

Abstract
The unfolded protein response (UPR) is constitutively active in yeast thioredoxin reductase mutants, suggesting a link between cytoplasmic thiol redox control and endoplasmic reticulum (ER) oxidative protein folding. The unique oxidative environment of the ER lumen requires tight regulatory control, and we show that the active UPR depends on the presence of oxidized thioredoxins rather than arising because of a loss of thioredoxin function. Preventing activation of the UPR by deletion of HAC1, encoding the UPR transcription factor, rescues a number of thioredoxin reductase mutant phenotypes, including slow growth, shortened longevity, and oxidation of the cytoplasmic GSH pool. This is because the constitutive UPR in a thioredoxin reductase mutant results in the generation of hydrogen peroxide. The oxidation of thioredoxins in a thioredoxin reductase mutant requires aerobic metabolism and the presence of the Tsa1 and Tsa2 peroxiredoxins, indicating that a complete cytoplasmic thioredoxin system is crucial for maintaining ER redox homeostasis.
Keywords: endoplasmic reticulum stress (ER stress), thioredoxin, peroxiredoxin, reactive oxygen species (ROS), redox regulation
Introduction
Thioredoxins are small, ubiquitous thiol proteins and are one of the most important regulators of cellular redox homeostasis (1, 2). In the thioredoxin system, thioredoxin reductase uses the reducing potential of NADPH to maintain thioredoxins in a reduced state so that thioredoxins can, in turn, reduce diverse substrate proteins that form a disulfide as part of their catalytic cycle. Thioredoxins contain two redox-active cysteine residues as part of a conserved CXXC motif that catalyzes the conversion of disulfide bonds in substrate proteins into thiols with high efficiency. This includes substrates that play diverse roles in numerous cellular processes, including cell proliferation, antioxidant defenses, metabolic regulation, and redox signaling (1, 2).
Yeast, like most eukaryotes, contains a complete cytoplasmic thioredoxin system, comprising two thioredoxins (Trx1 and Trx2) and a thioredoxin reductase (Trr1) (3, 4). Trx1 and Trx2 are highly homologous proteins containing the conserved dithiol active site sequence (Cys-Gly-Pro-Cys) and have similar redox potentials (5, 6). However, Trx2 appears to be more important as an antioxidant because strains deleted for TRX2 are sensitive to hydrogen peroxide (7), whereas strains deleted for TRX1 are unaffected in oxidant sensitivity (8). This difference in phenotype arises because of differences in the regulation of TRX1 and TRX2 gene expression, as overexpression of either isoform similarly increases resistance to oxidative stress (8). In agreement with this idea, TRX2 is a target of the Yap1 transcription factor, which is the major regulator of oxidative stress–related genes in Saccharomyces cerevisiae, and its expression is up-regulated in a Yap1-dependent manner following ROS3 exposure (7). In contrast, TRX1 gene expression is largely unaffected in response to oxidative stress conditions (8).
The main antioxidant function of the thioredoxin system is mediated through its activity with the peroxiredoxin family of proteins. Peroxiredoxins are thiol-specific proteins that have multiple functions in stress protection (9). Peroxiredoxins play key roles in redox signaling, acting as both hydrogen peroxide sensors and thiol oxidizers (10). Protein thiol oxidation and reduction are increasingly recognized as major reversible posttranslational modifications that regulate protein activity (11). Cysteine residues react relatively slowly with H2O2, but reactivity can be significantly enhanced by their ionization state, which depends on the local protein environment. This means that some redox-regulated proteins are directly oxidized by ROS, although it is thought that most regulatory thiol oxidation is mediated by protein catalysts such as peroxiredoxins (11, 12). Tsa1 is the major cytoplasmic 2-Cys peroxiredoxin in yeast and was originally characterized as an antioxidant in the detoxification of hydroperoxides (8, 13). The Tsa2 peroxiredoxin is highly homologous to Tsa1 (86% amino acid identity) and possesses similar peroxidase activity, although it is normally expressed at significantly lower levels compared with Tsa1 (13, 14).
Thioredoxin reductase is a member of the flavoprotein family of pyridine nucleotide disulfide oxidoreductases, which includes GSH reductase (15). It promotes catalysis via FAD and a redox-active disulfide and is the only known reductant for thioredoxin. Not surprisingly, therefore, yeast mutants lacking TRR1 are unable to recycle oxidized thioredoxins to their reduced forms and accumulate oxidized thioredoxins (16). Yeast trr1 mutants are sensitive to oxidative stress, presumably reflecting the requirement for the thioredoxin system to reduce oxidized peroxiredoxins (17, 18). One unexpected consequence of the loss of TRR1 is activation of the unfolded protein response (UPR) (19). The UPR is a signaling pathway that controls ER homeostasis and protein folding in eukaryotic cells. It transcriptionally regulates genes affecting multiple ER and secretory functions (20). An ER-localized kinase, Ire1, senses the accumulation of unfolded proteins and acts as a specific endoribonuclease in splicing the HAC1 mRNA. The translation product of spliced HAC1 mRNA then acts as a transcriptional activator for genes that contain an upstream UPR element.
In this study, we analyzed the physiological consequences of the constitutively high UPR in mutants lacking thioredoxin reductase. We show that the UPR generates ROS in a trr1 mutant and that ROS generation accounts for the well-known phenotypes that are displayed by thioredoxin reductase mutants, including slow growth and shortened chronological life span. Furthermore, UPR-generated ROS activate the Yap1 transcription factor, and Yap1 is required for ROS tolerance under conditions of ER stress.
Results
Oxidized thioredoxins activate the UPR
The UPR is a stress signaling pathway that is activated in response to an accumulation of unfolded proteins in the ER. It is therefore not clear why the UPR is activated in a trr1 mutant that lacks cytoplasmic Trr1 (16). We originally reported that deleting IRE1 in a trr1 mutant causes a slow growth phenotype (19). However, we subsequently found that the trr1 ire1 mutant had become a respiration-incompetent petite mutant, raising the concern that the trr1 ire1 mutant may have undergone other mutation events, accounting for its slow growth. Therefore, in this study, we constructed new trr1 ire1 and trr1 hac1 mutants, which were maintained as frozen stocks, and fresh isolates were routinely used in all experiments to avoid any subsequent reversion events. We first confirmed that the constitutively high UPR in a trr1 mutant depends on Ire1 and Hac1 using a UPR-lacZ reporter construct. As expected, deletion of HAC1 or IRE1 in a trr1 mutant abrogated the high UPR, confirming that the elevated UPR depends on its canonical regulators (Fig. 1A).
Figure 1.
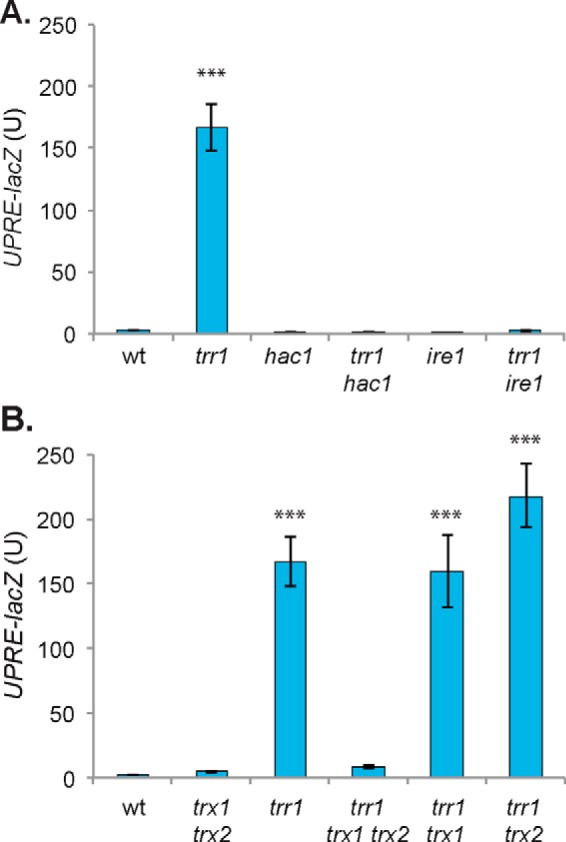
Oxidized thioredoxins activate the UPR. A and B, the UPR was assayed in WT, trr1, hac1, trr1 hac1, ire1, and trr1 ire (A) or WT, trx1 trx2, trr1, trr1 trx1 trx2, trr1 trx1, and trr1 trx2 (B) mutant strains containing a UPRE::lacZ fusion construct. β-Gal activity data are shown for exponential-phase calls grown in minimal SD medium and are the means of three independent biological repeat experiments ± S.D. Significance is shown compared with the WT strain (***, p < 0.001).
We next examined the UPR in thioredoxin mutants to determine whether the increased UPR arises because of loss of thioredoxin function per se or, alternatively, whether it is activated by oxidized thioredoxins. The UPR was modestly increased in a mutant deleted for both TRX1 and TRX2 but significantly lower than in a trr1 mutant. Interestingly however, deleting TRX1 and TRX2 in a trr1 mutant (trr1 trx1 trx2) abrogated its constitutively high UPR, consistent with thioredoxins, presumably in an oxidized form, activating the UPR (Fig. 1B). We found that the UPR was still elevated in trr1 trx1 and trr1 trx2 mutants, confirming that the presence of either Trx1 or Trx2 alone is sufficient to activate the UPR (Fig. 1B). Previous studies have shown that oxidized forms of Trx1 and Trx2 are present in trr1 trx2 and trr1 trx1 mutants, respectively (21).
The constitutive UPR in a trr1 mutant induces the Yap1 response
One well-established role for thioredoxins is in reduction of the Yap1 transcription factor. Yap1 is a redox-sensitive bZip transcription factor that is the main transcriptional regulator of the oxidative stress response in yeast (22). It is rapidly activated by oxidation in response to H2O2, mediated by peroxiredoxins (Gpx3/Orp1 and Tsa1) in a thiol-based relay system (23, 24). Switching off the Yap1 transcription factor by reduction is thought to be mediated by thioredoxins; thioredoxin mutants display constitutively high Yap1 activity, consistent with a requirement for thioredoxins to reduce oxidized Yap1 (25, 26). We tested whether oxidized thioredoxins activate the Yap1 response similar to the UPR. However, Yap1 activity measured using a Yap1-responsive reporter construct (YRE-lacZ) was similarly elevated in trr1 and trr1 trx1 trx2 mutants (Fig. 2A). This suggests that, in contrast to the UPR, it is the loss of thioredoxin-reducing activity that results in activation of the Yap1 transcription factor rather than being induced in response to oxidized thioredoxins.
Figure 2.

The constitutive UPR in a trr1 mutant generates ROS and induces the Yap1 response. A, the Yap1 response was assayed in WT, trr1, trx1 trx2, trr1 trx1 trx2, hac1, and trr1 hac1 mutant strains containing a YRE::lacZ fusion construct. β-Gal activity data are shown for exponential-phase calls grown in minimal SD medium and are the means of three independent biological repeat experiments ± S.D. B, hydrogen peroxide levels were measured using a peroxiredoxin-based roGFP2-Tsa2ΔCR H2O2 probe in the indicated strains. The degree of sensor OxD is shown with OxD = 1 for the fully oxidized probe and OxD = 0 for the fully reduced probe. Data shown are the means of three independent biological repeat experiments ± S.D. C, ROS generation was measured in the indicated strains using DHR123. D, cytosolic GSH redox potentials were measured in strains using a roGFP2 fluorescent probe that equilibrates with the local GSH pool. Data shown are the means of at least three independent biological repeat experiments ± S.D. E, the Yap1 response is induced in response to ER stress. YRE::lacZ activity was measured in the WT and yap1 mutant strains treated with 1 μg/ml Tm for 2 h. Data shown are the means of three independent biological repeat experiments ± S.D. F, mutants deleted for YAP1 are sensitive to ER stress induced by Tm exposure. Strains were grown to stationary phase, and the A600 was adjusted to 1, 0.1, 0.01, or 0.001 before spotting onto SD plates in the presence or absence of 0.4 μg/ml tunicamycin under aerobic or anaerobic growth conditions. Significance is shown compared with the WT strain: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Surprisingly, we found that the constitutively high Yap1 response observed in a trr1 mutant was abrogated by deletion of HAC1 (Fig. 2A). The UPR has previously been implicated in ROS generation via the process of disulfide bond formation (27), raising the possibility that Yap1 is activated in a trr1 mutant as an indirect effect of ROS generation. To test this possibility, we used the peroxiredoxin-based roGFP2-Tsa2ΔCR H2O2 probe (28) to measure whether the high UPR in a trr1 mutant generates ROS. Hydrogen peroxide was elevated in the trr1 mutant compared with a WT strain (Fig. 2B). This increase in hydrogen peroxide was unaffected by deletion of TRX1 and TRX2, whereas it was decreased by deletion of HAC1, consistent with the UPR generating ROS. Given that the roGFP2-Tsa2ΔCR probe is a peroxiredoxin-based system, as well as measuring H2O2 levels, it would also be influenced by the overall reducing power of the thioredoxin system. Although this would be unlikely to explain the decrease in roGFP2-Tsa2ΔCR activity in the trr1 hac1 mutant compared with a trr1 mutant, it may complicate the interpretation of the trr1, trx1 trx2, and trr1 trx1 trx2 mutant strains. We therefore measured ROS generation using dihydrorhodamine 123 (DHR123), which is widely used for in vivo detection of ROS (29, 30). Oxidation of DHR123 converts it into rhodamine, which emits a detectable fluorescent signal. A similar pattern of ROS generation data was obtained using DHR123 compared with the roGFP2-Tsa2ΔCR probe, further confirming that deletion of HAC1 abrogates ROS generation in a trr1 mutant (Fig. 2C).
To further test the consequences of the constitutive UPR in a trr1 mutant, we used a roGFP2 fluorescent probe to quantify the GSH cytosolic redox potential (31). The roGFP2 fluorescent probe equilibrates with the local GSH pool and registers thiol redox changes via disulfide bond formation. We calculated the degree of oxidation (OxD) (32, 33) and found that the OxD value was significantly increased in trr1, trx1 trx2, and trr1 trx1 trx2 mutants compared with the WT strain, as might be expected given the established links between the thioredoxin and GSH systems (16). Deletion of HAC1 in trr1 shifted its OxD value to a more reducing state, indicating that the constitutive UPR in a trr1 mutant causes GSH oxidation (Fig. 2D).
Given that Yap1 activity was induced in a trr1 mutant, dependent on the UPR, we examined whether Yap1 is also required for tolerance to ER stress. ER stress can readily be induced by chemicals such as the cytotoxic drug tunicamycin (Tm), which inhibits N-linked glycosylation (34, 35). We first confirmed that ER stress caused by tunicamycin could induce the Yap1 response. Exposure of exponential-phase cells to Tm resulted in an ∼2-fold induction of Yap1 activity, measured using a Yap1-responsive reporter construct (Fig. 2E). This induction depended on Yap1 because it was absent in a yap1 mutant. Furthermore, a mutant deleted for YAP1 was sensitive to growth in the presence of tunicamycin, confirming the requirement for Yap1 to respond to ER stress (Fig. 2F). If ROS generation accounts for this sensitivity, we reasoned, then anaerobic growth conditions in the absence of oxygen should rescue growth. In agreement with this idea, anaerobic growth conditions strongly rescued the tunicamycin sensitivity of a yap1 mutant (Fig. 2F).
Slow growth and decreased longevity in a trr1 mutant is caused by the constitutive UPR
Given that the constitutively high UPR generates ROS in a trr1 mutant, we examined whether ROS generation causes the slow growth normally observed in a trr1 mutant. Mutants lacking thioredoxin reductase are very slow-growing, which is somewhat surprising given that a significant proportion of the thioredoxin pool is still present in a reduced state in a trr1 mutant (16). However, thioredoxin reductase mutants are sensitive to oxidative stress conditions (17, 18, 36), and hence the ROS generated by the constitutively active UPR may account for this slow growth phenotype. We compared the growth rates of mutant strains, and, as expected, a trr1 mutant grew very slowly (doubling time, 681 min) compared with a WT strain (doubling time, 107 min) (Fig. 3A). Abrogating the UPR by deletion of HAC1 or IRE1 restored the doubling time of a trr1 mutant, similar to that of a WT strain (Fig. 3A). To further examine the dependence of the slow growth phenotype observed in a trr1 mutant on its constitute UPR, we examined the growth phenotype of thioredoxin mutants. Deletion of TRX1 or TRX2 alone in a trr1 mutant did not affect its growth rate, whereas simultaneous loss of TRX1 and TRX2 improved the slow growth of a trr1 mutant (Fig. 3A), consistent with the constitutively high UPR causing the slow growth of a trr1 mutant rather than loss of thioredoxin function.
Figure 3.
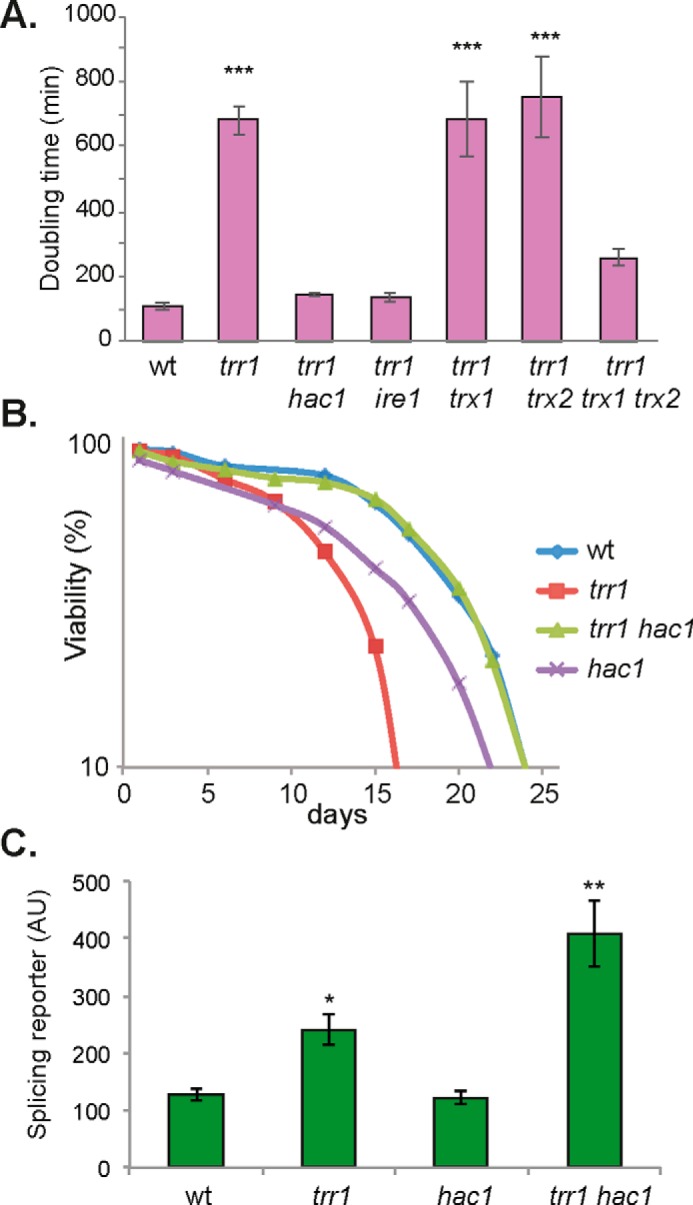
The constitutive UPR causes slow growth and shortened longevity in a thioredoxin reductase mutant. A, the growth rate (doubling time) of the indicated strains was determined in YEPD medium. Data shown are the means of three independent biological repeat experiments ± S.D. B, the chronological life span of the indicated strains was determined by propidium iodide staining and flow cytometry analysis. Data represent the percentage of live cells in stationary-phase cultures relative to day 0. C, Ire1 activity was monitored using an SR where the HAC1 ORF has been replaced with GFP (40). Fluorescence was measured by flow cytometry. Significance is shown compared with the WT strain: *, p < 0.05; **, p < 0.01; ***, p < 0.005. AU, arbitrary units.
An additional phenotype we thought might be explained by ROS generation is shortened longevity in a trr1 mutant. Yeast cells can survive for prolonged periods of time in culture and have been used as a model of the chronological life span (CLS) of mammals, particularly for tissues composed of nondividing populations (37, 38). In the CLS model, populations of stationary phase cells are maintained in liquid, and viability is measured over time. Using this model, yeast cells were shown to undergo a global redox collapse during CLS that is caused by decreased cellular NADPH concentrations and precedes cell death (39). Thioredoxin reductase was identified as a key protein that becomes oxidized prior to the general redox collapse, but its role in chronological aging was not directly examined. When we measured the yeast CLS, we found that a trr1 mutant has a shortened life span compared with a WT strain, and this was rescued by deletion of HAC1 (Fig. 3B). These data are consistent with the UPR accounting for the shortened chronological lifespan of a trr1 mutant, presumably because of ROS accumulation.
Our data indicate that deletion of HAC1 in a trr1 mutant rescues its slow growth and shortened longevity. We confirmed that it is the loss of the UPR itself that accounts for these phenotypes rather than any indirect effect that might arise from alterations in protein misfolding in the ER. We tested this using a splicing reporter (SR) where the HAC1 ORF has been replaced with GFP (40). This allows Ire1 activity to be monitored because it splices the Hac1 intron to derepress GFP expression. Higher levels of SR activation were observed in a trr1 mutant compared with a WT strain (Fig. 3C). Deletion of HAC1 did not affect this increased SR activation, indicating that ER stress still occurs in a trr1 hac1 mutant. The splicing reporter assay confirms that protein misfolding in the ER (ER stress) is still occurring in the trr1 hac1 mutant strain, but as Hac1 is absent, the ROS produced as a consequence of the constitutively activated UPR are also absent. This highlights again that what is detrimental for the trr1 mutant is not the protein misfolding in the ER but the inability to detoxify the ROS produced by the constitutively active UPR.
The Tsa1 and Tsa2 peroxiredoxins are required for thioredoxin oxidation in a trr1 mutant
Recent studies have shown that peroxiredoxins propagate hydrogen peroxide signaling by driving the oxidation of thioredoxins (41, 42). We therefore tested the requirement for peroxiredoxins in oxidizing thioredoxins and activating the UPR in a trr1 mutant. Tsa1 and Tsa2 are the two cytoplasmic 2-Cys peroxiredoxins in yeast, although Tsa2 is normally expressed at significantly lower levels compared with Tsa1 (13, 14). Deletion of TSA1 and TSA2 in a trr1 mutant reduced the UPR by more than 7-fold (Fig. 4A). Surprisingly however, this reduction depended more on the loss of TSA2, which reduced the activity of the UPR by ∼3-fold in a trr1 mutant, compared with the loss of TSA1, which did not alter the UPR in a trr1 mutant (Fig. 4A). The extent of the UPR also correlated with the growth rate of trr1 mutants because deletion of TSA2 rescued the slow growth of a trr1 mutant compared with deletion of TSA1 (Fig. 4B).
Figure 4.

The Tsa1 and Tsa2 peroxiredoxins are required for thioredoxin oxidation in a trr1 mutant. A, the UPR was assayed in WT, trr1, trr1 tsa1, trr1 tsa2, and trr1 tsa1 tsa2 mutant strains using a UPRE::lacZ fusion construct. β-Gal activity data are shown for exponential-phase cells and are the means of three independent biological repeat experiments ± S.D. B, the growth rate (doubling time) of the indicated strains was determined in YEPD medium. Data shown are the means of three independent biological repeat experiments ± S.D. C, redox state of thioredoxins. Proteins were precipitated with TCA and free thiols modified by reaction with AMS. Blots were probed with antibodies specific for Trx1 and Trx2 (Trx1/Trx2) or Trx2 (Trx2), and Pgk1 was included as a loading control. Fully oxidized and fully reduced proteins are indicated. Quantification of the oxidized to reduced form is shown where the oxidized form is detected. D, Western blot analysis of Tsa1 and Tsa2 in trr1 mutant strains. Proteins from the indicated strains were separated using reducing or nonreducing SDS-PAGE as indicated. Blots were probed with antibodies specific for HA (Tsa2) or Tsa1, and Pab1 was included as a loading control. Significance is shown compared with the WT strain: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To confirm that the changes in the activation of the UPR are mediated by thioredoxin oxidation, the redox state of Trx1 and Trx2 was measured in Prx mutant strains (Fig. 4C). We have previously described a rabbit anti-yeast thioredoxin antibody generated using purified Trx1 that recognizes both Trx1 and Trx2 (16) and an antibody generated using purified Trx2 that is specific for Trx2 (43). Trx1 is present at higher cellular concentrations compared with Trx2 in a WT strain (Fig. 4C). Loss of TRR1 elevated cellular thioredoxin concentrations, which predominantly appeared to arise because of increased concentrations of Trx2. The thioredoxin redox state was examined using the thiol-specific reagent 4-acetamido-4′maleimidyldystilbene-2,2′ disulfonic acid (AMS), which alkylates cysteine residues in a free -SH but not in an oxidized state (16, 36). Loss of Tsa1 and Tsa2 shifted the redox state of thioredoxins to a more reduced form, suggesting that Tsa1 and Tsa2 are required for Trx oxidation in a trr1 mutant.
Because the Tsa1/Tsa2 peroxiredoxins appear to be important for the oxidation of the Trx1/Trx2 thioredoxins, we next examined the protein levels of Tsa1 and Tsa2 in trr1 mutants (Fig. 4D). We found that Tsa2 is expressed at much lower levels than Tsa1 in a WT strain, in agreement with previous observations (14). Tsa1 and Tsa2 are both targets of the Yap1 transcription factor and were expressed at higher levels in a trr1 mutant, consistent with its constitutive activation of Yap1 (Fig. 4D). This increase in protein levels was most apparent for Tsa2, which was barely detectable in a WT strain and strongly increased in the trr1 mutant strain. This dramatic increase in the levels of Tsa2 in a trr1 mutant may explain why loss of TSA2 has a stronger effect on the UPR and growth rate in a trr1 mutant compared with loss of TSA1. Such an overexpression of Tsa2, in combination with the already high levels of Tsa1, requires a large amount of reducing power from the thioredoxin system for the recycling of peroxiredoxins, which may be detrimental in a trr1 mutant, where thioredoxins cannot be reduced. We also compared the relative levels of Tsa1 and Tsa2 that were present in dimeric (oxidized) versus monomeric (reduced) forms. Tsa1 and Tsa2 are typical 2-Cys peroxiredoxins that are active as a dimer. During catalysis, the peroxidatic cysteine residue of one subunit is oxidized to a sulfenic acid that condenses with the resolving cysteine from the other subunit to form a disulfide that is reduced by thioredoxin (9). Loss of TSA1 or TSA2 in a trr1 mutant did not alter the relative proportions of monomeric or dimeric Tsa1 or Tsa2 present in a trr1 tsa2 or trr1 tsa1 mutant, respectively. Tsa1 and Tsa2 are predominantly present in the oxidized dimeric form, even in a WT strain, and the monomeric form is increased most in a trr1 mutant. Thus, it may be interesting in future studies to examine the redox status of cysteine residues (Tsa1/Tsa2 have two Cys residues) that are not involved in the structural disulfide bond maintaining the dimeric forms of Tsa1 and Tsa2 to determine whether their oxidation state correlates with the oxidation state of the Trx1/2 thioredoxins and activation of the UPR.
Mitochondrial ROS oxidize thioredoxins to activate the UPR
Although a trr1 mutant lacks thioredoxin reductase activity, it is still able to synthesize thioredoxins that are made in a reduced form. We therefore questioned what might cause the appearance of oxidized thioredoxins in a trr1 mutant. The UPR was first measured under reduced oxygen conditions to confirm that the constitutive UPR requires aerobic metabolism. Anaerobic growth reduced the high UPR observed in a trr1 mutant, indicating that the elevated UPR depends on oxidizing conditions (Fig. 5A). Mitochondrial respiration is thought to provide a main source of ROS in eukaryotic cells via the process of oxidative phosphorylation (44). We measured the UPR in mutants lacking mitochondrial function, [rho0], and found that the UPR was also significantly decreased in a [rho0] version of the trr1 mutant, consistent with mitochondrially derived ROS oxidizing thioredoxins (Fig. 5A). Thioredoxin-specific antibodies were again used to confirm that the oxidation state of thioredoxins is altered by mitochondrial function. This analysis confirmed that the redox balance of both Trx1 and Trx2 was shifted to a more reduced state in the [rho0] version of the trr1 mutant (Fig. 5B).
Figure 5.

Mitochondrial ROS oxidize thioredoxins to activate the UPR. A, the UPR was assayed in WT and trr1 mutant strains using a UPRE::lacZ fusion construct. Data are shown for strains grown under aerobic or anaerobic conditions and for [rho0] mutants. β-Gal activity data are shown for exponential-phase calls and are the means of three independent biological repeat experiments ± S.D. Significance is shown compared with the WT strain: ***, p < 0.001. B, redox state of thioredoxins. Proteins were precipitated with TCA, and free thiols were modified by reaction with AMS. Blots were probed with antibodies specific for Trx1 and Trx2 (Trx1/Trx2) or Trx2 (Trx2), and Pgk1 was included as a loading control. Fully oxidized and fully reduced proteins are indicated. Quantification of the oxidized to reduced form is shown where the oxidized form is detected. C, strains deleted for TRR1 are resistant to tunicamycin. WT, [rho0], trr1, [rho0] trr1, trx1 trx2, [rho0] trx1 trx2, trr1 trx1 trx2, and [rho0] trr1 trx1 trx2 mutant strains were grown to stationary phase and adjusted to an A600 of 1.0, 0.1, and 0.001 before spotting onto SD plates containing 1.2 μg/ml Tm. D, the Yap1-response in a trr1 mutant is reduced in a [rho0] version. YRE::lacZ activity was measured in the WT, [rho0], trr1, and [rho0] trr1 strains. Data shown are the means of three independent biological repeat experiments ± S.D. Significance is shown compared with the WT strain and comparing the trr1 with [rho0] trr1 strains: ***, p < 0.001. E, Western blot analysis of Tsa1 in [rho0] mutants. Proteins from the indicated strains were separated using reducing or nonreducing SDS-PAGE as indicated. Blots were probed with antibodies specific for Tsa1, and Pab1 was included as a loading control.
Mutants deleted for TRR1 are resistant to tunicamycin, which is thought to arise because of their constitutive UPR (19). Because loss of mitochondrial function abrogated the UPR in a trr1 mutant, we reasoned that a [rho0] version of the trr1 mutant should also become tunicamycin-sensitive. This was indeed the case, and a [rho0] trr1 mutant was more sensitive to tunicamycin compared with its isogenic respiration-competent version (Fig. 5C). We also measured Yap1 activation in the [rho0] trr1 mutant. Consistent with the more reduced state of Trx1 and Trx2 in the [rho0] version of the trr1 mutant, significantly less activation of Yap1 was detected compared with the respiration-competent trr1 mutant strain (Fig. 5D).
Finally, we tested whether we could detect any alterations in the protein levels of Tsa1 in the [rho0] version of the trr1 mutant. There was no decrease in the cellular concentrations of Tsa1 in the [rho0] version of the trr1 mutant, which may not be surprising because the Yap1 response is still significantly activated in a [rho0] trr1 mutant compared with the WT strain (Fig. 5E). No differences in the relative proportions of Tsa1 present in its dimeric versus its monomeric forms were detected in a respiration-competent versus a respiration-incompetent trr1 mutant.
Discussion
Protein homeostasis in the ER represents a fine balance between the synthesis, folding/refolding, and degradation of damaged or misfolded proteins. An accumulation of misfolded proteins triggers the highly conserved UPR, which alters the expression of many genes involved in ER quality control. In yeast, the UPR transcriptionally regulates many genes affecting multiple ER and secretory functions (20, 45, 46). An ER-localized kinase, Ire1, senses the accumulation of unfolded proteins and acts as a specific endoribonuclease in splicing the HAC1 mRNA. The translation product of the spliced HAC1 mRNA then acts as a transcriptional activator for genes that contain an upstream UPR element. The unique oxidative environment of the ER lumen requires tight regulatory control, and the finding that the UPR is constitutively activated in yeast thioredoxin reductase mutants suggests a link between cytoplasmic thiol redox control and ER oxidative protein folding.
The elevated UPR observed in a trr1 mutant indicates that cytoplasmic thioredoxin reductase is required to maintain oxidative protein folding in the ER. However, thioredoxin activity itself is not required to maintain ER homeostasis because the UPR is not significantly altered in a strain lacking cytoplasmic thioredoxins. It is the presence of oxidized thioredoxins in a trr1 mutant that appears to mediate the cross-talk between the cytoplasmic and ER redox-regulatory systems, resulting in activation of the UPR. Our data show that the UPR is increased in response to the formation of oxidized thioredoxins because it can be prevented by growth under anaerobic conditions and in [rho0] and peroxiredoxin mutants, conditions that prevent oxidized thioredoxins from forming in thioredoxin reductase mutants. Given the cytoplasmic localization of thioredoxins, it seems unlikely that they can directly participate in protein folding in the ER, raising the question of how the UPR is activated in thioredoxin mutants.
The thioredoxin and GSH systems represent the major cellular redox-regulatory systems and are thermodynamically linked because each uses NADPH as a source of reducing equivalents (47). Loss of the thioredoxin system results in elevated levels of both GSH and GSSG, indicating a link between the thioredoxin system and GSH homeostasis in cells (8, 48). GSH provides a buffer against hyperoxidizing conditions in the ER and may function in the reductive pathway for the formation of native disulfide bonds (49, 50). It also competes for oxidizing equivalents in the ER because elevated GSH disrupts ER function and activates the UPR because of an accumulation of misfolded proteins (49). However, the elevated UPR in a trr1 mutant does not appear to arise as an indirect consequence of GSH alterations in a trr1 mutant because loss of GLR1, encoding GSH reductase, significantly increases the cellular concentrations of oxidized GSH but does not affect the UPR (19). This suggests that the elevated UPR is a direct response to oxidized thioredoxins.
In eukaryotic cells, disulfide bonds are formed in nascent proteins within the lumen of the ER and are essential for the folding of proteins that are secreted or localized through the secretory pathway. Genetic evidence has identified a pathway for disulfide formation where oxidizing equivalents are transferred between thiol-containing proteins and secretory proteins (51, 52). The formation of disulfide bonds is catalyzed by the Pdi1 family of proteins in a redox relay with Ero1 and oxygen acting as a terminal electron acceptor (53). To attain their final native structures, nonnative disulfide bonds must be reduced and isomerized (54). In mammalian cells, cytosolic thioredoxin reductase has been implicated in a pathway transferring reducing equivalents from the cytosol to the ER, which provides the reducing power for disulfide isomerization (55). This may therefore provide the link between the yeast cytosolic thioredoxin reductase and the UPR. One possibility is that an unknown redox-regulatory protein in the ER membrane may act to mediate the cross-talk between cytoplasmic thioredoxins and the lumen of the ER (Fig. 6). In this model, thioredoxins would reduce this unknown factor to provide reducing equivalents for disulfide bond isomerization. Oxidation of the cytoplasmic thioredoxin pool would oxidize this factor, preventing the resolution of non-native disulfide bonds, promoting protein misfolding and activation of the UPR. This may explain why simple loss of thioredoxin activity does not influence ER homeostasis, similar to thioredoxin oxidation, because loss of thioredoxin activity would not actively oxidize this unknown factor. More work will be required to identify the factors that mediate the cross-talk between the cytoplasmic thioredoxin system and the ER and why they are crucial for maintaining ER redox homeostasis.
Figure 6.

Graphical representation of possible models via which cytoplasmic thioredoxin reductase is required for correct disulfide bond formation in the ER. In the absence of Trr1, oxidized thioredoxins form in the cytosol and cause misregulation of the cross-talk between the cytoplasmic and ER redox-regulatory systems, leading to up-regulation of the UPR via activation of Ire1. Given the cytoplasmic localization of thioredoxins, it seems unlikely that they can directly participate in protein folding in the ER, and hence an unknown protein (X) with the ability to transfer reducing equivalents across the ER membrane may mediate this cross-talk. In the absence of thioredoxin reductase, oxidized thioredoxins cannot fully reduce protein X, leading to its inability to transfer reducing equivalents across the membrane, and protein misfolding occurs in the ER lumen, which leads to a constitutively active UPR. The constitutively active UPR results in ROS generation, which causes slow growth, shortened longevity, and oxidation of the cytoplasmic GSH pool in mutants with compromised thioredoxin reductase activity. The production of ER-derived ROS activates the Yap1 system to protect it against damaging effects. The Tsa1 and Tsa2 peroxiredoxins are required to oxidize thioredoxins in response to ROS generated because of ER protein misfolding. This also highlights a possible role for peroxiredoxins in mediating the oxidation of protein X or other proteins that maintain correct ER redox homeostasis.
The ER has long been known to act as a potential source of ROS, which can cause oxidative stress (56, 57). For example, the UPR can contribute to cell death, following protein misfolding, through the accumulation of ROS (27, 58). The use of oxygen as a terminal electron acceptor implicated Ero1 as a source of ROS, and overexpression of Ero1 is known to generate ROS (27, 59–61). We found that loss of thioredoxin reductase resulted in hydrogen peroxide accumulation, which depended on the UPR. An unexpected consequence of UPR-generated ROS was the activation of the Yap1 transcription factor. The Yap1 transcription factor is the main regulator of the oxidative stress response in yeast and, hence, would normally act to regulate the protective response against ER stress–derived ROS (Fig. 6). This would be important, as ER-derived ROS have been proposed to account for a major fraction of intracellular ROS generation (56). We also found that the constitutive UPR causes a number of the well-known phenotypes normally observed in thioredoxin reductase mutants. For example, our roGFP2 probe data showed that abrogating the UPR in a trr1 mutant by deletion of HAC1 restored the cytosolic OxD value, indicating that the constitute UPR accounts for cytosolic GSH oxidation in a trr1 mutant. Thioredoxin reductase mutants are also very slow-growing and show a shortened chronological life span, which is somewhat surprising given the relatively high levels of reduced thioredoxins that are still present in a trr1 mutant (21, 36). However, these phenotypes can also be rescued by preventing the UPR, further reinforcing the idea that an uncontrolled UPR can be detrimental to cells and, for example, is implicated in diseases such as diabetes (62).
Most regulatory thiol oxidation is mediated by protein catalysts such as the peroxiredoxin family of enzymes, which act as both hydrogen peroxide sensors and thiol oxidizers (10, 11, 41). Peroxiredoxins are efficient oxidases that can, under certain conditions, result in an accumulation of oxidized thioredoxins, and oxidized thioredoxins can, in turn, introduce disulfide bonds into target proteins (63, 64). We found that the oxidation of thioredoxins in a thioredoxin reductase mutant required aerobic conditions and the presence of the Tsa1 and Tsa2 peroxiredoxins. Hence, any oxidative stress conditions that cause oxidation of Tsa1/2 may potentially activate the UPR. Presumably, the up-regulation of the UPR is important under these conditions to buffer against any potential ER stress. The production of ER-derived ROS would be an unwanted side effect of the UPR, although the Yap1 system is normally sufficient to protect against detrimental damaging effects (Fig. 6). Taken together, our data highlight an unanticipated link between the cytoplasmic and ER redox status, which is important for cell growth and longevity.
Experimental procedures
Yeast strains and plasmids
The WT strain W303 (MATa ura3–52 leu2–3 leu2–112 trp1–1 ade2–1 his3–11 can1–100) and its isogenic derivatives deleted for TRX1 (trx1::TRP1), TRX2 (trx2::URA3), TRR1 (trr1::HIS3), TSA1 (tsa1::LEU2, tsa1::URA3), TSA2 (tsa2::KANMX), GLR1 (glr1::TRP1), YAP1 (yap1::HIS3), IRE1 (ire1::KanMX4), and HAC1 (hac1::KanMX4) have all been described previously (8, 19, 43, 65–68). Strains deleted for multiple genes were constructed using standard yeast techniques. Respiratory incompetent [rho0] mutants were created using ethidium bromide treatment. The Tsa2 coding region along with 500 bp of 5′ UTR and 3′ UTR sequences was commercially synthesized (Epoch Life Science Inc.), including the insertion of a single HA tag at the N terminus of the protein. The Tsa2 construct was cloned into the pRS414 vector using BamHI and EagI restriction sites. The centromeric plasmid pFL-TSA1 has been described previously (69).
Growth conditions
Strains were grown in rich YEPD medium (2% (w/v) glucose, 2% (w/v) bactopeptone, and 1% (w/v) yeast extract) or minimal SD medium (0.17% (w/v) yeast nitrogen base without amino acids, 5% (w/v) ammonium sulfate, and 2% (w/v) glucose) supplemented with appropriate amino acids and bases at 30 °C and 180 rpm. Media were solidified by addition of 2% (w/v) agar. For anaerobic growth conditions, medium was supplemented with 0.1% (v/v) Tween 80 and 30 mg/liter ergosterol. Stress sensitivity was determined by growing cells to stationary phase and spotting onto agar plates containing various concentrations of tunicamycin.
β-gal assays
Plasmids were used containing a lacZ reporter construct under the control of the UPR element (70) or the Yap1 response element (71). For the determination of β-gal activity, transformants were assayed essentially as described previously (72). Activity is expressed as nanomoles of O-nitrophenyl-β-d-galactopyranoside hydrolyzed per minute per microgram of total protein (units). Values shown are the means of at least three independent determinations.
Western blotting analyses
Protein extracts were electrophoresed under reducing or nonreducing conditions on SDS-PAGE minigels and electroblotted onto polyvinylidene difluoride membranes (Amersham Biosciences). Bound antibody was visualized using WesternSure® chemiluminescent reagents (LI-COR). Protein concentrations were measured using a NanoDrop ND-8000 spectrophotometer. Primary antibodies used were Pgk1 (Thermo Fisher Scientific), Trx1 (16), Trx2 (43), Tsa1 (73), HA (Sigma), and Pab1 (74).
Redox state measurements
The redox state of Trx1 and Trx2 was measured by covalent modification with the thiol-reactive probe AMS (Molecular Probes) as described previously (16). For nonreducing Western blot analysis, proteins were extracted in the presence of N-ethylmaleimide to prevent any nonspecific thiol oxidation during sample preparation. A redox-responsive fluorescent protein probe (roGFP2) was used to quantify cytosolic redox potentials (75), and a peroxiredoxin-based probe (roGFP2-Tsa2ΔCR) was used to measure H2O2 concentrations based on the methodology described by Morgan et al. (28). To obtain measurements of the fully oxidized and fully reduced probes, cells were treated with 20 mm H2O2 or 100 mm DTT for 30 min, respectively. Ten thousand cells were counted for each sample using a BD Biosciences LSRFortessaTM cell analyzer, and data were analyzed using BD Biosciences FACSDiva 8.0.1 software. The degree of probe OxD was calculated as described previously (32).
DHR123 was used to measure intracellular ROS, using a protocol modified from Ref. 65. Briefly, 5 μg/ml DHR123 was added to mid-exponentially grown cells and incubated for 1 h at 30 °C in the dark. Fluorescence was measured by flow cytometry at 485/528 nm. Ten thousand cells were counted for each sample using a BD Biosciences LSRFortessaTM cell analyzer.
Chronological life span experiments were performed according to Ref. 76. Briefly, cells were cultured in liquid synthetic complete (SCD) medium (180 rpm, 30 °C), with aliquots taken every 2–3 days for flow cytometry analysis (501 nm excitation, 586 nm/15 nm emission detection on a BD Biosciences LSRFortessaTM cell analyzer and BD FACSDiva 8.0.1 software) after propidium iodide staining.
Splicing reporter
The GFP SR used to monitor Ire1 activity has been described previously (40). 10,000 cells were analyzed in a BD Biosciences LSRFortessa flow cytometer with excitation at 488 nm and emission at 530/30 nm. The average median value of the intensity of fluorescence of three independent biological repeats was calculated.
Author contributions
P. K. and C. M. G. conceptualization; P. K. and C. M. G. formal analysis; P. K. and C. M. G. supervision; P. K., J. D. R., A. J. W., X. W., and C. J. K. investigation; P. K., J. D. R., A. J. W., and C. M. G. methodology; P. K. and C. M. G. writing-original draft; P. K. and C. M. G. writing-review and editing; C. M. G. data curation; C. M. G. funding acquisition; C. M. G. project administration.
Acknowledgments
We thank Anita Ayer, Ian Dawes, Tobias Dick, Bruce Morgan, Peter Walter, David Pincus, David Eide, and Scott Moye-Rowley for kindly supplying resources used in this study. We also thank Mike Jackson (University of Manchester Flow Cytometry Core Facility) for help with flow cytometry analysis.
This work was funded by Biotechnology and Biological Sciences Research (BBSRC) Council Project Grants BB/M020770/1 and BB/P005594/1 (to C. M. G.) and a BBSRC funded studentship (to A. J. W.). The authors declare that they have no conflicts of interest with the contents of this article.
- ROS
- reactive oxygen species
- UPR
- unfolded protein response
- ER
- endoplasmic reticulum
- DHR123
- dihydrorhodamine 123
- OxD
- oxidation
- Tm
- tunicamycin
- CLS
- chronological life span
- SR
- splicing reporter
- AMS
- 4-acetamido-4′maleimidyldystilbene-2,2′ disulfonic acid
- HA
- hemagglutinin.
References
- 1. Rietsch A., and Beckwith J. (1998) The genetics of disulfide bond metabolism. Annu. Rev. Genet. 32, 163–184 10.1146/annurev.genet.32.1.163 [DOI] [PubMed] [Google Scholar]
- 2. Lu J., and Holmgren A. (2014) The thioredoxin antioxidant system. Free Radic. Biol. Med. 66, 75–87 10.1016/j.freeradbiomed.2013.07.036 [DOI] [PubMed] [Google Scholar]
- 3. Muller E. G. (1991) Thioredoxin deficiency in yeast prolongs S phase and shortens the G1 interval of the cell cycle. J. Biol. Chem. 266, 9194–9202 [PubMed] [Google Scholar]
- 4. Chae H. Z., Chung S. J., and Rhee S. G. (1994) Thioredoxin-dependent peroxide reductase from yeast. J. Biol. Chem. 269, 27670–27678 [PubMed] [Google Scholar]
- 5. Mason J. T., Kim S. K., Knaff D. B., and Wood M. J. (2006) Thermodynamic basis for redox regulation of the Yap1 signal transduction pathway. Biochemistry 45, 13409–13417 10.1021/bi061136y [DOI] [PubMed] [Google Scholar]
- 6. Gan Z.-R. (1991) Yeast thioredoxin genes. J. Biol. Chem. 266, 1692–1696 [PubMed] [Google Scholar]
- 7. Kuge S., and Jones N. (1994) YAP1 dependent activation of TRX2 is essential for the response of Saccharomyces cerevisiae to oxidative stress by hydroperoxides. EMBO J. 13, 655–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Garrido E. O., and Grant C. M. (2002) Role of thioredoxins in the response of Saccharomyces cerevisiae to oxidative stress induced by hydroperoxides. Mol. Microbiol. 43, 993–1003 10.1046/j.1365-2958.2002.02795.x [DOI] [PubMed] [Google Scholar]
- 9. Perkins A., Nelson K. J., Parsonage D., Poole L. B., and Karplus P. A. (2015) Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 40, 435–445 10.1016/j.tibs.2015.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rhee S. G., Woo H. A., Kil I. S., and Bae S. H. (2012) Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 287, 4403–4410 10.1074/jbc.R111.283432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leichert L. I., and Dick T. P. (2015) Incidence and physiological relevance of protein thiol switches. Biol. Chem. 396, 389–399 [DOI] [PubMed] [Google Scholar]
- 12. Deponte M., and Lillig C. H. (2015) Enzymatic control of cysteinyl thiol switches in proteins. Biol. Chem. 396, 401–413 [DOI] [PubMed] [Google Scholar]
- 13. Wong C. M., Siu K. L., and Jin D. Y. (2004) Peroxiredoxin-null yeast cells are hypersensitive to oxidative stress and are genomically unstable. J. Biol. Chem. 279, 23207–23213 10.1074/jbc.M402095200 [DOI] [PubMed] [Google Scholar]
- 14. Jang H. H., Lee K. O., Chi Y. H., Jung B. G., Park S. K., Park J. H., Lee J. R., Lee S. S., Moon J. C., Yun J. W., Choi Y. O., Kim W. Y., Kang J. S., Cheong G. W., Yun D. J., et al. (2004) Two enzymes in one: two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 117, 625–635 10.1016/j.cell.2004.05.002 [DOI] [PubMed] [Google Scholar]
- 15. Williams C. H., Arscott L. D., Müller S., Lennon B. W., Ludwig M. L., Wang P. F., Veine D. M., Becker K., and Schirmer R. H. (2000) Thioredoxin reductase two modes of catalysis have evolved. Eur. J. Biochem. 267, 6110–6117 10.1046/j.1432-1327.2000.01702.x [DOI] [PubMed] [Google Scholar]
- 16. Trotter E. W., and Grant C. M. (2003) Non-reciprocal regulation of the redox state of the glutathione/glutaredoxin and thioredoxin systems. EMBO Rep. 4, 184–188 10.1038/sj.embor.embor729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Machado A. K., Morgan B. A., and Merrill G. F. (1997) Thioredoxin reductase-dependent inhibition of MCB cell cycle box activity in Saccharomyces cerevisiae. J. Biol. Chem. 272, 17045–17054 10.1074/jbc.272.27.17045 [DOI] [PubMed] [Google Scholar]
- 18. Pearson G. D., and Merrill G. F. (1998) Deletion of the Saccharomyces cerevisiae TRR1 gene encoding thioredoxin reductase inhibits p53-dependent reporter gene expression. J. Biol. Chem. 273, 5431–5434 10.1074/jbc.273.10.5431 [DOI] [PubMed] [Google Scholar]
- 19. Trotter E. W., and Grant C. M. (2002) Thioredoxins are required for protection against a reductive stress in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 46, 869–878 10.1046/j.1365-2958.2002.03216.x [DOI] [PubMed] [Google Scholar]
- 20. Travers K. J., Patil C. K., Wodicka L., Lockhart D. J., Weissman J. S., and Walter P. (2000) Functional and genomic analysis reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101, 249–258 10.1016/S0092-8674(00)80835-1 [DOI] [PubMed] [Google Scholar]
- 21. Ragu S., Dardalhon M., Sharma S., Iraqui I., Buhagiar-Labarchède G., Grondin V., Kienda G., Vernis L., Chanet R., Kolodner R. D., Huang M. E., and Faye G. (2014) Loss of the thioredoxin reductase Trr1 suppresses the genomic instability of peroxiredoxin tsa1 mutants. PLoS ONE 9, e108123 10.1371/journal.pone.0108123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Toledano M. B., Delaunay A., Monceau L., and Tacnet F. (2004) Microbial H2O2 sensors as archetypical redox signaling modules. Trends Biochem. Sci. 29, 351–357 10.1016/j.tibs.2004.05.005 [DOI] [PubMed] [Google Scholar]
- 23. Delaunay A., Pflieger D., Barrault M. B., Vinh J., and Toledano M. B. (2002) A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell 111, 471–481 10.1016/S0092-8674(02)01048-6 [DOI] [PubMed] [Google Scholar]
- 24. Okazaki S., Naganuma A., and Kuge S. (2005) Peroxiredoxin-mediated redox regulation of the nuclear localization of Yap1, a transcription factor in budding yeast. Antioxid. Redox Signal. 7, 327–334 10.1089/ars.2005.7.327 [DOI] [PubMed] [Google Scholar]
- 25. Carmel-Harel O., Stearman R., Gasch A. P., Botstein D., Brown P. O., and Storz G. (2001) Role of thioredoxin reductase in the Yap-1p-dependent response to oxidative stress in Saccharomyces cerevisiae. Mol. Microbiol. 39, 595–605 10.1046/j.1365-2958.2001.02255.x [DOI] [PubMed] [Google Scholar]
- 26. Izawa S., Maeda K., Sugiyama K., Mano J., Inoue Y., and Kimura A. (1999) Thioredoxin deficiency causes the constitutive activation of Yap1, an AP-1-like transcription factor in Saccharomyces cerevisiae. J. Biol. Chem. 274, 28459–28565 10.1074/jbc.274.40.28459 [DOI] [PubMed] [Google Scholar]
- 27. Haynes C. M., Titus E. A., and Cooper A. A. (2004) Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 15, 767–776 10.1016/j.molcel.2004.08.025 [DOI] [PubMed] [Google Scholar]
- 28. Morgan B., Van Laer K., Owusu T. N., Ezeriņa D., Pastor-Flores D., Amponsah P. S., Tursch A., and Dick T. P. (2016) Real-time monitoring of basal H2O2 levels with peroxiredoxin-based probes. Nat. Chem. Biol. 12, 437–443 10.1038/nchembio.2067 [DOI] [PubMed] [Google Scholar]
- 29. Swalwell H., Latimer J., Haywood R. M., and Birch-Machin M. A. (2012) Investigating the role of melanin in UVA/UVB- and hydrogen peroxide-induced cellular and mitochondrial ROS production and mitochondrial DNA damage in human melanoma cells. Free Radic. Biol. Med. 52, 626–634 10.1016/j.freeradbiomed.2011.11.019 [DOI] [PubMed] [Google Scholar]
- 30. Honda F., Kano H., Kanegane H., Nonoyama S., Kim E. S., Lee S. K., Takagi M., Mizutani S., and Morio T. (2012) The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat. Immunol. 13, 369–378 10.1038/ni.2234 [DOI] [PubMed] [Google Scholar]
- 31. Ayer A., Tan S. X., Grant C. M., Meyer A. J., Dawes I. W., and Perrone G. G. (2010) The critical role of glutathione in maintenance of the mitochondrial genome. Free Radic. Biol. Med. 49, 1956–1968 10.1016/j.freeradbiomed.2010.09.023 [DOI] [PubMed] [Google Scholar]
- 32. Kojer K., Bien M., Gangel H., Morgan B., Dick T. P., and Riemer J. (2012) Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J. 31, 3169–3182 10.1038/emboj.2012.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morgan B., Sobotta M. C., and Dick T. P. (2011) Measuring E(GSH) and H2O2 with roGFP2-based redox probes. Free Radic. Biol. Med. 51, 1943–1951 10.1016/j.freeradbiomed.2011.08.035 [DOI] [PubMed] [Google Scholar]
- 34. Cox J. S., Shamu C. E., and Walter P. (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206 10.1016/0092-8674(93)90648-A [DOI] [PubMed] [Google Scholar]
- 35. Kohno K., Normington K., Sambrook J., Gething M.-J., and Mori K. (1993) The promoter region of the yeast KAR2 (BiP) gene contains a regulatory region domain that responds to the presence of unfolded proteins in the endoplasmic reticulum. Cell 13, 877–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Trotter E. W., and Grant C. M. (2005) Overlapping roles of the cytoplasmic and mitochondrial redoc regulatory systems in the yeast Saccharomyces cerevisiae. Eukaryot. Cell 4, 392–400 10.1128/EC.4.2.392-400.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kaeberlein M. (2010) Lessons on longevity from budding yeast. Nature 464, 513–519 10.1038/nature08981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pan Y., Schroeder E. A., Ocampo A., Barrientos A., and Shadel G. S. (2011) Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab. 13, 668–678 10.1016/j.cmet.2011.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brandes N., Tienson H., Lindemann A., Vitvitsky V., Reichmann D., Banerjee R., and Jakob U. (2013) Time line of redox events in aging postmitotic cells. eLife 2, e00306 10.7554/eLife.00306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pincus D., Chevalier M. W., Aragón T., van Anken E., Vidal S. E., El-Samad H., and Walter P. (2010) BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 8, e1000415 10.1371/journal.pbio.1000415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown J. D., Day A. M., Taylor S. R., Tomalin L. E., Morgan B. A., and Veal E. A. (2013) A peroxiredoxin promotes H2O2 signaling and oxidative stress resistance by oxidizing a thioredoxin family protein. Cell Rep. 5, 1425–1435 10.1016/j.celrep.2013.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Day A. M., Brown J. D., Taylor S. R., Rand J. D., Morgan B. A., and Veal E. A. (2012) Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Mol. Cell 45, 398–408 10.1016/j.molcel.2011.11.027 [DOI] [PubMed] [Google Scholar]
- 43. Rand J. D., and Grant C. M. (2006) The thioredoxin system protects ribosomes against stress-induced aggregation. Mol. Biol. Cell 17, 387–401 10.1091/mbc.e05-06-0520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 10.1042/BJ20081386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pincus D., Aranda-Díaz A., Zuleta I. A., Walter P., and El-Samad H. (2014) Delayed Ras/PKA signaling augments the unfolded protein response. Proc. Natl. Acad. Sci. U.S.A. 111, 14800–14805 10.1073/pnas.1409588111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kimata Y., Ishiwata-Kimata Y., Yamada S., and Kohno K. (2006) Yeast unfolded protein response pathway regulates expression of genes for anti-oxidative stress and for cell surface proteins. Genes Cells 11, 59–69 [DOI] [PubMed] [Google Scholar]
- 47. Holmgren A. (1989) Thioredoxin and glutaredoxin systems. J. Biol. Chem. 264, 13963–13966 [PubMed] [Google Scholar]
- 48. Muller E. G. (1996) A glutathione reductase mutant of yeast accumulates high levels of oxidized glutathione and requires thioredoxin for growth. Mol. Biol. Cell 7, 1805–1813 10.1091/mbc.7.11.1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cuozzo J. W., and Kaiser C. A. (1999) Competition between glutathione and protein thiols for disulphide bond formation. Nat. Cell Biol. 1, 130–135 10.1038/11047 [DOI] [PubMed] [Google Scholar]
- 50. Jessop C. E., and Bulleid N. J. (2004) Glutathione directly reduces an oxidoreductase in the endoplasmic reticulum of mammalian cells. J. Biol. Chem. 279, 55341–55347 10.1074/jbc.M411409200 [DOI] [PubMed] [Google Scholar]
- 51. Gross E., Sevier C. S., Vala A., Kaiser C. A., and Fass D. (2002) A new FAD-binding fold and intersubunit disulfide shuttle in the thiol oxidase Erv2p. Nat. Struct. Biol. 9, 61–67 10.1038/nsb740 [DOI] [PubMed] [Google Scholar]
- 52. Margittai E., and Sitia R. (2011) Oxidative protein folding in the secretory pathway and redox signaling across compartments and cells. Traffic 12, 1–8 10.1111/j.1600-0854.2010.01108.x [DOI] [PubMed] [Google Scholar]
- 53. Bulleid N. J., and Ellgaard L. (2011) Multiple ways to make disulfides. Trends Biochem. Sci. 36, 485–492 10.1016/j.tibs.2011.05.004 [DOI] [PubMed] [Google Scholar]
- 54. Ellgaard L., Sevier C. S., and Bulleid N. J. (2018) How are proteins reduced in the endoplasmic reticulum? Trends Biochem. Sci. 43, 32–43 10.1016/j.tibs.2017.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Poet G. J., Oka O. B., van Lith M., Cao Z., Robinson P. J., Pringle M. A., Arnér E. S., and Bulleid N. J. (2017) Cytosolic thioredoxin reductase 1 is required for correct disulfide formation in the ER. EMBO J. 36, 693–702 10.15252/embj.201695336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tu B. P., and Weissman J. S. (2004) Oxidative protein folding in eukaryotes: mechanisms and consequences. J. Cell Biol. 164, 341–346 10.1083/jcb.200311055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eletto D., Chevet E., Argon Y., and Appenzeller-Herzog C. (2014) Redox controls UPR to control redox. J. Cell Sci. 127, 3649–3658 10.1242/jcs.153643 [DOI] [PubMed] [Google Scholar]
- 58. Hauptmann P., Riel C., Kunz-Schughart L. A., Fröhlich K. U., Madeo F., and Lehle L. (2006) Defects in N-glycosylation induce apoptosis in yeast. Mol. Microbiol. 59, 765–778 10.1111/j.1365-2958.2005.04981.x [DOI] [PubMed] [Google Scholar]
- 59. Harding H. P., Zhang Y., Zeng H., Novoa I., Lu P. D., Calfon M., Sadri N., Yun C., Popko B., Paules R., Stojdl D. F., Bell J. C., Hettmann T., Leiden J. M., and Ron D. (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633 10.1016/S1097-2765(03)00105-9 [DOI] [PubMed] [Google Scholar]
- 60. Tu B. P., and Weissman J. S. (2002) The FAD- and O2-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol. Cell 10, 983–994 10.1016/S1097-2765(02)00696-2 [DOI] [PubMed] [Google Scholar]
- 61. Gross E., Sevier C. S., Heldman N., Vitu E., Bentzur M., Kaiser C. A., Thorpe C., and Fass D. (2006) Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. U.S.A. 103, 299–304 10.1073/pnas.0506448103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Back S. H., and Kaufman R. J. (2012) Endoplasmic reticulum stress and type 2 diabetes. Annu. Rev. Biochem. 81, 767–793 10.1146/annurev-biochem-072909-095555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Stöcker S., Van Laer K., Mijuskovic A., and Dick T. P. (2018) The conundrum of hydrogen peroxide signaling and the emerging role of peroxiredoxins as redox relay hubs. Antioxid. Redox. Signal. 28, 558–573 10.1089/ars.2017.7162 [DOI] [PubMed] [Google Scholar]
- 64. Netto L. E., and Antunes F. (2016) The roles of peroxiredoxin and thioredoxin in hydrogen peroxide sensing and in signal transduction. Mol. Cell 39, 65–71 10.14348/molcells.2016.2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Weids A. J., and Grant C. M. (2014) The yeast peroxiredoxin Tsa1 protects against protein-aggregate-induced oxidative stress. J. Cell Sci. 127, 1327–1335 10.1242/jcs.144022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Grant C. M., Collinson L. P., Roe J.-H., and Dawes I. W. (1996) Yeast glutathione reductase is required for protection against oxidative stress and is a target gene for yAP-1 transcriptional regulation. Mol. Microbiol. 21, 171–179 10.1046/j.1365-2958.1996.6351340.x [DOI] [PubMed] [Google Scholar]
- 67. Draculic T., Dawes I. W., and Grant C. M. (2000) A single glutaredoxin or thioredoxin is essential for viability in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 36, 1167–1174 10.1046/j.1365-2958.2000.01948.x [DOI] [PubMed] [Google Scholar]
- 68. Sideri T. C., Stojanovski K., Tuite M. F., and Grant C. M. (2010) Ribosome-associated peroxiredoxins suppress oxidative stress-induced de novo formation of the [PSI+] prion in yeast. Proc. Natl. Acad. Sci. U.S.A. 107, 6394–6399 10.1073/pnas.1000347107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wu C. Y., Bird A. J., Winge D. R., and Eide D. J. (2007) Regulation of the yeast TSA1 peroxiredoxin by ZAP1 is an adaptive response to the oxidative stress of zinc deficiency. J. Biol. Chem. 282, 2184–2195 10.1074/jbc.M606639200 [DOI] [PubMed] [Google Scholar]
- 70. Sidrauski C., Cox J. S., and Walter P. (1996) tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 87, 405–413 10.1016/S0092-8674(00)81361-6 [DOI] [PubMed] [Google Scholar]
- 71. Wu A. L., and Moye-Rowley W. S. (1994) GSH1, which encodes γ-glutamylcysteine synthetase, is a target gene for yAP-1 transcriptional regulation. Mol. Cell Biol. 14, 5832–5839 10.1128/MCB.14.9.5832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rose M., and Botstein D. (1983) Construction and use of gene fusions to lacZ (β-galactosidase) which are expressed in yeast. Methods Enzymol. 101, 167–180 10.1016/0076-6879(83)01012-5 [DOI] [PubMed] [Google Scholar]
- 73. Trotter E. W., Rand J. D., Vickerstaff J., and Grant C. M. (2008) The yeast Tsa1 peroxiredoxin is a ribosome-associated antioxidant. Biochem. J. 412, 73–80 10.1042/BJ20071634 [DOI] [PubMed] [Google Scholar]
- 74. Costello J., Castelli L. M., Rowe W., Kershaw C. J., Talavera D., Mohammad-Qureshi S. S., Sims P. F. G., Grant C. M., Pavitt G. D., Hubbard S. J., and Ashe M. P. (2015) Global mRNA selection mechanisms for translation initiation. Genome Biol. 16, 10 10.1186/s13059-014-0559-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ayer A., Fellermeier S., Fife C., Li S. S., Smits G., Meyer A. J., Dawes I. W., and Perrone G. G. (2012) A genome-wide screen in yeast identifies specific oxidative stress genes required for the maintenance of sub-cellular redox homeostasis. PLoS ONE 7, e44278 10.1371/journal.pone.0044278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ocampo A., and Barrientos A. (2011) Quick and reliable assessment of chronological life span in yeast cell populations by flow cytometry. Mech. Ageing Dev. 132, 315–323 10.1016/j.mad.2011.06.007 [DOI] [PubMed] [Google Scholar]