

Abstract
We have used a novel time-resolved FRET (TR-FRET) assay to detect small-molecule modulators of actin–myosin structure and function. Actin–myosin interactions play crucial roles in the generation of cellular force and movement. Numerous mutations and post-translational modifications of actin or myosin disrupt muscle function and cause life-threatening syndromes. Here, we used a FRET biosensor to identify modulators that bind to the actin–myosin interface and alter the structural dynamics of this complex. We attached a fluorescent donor to actin at Cys-374 and a nonfluorescent acceptor to a peptide containing the 12 N-terminal amino acids of the long isoform of skeletal muscle myosin's essential light chain. The binding site on actin of this acceptor-labeled peptide (ANT) overlaps with that of myosin, as indicated by (a) a similar distance observed in the actin–ANT complex as in the actin–myosin complex and (b) a significant decrease in actin–ANT FRET upon binding myosin. A high-throughput FRET screen of a small-molecule library (NCC, 727 compounds), using a unique fluorescence lifetime readout with unprecedented speed and precision, showed that FRET is significantly affected by 10 compounds in the micromolar range. Most of these “hits” alter actin-activated myosin ATPase and affect the microsecond dynamics of actin detected by transient phosphorescence anisotropy. We conclude that the actin–ANT TR-FRET assay enables detection of pharmacologically active compounds that affect actin structural dynamics and actomyosin function. This assay establishes feasibility for the discovery of allosteric modulators of the actin–myosin interaction, with the ultimate goal of developing therapies for muscle disorders.
Keywords: actin, myosin, fluorescence resonance energy transfer (FRET), peptide, high-throughput screening (HTS)
Introduction
The structural transition of the actin–myosin complex from the weak (lever arm up) to the strong (lever arm down) binding states during myosin's ATPase cycle produces the force of muscle contraction (1). The molecular mechanism involves structural transitions at the interface between actin and myosin's catalytic domain and within myosin's light chain domain, which contains binding sites for essential (ELC)2 and regulatory light chains. Both light chains play important roles in regulating the actin–myosin interaction (2, 3). ELCs are highly conserved and are expressed as two isoforms, A1 and A2. The principal difference between them is that A1 contains an N-terminal extension (NTE) of 40–45 additional amino acids. Our previous work showed that when myosin binds to actin, the NTE of fast skeletal or cardiac ELC is in close proximity to actin (Fig. 1) (4, 5). N-terminal amino acids of NTE, specifically the first four amino acids (APKK), are highly conserved among skeletal and cardiac muscles and also across species (6). These positively charged lysine residues interact with the negatively charged C terminus glutamates of actin (7). The actin–NTE interaction is functionally relevant, because myosin isoforms having NTEs show higher catalytic efficiency and slower motility on actin (3, 8, 9).
Figure 1.

Model of actin–myosin complex with skeletal myosin S1 (heavy chain (blue), ELC (green), and regulatory light chain (red)) strongly bound to actin (gray) (19). Orange spheres show the labeling site on actin. The green circle indicates the location of the dabcyl-labeled N-terminal peptide (ANT).
Recent studies from our laboratory have explored the structural basis of ELC-mediated modulation of the actin–myosin interaction (4, 5, 10). Time-resolved FRET (TR-FRET), using a donor on actin and an acceptor on the A1 NTE of skeletal myosin subfragment 1 (S1), showed that the NTE plays an important role modulating myosin's force-producing powerstroke (4). Similar studies with cardiac (ventricular) myosin S1 provided direct insight into the mechanism for perturbation of actin–myosin interactions by a cardiomyopathy mutation in the light chain domain (5). Many mutations or post-translational modifications in both actin and myosin cause life-threatening muscle disorders, and treatment options remain limited (11). We hypothesize that this TR-FRET approach can be used as a tool to screen for compounds that rescue defects in actomyosin structure and function. Small-molecule modulators of actomyosin structural dynamics represent potential leads for future drug development, and this search is greatly facilitated by recent developments in high-throughput FRET-based screening methods, which measure the effects of compounds on the distance between donor and acceptor probes on interacting proteins (1, 12–15).
In the present study, we labeled actin at Cys-374 with a fluorescent donor, fluorescein 5-maleimide (FM), and attached the nonfluorescent acceptor probe dabcyl to the N terminus of a 12-amino acid peptide derived from the N terminus of NTE of rabbit skeletal muscle A1 (Fig. 1). Use of this dabcyl-labeled peptide, designated ANT, was inspired by previous reports showing that the first 13 amino acid residues of NTE are important regulators of the actin–myosin interaction (16) and affect contractility of muscle cells (17). A key advantage of ANT, over our previously used acceptor-labeled myosin (4), is that it can be synthesized and purified in large quantity, thus facilitating large-scale high-throughput screening (HTS). We hypothesized that compounds affecting the actin–ANT interaction are likely to perturb structural and enzymatic properties of actin–myosin. Here, we measured TR-FRET from actin to ANT with a high-precision fluorescence lifetime plate reader (FLTPR) (18) in the presence and absence of compounds from a small-molecule library. Hits from this assay, defined as compounds producing effects more than 4 S.D. from the mean, were analyzed further to determine their effects on actin-activated myosin ATPase activity, to evaluate the potential of this TR-FRET approach for drug discovery.
Results
Actin–ANT FRET biosensor
Time-resolved fluorescence decays of donor-labeled actin in the presence of increasing concentrations of acceptor-labeled peptide (ANT) (Fig. 2, A and B) were analyzed as indicated in Equation 1 (see “Experimental procedures) to determine the mole fraction of actin containing bound ANT (XDA) (Fig. 2A). The fluorescence lifetime, τDA, of the bound complex did not change with [ANT], indicating that FRET was not affected by the interaction of the donor with multiple acceptors. Analysis revealed a Kd of 16.0 ± 1.2 μm (Fig. 2B), consistent with a previous report for the unlabeled peptide (17). This peptide had no effect on Vmax and Km of actin-activated ATPase of purified skeletal muscle acto-S1A1 (with NTE) (Fig. 2C) or the ATPase activity of rabbit skeletal myofibrils in activating or relaxing conditions (Fig. 2D).
Figure 2.

A, TR-FRET. Shown is time-resolved fluorescence of 2 μm donor-labeled actin (D-actin) in the absence (black) and presence (red) of 50 μm acceptor-labeled N-terminal peptide (ANT). B, binding of ANT to D-actin, calculated from TR-FRET data in A. C, actin-activated ATPase activity of skeletal muscle S1A1 (100% A1) in the absence (black) and in the presence of 50 μm ANT (S1A1 = 0.15 μm). D, myofibrillar ATPase activity versus [ANT] during relaxation (open circles) and contraction (closed circles). Error bars, S.D.
The addition of ANT to FM actin decreased the donor lifetime (Fig. 3A, black to red), indicating FRET. Analysis of the FRET decay showed that the mean distance (R) between the two probes was 3.3 ± 0.2 nm, in very good agreement with the distance detected for probes at Cys-374 on actin and on Cys-16 at the NTE of A1 (4). The addition of NTE-containing S1s to the actin and ANT complex increased the lifetime (Fig. 3A, representative decay of S1s having NTE, red to blue), indicating that S1 either displaced or dissociated the actin-bound ANT. The analysis of FRET waveforms could not resolve the two possibilities. The FRET efficiency 〈E〉, calculated from the average lifetime in a model-independent analysis, is summarized in Fig. 3B. The addition of S1 isoforms with NTEs (skeletal S1, 75% of A1 + 25% of A2; skeletal S1A1, 100% A1; cardiac S1, 100% A1) had a significant effect on FRET, whereas the addition of an S1 isoform (S1A2, 0% A1) that does not contain an NTE had no effect on FRET between actin and ANT (Fig. 3B). These results indicate that ANT shares an overlapping binding site on actin with all tested ELC NTEs. The addition of ATP to a nucleotide-free mixture of actin, ANT, and S1 increased FRET between actin and ANT for the NTE-containing S1 isoforms, but not for S1A2 (Fig. 3B). This result serves as further evidence for overlapping ANT and NTE binding sites, as ATP causes dissociation of all S1 constructs from actin. The similarity of actin–ANT and actin–NTE interactions is further supported by the observation that both are sensitive to ionic strength; the addition of 0.1 m KCl to the actin–ANT complex also significantly decreased the rate of donor decay (Fig. 3A, red to green). Such sensitivity to the ionic strength specifies the electrostatic nature of this interaction and mirrors the model-based proposal that a positively charged NTE binds to a cluster of negatively charged residues in the C-terminal region of actin (19). These results clearly show that actin–ANT FRET is sensitive to the association of S1 NTEs with actin and also to ionic strength; both factors are known to be strong modulators of the actin–myosin interaction (19). Thus, we conclude that our actin–ANT FRET sensor has sufficient sensitivity for employment in a search for modulators of actin–ANT FRET in an HTS assay.
Figure 3.
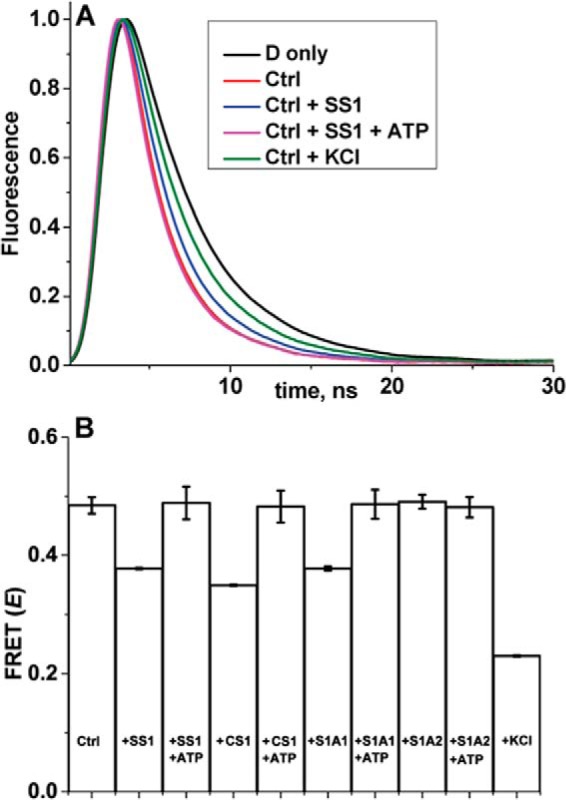
Actin–ANT FRET. A, representative fluorescence decays of donor-labeled actin (2 μm) in the absence (D only, black, same as D-actin in Fig. 2A) and presence of 50 μm ANT (Ctrl, red, same as D-actin + 50 μm ANT in Fig. 2A) and after the addition to Ctrl of saturating (5 μm) skeletal S1 (blue), S1 + saturating (3 mm) ATP (magenta), or 0.1 m KCl (green). The D only (black) and Ctrl (red) decays are included in the present figure for visualization and comparison purposes. B, FRET between actin and ANT in the absence (Ctrl) and presence of different isoforms of S1 (skeletal SS1, cardiac CS1, skeletal S1A1, skeletal S1A2), without and with 3 mm ATP and 0.1 m KCl. Error bars, S.D.
High-throughput screening of National Institutes of Health Clinical Collection (NCC) library to identify compounds that modulate actin–ANT FRET
Using the actin–ANT FRET sensor, we performed HTS of the NCC library (727 compounds). The NCC library is a collection of small molecules that have previously been tested in clinical trials and therefore have known safety profiles. The complete NCC library was applied to three 384-well black-wall/black-bottom Greiner plates with drug-free control (50 nl of DMSO) wells on each side of the individual plates (see “Experimental procedures”). For each screen, one set of drug plates was loaded with 2 μm FM-labeled actin (donor only), and the other set was loaded with 2 μm FM actin and 50 μm ANT (donor-acceptor). All plates were incubated for 20 min at 25 °C before reading. FM actin was excited with a 473-nm laser, and time-resolved fluorescence decay waveforms were read in the FLTPR over a time course of 20 and 120 min. A two-exponential fit was used to obtain the lifetime of donor-only (τD) and donor-acceptor samples (τDA). Interfering (intrinsically fluorescent) compounds were removed using a control screen with 2 μm unlabeled actin. A histogram of average FRET distribution from the NCC compounds was plotted and fitted to a Gaussian curve (Fig. 4A) to obtain a mean and S.D. FRET efficiency, 〈E〉, was computed on a well-to-well basis using Equation 1 (Fig. 4B). The Z′ factor for this screen was calculated as 0.8 ± 0.1 using DMSO-only controls, which validates the robustness of this HTS screen (14, 20). This screen was performed in triplicate with three different preparations of donor and acceptor samples. Excellent agreement was observed in all three screens. Of 27, 21, and 23 hits across the three screens, a total of 10 compounds (Fig. 4B, red) reproducibly altered the average FRET by more than 4 S.D. greater than the mean of the control samples. This is a true hit rate of 35–45%. Compounds that did not reproducibly alter average FRET were considered false positives. The 10 remaining hit compounds were further tested in concentration–response FRET assays.
Figure 4.

TR-FRET–based HTS of NCC library for compounds that modulate actin–ANT FRET. A, histogram plots of all compounds from the NCC screen after removal of fluorescent compounds show an average FRET efficiency of 0.45 ± 0.003. B, FRET from a representative NCC screen with hit threshold (>4 S.D. of mean) indicated by magenta lines. Reproducible hits from triplicate screens are shown in red.
FRET concentration–response assay
Using the same condition as in the primary screen, we examined the dependence of FRET on the concentration of each reproducible hit compound (0.5–100 μm). Significant concentration-dependent effects on FRET were observed for most of the identified hits (Fig. 5). Actin–ANT FRET (E) for each compound was determined at each concentration and was subsequently normalized to the DMSO-only control (E0). Concentration dependence of the normalized FRET change (E/E0) was fitted using the Hill equation, which is routinely used in pharmacological studies to analyze concentration–response data of the drugs (21). All hit compounds decreased actin–ANT FRET at micromolar concentrations with notable differences in the apparent EC50 (concentration needed for half-maximal effect) of the FRET curve (Fig. 5, A and B). EC50 values are summarized in Table 1. Concentration dependence indicates that these compounds are interacting with actin. Concentration–response of two known myosin-binding drugs, OM and Myk 461, did not affect actin–ANT FRET (Fig. 5C). This control experiment further indicates the structural specificity of the identified hit compounds for actin. Future studies with compounds known to interact with actin will also be informative.
Figure 5.

Concentration response of reproducible hit compounds on FRET. A and B, hit compounds decreased FRET in a concentration- dependent manner. C, myosin-specific compounds, OM and MYK461, did not show concentration dependence (control experiment). Error bars, S.D.
Table 1.
EC50 values
| Compound | EC50(FRET) | EC50(ATPase) skeletal acto-S1 | EC50(ATPase) cardiac acto-S1 |
|---|---|---|---|
| μm | μm | μm | |
| Honokiol | 44 ± 2 | >>100 | >>100 |
| Tegaserod | 29 ± 2 | >>80 | 20.2 ± 5.1 |
| Thioridazine | 21 ± 3 | 25 ± 3.0 | 29 ± 6 |
| Flutamide | 15 ± 2 | >NDa | >ND |
| Phenothiazine | 7 ± 2 | >ND | >ND |
| Fluphenazine | 25 ± 4 | 12.9 ± 3.9 | 11.0 ± 3.2 |
| Dantrolene | 48 ± 4 | >ND | >ND |
| Novantrone | 5.4 ± 0.7 | 11.3 ± 3.5 | 11.0 ± 2.0 |
| Carvedilol | 48 ± 5 | >ND | >ND |
| Mefloquine | 21 ± 2 | >ND | >ND |
a ND, not determined.
Functional characterization of FRET hits on actomyosin ATPase activity
Functional effects of the 10 hit compounds on actin-activated myosin ATPase (Fig. 6) were measured in a concentration-dependent manner. The concentration of actin (2 μm) and myosin was chosen to be the same as in the FRET measurements for consistency. None of the compounds altered Mg-ATPase of skeletal or cardiac S1 in the absence of actin (0.007 ± 0.002 S−1 in the absence of compound and 0.009 ± 0.002 S−1 in the presence of compounds). However, most of the Hit compounds affected the actin-activated ATPase of skeletal S1 (75% A1 and 25% A2) as well as cardiac S1 (100% A1) in a concentration-dependent manner. This is not surprising, because both skeletal and cardiac S1 contain predominantly the A1 isoform. Significant inhibition of actin-activated ATPase for both skeletal and cardiac myosin was observed for three compounds: fluphenazine, thioradizine, and novantrone (Fig. 6). Honokiol activated both. Flutamide, dantrolene, and carvediol had small and similar effects on both ATPases.
Figure 6.

Concentration dependence of the ATPase activity of acto-S1. A and B, skeletal acto-S1. C and D, cardiac acto-S1. Data are normalized to the value of the ATPase in the absence of the compounds. Error bars, S.D.
Some compounds showed significantly different effects on skeletal and cardiac myosin, suggesting the potential to identify isoform-specific effectors. Tegaserod had biphasic effects on cardiac acto-S1, and its inhibitory effects on skeletal acto-S1 were much more significant. Mefloquine activated cardiac acto-S1 at low concentrations but had no activating effect on skeletal acto-S1. Phenothiazine inhibited cardiac acto-S1, while slightly activating skeletal acto-S1.
Significant effectors show a good correlation of EC50 values determined from FRET and ATPase assays (Table 1). Fluphenazine, thioradizine, and novantrone reduced actin-activated ATPase of both skeletal and cardiac S1 by 50% at their EC50 values of 10–25 μm (Table 1). The decrease in ATPase activity induced by these three compounds was proportional to the decrease in FRET (Fig. 8, A and B), indicating that the functional effects are associated with structural changes in both species of the acto-S1 complex.
Figure 8.

Structure–function correlation. A, ATPase versus FRET change for skeletal acto-S1. B, ATPase versus FRET change for cardiac acto-S1. Significant inhibitors show that the FRET change is proportional to ATPase change. C, FRET change versus final anisotropy. D, FRET change versus phosphorescence lifetime. Compound-associated change in FRET is proportional to actin anisotropy, suggesting the compound-related change in actin structure. Error bars, S.D.
Transient phosphorescent anisotropy
Compound-induced changes in FRET (Fig. 5) suggested that these Hit compounds bind actin and modulate its structural properties. To test this possibility, we measured compound-induced changes in actin's structural dynamics using transient phosphorescent anisotropy (TPA) of actin labeled at Cys-374 with erythrosine iodoacetamide (ErIA) (22–24). Two parameters of TPA decay characterize actin's dynamics are (a) final anisotropy, related to the large-scale torsional flexibility of the filament (Fig. 7, A–D), and (b) lifetime, related to the probe environment near the C terminus of actin (Fig. 7, A, E, and F). Representative phosphorescence anisotropy decays in the presence of one of the Hit compounds, thioridazine (Fig. 7B), show concentration-dependent increase in the final anisotropy and decrease in signal intensity due to decrease in the phosphorescence lifetime. The effects of all hit compounds on both parameters of TPA decays are summarized in Fig. 7 (C–F). Most significant changes were observed in the presence of fluphenazine (Fig. 7, C and E), thioridazine, and novantrone (Fig. 7, D and F). The increase in actin anisotropy in the presence of fluphenazine and thioridazine suggests significant decrease in torsional flexibility of the filament. Novantrone-induced dramatic decrease in the anisotropy suggests large increase in torsional flexibility and/or fragmentation or depolymerization of actin filaments. Compounds that affected the anisotropy also decreased the erythrosine probe lifetime, suggesting that that changes in actin's flexibility are associated with changes in the probe environment, increasing exposure to the quenching effect of solvent oxygen. The changes in both anisotropy and phosphorescence lifetime were roughly proportional to changes in FRET efficiency (Fig. 8, C and D), suggesting that the compound-induced changes in FRET are associated with changes in actin structural dynamics. The compounds most effective in their effects on TPA and FRET were also most effective in changing actin-activated myosin ATPase (Fig. 6). Seven other compounds induced also significant but less pronounced changes in TPA and in actomyosin ATPase (Fig. 6), supporting previously observed correlations between actin dynamics and the mechanism of activation of myosin ATPase (10, 22, 23).
Figure 7.

TPA of ErIA-labeled actin. A, schematic representation of actin's microsecond dynamics detection by TPA measurements. IV and IH are vertically and horizontally polarized components of the emitted light used to calculate anisotropy (see “Experimental procedures”). B, TPA decays of ErIA-actin in the presence of increasing concentrations of thioridazine (0–100 μm), a representative compound. C and D, final anisotropy in the presence of hit compounds, which decreases with the amplitude of reflecting global rotational dynamics of the actin filament. E and F, phosphorescence lifetime of decays measured in the presence of compounds, reflecting local structural change near the probe binding site. Error bars, S.D.
Discussion
Time-resolved FRET showed that a fluorescently labeled 12-amino acid peptide (ANT), corresponding to the NTE of A1 ELC (Fig. 1), binds to the C-terminal region of actin with Kd = 16 μm. This actin–ANT interaction is affected by strong binding of S1 isoforms and increasing ionic strength (Fig. 3), suggesting overlap between ANT and myosin-binding regions on actin. ANT itself did not alter actin-activated or myofibrillar ATPase (Fig. 2, C and D). The lack of ANT effect on Km has two possible explanations: (a) although ANT and S1 compete for actin binding in the absence of ATP (Fig. 3B), the 16 μm affinity of ANT (Fig. 2B) for actin is about 2 orders of magnitude lower than that the submicromolar affinity of S1 for actin (4), and (b) ANT and S1 do not compete in the presence of ATP (Fig. 3B). The advantage of using low-affinity ANT to probe the actomyosin interface is that it is more likely to be susceptible to compete with low-concentration pharmaceutically active compounds, increasing the probability of detecting more “hit” compounds. Therefore, FRET between probes on actin (donor) and ANT (acceptor) was utilized as a sensor in a high-throughput screening assay to identify compounds that alter the actin–myosin interface. We identified 10 compounds from a small (NCC) library that affected FRET (Figs. 4 and 5) in a concentration-dependent manner. Most of them also altered the actin-activated myosin ATPase of both skeletal and cardiac S1 as well as the structural states of actin filament. Thus, we developed a actin-based high-throughput assay where FRET changes are associated with structural and functional changes in the whole acto-S1 complex.
Validity of actin–ANT FRET sensor
The detected distance (R = 3.3 ± 0.2 nm) between probes on actin and ANT in the actin–ANT complex is in good agreement with the distance observed between Cys-16 of A1 ELC (skeletal, 2.9 ± 0.2 nm; cardiac, 3.5 ± 0.2 nm) and Cys-374 of actin in the strongly bound acto-S1 complex (4, 5), indicating that ANT binds to the A1 ELC-binding regions of actin. This possibility is further supported by the effects of S1 binding on FRET signal between actin and ANT (Fig. 3). In the absence of ATP, binding of skeletal S1 (75% of A1 + 25% of A2), skeletal S1A1 (100% A1), and cardiac S1 (100% A1) to the actin–ANT complex significantly decreased FRET, whereas the addition of S1A2 (0% A1) did not alter the FRET signal. Thus, only muscle S1s having NTEs were capable of displacing the peptide from actin C-terminal region. On the other hand, FRET was not affected in the presence of ATP, where S1 binds weakly and is rotationally disordered, and the distance between A1NTE and actin significantly increases compared with that in the strongly bound complex (25, 26). FRET of actin–ANT complex was also significantly decreased by an increase in ionic strength (0.1 m KCl), further supporting the similarity between ANT–actin and actin–A1NTE interaction; a structural model of the acto-S1A1 complex (19) shows that the positively charged N terminus of A1 binds to a cluster of negatively charged residues in the C-terminal region of actin (19). Thus, we conclude that our actin–ANT FRET sensor (a) mimics well actin–A1NTE binding, (b) has potential as a platform for the discovery of allosteric modulators of the actin–myosin interaction, and (c) can ultimately help in developing therapies to treat muscle disorders.
Hit compounds identified in the HTS assay
The principal goal of this HTS is to identify novel small-molecule effectors with therapeutic potential for disorders associated with mutations in actin or myosin. Small-molecule effectors designed to target and modulate striated and smooth muscle myosin isoforms for the treatment of disease are showing promise in preclinical and clinical trials (11, 27). In our work, we focus on modulation of actin. We identified 10 pharmacologically active compounds that affected both actin's structure and functional interaction with myosin. The most pronounced effects were observed with fluphenazine, thioridazine, and novantrone. Fluphenazine and thioridazine are used as antipsychotic medicines (28), whereas novantrone is used to treat multiple sclerosis (29) (Fig. 8). The effects of other identified compounds are less pronounced but still significant, particularly at higher concentrations. Tegaserod (30) and mefloquine (31) are used to treat conditions like irritable bowel syndrome and malaria. Dantrolene is a post-synaptic muscle relaxant and works on the ryanodine receptor (32). Carvedilol is already known as a β-blocker and is used to treat high blood pressure and heart failure (33). Honokiol, a traditional medicine, has several applications (34). Flutamide is an antagonist for androgen receptor and has reported anti-cancer activity (35).
All identified compounds are currently used as medications. Whereas these medicines are therapeutically effective, they also have significant side effects on muscle function, such as cardiac arrhythmia (29, 33). Thus, it is not surprising that these compounds were identified as hits, as they are truly related to the alteration of muscle function. Some of the undesirable side effects may be associated with the effects reported here on actin structure and interaction with myosin (Fig. 8). An effective drug must achieve a balance between therapeutic benefit and undesired side effects. This balance probably depends on the relative binding affinities to actin and other cellular targets. Our hit compounds affect actin when present at micromolar concentrations, indicating moderate actin binding affinity. If binding to therapeutically desirable targets is much stronger, therapeutic effects may be achieved at low enough doses to avoid effects on actin structure. On the other hand, drug-induced alteration of actin structure can be beneficial in cancer research as a method of inhibiting actin cytoskeleton function in malignant cells; for example, Novantrone is used in chemotherapy of certain cancers (31).
Our results have pharmacological implications, aiding understanding of the undesirable side effects of many medications. Actin is present in every human cell, and its interaction with multiple myosin isoforms and multiple actin-binding proteins is essential for cellular viability. Although actin is a very conserved protein, different actin isoforms cannot be substituted in living tissues (36, 37). Our HTS assay uses the muscle actin–myosin system for detection of compounds, but these compounds may also bind to cellular actin. Our future goals are to screen larger libraries in search of drugs that are specific for actin isoforms (i.e. skeletal muscle versus smooth muscle versus cardiac muscle versus nonmuscle), as desired for safe clinical applications. Subsequent efforts in medicinal chemistry will be needed to enhance specificity and reduce side effects.
Mechanism of action
Compound-specific structural and functional changes in actin–ANT FRET, phosphorescence, and actin-activated ATPase (Figs. 6 and 7) suggest that different hit compounds bind at different regions of actin. The binding and functional effects of each compound depend on the structure of the compound and the structure of the targeted region of actin. For example, two of the identified compounds, fluphenazine and thioridazine, are structurally similar to trifluoperazine, a known myosin ATPase inhibitor (38), and have very similar effects on the actomyosin ATPase and TPA decays (Figs. 6 and 7). A few compounds had different effects on skeletal and cardiac myosin (Fig. 6), suggesting interaction with myosin isoform–specific regions of the actomyosin interface. This interface is probably different for skeletal and cardiac S1, as indicated by significant differences in actin-activated ATPase. We consider several possible mechanisms by which binding of hit compounds alters the interaction between the C terminus of actin and the NTE-binding site. Some compounds may bind to the C terminus of actin and alter its local structure. Other compounds may bind to other regions of actin and have allosteric effects. Other compounds may alter the microsecond flexibility of actin (22–24). Each mode of binding could result in displacement of ANT, as indicated by the decrease in FRET (Fig. 5), decrease in phosphorescence lifetime (Fig. 7) due to increased exposure of the probe to buffer oxygen, and/or changes in flexibility of actin due to local and allosteric changes in the monomers. Examples of long-range allosteric changes in actin were previously documented using site-specific modifications (39). Our previous studies on actin-binding proteins demonstrated coupling between changes in actin flexibility (measured by TPA) and functional interactions with myosin and other actin-binding proteins (22–24). This kind of coupling is also observed in the present studies (Fig. 8).
None of these compounds has any effect on basal Mg-ATPase (without actin) of either skeletal or cardiac S1, confirming their actin specificity. In addition, FRET concentration–response with two known myosin-specific compounds (11, 27) showed no effects (Fig. 5), providing further indication that compounds identified in this screen act on actin to affect actomyosin function. This is unique, because previous drug discovery campaigns in this field used myosin as the primary target (11, 27). Our future screening of larger libraries will focus on finding compounds that are specific for skeletal or cardiac muscle.
Conclusion
Using the high precision of our TR-FRET technology, coupled with structure-based design of an intermolecular FRET biosensor involving actin and ANT, we have established and validated an HTS platform that can detect small-molecule effectors that affect the functional interaction of actin and myosin. This tool can be used in the future to screen larger libraries and sets the groundwork for the discovery of allosteric modulators of other actin-binding proteins of interest where protein-protein interactions are targeted.
Experimental procedures
Protein preparations and labeling
Actin was prepared from rabbit skeletal muscle by extracting acetone powder in cold water, as described previously (23). Cys-374 in F-actin was labeled with the FRET donor/phosphorescent probe as follows. FM/ErIA (Life Technologies, Inc.), freshly dissolved in N,N-dimethylformamide (500 μm), was mixed with 50 μm F-actin and incubated for 1 h at 25 °C, followed by 18 h at 4 °C. Labeling was terminated by adding 10 mm DTT. After 30 min of sedimentation at 350,000 × g, the F-actin pellet was suspended in G-Mg buffer (5 mm Tris, 0.5 mm ATP, 0.2 mm MgCl2, pH 7.5) followed by clarification at 300,000 × g for 10 min. Actin was again polymerized for 30 min at 25 °C in the presence of 3 mm MgCl2 and centrifuged at 300,000 × g for 10 min, and the pellet was suspended in F-Mg buffer (3 mm MgCl2, 10 mm Tris, pH 7.5) containing 0.2 mm ATP. The labeled F-actin was immediately stabilized against depolymerization and denaturation by adding 1 molar eq of phalloidin. Skeletal muscle S1 was obtained by α-chymotryptic digestion of rabbit skeletal muscle myosin (10), and β-cardiac S1 was prepared by α-chymotryptic digestion of bovine β-cardiac myosin (5) as described previously. Isoforms of skeletal muscle S1, S1A1 and S1A2, were purified as reported in our earlier study (10).
The N-terminal peptide
The N-terminal peptide of rabbit skeletal A1 ELC was synthesized and purified by the LifeTein Co. Unlabeled peptide was acetylated at the N terminus and amidated at the C terminus: Ac-APKKDVKKPVAA-NH2 (Mr 1292.6). The labeled peptide had dabcyl conjugated to the first A at the N terminus, dabcyl-APKKDVKKPVAA-NH2 (Fig. 1), and amidated C terminus (Mr 1501.85). The synthetized peptides were purified and converted to HCl salts to remove residual salts from the synthesis procedure and were provided as lyophilized powder. The peptide was >99% pure, as determined by HPLC and MS. Before experiments, peptides were dissolved in H2O (added directly to the vial), aliquoted, and stored at −20 °C. The final concentration of unlabeled peptide was determined using the provided molecular weight, and the concentration of dabcyl-labeled peptide (ANT) was calculated by measuring absorbance at 434 nm, using the molar extinction coefficient of dabcyl, 22,850 cm−1 m−1.
Fluorescence data acquisition
Fluorescence lifetime measurements were carried out by a high-precision FLTPR (Fluorescence Innovations, Inc., Minneapolis, MN) (13–15). FM-labeled donor actin was excited with a 473-nm microchip laser (Bright Solutions, Cura Carpignano, Italy), and emission was filtered with 488-nm long pass and 517/20-nm band pass filters (Semrock, Rochester, NY) (13, 18). This instrument enables high-throughput fluorescence lifetime detection at high precision by utilizing a unique direct waveform-recording technology (40). The performance of this FLTPR has been previously demonstrated with FRET-based HTS that targets sarcoplasmic reticulum Ca-ATPase (SERCA) and the ryanodine receptor (14, 15).
Pilot screen with NCC library
The NCC compounds (727 compounds) were received in 96-well plates and were reformatted into 384-well polypropylene intermediate plates (Greiner Bio-One, Kremsmunster, Austria) using a multichannel liquid handler, BioMek FX (Beckman Coulter, Miami, FL). Assay plates were prepared by transferring 50 nl of the 10 mm compound stocks or DMSO from the source plates to 384-well black polypropylene plates (Greiner), using an Echo 550 acoustic dispenser (Labcyte). NCC compounds were formatted in three plates, with the first two and last two columns loaded with 50 nl of DMSO and used for drug-free controls. Final concentration of the compounds was 10 μm. These assay plates were then heat-sealed using a PlateLoc Thermal Microplate Sealer (Agilent Technologies, Santa Clara, CA) and stored at −20 °C. Before screening, compound plates were equilibrated to room temperature (25 °C). Before screening, 2 μm FM (donor)-labeled actin without or with 50 μm acceptor labeled (ANT) peptide (50 μl/well) was dispensed by a Multidrop Combi Reagent Dispenser (Thermo Fisher Scientific) into the 384-well assay plates containing the compounds. Plates were incubated at room temperature for 20 min before recording data with the FLTPR.
TR-FRET and HTS data analysis
TR-FRET waveforms were analyzed globally by model-independent and -dependent methods as described previously (4, 5). The observed donor-only (FM actin) waveform FDobs(t) was fitted by a simulation FDsim(t), consisting of a multiexponential decay FD(t) (Fig. 3A, black) convolved with the instrument response function IRF(t). The observed donor + acceptor waveform (FM actin + ANT) FD+Aobs(t) (Fig. 3A, red, blue, magenta, and green) was fitted by a multiexponential function using the same approach. The model-independent ensemble-average FRET efficiency 〈E〉, which is equivalent to the result of a steady-state fluorescence measurement, is given by the equation, 〈E〉 = 1 − 〈τDA〉/〈τD〉, where brackets indicate average. The model-dependent structural state was resolved using the following,
| (Eq. 1) |
| (Eq. 2) |
| (Eq. 3) |
| (Eq. 4) |
where FDA(t) is the time-resolved fluorescence waveform of the donor-acceptor complex, XDA is the fraction of actin-attached donors that are bound to and transferring energy to acceptor-labeled ANT, R is the effective donor-acceptor distance in the complex, and R0 is the distance (3.6 nm) at which FRET is 50% for this donor–acceptor pair (4).
Waveforms for each well in HTS were fitted by a two-exponential decay function using least-squares minimization. The FRET efficiency E was determined as the fractional decrease in donor fluorescence lifetime due to the acceptor, E = 1 − 〈τDA〉/〈τDA〉.
Assay quality was determined based on controls (DMSO-only samples) on each plate, as indexed by the Z′ factor, a value of 0.5 or higher indicating excellent assay quality (14, 20). A compound was considered a hit if it changed E by >4 S.D. relative to that of control samples (E0) that were exposed to 0.1% DMSO. The threshold may be further adjusted to constrain the number of hits according to the resources available for evaluation via secondary (orthogonal) assays.
FRET concentration–response assay
The hit compounds were dissolved in DMSO to make a 10 mm stock solution, which was serially diluted in 96-well mother plates. Hits were screened at eight concentrations (0.5–100 μm). Compounds (1 μl) were transferred from the mother plates into 384-well assay plates using a Mosquito HV liquid handler (TTP Labtech Ltd., Hertfordshire, UK). The same procedure of dispensing as for the pilot screening was applied in the FRET concentration–response assays. Concentration dependence of the FRET change was fitted using the Hill equation (21),
| (Eq. 5) |
where E and E0 are FRET in the presence and in the absence of the compound, Emax is the maximum effect, C is the drug concentration, EC50 is the drug concentration for which 50% of maximum effect is obtained, and α is the Hill coefficient of sigmoidicity. Concentration–response assays of two known myosin-specific compounds, OM and Myk 461, were also done as controls to validate the structural specificity of the compounds.
Actin-activated ATPase
Actin-activated S1 ATPase activity was measured at 25 °C in F-Mg buffer containing 3 mm ATP with 2 μm F-actin and 5 μm skeletal/cardiac S1. Concentration–response of drugs (0–100 μm) on ATPase was measured in the presence of the compounds, and the EC50 was determined by fitting the data to a Hill plot. The ATPase concentration–response data were plotted as normalized with reference to the ATPase of actomyosin in the absence of compounds (Fig. 6). Basal Mg-ATPase of S1 was measured under the same conditions in the absence of actin.
Transient phosphorescent anisotropy
Phalloidin-stabilized ErIA–F-actin was diluted in F-Mg buffer to 2.0 μm, and compounds were added at specified concentrations (0–100 μm). To maximize phosphorescence signals and prevent photobleaching of the dye, oxygen was removed from the sample by a 5-min incubation with glucose oxidase (55 μg/ml), catalase (36 μg/ml), and glucose (45 μg/ml). Phosphorescence was measured at 25 °C as described previously (10). ErIA actin was excited with a vertically polarized 1.2-ns pulse from an FDSS 532-150 laser (CryLas) at 532 nm, operating at a repetition rate of 100 Hz. Phosphorescence emission was selected by a 670-nm glass cut-off filter (Corion), detected by a photomultiplier (R928, Hamamatsu), and digitized by a transient digitizer (CompuScope 14100, GaGe) at a time resolution of 1 μs/channel. The TPA decay was calculated as r(t) = (IV(t) − GIH(t))/(IV(t) + 2GIH(t)), where IV(t) and IH(t) are vertically and horizontally polarized components of the emission signal, detected at 90° with a single detector and a polaroid sheet polarizer that alternated between the two orientations every 500 laser pulses. G is an instrumental correction factor, determined by performing the measurement with horizontally polarized excitation, for which the corrected anisotropy value is set to zero. TPA decays were obtained by recording 10 cycles of 1000 pulses (500 in each orientation of the polarizer), corresponding to a total acquisition time of about 2 min. TPA decays were analyzed by calculating the final anisotropy (r∞), defined as the average value of r in the time window from 400 to 500 μs, which has been shown previously to provide the most sensitive and precise measurement of actin's microsecond rotational dynamics (10). The phosphorescence intensity and mean lifetime were calculated as described previously (41).
Author contributions
P. G., E. P., and D. D. T. designed the research. P. G. and E. P. prepared samples, performed biochemical experiments, and analyzed the data. K. C. P. designed, constructed, and maintained the FLTPR. P. G., E. P., and B. D. G. acquired and analyzed fluorescence data. P. G., E. P., and D. D. T. wrote the paper.
Acknowledgments
Fluorescence experiments were performed at the Biophysical Technology Center, University of Minnesota. We thank Samantha Yuen for technical assistance, Dr. Ben Binder for critical review, and Octavian Cornea for preparation of the manuscript.
This work was supported by National Institutes of Health Grants R01 HL129814, R01 AR32961, and R37 AG26160 (to D. D. T.) and R42 DA037622 (to Fluorescence Innovations, Inc.; subcontract to the University of Minnesota). D. D. T. holds equity in, and serves as President of, Photonic Pharma LLC. This relationship has been reviewed and managed by the University of Minnesota. Photonic Pharma had no role in this study. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
- ELC
- essential light chain
- NTE
- N-terminal extension
- TR-FRET
- time-resolved FRET
- S1
- subfragment 1
- FM
- fluorescein 5-maleimide
- HTS
- high-throughput screening
- FLTPR
- fluorescence lifetime plate reader
- TPA
- transient phosphorescent anisotropy
- ErIA
- erythrosine iodoacetamide
- NCC
- National Institutes of Health Clinical Collection.
References
- 1. Thomas D. D., Muretta J. M., Colson B. A., Mello R. N., and Kast D. (2012) Spectroscopic probes of muscle proteins. in Comprehensive Biophysics (Edward H. E., ed) pp. 226–250, Elsevier, Amsterdam [Google Scholar]
- 2. Sweeney H. L., Bowman B. F., and Stull J. T. (1993) Myosin light chain phosphorylation in vertebrate striated muscle: regulation and function. Am. J. Physiol. 264, C1085–C1095 10.1152/ajpcell.1993.264.5.C1085 [DOI] [PubMed] [Google Scholar]
- 3. Lowey S., Waller G. S., and Trybus K. M. (1993) Skeletal muscle myosin light chains are essential for physiological speeds of shortening. Nature 365, 454–456 10.1038/365454a0 [DOI] [PubMed] [Google Scholar]
- 4. Guhathakurta P., Prochniewicz E., and Thomas D. D. (2015) Amplitude of the actomyosin power stroke depends strongly on the isoform of the myosin essential light chain. Proc. Natl. Acad. Sci. U.S.A. 112, 4660–4665 10.1073/pnas.1420101112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guhathakurta P., Prochniewicz E., Roopnarine O., Rohde J. A., and Thomas D. D. (2017) A cardiomyopathy mutation in the myosin essential light chain alters actomyosin structure. Biophys. J. 113, 91–100 10.1016/j.bpj.2017.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hernandez O. M., Jones M., Guzman G., and Szczesna-Cordary D. (2007) Myosin essential light chain in health and disease. Am. J. Physiol. Heart Circ. Physiol. 292, H1643–H1654 10.1152/ajpheart.00931.2006 [DOI] [PubMed] [Google Scholar]
- 7. Sweeney H. L. (1995) Function of the N terminus of the myosin essential light chain of vertebrate striated muscle. Biophys. J. 68, 112S–118S; discussion 118S–119S [PMC free article] [PubMed] [Google Scholar]
- 8. Wagner P. D., Slater C. S., Pope B., and Weeds A. G. (1979) Studies on the actin activation of myosin subfragment-1 isoezymes and the role of myosin light chains. Eur. J. Biochem. 99, 385–394 10.1111/j.1432-1033.1979.tb13267.x [DOI] [PubMed] [Google Scholar]
- 9. Bottinelli R., Betto R., Schiaffino S., and Reggiani C. (1994) Unloaded shortening velocity and myosin heavy chain and alkali light chain isoform composition in rat skeletal muscle fibres. J. Physiol. 478, 341–349 10.1113/jphysiol.1994.sp020254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prochniewicz E., Guhathakurta P., and Thomas D. D. (2013) The structural dynamics of actin during active interaction with myosin depends on the isoform of the essential light chain. Biochemistry 52, 1622–1630 10.1021/bi3014467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Malik F. I., Hartman J. J., Elias K. A., Morgan B. P., Rodriguez H., Brejc K., Anderson R. L., Sueoka S. H., Lee K. H., Finer J. T., Sakowicz R., Baliga R., Cox D. R., Garard M., Godinez G., et al. (2011) Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science 331, 1439–1443 10.1126/science.1200113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cornea R. L., Gruber S. J., Lockamy E. L., Muretta J. M., Jin D., Chen J., Dahl R., Bartfai T., Zsebo K. M., Gillispie G. D., and Thomas D. D. (2013) High-throughput FRET assay yields allosteric SERCA activators. J. Biomol. Screen. 18, 97–107 10.1177/1087057112456878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gruber S. J., Cornea R. L., Li J., Peterson K. C., Schaaf T. M., Gillispie G. D., Dahl R., Zsebo K. M., Robia S. L., and Thomas D. D. (2014) Discovery of enzyme modulators via high-throughput time-resolved FRET in living cells. J. Biomol. Screen. 19, 215–222 10.1177/1087057113510740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rebbeck R. T., Essawy M. M., Nitu F. R., Grant B. D., Gillispie G. D., Thomas D. D., Bers D. M., and Cornea R. L. (2017) High-throughput screens to discover small-molecule modulators of ryanodine receptor calcium release channels. SLAS Discov. 22, 176–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schaaf T. M., Peterson K. C., Grant B. D., Bawaskar P., Yuen S., Li J., Muretta J. M., Gillispie G. D., and Thomas D. D. (2017) High-throughput spectral and lifetime-based FRET screening in living cells to identify small-molecule effectors of SERCA. SLAS Discov. 22, 262–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hayashibara T., and Miyanishi T. (1994) Binding of the amino-terminal region of myosin alkali 1 light chain to actin and its effect on actin-myosin interaction. Biochemistry 33, 12821–12827 10.1021/bi00209a013 [DOI] [PubMed] [Google Scholar]
- 17. Haase H., Dobbernack G., Tünnemann G., Karczewski P., Cardoso C., Petzhold D., Schlegel W. P., Lutter S., Pierschalek P., Behlke J., and Morano I. (2006) Minigenes encoding N-terminal domains of human cardiac myosin light chain-1 improve heart function of transgenic rats. FASEB J. 20, 865–873 10.1096/fj.05-5414com [DOI] [PubMed] [Google Scholar]
- 18. Petersen K. J., Peterson K. C., Muretta J. M., Higgins S. E., Gillispie G. D., and Thomas D. D. (2014) Fluorescence lifetime plate reader: resolution and precision meet high-throughput. Rev. Sci. Instrum. 85, 113101 10.1063/1.4900727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aydt E. M., Wolff G., and Morano I. (2007) Molecular modeling of the myosin-S1(A1) isoform. J. Struct. Biol. 159, 158–163 10.1016/j.jsb.2007.04.002 [DOI] [PubMed] [Google Scholar]
- 20. Zhang J. H., Chung T. D., and Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73 10.1177/108705719900400206 [DOI] [PubMed] [Google Scholar]
- 21. Goutelle S., Maurin M., Rougier F., Barbaut X., Bourguignon L., Ducher M., and Maire P. (2008) The Hill equation: a review of its capabilities in pharmacological modelling. Fundam. Clin. Pharmacol. 22, 633–648 10.1111/j.1472-8206.2008.00633.x [DOI] [PubMed] [Google Scholar]
- 22. Prochniewicz E., Chin H. F., Henn A., Hannemann D. E., Olivares A. O., Thomas D. D., and De La Cruz E. M. (2010) Myosin isoform determines the conformational dynamics and cooperativity of actin filaments in the strongly bound actomyosin complex. J. Mol. Biol. 396, 501–509 10.1016/j.jmb.2009.11.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prochniewicz E., Walseth T. F., and Thomas D. D. (2004) Structural dynamics of actin during active interaction with myosin: different effects of weakly and strongly bound myosin heads. Biochemistry 43, 10642–10652 10.1021/bi049914e [DOI] [PubMed] [Google Scholar]
- 24. Prochniewicz E., Zhang Q., Howard E. C., and Thomas D. D. (1996) Microsecond rotational dynamics of actin: spectroscopic detection and theoretical simulation. J. Mol. Biol. 255, 446–457 10.1006/jmbi.1996.0037 [DOI] [PubMed] [Google Scholar]
- 25. Thomas D. D., and Cooke R. (1980) Orientation of spin-labeled myosin heads in glycerinated muscle fibers. Biophys. J. 32, 891–906 10.1016/S0006-3495(80)85024-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walker M., Zhang X. Z., Jiang W., Trinick J., and White H. D. (1999) Observation of transient disorder during myosin subfragment-1 binding to actin by stopped-flow fluorescence and millisecond time resolution electron cryomicroscopy: evidence that the start of the crossbridge power stroke in muscle has variable geometry. Proc. Natl. Acad. Sci. U.S.A. 96, 465–470 10.1073/pnas.96.2.465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Green E. M., Wakimoto H., Anderson R. L., Evanchik M. J., Gorham J. M., Harrison B. C., Henze M., Kawas R., Oslob J. D., Rodriguez H. M., Song Y., Wan W., Leinwand L. A., Spudich J. A., McDowell R. S., Seidman J. G., and Seidman C. E. (2016) A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351, 617–621 10.1126/science.aad3456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stöllberger C., Huber J. O., and Finsterer J. (2005) Antipsychotic drugs and QT prolongation. Int. Clin. Psychopharmacol. 20, 243–251 10.1097/01.yic.0000166405.49473.70 [DOI] [PubMed] [Google Scholar]
- 29. Martinelli Boneschi F., Vacchi L., Rovaris M., Capra R., and Comi G. (2013) Mitoxantrone for multiple sclerosis. Cochrane Database Syst. Rev. CD002127 10.1002/14651858.CD002127.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Layer P., Keller J., Loeffler H., and Kreiss A. (2007) Tegaserod in the treatment of irritable bowel syndrome (IBS) with constipation as the prime symptom. Ther. Clin. Risk Manag. 3, 107–118 10.2147/tcrm.2007.3.1.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tickell-Painter M., Maayan N., Saunders R., Pace C., and Sinclair D. (2017) Mefloquine for preventing malaria during travel to endemic areas. Cochrane Database Syst. Rev. 10, CD006491 10.1002/14651858.CD006491.pub4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao F., Li P., Chen S. R., Louis C. F., and Fruen B. R. (2001) Dantrolene inhibition of ryanodine receptor Ca2+ release channels: molecular mechanism and isoform selectivity. J. Biol. Chem. 276, 13810–13816 10.1074/jbc.M006104200 [DOI] [PubMed] [Google Scholar]
- 33. Dézsi C. A., and Szentes V. (2017) The real role of β-blockers in daily cardiovascular therapy. Am. J. Cardiovasc. Drugs 17, 361–373 10.1007/s40256-017-0221-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Woodbury A., Yu S. P., Wei L., and García P. (2013) Neuro-modulating effects of honokiol: a review. Front. Neurol. 4, 130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Elzoghby A. O., Saad N. I., Helmy M. W., Samy W. M., and Elgindy N. A. (2013) Ionically-crosslinked milk protein nanoparticles as flutamide carriers for effective anticancer activity in prostate cancer-bearing rats. Eur. J. Pharm. Biopharm. 85, 444–451 10.1016/j.ejpb.2013.07.003 [DOI] [PubMed] [Google Scholar]
- 36. Kumar A., Crawford K., Close L., Madison M., Lorenz J., Doetschman T., Pawlowski S., Duffy J., Neumann J., Robbins J., Boivin G. P., O'Toole B. A., and Lessard J. L. (1997) Rescue of cardiac α-actin-deficient mice by enteric smooth muscle γ-actin. Proc. Natl. Acad. Sci. U.S.A. 94, 4406–4411 10.1073/pnas.94.9.4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jaeger M. A., Sonnemann K. J., Fitzsimons D. P., Prins K. W., and Ervasti J. M. (2009) Context-dependent functional substitution of α-skeletal actin by γ-cytoplasmic actin. FASEB J. 23, 2205–2214 10.1096/fj.09-129783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sellers J. R., Wang F., and Chantler P. D. (2003) Trifluoperazine inhibits the MgATPase activity and in vitro motility of conventional and unconventional myosins. J. Muscle Res. Cell Motil. 24, 579–585 10.1023/B:JURE.0000009969.04562.58 [DOI] [PubMed] [Google Scholar]
- 39. Crosbie R. H., Miller C., Cheung P., Goodnight T., Muhlrad A., and Reisler E. (1994) Structural connectivity in actin: effect of C-terminal modifications on the properties of actin. Biophys. J. 67, 1957–1964 10.1016/S0006-3495(94)80678-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muretta J. M., Kyrychenko A., Ladokhin A. S., Kast D., Gillispie G. D., and Thomas D. D. (2010) High-performance time-resolved fluorescence by direct waveform recording. Rev. Sci. Instrum. 81, 103101 10.1063/1.3480647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Prochniewicz E., Pierre A., McCullough B. R., Chin H. F., Cao W., Saunders L. P., Thomas D. D., and De La Cruz E. M. (2011) Actin filament dynamics in the actomyosin VI complex is regulated allosterically by calcium-calmodulin light chain. J. Mol. Biol. 413, 584–592 10.1016/j.jmb.2011.08.058 [DOI] [PMC free article] [PubMed] [Google Scholar]