kg. a-CD147 = mAb of CD147.
APE increased the protein expression of CyPA and CD147
Western blot analysis was performed to assess the expression of CyPA and CD147 in the RV myocardium after APE. Compared with the sham group, the protein levels of CyPA and CD147 in RV tissue from the APE groups increased and peaked at 24 h and then decreased. However, the protein levels of CyPA and CD147 were still higher than that in the sham group, even 72 h after APE (Fig. 2, A–D). Results of immunofluorescence analysis for CyPA showed that the expression trend was similar to that of Western blotting (Fig. 2, E and F). In addition, Western blot analysis of the rat plasma showed that APE could also increase the level of CyPA in plasma (Fig. 2, G and H). These results indicated that CyPA and CD147 might participate in the progression of RV cardiomyocyte injury, and 24 h after APE may be a suitable time point for further research.
Figure 2.

Expressions of CyPA and CD147 after APE. A, Western blot analysis showed the expression of CyPA in RV tissue at 6, 12, 24, 48, and 72 h after APE. B, quantification of the CyPA protein levels as shown in A. Western blot analysis and quantification of the expression of CD147 in RV tissue are shown in C and D. Protein levels were normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). E, immunofluorescence analysis was performed with antibody for CyPA. Nuclei were fluorescently labeled with 4,6-diamino-2-phenylindole (DAPI) (blue). Scale bar, 50 μm. F, relative fluorescent intensity of CyPA in myocardial cell. Western blot analysis and quantification of the expression of CyPA in plasma are shown in G and H. B, D, F, and H, mean values for sham group were normalized to 1.0. Data are mean ± S.D. *, p < 0.05; **, p < 0.01 versus sham group (n = 6, each group).
Treatment with CsA and anti-CD147 ameliorated APE-induced increase in RVSP
The results of RV systolic pressure (RVSP) were similar across the three sham groups. APE significantly increased RVSP in the four APE groups (Fig. 3). However, this increase was much smaller in the APE groups treated with CsA or anti-CD147, and this protective effect seemed more remarkable when both CsA and anti-CD147 were administered.
Figure 3.

RVSP at 24 h after APE in all experimental groups. Data are mean ± S.D. *, p < 0.05; **, p < 0.01 versus sham group; #, p < 0.05; ##, p < 0.01 versus APE control group (n = 6, each group). a-CD = mAb of CD147.
Treatment with CsA and anti-CD147 reduced APE-induced neutrophil accumulation, cardiomyocyte injury, and RV dysfunction
A significant increase in the myeloperoxidase activity of the RV tissue was observed in the APE group, and treatment with CsA, anti-CD147, or both profoundly attenuated this increase (Fig. 4B). This outcome was also confirmed by histopathological examination of RV tissue (Fig. S1). To evaluate RV cardiomyocyte injury after APE, we measured plasma cTnI. We found that the level of cTnI increased significantly 24 h after APE, and this increase was attenuated by treatment with CsA or anti-CD147 or both (Fig. 4A). By echocardiography, a much higher RV/left ventricle (RV/LV) ratio and Tei index were observed in embolized animals, and treatment with CsA or anti-CD147 or both was associated with significantly lower increases in these two measurements (Fig. 5, A–D). There were no significant differences in the main pulmonary artery diameter between any of the experimental groups, although the diameters in the APE control group seemed larger (Fig. 5, E and F).
Figure 4.

Plasma cTnI and myeloperoxidase activity of unit weight right ventricular tissue at 24 h after APE in all experimental groups are shown in A and B. Data are mean ± S.D. *, p < 0.05; **, p < 0.01 versus sham group; ##, p < 0.01 versus APE control group (n = 6, each group). a-CD = mAb of CD147.
Figure 5.

Results of echocardiography at 24 h after APE in all experimental groups. Measurements of LV diameter, RV diameter, and pulmonary artery diameter (white arrow) on echocardiography image are shown in A, B, and E, respectively. Results of RV/LV ratio, Tei index, and pulmonary artery diameter in all experimental groups are shown in C, D, and F, respectively. Data are mean ± S.D. *, p < 0.05; **, p < 0.01 versus sham group; ##, p < 0.01 versus APE control group (n = 6, each group). a-CD = mAb of CD147.
Effects of CsA and anti-CD147 treatments on the protein levels of CyPA and CD147 in RV tissue
Consistent with the results in the first part of the experiment, Western blotting and immunofluorescence analysis of RV tissue showed that CyPA protein levels increased significantly at 24 h after APE when compared with those in the sham group. This increase was significantly attenuated by CsA treatment but not anti-CD147 (Fig. 6, A, B, D, and E). Also, Western blot analysis showed that CD147 protein levels were much higher in the APE control group than in the sham group, and this increase was attenuated by both CsA and anti-CD147 treatments (Fig. 6, A and C). These results suggest that CyPA may be an upstream regulator of CD147 expression.
Figure 6.

Effects of CsA and mAb of CD147 (a-CD) on the expressions of CyPA and CD147. A, Western blot analysis of the protein levels of CyPA and CD147 in RV tissues in rats of each group. B and C, quantification of the protein levels of CyPA and CD147. Protein levels were normalized to that of GAPDH. D, immunofluorescence analysis was performed using CyPA antibodies, and nuclei were fluorescently labeled with 4,6-diamino-2-phenylindole (DAPI) (blue). Scale bar, 50 μm. E, relative fluorescent intensity of CyPA. B, C, and E, mean values for sham group were normalized to 1.0. Data are mean ± S.D. *, p < 0.05; **, p < 0.01 versus sham group; ##, p < 0.01 versus APE control group; ns, no statistical significance (n = 6, each group).
Effects of CsA and anti-CD147 treatments on the ERK–NF-κB pathway
The phosphorylation of ERK1/2 was evaluated by Western blot analysis (Fig. 7, A and B). The phosphorylation level of ERK1/2 in the RV tissue of the APE group was much higher than that in the sham group. APE + CsA, APE + anti-CD147, and APE + CsA + anti-CD147 showed significantly lower ERK1/2 phosphorylation levels than that in the APE control group. The Western blot analysis of the p65 phosphorylation level is shown in Fig. 7, C and D. Relative to the sham group, the phosphorylation level of p65 in the RV tissue from the APE control group was significantly higher, and treatment with CsA or anti-CD147 or both suppressed the expression of phosphorylation of p65. These results suggest that CyPA–CD147 may participate in RV injury and dysfunction after APE by regulation of the ERK–NF-κB pathway.
Figure 7.

Effects of CyPA/CD147 on the phosphorylation of ERK1/2 and NF-κB activity under APE conditions. A, Western blot analysis of the phosphorylation level of ERK1/2. B, quantification of the phosphorylation level of ERK1/2 shown in A. C, Western blot analysis of the phosphorylation level of p65. D, quantification of the phosphorylation level of p65 shown in C. Protein levels were normalized to that of GAPDH. B and D, mean values for sham group were normalized to 1.0. Data are mean ± S.D. *, p < 0.05; **, p < 0.01 versus sham group; ##, p < 0.01 versus APE control group (n = 6, each group). a-CD = mAb of CD147.
Effects of CsA and anti-CD147 treatments on MMP-2 and MMP-9
A representative zymogram of RV samples showing the MMP-9 and MMP-2 bands is presented in Fig. 8. Both MMP-2 and MMP-9 levels increased significantly in the APE control group. However, this increase was attenuated by CsA or anti-CD147, and this attenuation effect seemed more remarkable when CsA and anti-CD147 were administered simultaneously.
Figure 8.

Effects of CyPA/CD147 on the level of MMP-9 and MMP-2. A, representative zymogram of RV tissue extract showing MMP-9 and MMP-2 bands in sham, APE, APE + CsA, APE + mAb of CD147 (a-CD), and APE + CsA + a-CD groups. B and C, quantification of the levels of MMP-9 and MMP-2. Mean values for sham group were normalized to 1.0. Data are mean ± S.D. *, p < 0.05; **, p < 0.01 versus sham group; ##, p < 0.01 versus APE control group (n = 6, each group).
Effects of FK506 on APE rats
There was no significant difference in cTnI, the RV/LV diameter ratio, the Tei index, or the myeloperoxidase activity of RV tissue between FK506 and APE groups (Fig. 9).
Figure 9.

Effects of FK506 on cTnI (A), ratio of the diameters of the right and left ventricles (B), Tei index (C), and the myeloperoxidase activity of unit weight right ventricular tissue (D) at 24 h after APE in rats. Data are mean ± S.D. **, p < 0.01 versus sham group; ns, no statistical significance (n = 6, each group). a-CD = mAb of CD147.
Discussion
To our knowledge, this study is the first to demonstrate that the levels of CyPA and CD147 in RV tissue increase synchronously following APE. Furthermore, our results also suggest that the inhibition of CyPA–CD147 interactions attenuates APE-increased RVSP, ameliorates APT-associated cardiomyocyte injury, and prevents APE-induced RV dilatation and impairment in function 24 h after the model's establishment. Thus, interactions of CyPA–CD147 likely contribute to cardiomyocyte injury and RV dysfunction following APE.
RV dysfunction is commonly observed in patients with APE and often predicts a poor prognosis. From a clinical perspective, the RV/LV diameter ratio is a good indicator of RV dilation (27), and the RV Tei index, measured by echocardiography, is associated with decreased RV contractile function (28) and mortality (29) following APE. In line with clinical findings, we observed a significantly increased RV/LV diameter ratio and Tei index after the induction of an experimental APE, which indicates the presence of RV dilatation and dysfunction in our models. Also, the level of plasma cTnI in the APE control group also increased significantly, demonstrating cardiomyocyte damage caused by APE. Apart from elevated pulmonary vascular resistance, compelling evidence also suggests that inflammation plays a deterministic role in the development of RV damage and dysfunction following APE (3, 5, 11, 30). However, the specific mechanism(s) that cause the recruitment of inflammatory cells has yet to be fully clarified. Our results confirm recent evidence that following APE, there is an influx of neutrophils into the RV, which results in increased activation of MMP-2 and MMP-9 in the RV. Importantly, we also found that the levels of CyPA and CD147 increased in RV tissue following APE, and inhibition of the interactions between them prevents myocardial injury and RV dilation and dysfunction. These beneficial effects were associated with lower myeloperoxidase activity and decreased MMP-9 and MMP-2 levels, indicating that inflammatory responses in the RV following APE may be modulated by CyPA–CD147 interactions.
Increased expression of CyPA and its interaction with CD147 have been implicated in many human diseases, such as cancer, cardiovascular diseases, and neurological disorders (13). Various cell types have been described as secreting CyPA into the extracellular space, such as leukocytes, endothelial cells (17), vascular smooth muscle cells (22), and activated platelets (31). Also, previous work has demonstrated that cultured rat cardiac myocytes can secrete CyPA under the condition of hypoxia/reoxygenation (32). The results of our study and previous ones indicate that the abrupt rise in pulmonary vascular resistance following APE increases the afterload and enlarges the RV (1). These changes are thought to produce an increased demand for oxygen that exceeds the supply, resulting in functional RV ischemia (1). Mounting evidence also suggests that a mass of reactive oxygen species is generated in RV tissue after APE (6, 11, 30). Based on this evidence, it can be concluded that CyPA in this study was secreted from RV cardiomyocytes under the condition of functional ischemia and increased oxidative stress post-APE.
Results from the first part of the experiment suggest that the levels of CyPA and CD147 in RV tissue and the level of CyPA in plasma increased after APE, and the most significant time point was 24 h post-APE. In the second part of the experiment, CsA attenuated APE-induced increases in CyPA and CD147 levels, and anti-CD147 eliminated the elevation of CD147 in RV but not the elevation of CyPA; treatment with CsA or anti-CD147 or both reduced the APE-induced elevation of cTnI concentrations and preserved RV function 24 h after the model's establishment. Nevertheless, the mechanism underlying this is not clear. Previous studies have shown that CyPA–CD147 interactions could mediate ERK–NF-κB pathways in leukocytes and endothelial cells to stimulate cytokine release, accelerate leukocyte recruitment, and activate MMPs in the pathogenesis of inflammatory diseases (16, 17, 33). Similar results were observed in this study, in which inhibition of CyPA–CD147 reduced p-ERK1/2 and NF-κB activity, decreased neutrophil accumulation, and suppressed MMP-9 and MMP-2 activation in RV tissue. Based on these results, we suggest that the interactions between extracellular CyPA and CD147 activate ERK1/2 and NF-κB, and then increase neutrophil accumulation and MMP activation in RV tissue, thus aggravating RV injury and dysfunction post-APE. Another notable finding of this investigation was that treating animals with a combination of CsA and anti-CD147 seemed to attenuate cardiomyocyte injury and RV dysfunction more obviously than that induced by the individual intervention, indicating that CyPA or CD147 may contribute to post-APE RV injury and dysfunction via other pathways.
There are several limitations to this investigation that warrant discussion. Although CsA can bind tightly to the CyPA ligand and has been reported as the most well-studied inhibitor of CyPA, it is also an immune suppressor. We thus administered another immune suppressor, FK506, to the rats with APE, and we found that FK506 had no effects on cTnI, the morphology and function of RV, or the myeloperoxidase activity of RV tissue. Because both CsA and FK506 exert their immunosuppressive effect by inhibiting the activation of T-cells (34, 35), we thus confirmed that the protective effect of CsA for RV injury observed in our study was attributable to inhibition of CyPA instead of its immune suppressive effects. Besides, CsA binds both to extracellular and intracellular CyPA (13). It is appropriate to test selective inhibitors that directly block the proinflammatory and chemotactic functions of extracellular CyPA in the future. We also did not investigate the mechanism(s) by which inhabitation of CyPA or CD147 ameliorates APE-increased RVSP. Previous studies indicate that inflammation participates in the vasoconstriction of pulmonary vessels, thus aggravating pulmonary hypertension after APE (4, 36), suggesting that CyPA–CD147 may also affect hemodynamic changes by suppressing inflammation. Moreover, it is important to mention that part of the protective effects against the RV dysfunction observed in this study may be a consequence of a reduced RV afterload.
In summary, this study indicates that the inflammatory response induced by the CyPA–CD147–ERK1/2–NF-κB pathway might be partially responsible for RV injury and dysfunction following APE. Inhibition of CyPA–CD147 interactions attenuates the RV damage and dysfunction via an anti-inflammatory mechanism. We thereby provide a novel and promising therapeutic target for RV dysfunction post-APE.
Experimental procedures
All animal experiments were approved by the institutional ethics committee of Nanjing Medical University (ethical code: IACUC-1701004) and were performed by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animals were maintained under a 12-h light/dark cycle at a temperature of 22–25 °C with free access to food and water.
APE model
One hundred and forty nine adult male Sprague-Dawley rats (300–320 g) were purchased from the Beijing Vital River Laboratory of Animal Technology, Beijing, China. After the rats were anesthetized with 10% chloral hydrate (400 mg/kg body weight) by intraperitoneal injection, they were fixed in a supine position on a constant temperature workbench. An intravenous cannula was inserted into the right femoral vein under a stereomicroscope, and as described by previous studies (4, 6), 12 mg/kg of a suspension (40 mg/ml) of microspheres (Sephadex G-50, 300 μm in diameter, Pharmacia Biotech, Freiburg, Germany) was injected into the inferior vena cava to produce the experimental APE model. Sham group animals underwent the same procedure as described, but an equivalent volume of normal saline was injected into the vein instead of microspheres. The animals were allowed to recover for 60 min after APE and then were returned to their cages; the temperature was maintained at 22–25 °C.
In a preliminary experiment, we investigated the effects of various degrees (from 9 to 15 mg/kg microspheres) of embolization. We selected the dose of 12 mg/kg as it produced definite severe APE with an acceptable mortality rate (about 20%). The presence of APE was confirmed by the gross appearance and histological examination (Fig. 1A).
Study design and experimental groups
In the first part of the experiment (Fig. 1B), 36 rats (44 rats were used, and 36 rats survived after the procedure) were randomly divided into six groups, including six rats each, a sham group, and five experimental groups arranged by time: 6, 12, 24, 48, and 72 h after APE. All the animals were euthanized at the above-mentioned post-APE time points. Plasma samples were collected, and CyPA levels were ascertained by Western blotting, and RV tissue was excised and divided into two samples. One RV tissue sample was fixed with 10% formalin and used for immunofluorescence analysis; another sample was stored at −70 °C until used for Western blot analysis.
In the second part of the experiment (Fig. 1C), 84 rats (97 rats were used, and 84 rats survived) were randomly assigned to seven groups, which included 12 rats each: a sham group, a sham + CsA group (10 mg/kg, i.v.), a sham + anti-CD147 group (3 mg/kg, i.v.), an APE control group, an APE + CsA group, an APE + anti-CD147 group, and an APE + CsA + anti-CD147 group. At 24 h post-APE, which was chosen based on the first part of the experiment, morphology and RV function were assessed via echocardiography, and RVSP was measured via catheterization; plasma samples were collected to detect the levels of cTnI; the RV tissue of six rats was extracted for Western blot analysis, quantification of neutrophil accumulation, and gelatin zymography of MMPs; and the RV tissue of the other six rats was used for pathological examination and immunofluorescence analysis.
Because CsA is also an immune suppressor, we administered FK506 in another six APE rats (eight rats were used, and six rats survived) to determine whether immunosuppression had any effects on cTnI, morphology, and RV function, and the myeloperoxidase activity of RV tissue was compared with that of tissue from the group given APE alone.
Drug administration
A 5-ml (250 mg) sterile ampule of CsA (Sandimmune Injection) was purchased from the Jiangsu Province Hospital pharmacy. On the day of the experiment, 1 ml of stock solution was diluted with 9 ml of sterile physiological saline to yield a total volume of 10 ml at 5 mg/ml (1:10 dilution). For the rats treated with CsA, 15 min before surgery, the diluted solution was administered at 2 ml/kg by slow i.v. infusion from the right femoral vein for 5 min (37, 38). The mAb of CD147 (Santa Cruz Biotechnology, Santa Cruz, CA; sc-46700) was used to inhibit CD147. For the rats receiving anti-CD147, immediately after the APE model was established, anti-CD147 was administered by i.v. infusion at a dose of 3 mg/kg, which was determined based on a previous study (21). FK506 was purchased from Selleck Chemicals (Shanghai, China) and was intragastrically administered to rats at dose of 0.2 mg/kg body weight 1 h before the operation (39).
Evaluation of RV using echocardiography
Echocardiographic examinations were performed in six rats from each group 24 h following APE. Following the induction of anesthesia, the animals were placed supine on a heated pad and were connected to the simulated electrocardiogram. With the fur on the chest shaved, a sequential examination of the left and right ventricles was performed using a Vevo2100 (VisualSonics, Canada), with an MS250 sectorial transducer. Measured variables in this study included the RV/LV ratio (RV end-diastolic diameter/LV end-diastolic diameter), the main pulmonary artery diameter, and the RV Tei index (myocardial performance index). The RV Tei index was calculated as described previously: (isovolumic contraction time + isovolumic relaxation time)/RV ejection time (28). Three consecutive measures of all variables were performed and then averaged.
Hemodynamic measurements
The measurement of RVSP was performed in six rats from each group at 24 h after the establishment of the model as described previously (22). Briefly, under anesthesia, the right jugular vein of the rat was exposed, and a 2.7-F microcatheter (inner diameter 0.9 mm; Terumo, Tokyo, Japan), filled with heparinized saline (10 units/ml of heparin in 0.9% saline), was passed via a small transverse cut and then advanced into the RV until a satisfactory RV pressure waveform was obtained. The catheter was connected to a miniature pressure transducer, and RVSP was recorded and averaged for 10 sequential beats by a PowerLab data acquisition system (AD Instruments, Colorado Springs, CO).
Enzyme-linked immunosorbent assay
Arterial blood samples (5 ml) from the left ventricle were collected in tubes containing EDTA and were immediately centrifuged at 3000 rpm at 4 °C for 10 min. Plasma samples were collected and stored at −70 °C. The plasma level of cTnI was analyzed using commercial ELISA kits (Feiya Biotechnology Co. Ltd., Jiangsu, China) according to the manufacturer's instructions, and assays were performed in duplicate.
Myeloperoxidase activity measurement in RV tissue
The extent of neutrophil accumulation in the RV tissue was qualified by assaying myeloperoxidase activity. The operating steps and the reagents used were previously described (5, 6). The results were expressed as the units of enzyme activity/g wet tissue.
Western blot analysis
The RV tissues were weighed, cut into pieces, and homogenized in ice-cold lysis buffer. Western blot analysis was performed as described previously (21). The protein concentrations were quantified using a BCA protein assay kit (Beyotime Institute of Biotechnology, Shanghai, China). Forty micrograms of protein from each sample were loaded onto a 12% SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA), and blocked for 2 h at room temperature in TBS with Tween (TBST) containing 5% skim milk. The membrane was then incubated overnight at 4 °C with primary antibodies. The primary antibodies were CyPA (Abcam, Cambridge, UK), CD147 (Abcam), phospho-ERK1/2 (Affinity Biosciences), total-ERK1/2 (Affinity), and p65 and phospho-p65 (Cell Signaling Technology). The primary antibodies against glyceraldehyde-3-phosphate dehydrogenase (Santa Cruz Biotechnology) were used as a loading control. After washing, the membrane was incubated with a horseradish peroxidase-conjugated secondary antibody (Boster Biological Technology, Ltd., Wuhan, China) for 2 h. The band signal was visualized by enhanced chemiluminescence (ThermoFisher Scientific). Densitometric analysis was performed with ImageJ software (National Institutes of Health).
Immunofluorescence analysis
Immunofluorescence analysis was performed for CyPA to detect its expression in the myocardium. Briefly, the sections of RV tissue were incubated with a primary antibody for CyPA overnight at 4 °C. Secondary antibodies, including IFKineTM green donkey anti-rabbit IgG antibody and IFKineTM green donkey anti-mouse IgG antibody (Abbkine, California), were added for 1 h at 37 °C, and then the samples were washed three times. After the final washing, sections were covered with coverslips with an antifoaming mounting medium containing 4,6-diamino-2-phenylindole (Beyotime Institute of Biotechnology) for 5 min. Images were observed by a fluorescence microscope (Olympus DP73; Olympus Co., Tokyo, Japan). Phosphate-buffered saline (PBS) was used as a negative control for the immunofluorescence assay. Relative fluorescence intensity analysis was performed using ImageJ Software (National Institutes of Health).
Gelatin zymography of MMP-2 and MMP-9
A gelatin zymography was performed to assess MMP-2 and MMP-9 in the RV tissue, as described previously (5, 11). Briefly, supernatants obtained from homogenized RV tissues were subjected to electrophoresis on a 12% SDS-polyacrylamide gel co-polymerized with 1% of gelatin as the substrate. After complete electrophoresis, the gel was incubated for 40 min twice in a 2.5% Triton X-100 solution, washed two times with water, and then incubated at 37 °C for 42 h in Tris-HCl buffer, pH 7.6, containing 5 mmol CaCl2. The gels were stained with 0.05% Coomassie Brilliant Blue R-250 for 3 h and then destained with methanol and acetic acid. Gelatinolytic activities were detected as unstained bands against the background of Coomassie Blue-stained gelatin. Gelatinolytic activities were analyzed by densitometry using ImageJ Software (National Institutes of Health). MMP-2 and MMP-9 were identified as bands at 72 and 92 kDa, respectively.
Statistical analysis
Data were expressed as means ± S.D. Between-group comparisons were performed using one-way analysis of variance followed by a Tukey post hoc test to compare across multiple groups. The SPSS 24.0 software (Chicago, IL) was used for data analysis, and a p value less than 0.05 was considered statistically significant.
Author contributions
G. L., Z. J., and H. S. conceptualization; G. L., Z. J., and J. Z. data curation; G. L., Z. J., and J. Z. software; G. L. and Q. Z. formal analysis; G. L., Z. J., and J. Z. investigation; G. L., Z. J., Q. Z., and J. Z. methodology; G. L., Z. J., Q. Z., and J. Z. writing-original draft; Q. Z. and L. Z. project administration; L. Z. and H. S. supervision; L. Z. and H. S. writing-review and editing; H. S. funding acquisition.
Supplementary Material
Acknowledgments
We thank LetPub for providing linguistic assistance during the preparation of this manuscript.
Note added in proof
In the version of this article that was published as a Paper in Press on June 18, 2018, the wrong set of images was used in Fig 2E, 48 h. This error has now been corrected and does not affect the results or conclusions of this work.
This work was supported by National Natural Science Foundation of China Grant 81271678. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Fig. S1.
- APE
- acute pulmonary embolism
- RV
- right ventricle
- LV
- left ventricle
- cTnI
- cardiac troponin-I
- CyPA
- cyclophilin A
- MMP
- matrix metalloproteinase
- EMMPRIN
- extracellular matrix metalloproteinase inducer
- RVSP
- RV systolic pressure
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- CsA
- cyclosporine A
- mAb
- monoclonal antibody.
References
- 1. Konstantinides S. V., Torbicki A., Agnelli G., Danchin N., Fitzmaurice D., Galiè N., Gibbs J. S., Huisman M. V., Humbert M., Kucher N., Lang I., Lankeit M., Lekakis J., Maack C., Mayer E., et al. (2014) 2014 ESC guidelines on the diagnosis and management of acute pulmonary embolism. Eur. Heart J. 35, 3033–3069, 3069a–3069k 10.1093/eurheartj/ehu283 [DOI] [PubMed] [Google Scholar]
- 2. Konstantinides S. V., Barco S., Lankeit M., and Meyer G. (2016) Management of pulmonary embolism: an update. J. Am. Coll. Cardiol. 67, 976–990 10.1016/j.jacc.2015.11.061 [DOI] [PubMed] [Google Scholar]
- 3. Watts J. A., Zagorski J., Gellar M. A., Stevinson B. G., and Kline J. A. (2006) Cardiac inflammation contributes to right ventricular dysfunction following experimental pulmonary embolism in rats. J. Mol. Cell. Cardiol. 41, 296–307 10.1016/j.yjmcc.2006.05.011 [DOI] [PubMed] [Google Scholar]
- 4. Souza-Costa D. C., Figueiredo-Lopes L., Alves-Filho J. C., Semprini M. C., Gerlach R. F., Cunha F. Q., and Tanus-Santos J. E. (2007) Protective effects of atorvastatin in rat models of acute pulmonary embolism: involvement of matrix metalloproteinase-9. Crit. Care Med. 35, 239–245 10.1097/01.CCM.0000251638.67104.C3 [DOI] [PubMed] [Google Scholar]
- 5. Neto-Neves E. M., Dias-Junior C. A., Rizzi E., Castro M. M., Sonego F., Gerlach R. F., and Tanus-Santos J. E. (2011) Metalloproteinase inhibition protects against cardiomyocyte injury during experimental acute pulmonary thromboembolism. Crit. Care Med. 39, 349–356 10.1097/CCM.0b013e3181fa3dfe [DOI] [PubMed] [Google Scholar]
- 6. Cau S. B., Barato R. C., Celes M. R., Muniz J. J., Rossi M. A., and Tanus-Santos J. E. (2013) Doxycycline prevents acute pulmonary embolism-induced mortality and right ventricular deformation in rats. Cardiovasc. Drugs Ther. 27, 259–267 10.1007/s10557-013-6458-9 [DOI] [PubMed] [Google Scholar]
- 7. Kandasamy A. D., Chow A. K., Ali M. A., and Schulz R. (2010) Matrix metalloproteinase-2 and myocardial oxidative stress injury: beyond the matrix. Cardiovasc. Res. 85, 413–423 10.1093/cvr/cvp268 [DOI] [PubMed] [Google Scholar]
- 8. Lalu M. M., Pasini E., Schulze C. J., Ferrari-Vivaldi M., Ferrari-Vivaldi G., Bachetti T., and Schulz R. (2005) Ischaemia-reperfusion injury activates matrix metalloproteinases in the human heart. Eur. Heart J. 26, 27–35 10.1093/eurheartj/ehi007 [DOI] [PubMed] [Google Scholar]
- 9. Sawicki G., Leon H., Sawicka J., Sariahmetoglu M., Schulze C. J., Scott P. G., Szczesna-Cordary D., and Schulz R. (2005) Degradation of myosin light chain in isolated rat hearts subjected to ischemia-reperfusion injury: a new intracellular target for matrix metalloproteinase-2. Circulation 112, 544–552 10.1161/CIRCULATIONAHA.104.531616 [DOI] [PubMed] [Google Scholar]
- 10. Jones A. E., Watts J. A., Debelak J. P., Thornton L. R., Younger J. G., and Kline J. A. (2003) Inhibition of prostaglandin synthesis during polystyrene microsphere-induced pulmonary embolism in the rat. Am. J. Physiol. Lung Cell. Mol. Physiol. 284, L1072–L1081 10.1152/ajplung.00283.2002 [DOI] [PubMed] [Google Scholar]
- 11. Neto-Neves E. M., Sousa-Santos O., Ferraz K. C., Rizzi E., Ceron C. S., Romano M. M., Gali L. G., Maciel B. C., Schulz R., Gerlach R. F., and Tanus-Santos J. E. (2013) Matrix metalloproteinase inhibition attenuates right ventricular dysfunction and improves responses to dobutamine during acute pulmonary thromboembolism. J. Cell. Mol. Med. 17, 1588–1597 10.1111/jcmm.12163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seizer P., Gawaz M., and May A. E. (2014) Cyclophilin A and EMMPRIN (CD147) in cardiovascular diseases. Cardiovasc. Res. 102, 17–23 10.1093/cvr/cvu035 [DOI] [PubMed] [Google Scholar]
- 13. Nigro P., Pompilio G., and Capogrossi M. C. (2013) Cyclophilin A: a key player for human disease. Cell Death Dis. 4, e888 10.1038/cddis.2013.410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jin Z. G., Melaragno M. G., Liao D. F., Yan C., Haendeler J., Suh Y. A., Lambeth J. D., and Berk B. C. (2000) Cyclophilin A is a secreted growth factor induced by oxidative stress. Circ. Res. 87, 789–796 10.1161/01.RES.87.9.789 [DOI] [PubMed] [Google Scholar]
- 15. Shimokawa H., Sunamura S., and Satoh K. (2016) RhoA/Rho-kinase in the cardiovascular system. Circ. Res. 118, 352–366 10.1161/CIRCRESAHA.115.306532 [DOI] [PubMed] [Google Scholar]
- 16. Dawar F. U., Xiong Y., Khattak M. N. K., Li J., Lin L., and Mei J. (2017) Potential role of cyclophilin A in regulating cytokine secretion. J. Leukoc. Biol. 102, 989–992 10.1189/jlb.3RU0317-090RR [DOI] [PubMed] [Google Scholar]
- 17. Dawar F. U., Wu J., Zhao L., Khattak M. N., Mei J., and Lin L. (2017) Updates in understanding the role of cyclophilin A in leukocyte chemotaxis. J. Leukoc. Biol. 101, 823–826 10.1189/jlb.3RU1116-477R [DOI] [PubMed] [Google Scholar]
- 18. Sun S., Guo M., Zhang J. B., Ha A., Yokoyama K. K., and Chiu R. H. (2014) Cyclophilin A (CypA) interacts with NF-κB subunit, p65/RelA, and contributes to NF-κB activation signaling. PLoS ONE 9, e96211 10.1371/journal.pone.0096211 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19. Malesevic M., Gutknecht D., Prell E., Klein C., Schumann M., Nowak R. A., Simon J. C., Schiene-Fischer C., and Saalbach A. (2013) Anti-inflammatory effects of extracellular cyclosporins are exclusively mediated by CD147. J. Med. Chem. 56, 7302–7311 10.1021/jm4007577 [DOI] [PubMed] [Google Scholar]
- 20. Jin R., Xiao A. Y., Chen R., Granger D. N., and Li G. (2017) Inhibition of CD147 (Cluster of Differentiation 147) ameliorates acute ischemic stroke in mice by reducing thromboinflammation. Stroke 48, 3356–3365 10.1161/STROKEAHA.117.018839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dang B., Li H., Xu X., Shen H., Wang Y., Gao A., He W., Wang Z., and Chen G. (2015) Cyclophilin A/cluster of differentiation 147 interactions participate in early brain injury after subarachnoid hemorrhage in rats. Crit. Care Med. 43, e369–e381 10.1097/CCM.0000000000001146 [DOI] [PubMed] [Google Scholar]
- 22. Satoh K., Satoh T., Kikuchi N., Omura J., Kurosawa R., Suzuki K., Sugimura K., Aoki T., Nochioka K., Tatebe S., Miyamichi-Yamamoto S., Miura M., Shimizu T., Ikeda S., Yaoita N., et al. (2014) Basigin mediates pulmonary hypertension by promoting inflammation and vascular smooth muscle cell proliferation. Circ. Res. 115, 738–750 10.1161/CIRCRESAHA.115.304563 [DOI] [PubMed] [Google Scholar]
- 23. Heinzmann D., Bangert A., Müller A. M., von Ungern-Sternberg S. N., Emschermann F., Schönberger T., Chatterjee M., Mack A. F., Klingel K., Kandolf R., Malesevic M., Borst O., Gawaz M., Langer H. F., Katus H., et al. (2015) The novel extracellular cyclophilin A (CyPA)–inhibitor MM284 reduces myocardial inflammation and remodeling in a mouse model of troponin I-induced myocarditis. PLoS ONE 10, e0124606 10.1371/journal.pone.0124606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seizer P., Ochmann C., Schönberger T., Zach S., Rose M., Borst O., Klingel K., Kandolf R., MacDonald H. R., Nowak R. A., Engelhardt S., Lang F., Gawaz M., and May A. E. (2011) Disrupting the EMMPRIN (CD147)-cyclophilin A interaction reduces infarct size and preserves systolic function after myocardial ischemia and reperfusion. Arterioscler. Thromb. Vasc. Biol. 31, 1377–1386 10.1161/ATVBAHA.111.225771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Piot C., Croisille P., Staat P., Thibault H., Rioufol G., Mewton N., Elbelghiti R., Cung T. T., Bonnefoy E., Angoulvant D., Macia C., Raczka F., Sportouch C., Gahide G., Finet G., et al. (2008) Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 359, 473–481 10.1056/NEJMoa071142 [DOI] [PubMed] [Google Scholar]
- 26. Cuadrado I., Piedras M. J., Herruzo I., Turpin Mdel C., Castejón B., Reventun P., Martin A., Saura M., Zamorano J. L., and Zaragoza C. (2016) EMMPRIN-targeted magnetic nanoparticles for in vivo visualization and regression of acute myocardial infarction. Theranostics 6, 545–557 10.7150/thno.13352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jaff M. R., McMurtry M. S., Archer S. L., Cushman M., Goldenberg N., Goldhaber S. Z., Jenkins J. S., Kline J. A., Michaels A. D., Thistlethwaite P., Vedantham S., White R. J., Zierler B. K., American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation, American Heart Association Council on Peripheral Vascular Disease, American Heart Association Council on Arteriosclerosis, and Thrombosis and Vascular Biology. (2011) Management of massive and submassive pulmonary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonary hypertension: a scientific statement from the American Heart Association. Circulation 123, 1788–1830 10.1161/CIR.0b013e318214914f [DOI] [PubMed] [Google Scholar]
- 28. Park J. H., Park Y. S., Park S. J., Lee J. H., Choi S. W., Jeong J. O., and Seong I. W. (2008) Midventricular peak systolic strain and Tei index of the right ventricle correlated with decreased right ventricular systolic function in patients with acute pulmonary thromboembolism. Int. J. Cardiol. 125, 319–324 10.1016/j.ijcard.2007.02.030 [DOI] [PubMed] [Google Scholar]
- 29. Dahhan T., Siddiqui I., Tapson V. F., Velazquez E. J., Sun S., Davenport C. A., Samad Z., and Rajagopal S. (2016) Clinical and echocardiographic predictors of mortality in acute pulmonary embolism. Cardiovasc. Ultrasound 14, 44 10.1186/s12947-016-0087-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sousa-Santos O., Neto-Neves E. M., Ferraz K. C., Ceron C. S., Rizzi E., Gerlach R. F., and Tanus-Santos J. E. (2012) Antioxidant treatment protects against matrix metalloproteinase activation and cardiomyocyte injury during acute pulmonary thromboembolism. Naunyn Schmiedebergs Arch. Pharmacol. 385, 685–696 10.1007/s00210-012-0748-9 [DOI] [PubMed] [Google Scholar]
- 31. Ramachandran S., Venugopal A., Sathisha K., Reshmi G., Charles S., Divya G., Chandran N. S., Mullassari A., Pillai M. R., and Kartha C. C. (2012) Proteomic profiling of high glucose primed monocytes identifies cyclophilin A as a potential secretory marker of inflammation in type 2 diabetes. Proteomics 12, 2808–2821 10.1002/pmic.201100586 [DOI] [PubMed] [Google Scholar]
- 32. Seko Y., Fujimura T., Taka H., Mineki R., Murayama K., and Nagai R. (2004) Hypoxia followed by reoxygenation induces secretion of cyclophilin A from cultured rat cardiac myocytes. Biochem. Biophys. Res. Commun. 317, 162–168 10.1016/j.bbrc.2004.03.021 [DOI] [PubMed] [Google Scholar]
- 33. Yang Y., Lu N., Zhou J., Chen Z. N., and Zhu P. (2008) Cyclophilin A up-regulates MMP-9 expression and adhesion of monocytes/macrophages via CD147 signalling pathway in rheumatoid arthritis. Rheumatology 47, 1299–1310 10.1093/rheumatology/ken225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Beauchesne P. R., Chung N. S., and Wasan K. M. (2007) Cyclosporine A: a review of current oral and intravenous delivery systems. Drug Dev. Ind. Pharm. 33, 211–220 10.1080/03639040601155665 [DOI] [PubMed] [Google Scholar]
- 35. Kalt D. A. (2017) Tacrolimus: a review of laboratory detection methods and indications for use. Lab. Med. 48, e62–e65 10.1093/labmed/lmx056 [DOI] [PubMed] [Google Scholar]
- 36. Fortuna G. M., Figueiredo-Lopes L., Dias-Junior C. A., Gerlach R. F., and Tanus-Santos J. E. (2007) A role for matrix metalloproteinase-9 in the hemodynamic changes following acute pulmonary embolism. Int. J. Cardiol. 114, 22–27 10.1016/j.ijcard.2005.11.109 [DOI] [PubMed] [Google Scholar]
- 37. Dixon C. E., Bramlett H. M., Dietrich W. D., Shear D. A., Yan H. Q., Deng-Bryant Y., Mondello S., Wang K. K., Hayes R. L., Empey P. E., Povlishock J. T., Tortella F. C., and Kochanek P. M. (2016) Cyclosporine treatment in traumatic brain injury: operation brain trauma Therapy. J. Neurotrauma 33, 553–566 10.1089/neu.2015.4122 [DOI] [PubMed] [Google Scholar]
- 38. Dai Y., Sun Q., Zhang X., Hu Y., Zhou M., and Shi J. (2014) Cyclosporin A ameliorates early brain injury after subarachnoid hemorrhage through inhibition of a Nur77 dependent apoptosis pathway. Brain Res. 1556, 67–76 10.1016/j.brainres.2014.01.052 [DOI] [PubMed] [Google Scholar]
- 39. Zhang W., Li F., Ye Y., Liu Y., Yu S., Cen C., Chen X., Zhou L., Tang X., Yu J., and Zheng S. (2017) Isoglycyrrhizinate magnesium enhances hepatoprotective effect of FK506 on ischemia-reperfusion injury through HMGB1 inhibition in a rat model of liver transplantation. Transplantation 101, 2862–2872 10.1097/TP.0000000000001941 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.