

Abstract
Certain dysregulated chondrocyte metabolic adaptive responses such as decreased activity of the master regulator of energy metabolism AMP-activated protein kinase (AMPK) promote osteoarthritis (OA). Metabolism intersects with epigenetic and transcriptional responses. Hence, we studied chondrocyte ATP-citrate lyase (ACLY), which generates acetyl-CoA from mitochondrial-derived citrate, and modulates acetylation of histones and transcription factors. We assessed ACLY in normal and OA human knee chondrocytes and cartilages by Western blotting and immunohistochemistry, and quantified acetyl-CoA fluorometrically. We examined histone and transcription factor lysine acetylation by Western blotting, and assessed histone H3K9 and H3K27 occupancy of iNOS, MMP3, and MMP13 promoters by chromatin immunoprecipitation (ChIP) and quantitative PCR (qPCR). We analyzed iNOS, MMP3, MMP13, aggrecan (ACAN), and Col2a1 gene expression by RT-qPCR. Glucose availability regulated ACLY expression and function, nucleocytosolic acetyl-CoA, and histone acetylation. Human knee OA chondrocytes exhibited increased ACLY activation (assessed by Ser-455 phosphorylation), associated with increased H3K9 and H3K27 acetylation. Inhibition of ACLY attenuated IL-1β–induced transcription of iNOS, MMP3, and MMP13 by suppressing acetylation of p65 NF-κB, H3K9, and H3K27, blunted release of NO, MMP3, and MMP13, and also reduced SOX9 acetylation that promoted SOX9 nuclear translocation, leading to increased aggrecan and Col2a1 mRNA expression. ACLY is a novel player involved in regulation of cartilage matrix metabolism. Increased ACLY activity in OA chondrocytes increased nucleocytosolic acetyl-CoA, leading to increased matrix catabolism via dysregulated histone and transcription factor acetylation. Pharmacologic ACLY inhibition in OA chondrocytes globally reverses these changes and stimulates matrix gene expression and AMPK activation, supporting translational investigation in OA.
Keywords: acetyl coenzyme A (acetyl-CoA), acetylation, AMP-activated kinase (AMPK), epigenetics, chondrocyte, ATP-citrate lyase, hydroxycitric acid, nucleocytosolic acetyl-CoA
Introduction
Osteoarthritis (OA)2 is a major and growing public health problem (1). The disease is promoted by factors including aging, biomechanical joint trauma, and changes in systemic metabolism, including obesity and insulin resistance (1–4). As OA progresses, failure of the synovial joint organ frequently develops, with degeneration of articular cartilage as a core disease feature (1). Chondrocytes, the sole cells in articular hyaline cartilage, are responsible for maintaining the homeostatic balance between extracellular matrix anabolism and catabolism (1). Dysfunction of chondrocytes in OA, amplified by local inflammatory and biomechanical injury processes, leads to an excess of matrix catabolic activity, medicated by factors including MMPs and aggrecanases (1, 3).
The avascular, hypoxic environment that articular chondrocytes reside in renders cartilage homeostasis more challenging due to cellular nutritional and metabolic demands (3). In OA, chondrocyte responses to changes in glucose and other available nutrients include transition from resting to a highly metabolically active state (3), associated with development of excess glycolysis, and decreased mitochondrial function with less oxidative phosphorylation, and consequent pro-inflammatory effects (3, 4). As OA advances, altered chondrocyte energy balance is compounded by changes in physiologic chondrocyte nutritional biosensing responses (3, 4). A prime example is decrease in aging, biomechanically injured, and OA chondrocytes in vitro and in vivo of chondrocyte activity of AMPK (5, 6), a master regulator of energy balance (7). AMPK activation inhibits inflammatory and matrix catabolic responses in chondrocytes, partly mediated by inhibition of the master inflammation transcription factor NF-κB (5). Inducible chondrocyte-specific AMPKα knockout (KO) mice develop accelerated mechanical injury–induced and age–related spontaneous knee OA (8).
Metabolism intersects with certain epigenetic and transcription factor modifications mediating cellular re-programming (9). In this light, epigenetic changes that promote OA include not only altered chondrocyte gene methylation (10), but also modification of certain transcriptional responses by histone deacetylases (10). Deacetylation of nuclear histones and transcription factors that impacts gene expression (11, 12), as well as cytosolic protein deacetylation, are countered by acetylation utilizing donor acetyl-CoA, a key metabolic intermediate (11, 12). Mitochondria generate acetyl-CoA via glucose, fatty acid, and amino acid catabolism (11, 12). Acetyl-CoA conveys carbon atoms into the tricarboxylic acid (TCA) cycle for ATP production (11, 12). The TCA cycle intermediate citrate can be exported from mitochondria into the cytosol and nucleus, where ATP citrate lyase (ACLY) converts citrate back to acetyl-CoA (11–13).
Acetyl-CoA abundance in distinct subcellular compartments reflects, in part, the bioenergetic state of the cell (11, 12), rising in nucleus and cytosol when glucose and other carbon sources are abundant (11, 12). Conversely, nutrient deprivation imposes increased requirement for acetyl-CoA residence in the mitochondria for ATP synthesis (11, 12), and nucleocytosolic acetyl-CoA levels decrease (11, 12). ACLY is shown to be responsible for generation of the majority of nucleocytosolic acetyl-CoA in mammalian cells (11–13). Because histone acetylation alters accessibility of chromatin and allows DNA-binding proteins to interact with exposed sites to activate gene transcription and downstream cellular functions (13, 14), ACLY plays a critical role in integrating the cell metabolic state to gene transcription (14). ACLY is essential for growth and development (15). However, ACLY expression and activity are increased in tumor cells and also can be induced by inflammatory mediators in immune cells (16, 17). Moreover, inhibition of ACLY prevents tumor growth (18) and suppresses inflammatory responses (17). As such, overactive ACLY may have a detrimental effect after growth and development.
Altered ACLY activity has also been observed in some metabolic disorders, including dyslipidemia and impaired glucose tolerance (13, 19). Several core features of metabolic syndrome (dyslipidemia, obesity, hyperglycemia, and insulin resistance) have been linked to OA (2–4). Because OA chondrocytes exhibit altered metabolism (3, 4), we studied expression, activity, and the role of ACLY in human knee OA articular chondrocytes. We included translationally pertinent studies of pharmacologic ACLY inhibition in OA chondrocytes in vitro, using the naturally occurring and potent ACLY inhibitor (−)-hydroxycitric acid (HCA), an orally bioavailable citrate analog and caloric restriction mimetic (20). We identified ACLY as a potentially novel metabolic target for OA, as our results suggest a potential role of increased ACLY activity in chondrocytes in OA pathogenesis, and demonstrate that pharmacologically targeting ACLY reverses global changes in chondrocyte from human knee OA cartilages.
Results
Glucose availability in articular chondrocytes influenced the amount of nucleocytosolic acetyl-CoA and histone acetylation in an ACLY-dependent manner
Because ACLY plays an important role linking glucose metabolism to chromatin modification (14), we examined how ACLY expression and function, and epigenetic changes, intersected with glucose availability in articular chondrocytes. To do so, we first glucose-deprived and then glucose-replenished cultured normal primary human knee chondrocytes. Glucose starvation induced a marked decrease in total cellular acetyl-CoA, driven mostly by loss of nucleocytosolic acetyl-CoA. No significant reduction in mitochondrial acetyl-CoA level was seen (Fig. 1A); similar to previous findings that the mitochondrial pool of acetyl-CoA is less susceptible to variations than its nucleocytosolic counterpart (20, 21). Glucose re-feeding rapidly drove an increase of total cellular acetyl-CoA (Fig. 1A). Again, this was mostly due to an increase of nucleocytosolic acetyl-CoA (Fig. 1A). Under the conditions of glucose deprivation, glycolysis was inhibited, indicated by blunted expression of PKM2 (pyruvate kinase isoenzyme M2), which catalyzes the pyruvate-generating final step in glycolysis (Fig. 1B). In parallel, glucose deprivation decreased ACLY expression (Fig. 1B) and lowered acetylation of specific lysine residues 9 and 27 of histone 3 (Ac-H3K9 and Ac-H3K27) (Fig. 1B). Each of these responses was reversed after glucose re-feeding (Fig. 1B). Because acetylation of H3K9 and H3K27 are generally associated with increased gene transcription (22, 23), changes in levels of acetylation of H3K9 and H3K27 in chondrocytes could have an impact on gene expression. We also observed that re-feeding the glucose-starved chondrocytes with glucose at 10 mm, but not at 1 mm, sufficiently rescued total acetyl-CoA (Fig. S1). Similarly, this was mostly due to recovery of nucleocytosolic acetyl-CoA (Fig. S1). Pharmacological ACLY inhibitor HCA at 20 mm concentration has been shown to inhibit ACLY in other cell types (20, 21). We found that HCA at the same concentration, which was confirmed to have no effect on chondrocyte cytotoxicity (data not shown), significantly blocked glucose re-feeding–induced recovery of nucleocytosolic and total acetyl-CoA (Fig. 1C), and Ac-H3K9 and Ac-H3K27 levels in glucose-starved chondrocytes (Fig. 1D). This was associated with attenuated ACLY phosphorylation at Ser-455, which enhances ACLY catalytic activity (13), and ACLY protein expression (Fig. 1D), suggesting ACLY-dependent histone acetylation in chondrocytes.
Figure 1.

Glucose availability in articular chondrocytes influenced nucleocytosolic acetyl-CoA and histone acetylation in an ACLY-dependent manner. Cultured normal primary human knee chondrocytes in DMEM containing 25 mm glucose were subjected to glucose starvation in DMEM without glucose for 5 h, and then placed back in the DMEM containing 25 mm glucose for 1 and 5 h (A and B) or the DMEM containing 10 mm glucose for 5 h in the presence or absence of HCA (20 mm) (C and D). The amounts of total and compartmental (mitochondrial and nucleocytosolic) acetyl-CoA were measured (A and C). Whole cell lysates and nuclear proteins from these cells were studied by Western blotting for expression of PKM2, ACLY, phosphor-ACLY (Ser-455), Ac-H3K9, and Ac-H3K27 (C and D). β-Actin and histone H3 served as loading controls. Semi-quantitative densitometry analysis of each Western blotting was performed over 3 individual experiments with 3 different donors (B and D). Two-way ANOVA (A and C) and one-way ANOVA (B and D) followed by Bonferroni's post hoc test were used for statistical data analysis. p values represent comparisons of the mean ± S.D.
ACLY is up-regulated in human knee OA chondrocytes/cartilage
Because human OA chondrocytes have an increased capacity of glycolysis (24), we next examined ACLY phosphorylation and expression in either human knee cartilage sections or primary human knee chondrocytes from both normal (grade 0–1) and OA donors (grade 4). Immunohistochemistry (IHC) analysis of cartilage sections demonstrated that ACLY phosphorylation (Ser-455) and expression were low in normal donors, but increased in OA donors, evidenced by in situ positive staining in almost all chondrocytes, particularly prominent in chondrocyte clusters (Fig. 2). Notably, more cells stained positively for both phosphorylated ACLY (Ser-455) and ACLY in donor 2 (age 45), compared with donor 1 (age 24) in normal cartilage sections (Fig. 2). Western blot analysis of primary human knee chondrocytes also showed markedly increased phosphorylation of ACLY (Ser-455) in all 5 OA donors, when compared with 5 normal donors (Fig. 3, top panel). This was correlated with increased levels of total acetyl-CoA (data not shown) and acetylation of H3K9 and H3K27 in OA chondrocytes, compared with normal chondrocytes (Fig. 3A). Semi-quantitative densitometry analysis of Western blots of 28 normal donors (20 male and 8 female, age 42.3 ± 10.6) and 28 OA donors (10 male and 18 female, age 67.8 ± 7.9) of primary human knee chondrocytes further confirmed that ACLY phosphorylation (Ser-455) and acetylation of H3K9 and H3K27 were significantly increased in OA chondrocytes (Fig. 3B). Levels of ACLY phosphorylation (Ser-455) and expression were not much different between male and female in either normal or OA donors studied, but seemed to increase slightly in normal donors with age. This was in agreement with the IHC results, suggesting a potential age-related effect on ACLY.
Figure 2.
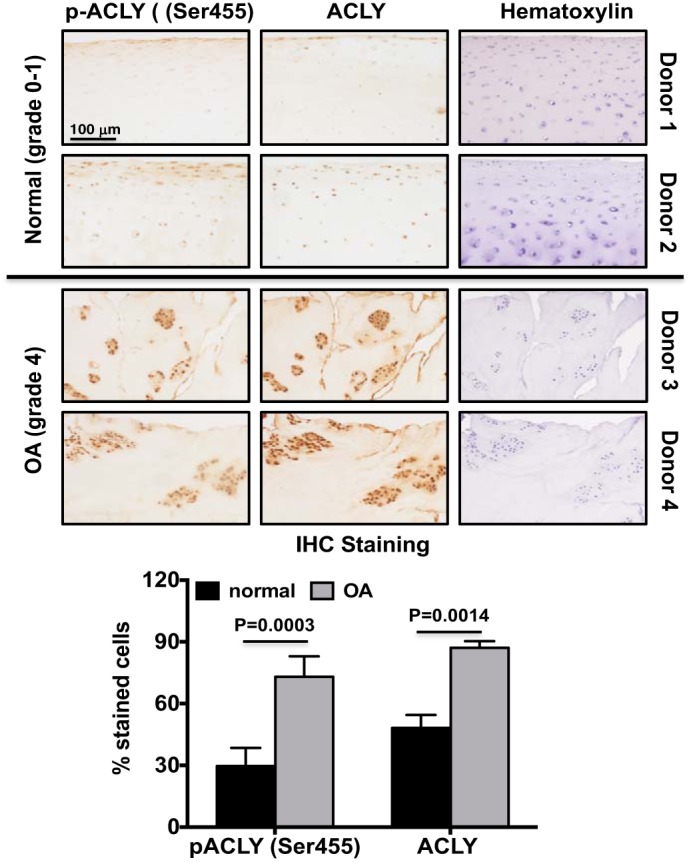
ACLY was up-regulated in human knee OA cartilage in situ. Human knee cartilage serial sections were analyzed for ACLY phosphorylation (Ser-455) and expression by immunohistochemistry. Hematoxylin staining verified cellularity in each section. Data are representative of 2 normal donors (24- and 45-year-old male) and 2 OA donors (84- and 90-year-old female) from 8 different donors (4 normal 4 OA) studied. The graph represents percentage of cells stained positively for phosphorylated ACLY (Ser-455) or ACLY. Student's t test was used for statistical data analysis. p values represent comparisons of the mean ± S.D.
Figure 3.

Concomitantly increased phosphorylation of ACLY (Ser-455) and acetylation of H3K9 and H3K27 in primary human knee OA chondrocytes. Whole cell lysates isolated from primary human knee chondrocytes from normal (grade 0-I) and OA (grade IV) donors were studied by Western blotting for ACLY expression and phosphorylation (Ser-455), and H3K9 and H3K27 acetylation. β-Actin and histone H3 served as loading controls. Data in A represent 5 normal and 5 OA donors. Age and gender for normal donors were 26 male, 28 male, 34 male, 44 male, 58 male, and age and gender for OA donors were 58 male, 68 female, 71 female, 75 female, 76 male. Semi-quantitative densitometry analyses of Western blots of primary human knee chondrocytes (B) from 28 normal donors (20 male and 8 female, age 42.3 ± 10.6) and 28 OA donors (10 male and 18 female, age 67.8 ± 7.9) were performed. Student's t test was used for statistical data analysis. p values represent comparison of the mean ± S.E. between normal and OA donors.
Inhibition of ACLY, mimicking glucose starvation, reduced nucleocytosolic acetyl-CoA and promoted phosphorylation of AMPK (Thr-172) in OA chondrocytes
AMPK is activated by glucose starvation in mammalian cells (25). We confirmed that phosphorylation of AMPKα at Thr-172, which is known to be critical for AMPK activity, was greatly increased in chondrocytes (Fig. 4A) under the glucose starvation condition shown in Fig. 1. However, phosphorylation of AMPKα (Thr-172) was reduced after glucose re-feeding (Fig. 4A), correlated with increased ACLY expression and function (Fig. 1), suggesting an inverse relationship between ACLY and AMPK activities. Treatment of OA chondrocytes with HCA resulted in a significant decrease in the amount of nucleocytosolic acetyl-CoA (Fig. 4B), which mimicked glucose starvation. A similar result was observed in OA chondrocytes in which ACLY was knockdown (Fig. 4B). Inhibition of ACLY by HCA or siRNA knockdown also led to a marked increase in phosphorylation of AMPKα (Thr-172) (Fig. 4C). Because activation of AMPK in chondrocytes inhibits catabolic responses to inflammatory cytokines (5), we examined the effect of inhibition of ACLY by HCA on IL-1β–induced nitric oxide (NO) release in AMPKα1 knockout (KO) and wildtype (WT) mouse chondrocytes. As expected, IL-1β–induced NO release was greatly enhanced in AMPKα1KO, compared with WT chondrocytes (Fig. 4D). HCA significantly inhibited IL-1β–induced NO release in not only WT but also AMPKα1KO chondrocytes (Fig. 4D), despite that inhibition was slightly less in AMPKα1KO compared with WT chondrocytes. There results suggest that the effect of HCA on chondrocyte responses to IL-1β was most likely independent of AMPK.
Figure 4.

Inhibition of ACLY, mimicking glucose starvation, reduced nucleocytosolic acetyl-CoA and promoted phosphorylation of AMPK (Thr-172) in OA chondrocytes. Western blot analysis of AMPKα phosphorylation (Thr-172) and expression was performed with the same cell lysates as described in the legend to Fig. 1B (A), or the cell lysates from cultured primary human knee OA chondrocytes either treated with HCA (20 mm) or in which ACLY was knocked down via transfection with ACLY siRNA (C). Semi-quantitative densitometry analysis of each Western blotting was performed over 3 individual experiments with 3 different donors (A and C). The amounts of total and compartmental acetyl-CoA were measured from cultured primary human knee OA chondrocytes treated with HCA (20 mm) for 5 h or ACLY knockdown chondrocytes (B). The conditioned media from mouse chondrocytes isolated from AMPKα1KO and WT control mice that were treated with IL-1β (2 ng/ml) in the presence or absence of HCA (20 mm) for 18 h were used to assess NO release (D). One-way ANOVA (A) and two-way ANOVA followed by Bonferroni's post hoc test (B and D) and Student's t test (C) were used for statistical data analysis. p values represent comparisons of the mean ± S.D.
Inhibition of ACLY by HCA attenuated specific histone acetylation and catabolic responses to IL-1β in OA chondrocytes
IL-1β treatment increased nucleocytosolic acetyl-CoA levels (by 31.5%) in OA chondrocytes (Fig. 5A). HCA treatment decreased basal and IL-1β–induced levels of nucleocytosolic acetyl-CoA by 40 and 45%, respectively (Fig. 5A). In addition, IL-1β increased phosphorylation of ACLY at Ser-455 (Fig. 5B) and H3K9 and K3K27 acetylation (Fig. 5B), effects were also attenuated by HCA (Fig. 5B). Moreover, ChIP-quantitative PCR (qPCR) analysis demonstrated that HCA diminished the IL-1β–induced Ac-H3K9 and Ac-H3K27 occupancy of genes that promote loss of chondrocyte extracellular matrix and whose expression is promoted by NF-κB activity, specifically iNOS, MMP3, and MMP13 (Fig. 5C). Furthermore, HCA inhibited IL-1β–induced acetylation of p65 NF-κB at Lys-310, a post-translational modification required for full transcriptional activity of NF-κB (26) (Fig. 5D). HCA also inhibited Ser-536 phosphorylation of p65 NF-κB, a change that enhances NF-κB transcriptional activity (26) (Fig. 5D). Quantitative RT-PCR confirmed that HCA attenuated IL-1β–induced iNOS, MMP3, and MMP13 mRNA expression (Fig. 6A). Moreover, HCA mitigated IL-1β–induced NO, MMP3, and MMP13 release in OA chondrocytes (Fig. 6B). Comparable results were seen in OA chondrocytes subjected to ACLY siRNA knockdown (Fig. 7). Furthermore, cartilage explant studies also demonstrated that HCA suppressed IL-1β–induced GAG, NO, MMP3, and MMP13 release (Fig. 6C).
Figure 5.

Inhibition of ACLY by HCA attenuated IL-1β–induced augmentation of acetylation H3K9 and H3K27 both globally and specifically in the iNOS, MMP3, and MMP13 promoters. Cultured primary human knee chondrocytes were treated with IL-1β (2 ng/ml) with or without HCA (20 mm) for 5 h. Nucleocytosolic acetyl-CoA was measured (A). Phosphorylation (Ser-455) and expression of ACLY and acetylation of H3K9 and H3K27 (B), and acetylation of p65 NF-κB (Lys-310) and phosphorylation of p65 NF-κB (Ser-536) (D) were examined by Western blotting, with β-actin and histone H3 as loading controls. Semi-quantitative densitometry analysis of each Western blotting was performed over 3 individual experiments with 3 different donors (B and D). ChIP-qPCR assessed Ac-H3K9 and Ac-H3K27 occupancy of iNOS, MMP3, and MMP13 promoters, and were presented as percentage of input of sonicated genomic DNA (C). Data in C represent the mean ± S.D. of 3 individual experiments with 3 different donors. Two-way ANOVA followed by Bonferroni's post hoc test (A and C) and Student's t test (B and D) were used for statistical data analysis. p values represent comparisons of the mean ± S.D.
Figure 6.

Inhibition of ACLY by HCA attenuated catabolic responses to IL-β in OA chondrocytes. Cultured primary human knee OA chondrocytes (A and B) or human knee cartilage explants (C) were treated with IL-1β (2 ng/ml) in the presence or absence of HCA (20 mm) for 5, 18, and 24 h, respectively. Total RNA was extracted from 5-h treated samples and subjected to quantitative RT-PCR analysis for mRNA expression of iNOS, MMP3, and MMP13 (A). Conditioned media from the remaining samples were analyzed for release of NO, MMP3, and MMP13 by Griess reaction and ELISA, respectively (B and C). Glycosaminoglycan (GAG) release was determined by dimethylmethylene blue assay (C). Data in A and B represent 3 individual experiments in OA chondrocytes from 3 different donors with replicates for each donor. Data in C represent 18 cartilage explants. Two-way ANOVA followed by Bonferroni's post hoc test was used for statistical data analysis. p values represent comparisons of the mean ± S.D.
Figure 7.

Knockdown of ACLY in OA chondrocytes led to inhibition of IL-1β–induced increase in H3K9 and H3K27 acetylation and catabolic responses. Cultured primary human knee OA chondrocytes were transfected with ACLY and nontargeted control siRNAs for 48 h. The cells were then treated with IL-1β (2 ng/ml) for 5 and 18 h. The 5-h treated samples were subjected to Western blot analysis for phosphorylation (Ser-455) and expression of ACLY and acetylation of H3K9 and H3K27, with β-actin and histone H3 as loading controls (A). Semi-quantitative densitometry analysis of Western blotting were performed over 3 individual experiments with 3 different donors (B). Conditioned media from 18-h treated samples were analyzed for release of NO and MMP13 by Griess reaction and ELISA, respectively (C and D). Two-way ANOVA followed by Bonferroni's post hoc test was used for statistical data analysis. p values represent comparisons of the mean ± S.D.
HCA increased matrix gene expression in OA chondrocytes by reducing SOX9 acetylation
Treatment of OA chondrocytes with HCA significantly increased basal levels of aggrecan (ACAN) and Col2a1 mRNA expression (Fig. 8, A and B. However, this was unlikely due to acetylation of histones, because HCA markedly reduced basal levels of Ac-H3K9 and Ac-H3K27 in OA chondrocytes (Fig. 5B). Because acetylation of SOX9, the chondrogenic master transcription factor, is recently shown to increase in human knee OA chondrocytes (27), and acetylation of SOX9 reduces its nuclear entry and subsequent ACAN gene expression (27), we examined the effect of HCA on SOX9 acetylation. HCA treatment blunted acetylation of SOX9 (Fig. 8C) in OA chondrocytes. In addition, HCA promoted SOX9 translocation from the cytosol to nucleus, indicated by decreased SOX9 expression in the cytosol and increased SOX9 expression in nucleus (Fig. 8D). These data indicate that ACLY regulated matrix anabolism in OA chondrocytes through modulation of acetylation of SOX9.
Figure 8.

Effects of pharmacologic inhibition of ACLY by HCA on SOX9 acetylation and matrix gene expression in OA chondrocytes. Cultured primary human knee OA chondrocytes were treated with HCA (20 mm) for 5 h. Total RNA was extracted and subjected to quantitative RT-PCR analysis for ACAN and Col2a1 mRNA (A and B). Data represent the mean ± S.D. of 3 different experiments in OA chondrocytes from 3 different donors. Whole cell lysates were subjected to immunoprecipitation with SOX9 antibody. Acetylation status of SOX9 was analyzed from SOX9 immunoprecipitates by Western blotting, using antibody to acetylated lysine (C). SOX9 in the cytosol and nucleus were examined by Western blotting, with tubulin and lamin B as the respective loading controls (D). Semi-quantitative densitometry analysis of each Western blot were performed over 3 individual experiments with 3 different donors (C and D). Student's t test was used for statistical data analysis. p values represent comparison of the mean ± S.D.
Discussion
The OA chondrocyte ACLY findings presented here are aligned with growing evidence that histone acetylation is tightly linked to the cellular metabolic state (11–14). The results place ACLY potentially at the hub of metabolic changes in OA chondrocytes that could promote disease progression. Glucose is required as metabolic fuel for chondrocytes, and as a structural precursor in extracellular matrix synthesis (3). However, heightened glycolysis can promote tissue inflammation (28). OA chondrocytes exposed to high glucose have increased glucose transport due to their inability to down-regulate GLUT-1 (29). This study reinforced the substantial role of ACLY in linking glucose metabolism to chromatin and transcription factor modification and consequent effects on gene transcription (14). In this light, glucose availability in chondrocytes was found to modulate global levels of acetylation of histone H3 at lysine residues 9 and 27 by regulating nucleocytosolic acetyl-CoA generation in large part via ACLY. Increased glucose availability in OA chondrocytes may augment the impact of ACLY on gene transcription through lysine acetylation of histones and transcription factors. Because IL-1β also promotes glucose uptake in chondrocytes (29, 30), we attribute increased glucose metabolism as a likely contributor to the herein demonstrated ability of IL-1β to increase ACLY activity and nucleocytosolic acetyl-CoA in chondrocytes.
Our findings revealed a regulatory role of ACLY activity in chondrocyte matrix homeostasis by modulation of the nucleocytosolic pool of acetyl-CoA, which impacted on catabolic and anabolic responses via post-translational and epigenetic modifications. Glucose availability in articular chondrocytes influenced nucleocytosolic acetyl-CoA generation and histone acetylation, which were largely dependent on ACLY. As illustrated in our putative model (Fig. 9), ACLY may become overactive in chondrocytes as a result of increased glycolysis and/or inflammatory response, causing an increase in the nucleocytosolic pool of acetyl-CoA. Subsequently, chondrocyte capacity to active AMPK is reduced. In addition, matrix gene expression (ACAN, Col2a1) is inhibited and genes that promote degradation of extracellular matrix (iNOS, MMP3, and MMP13) are activated. These effects are mediated by specific post-translational acetylation modifications of histones and transcription factors (increased acetylation of H3K9, H3K27, p65, NF-κB, and SOX9). Consequently, cartilage homeostasis was impaired due to imbalance between matrix anabolic and catabolic activities in chondrocytes, which could lead to cartilage degradation and OA development.
Figure 9.

Putative model of ACLY in cartilage matrix metabolism and OA. Shown is a schematic model that summarizes our results, in which pharmacologically targetable ACLY was increased in OA chondrocytes in situ and in response to increased glycolysis and IL-1β in vitro, with associated changes that could disrupt cartilage matrix homeostatic balance, leading to cartilage degradation, as discussed in detail in the text.
We do not yet know how ACLY is up-regulated in human knee OA chondrocytes. In cells other than chondrocytes, ACLY mRNA is regulated by the transcription factor sterol regulatory element-binding protein-1 (SREBP-1), via Akt signaling (13). However, ACLY protein levels are independent of SREBP-1 (13). The PI3K/Akt pathway stimulates ACLY catalytic activity predominantly through phosphorylation of ACLY at Ser-455, which also contributes to stabilization of ACLY protein (13). We speculate that IGF1 and other chondrocyte growth factors have the potential to modulate ACLY expression in OA cartilage chondrocytes. Specifically, growth factor IGF1 can induce phosphorylation of ACLY at Ser-455 via Akt signaling (31). In addition, IGF1 signaling is altered in OA cartilage (32), and OA chondrocytes exhibit decreased anabolic response to IGF-1 (32).
Depletion of the nucleocytosolic pool of acetyl-CoA was recently revealed to activate AMPK (11, 20). We observed the capacity of inhibition of ACLY in OA chondrocytes (by either siRNA knockdown or pharmacologic inhibition) to drop nucleocytosolic acetyl-CoA and promote AMPK activation. Thus, decreased AMPK activity in human and mouse knee OA cartilages (5, 6) may be partly due to increased ACLY activity in OA chondrocytes. Similarly, age-related loss of AMPK activity in knee cartilage (6) may potentially result in part from increased ACLY activity. Because chondrocyte-specific AMPKαKO mice develop accelerated mechanical injury–induced and age–related spontaneous knee OA (8), maintaining AMPK activity by preventing ACLY to become overactive in chondrocytes may potentially limit OA development and progression. Selective depletion of nucleocytosolic acetyl-CoA is also shown to induce autophagy through inhibition of mTOR via activation of AMPK (11, 33, 34). In addition, acetyl-CoA modulates transcription of core autophagy genes (11, 12). Future study would be of interest to determine whether inhibition of ACLY in OA chondrocytes activates autophagy, which is chondroprotective in vivo (35–37).
Putative translational significance of the work is the potential intersections of ACLY modulation with diet, nutrition, and metabolic syndrome, and respective impact of each of these factors on pathogenesis and progression of OA (1–4). Given that ACLY is emerging as a drug target for long-term use in metabolic disorders (19), it could provide an early opportunity to study potential impact of ACLY inhibition on OA onset, symptoms, and progression. Thus, further translational studies in OA, by testing the role of pharmacologic inhibition of ACLY on disease progression, are warranted.
Experimental procedures
Reagents
Chemical reagents, including HCA, tripotassium salt, were obtained from Sigma. Antibodies to phospho-AMPKα (Thr-172), AMPKα, phospho-p65 NF-κB (Ser-536), acetyl-p65 NF-κB (Lys-310), p65 NF-κB, ACLY, pyruvate kinase isoenzyme M2 (PKM2), H3 (acetyl-K9), H3 (acetyl-K27), and histone H3 were from Cell Signaling Technology, Inc. (Danvers, MA). Antibodies to SOX9, α-tubulin, and lamin B were from Abcam (Cambridge, MA). Antibody to phospho-ACLY (Ser-455) was from Aviva Systems Biology (San Diego, CA). Human ACLY siRNA and control siRNA were from Thermo Fisher Scientific (Waltham, MA).
Studies of human and mouse articular chondrocytes
Studies with human knee articular chondrocytes were performed in compliance with institutional IRB reviewed and approved human subjects protocols at the Scripps Research Institute and San Diego VA Medical Center, and abide by the Declaration of Helsinki principles. Studies with mouse knee articular chondrocytes were carried out in compliance with institutional IACUC reviewed animal protocols at the San Diego Veterans Affairs Medical Center. Human knee chondrocytes were isolated from autopsy donors that were graded macroscopically according to a modified Outerbridge scale (38) and cultured in Dulbecco's modified Eagle's (DMEM) high glucose medium with 10% fetal calf serum, 100 μg/ml of streptomycin, and 100 IU/ml of penicillin at 37 °C. Once the cells reached confluence (cultured primary chondrocytes), they were either collected and stored in −80 °C for later analyses, or were re-plated at 3 × 105 cells/well in 12-well plates or 7 × 105 cells/well in 6-well plates for all other experiments. Mouse articular chondrocytes were isolated from knee joints of 7–8–day-old pups of AMPKα1 KO and WT mice using previously described methods (39), and were cultured in the same media aforementioned in 6-well plates. They were then re-plated at 3 × 105 cells/well in 12-well plates for experiments.
Measurement of acetyl-CoA
Acetyl-CoA concentrations were determined using a PicoProbeTMAcetyl-CoA Fluorometric Assay Kit (Biovision). To measure cell compartmental acetyl-CoA concentrations, the Thermo Scientific Mitochondria Isolation Kit for Cultured Cells was used to separate mitochondria from the rest of the cytosolic and nuclear components according to the manufacturer's protocol. Isolated mitochondria and the remaining nucleus and cytosol fraction were then used to measure mitochondrial and nucleocytosolic acetyl-CoA, respectively.
SDS-PAGE/Western blotting
Total cell lysates were prepared using RIPA buffer with 2 mm sodium vanadate and protease inhibitor cocktails (Roche Applied Science). Cytosolic and nuclear proteins were prepared by lysing the cytosolic and nuclear fractions of cells extracted using a Cell Fractionation kit (Abcam, Cambridge, MA). Proteins (10–15 μg) were separated by gradient 4–20% SDS-PAGE and transferred onto nitrocellulose membranes (Bio-Rad), probed with antibodies, exposed to SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific), and visualized by autoradiography.
Immunohistochemistry
Slides of human knee cartilage sections from both normal and advanced OA donors were treated with 3% H2O2 for 10 min, then blocked with 10% goat serum for 2 h at room temperature. After washing with Tris-buffered saline, rabbit antibodies to phospho-ACLY (Ser-455), ACLY, and the negative control rabbit IgG (all in 1:100 dilution) were applied to the sections and incubated overnight at 4 °C. Next, sections were washed with Tris-buffered saline, incubated with biotinylated goat anti-rabbit IgG secondary antibody for 30 min, and then incubated for 30 min using the Histostain Plus kit (Invitrogen). Finally, sections were washed and incubated with 3,3′-diaminobenzidine substrate for 2–5 min.
Chromatin immunoprecipitation (ChIP) assay followed by quantitative PCR (ChIP-qPCR)
ChIP assay employed a commercial kit (EMD Millipore, Temecula, CA), according to the manufacturer's protocol. In brief, cells that were fixed with formaldehyde to cross-link histone and nonhistone proteins to DNA were lysed, and harvested chromatin was fragmented by sonication. Chromatin was immunoprecipitated with antibodies to Ac-H3K9 or Ac-H3K27. The nonimmune IgG was included as a control. The protein-DNA cross-links were then reversed and the DNA was purified. Enriched DNA sequences occupied by H3K9 and H3K27 in promoter regions of iNOS, MMP3, and MMP13 were detected by quantitative PCR using the primers listed in Table S1. The sonicated genomic DNA was included in qPCR as the input control. The PCR was carried out at 95 °C for 5 min for initial denaturation, followed by 40 cycles at 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 10 s.
Knockdown of ACLY expression in human knee articular chondrocytes
Cultured human knee articular chondrocytes were transfected with ACLY siRNA, and nontarget control using the X-tremeGene siRNA transfection reagent (Roche Applied Science, Penzberg, Germany) according to the manufacturer's protocol.
Quantitative RT-PCR
Total RNA was isolated from chondrocytes by a RNeasy mini kit (Qiagen), per the manufacturer's instructions. First strand cDNA was synthesized from 500 ng of total RNA using Transcript or First Strand cDNA Synthesis Kit (Roche). We performed quantitative RT-PCR (95 °C for 10 s, 60 °C for 10 s, and 72 °C for 10 s, total 40 cycles) using LightCycler 480 (Roche). The high efficiency ΔΔCT method calculated the expression level of target and control (18S rRNA) genes. Table S2 lists primers for target and control genes.
Assays of NO, MMP3, and MMP13 release, and SOX9 acetylation, distribution, and expression
Conditioned media were tested for NO generation by a Griess reaction assay to quantify nitrite, and MMP3 and MMP13 release was measured using Total MMP3 and MMP13 Quantikine ELISA Kits (R&D Systems). SOX9 was immunoprecipitated from cell lysates using a SOX9 antibody (Abcam). The SOX9 immunoprecipitates were analyzed by SDS-PAGE/Western blotting for acetylation status of SOX9 using acetyl-lysine antibody (Cell Signaling, Danvers, MA). To examine compartmental SOX9, cytosolic and nuclear proteins were analyzed by SDS-PAGE/Western blotting.
Statistical analyses
Most of data were expressed as mean ± S.D. Some data were expressed as mean ± S.E. Statistical analyses were performed by either Student's t test, one-way ANOVA, or two-way ANOVA with Bonferroni's post hoc test using GraphPad PRISM 6. p values less than 0.05 were considered significant.
Author contributions
L.-Y. C. and R. L.-B. conceptualization; L.-Y. C. data curation; L.-Y. C. validation; L.-Y. C. and R. L.-B. investigation; L.-Y. C. visualization; M. L. and R. A. T. resources; M. L., R. A. T., and R. L.-B. funding acquisition; M. L., R. A. T., and R. L.-B. writing-review and editing; R. A. T. and R. L.-B. supervision; R. L.-B. formal analysis; R. L.-B. methodology; R. L.-B. writing-original draft; R. L.-B. project administration.
Supplementary Material
Acknowledgment
We gratefully acknowledge the Sample Acquisition core of the National Institutes of Health Program Project Grant PAG007996 on Cartilage Aging and Osteoarthritis for human knee cartilage grading and chondrocyte isolation.
This work was supported by Department of Veterans Affairs Grants 1I01BX002234 and I01BX001660 and National Institutes of Health Grant AG007996. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1 and Tables S1 and S2.
- OA
- osteoarthritis
- ACLY
- ATP-citrate lyase
- AMPK
- AMP-activated protein kinase
- acetyl-CoA
- acetyl-coenzyme A
- Ac-H3K9
- acetylation of lysine residue 9 of histone 3
- ACAN
- aggrecan
- HCA
- hydroxycitric acid
- iNOS
- inducible nitric-oxide synthase
- MMP
- matrix metalloproteinase
- NO
- nitric oxide
- PKM2
- pyruvate kinase isoenzyme 2
- RT-qPCR
- reverse transcription quantitative PCR
- IHC
- immunohistochemistry
- IL
- interleukin
- DMEM
- Dulbecco's modified Eagle's
- ANOVA
- analysis of variance.
References
- 1. Loeser R. F., Goldring S. R., Scanzello C. R., and Goldring M. B. (2012) Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 64, 1697–1707 10.1002/art.34453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Courties A., Sellam J., and Berenbaum F. (2017) Metabolic syndrome-associated osteoarthritis. Curr. Opin. Rheumatol. 29, 214–222 10.1097/BOR.0000000000000373 [DOI] [PubMed] [Google Scholar]
- 3. Mobasheri A., Rayman M. P., Gualillo O., Sellam J., van der Kraan P., and Fearon U. (2017) The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 13, 302–311 10.1038/nrrheum.2017.50 [DOI] [PubMed] [Google Scholar]
- 4. Berenbaum F., Griffin T. M., and Liu-Bryan R. (2017) Review: metabolic regulation of inflammation in osteoarthritis. Arthritis Rheumatol. 69, 9–21 10.1002/art.39842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Terkeltaub R., Yang B., Lotz M., and Liu-Bryan R. (2011) Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to inflammatory cytokines IL-1β and TNFα. Arthritis Rheum. 63, 1928–1937 10.1002/art.30333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao X., Petursson F., Viollet B., Lotz M., Terkeltaub R., and Liu-Bryan R. (2014) PGC-1α and FOXO3a mediate chondroprotection by AMP-activated protein kinase. Arthritis Rheum. 66, 3073–3082 10.1002/art.38791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carling D. (2017) AMPK signalling in health and disease. Curr. Opin. Cell Biol. 45, 31–37 10.1016/j.ceb.2017.01.005 [DOI] [PubMed] [Google Scholar]
- 8. Zhou S., Lu W., Chen L., Ge Q., Chen D., Xu Z., Shi D., Dai J., Li J., Ju H., Cao Y., Qin J., Chen S., Teng H., and Jiang Q. (2017) AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci. Rep. 7, 43245 10.1038/srep43245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Keating S. T., and El-Osta A. (2015) Epigenetics metabolism. Circ. Res. 116, 715–736 10.1161/CIRCRESAHA.116.303936 [DOI] [PubMed] [Google Scholar]
- 10. Kim H., Kang D., Cho Y., and Kim J. H. (2015) Epigenetic regulation of chondrocyte catabolism and anabolism in osteoarthritis. Mol. Cells 38, 677–684 10.14348/molcells.2015.0200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pietrocola F., Galluzzi L., Bravo-San Pedro J. M., Madeo F., and Kroemer G. (2015) Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 21, 805–821 10.1016/j.cmet.2015.05.014 [DOI] [PubMed] [Google Scholar]
- 12. Shi L., and Tu B. P. (2015) Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr. Opin. Cell Biol. 33, 125–131 10.1016/j.ceb.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chypre M., Zaidi N., and Smans K. (2012) ATP-citrate lyase: a mini-review. Biochem. Biophys. Res. Commun. 422, 1–4 10.1016/j.bbrc.2012.04.144 [DOI] [PubMed] [Google Scholar]
- 14. Wellen K. E., Hatzivassiliou G., Sachdeva U. M., Bui T. V., Cross J. R., and Thompson C. B. (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 10.1126/science.1164097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beigneux A. P., Kosinski C., Gavino B., Horton J. D., Skarnes W. C., and Young S. G. (2004) ATP-citrate lyase deficiency in the mouse. J. Biol. Chem. 279, 9557–9564 10.1074/jbc.M310512200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khwairakpam A. D., Shyamananda M. S., Sailo B. L., Rathnakaram S. R., Padmavathi G., Kotoky J., and Kunnumakkara A. B. (2015) ATP citrate lyase (ACLY): a promising target for cancer prevention and treatment. Curr. Drug Targets 16, 156–163 10.2174/1389450115666141224125117 [DOI] [PubMed] [Google Scholar]
- 17. Infantino V., Iacobazzi V., Palmieri F., and Menga A. (2013) ATP-citrate lyase is essential for macrophage inflammatory response. Biochem. Biophys. Res. Commun. 440, 105–111 10.1016/j.bbrc.2013.09.037 [DOI] [PubMed] [Google Scholar]
- 18. Hatzivassiliou G., Zhao F., Bauer D. E., Andreadis C., Shaw A. N., Dhanak D., Hingorani S. R., Tuveson D. A., and Thompson C. B. (2005) ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 8, 311–321 10.1016/j.ccr.2005.09.008 [DOI] [PubMed] [Google Scholar]
- 19. Burke A. C., and Huff M. W. (2017) ATP-citrate lyase: genetics, molecular biology and therapeutic target for dyslipidemia. Curr. Opin. Lipidol. 28, 193–200 10.1097/MOL.0000000000000390 [DOI] [PubMed] [Google Scholar]
- 20. Madeo F., Pietrocola F., Eisenberg T., and Kroemer G. (2014) Caloric restriction mimetics: towards a molecular definition. Nat. Rev. Drug Discov. 13, 727–740 10.1038/nrd4391 [DOI] [PubMed] [Google Scholar]
- 21. Mariño G., Pietrocola F., Eisenberg T., Kong Y., Malik S. A., Andryushkova A., Schroeder S., Pendl T., Harger A., Niso-Santano M., Zamzami N., Scoazec M., Durand S., Enot D. P., Fernández Á., et al. (2014) Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell. 53, 710–725 10.1016/j.molcel.2014.01.016 [DOI] [PubMed] [Google Scholar]
- 22. Koch C. M., Andrews R. M., Flicek P., Dillon S. C., Karaöz U., Clelland G. K., Wilcox S., Beare D. M., Fowler J. C., Couttet P., James K. D., Lefebvre G. C., Bruce A. W., Dovey O. M., Ellis P. D., et al. (2007) The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 17, 691–707 10.1101/gr.5704207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Creyghton M. P., Cheng A. W., Welstead G. G., Kooistra T., Carey B. W., Steine E. J., Hanna J., Lodato M. A., Frampton G. M., Sharp P. A., Boyer L. A., Young R. A., and Jaenisch R. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 107, 21931–21936 10.1073/pnas.1016071107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rosa S. C., Gonçalves J., Judas F., Mobasheri A., Lopes C., and Mendes A. F. (2009) Impaired glucose transporter-1 degradation and increased glucose transport and oxidative stress in response to high glucose in chondrocytes from osteoarthritic versus normal human cartilage. Arthritis Res. Ther. 11, R80 10.1186/ar2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hardie D. G., and Lin S. C. (2017) AMP-activated protein kinase: not just an energy sensor. F1000 Res. 6, 1724 10.12688/f1000research.11960.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perkins N. D. (2006) Post-translational modifications regulating the activity and function of the nuclear factor κB pathway. Oncogene 25, 6717–6730 10.1038/sj.onc.1209937 [DOI] [PubMed] [Google Scholar]
- 27. Bar Oz M., Kumar A., Elayyan J., Reich E., Binyamin M., Kandel L., Liebergall M., Steinmeyer J., Lefebvre V., and Dvir-Ginzberg M. (2016) Acetylation reduces SOX9 nuclear entry and ACAN gene transactivation in human chondrocytes. Aging Cell 15, 499–508 10.1111/acel.12456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chang X., and Wei C. (2011) Glycolysis, rheumatoid arthritis. Int. J. Rheum Dis. 14, 217–222 10.1111/j.1756-185X.2011.01598.x [DOI] [PubMed] [Google Scholar]
- 29. Phillips T., Ferraz I., Bell S., Clegg P. D., Carter S. D., and Mobasheri A. (2005) Differential regulation of the GLUT1 and GLUT3 glucose transporters by growth factors and pro-inflammatory cytokines in equine articular chondrocytes. Vet. J. 169, 216–222 10.1016/j.tvjl.2004.01.026 [DOI] [PubMed] [Google Scholar]
- 30. Shikhman A. R., Brinson D. C., Valbracht J., and Lotz M. K. (2009) Differential metabolic effects of glucosamine and N-acetylglucosamine in human articular chondrocytes. Osteoarthritis Cartilage 17, 1022–1028 10.1016/j.joca.2009.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Das S., Morvan F., Jourde B., Meier V., Kahle P., Brebbia P., Toussaint G., Glass D. J., and Fornaro M. (2015) ATP citrate lyase improves mitochondrial function in skeletal muscle. Cell Metab. 21, 868–876 10.1016/j.cmet.2015.05.006 [DOI] [PubMed] [Google Scholar]
- 32. Martel-Pelletier J., Di Battista J. A., Lajeunesse D., and Pelletier J. P. (1998) IGF/IGFBP axis in cartilage and bone in osteoarthritis pathogenesis. Inflamm Res. 47, 90–100 10.1007/s000110050288 [DOI] [PubMed] [Google Scholar]
- 33. Eisenberg T., Schroeder S., Andryushkova A., Pendl T., Küttner V., Bhukel A., Mariño G., Pietrocola F., Harger A., Zimmermann A., Moustafa T., Sprenger A., Jany E., Büttner S., Carmona-Gutierrez D., et al. (2014) Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab. 19, 431–444 10.1016/j.cmet.2014.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schroeder S., Pendl T., Zimmermann A., Eisenberg T., Carmona-Gutierrez D., Ruckenstuhl C., Mariño G., Pietrocola F., Harger A., Magnes C., Sinner F., Pieber T. R., Dengjel J., Sigrist S. J., Kroemer G., and Madeo F. (2014) Acetyl-coenzyme A: a metabolic master regulator of autophagy and longevity. Autophagy 10, 1335–1337 10.4161/auto.28919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Caramés B., Taniguchi N., Otsuki S., Blanco F. J., and Lotz M. (2010) Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 62, 791–801 10.1002/art.27305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Caramés B., Taniguchi N., Seino D., Blanco F. J., D'Lima D., and Lotz M. (2012) Mechanical injury suppresses autophagy regulators and pharmacologic activation of autophagy results in chondroprotection. Arthritis Rheum. 64, 1182–1192 10.1002/art.33444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bohensky J., Leshinsky S., Srinivas V., and Shapiro I. M. (2010) Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr Nephrol. 25, 633–642 10.1007/s00467-009-1310-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamada K., Healey R., Amiel D., Lotz M., and Coutts R. (2002) Subchondral bone of the human knee joint in aging and osteoarthritis. Osteoarthritis Cartilage 10, 360–369 10.1053/joca.2002.0525 [DOI] [PubMed] [Google Scholar]
- 39. Cecil D. L., and Terkeltaub R. (2008) Transamidation by transglutaminase 2 transforms S100A11 calgranulin into a procatabolic cytokine for chondrocytes. J. Immunol. 180, 8378–8385 10.4049/jimmunol.180.12.8378 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.