

Abstract
In this paper we review the history of antidepressant (AD) development, since the discovery of imipramine in 1957 to the present day. Through this exploration we will show that the increasing placebo response is likely a red herring and that a higher magnitude of placebo response is not an adequate explanation for AD trials’ high failure rates. As a better explanation for their lack of success, we will examine some of the fundamental flaws of AD clinical trials and their origins in historical forces. We focus on underpowering, which occurs as a consequence of unrealistic expectations for AD performance. In addition, we describe the lack of precision in the depression outcome measurements for the past 40 years and show how these measures contrast with those used in clinical trials of other chronic diseases, which use simpler outcome measures. Finally, we describe the role of regulatory agencies in influencing clinical trial design and how the assumption that ‘one size fits all’ for the past 60 years has led to flawed design of AD clinical trials.
Keywords: antidepressants, clinical trials, depression, efficacy, measurements, sample size
Introduction
The function of the controlled clinical trial is not the ‘discovery’ of a new drug or therapy. Discoveries are made in the animal laboratory, by chance observation, or at the bedside by an [astute] clinician. The function of the formal controlled clinical trial is to separate the relative handful of discoveries which prove to be true advances in therapy from a legion of false leads and unverifiable clinical impressions, and to delineate in a scientific way the extent of and the limitations which attend the effectiveness of drugs. Affidavit of William Thomas Beaver (2007).
Randomized, placebo-controlled, double blind trials came into vogue as a potentially definitive tool to assess the effectiveness of a putative treatment soon after the Second World War, when a plethora of new pharmacological agents were serendipitously discovered.
The often-cited harbinger of the modern day clinical trial was developed in 1948 by the British Medical Research Council as a method for eliminating bias while evaluating the effectiveness of streptomycin (Chalmers, 2010). Following the publication of the paper by Beecher (1955) documenting the prevalence of the placebo response, clinical researchers assumed that in trials of major depression one-third of patients get better with placebo treatment. This concept has held sway for over 60 years and has had significant impact on drug assessment models.
Modern clinical trial design is far from perfect and the historical assumptions that such trial models are based on may not fit with all conditions and classes of drugs. Nowhere is this more evident than in the development of psychopharmacological agents, particularly in the history of antidepressant (AD) programs. We aim to explore how this came to be.
In this paper we review the history of AD development, since the discovery of imipramine in 1957 to the present day. Through this exploration we will show that the focus on the increasing placebo response as a source of trial failure is misplaced and that a higher magnitude of placebo response is not an adequate explanation for AD trials’ high failure rates. As a better explanation for their lack of success, we will examine some of the fundamental flaws of AD clinical trials and their origins in historical forces.
We focus on underpowering, which occurs as a consequence of unrealistic expectations for AD performance. In addition, we describe the lack of precision in the depression outcome measurements for the past 40 years and show how these measures contrast with those used in clinical trials of other chronic diseases, which use simpler outcome measures. Finally, we describe the role of regulatory agencies in influencing clinical trial design and how the assumption that ‘one size fits all’ for the past 60 years has led to flawed design of AD clinical trials.
History of antidepressant clinical trials
In the 1950s Roland Kuhn, a Swiss psychiatrist, obtained samples of the compound G22355 (later named imipramine) from Geigy pharmaceuticals (Basel, Switzerland). As was common practice at the time, he gave it as a test-drug for his patients with schizophrenia as it was suspected to treat psychotic symptoms (Brown and Rosdolsky, 2015). Although this drug appeared to have minimal effect on the symptoms of schizophrenia, unexpectedly, some patients with schizophrenia appeared to recover from their depressive symptoms.
Noting this, Kuhn decided to give the compound to patients with severe primary depression to see how it would help them. After administering the drug to a series of 100 depressed patients under his care, Kuhn wrote up detailed clinical histories and vignettes of what appeared to be impressive recovery. He later published these writings in Swiss Medical Weekly in 1957 – within a year, Geigy was able to successfully market imipramine in the USA and internationally as an antidote for depression.
The discovery of amitriptyline followed shortly after. On the basis of clinical impressions, amitriptyline was deemed effective for use with depressed patients. Several more compounds followed suit. In the 1960s, six or seven ADs were in routine use by physicians treating depressed patients. At this time, none of these ADs had labels and none had been systematically evaluated for safety or efficacy. The information we had was based entirely on collections of clinician observations.
Intrigued by this new class of ‘antidepressant’ drugs, Hamilton (1960) put together a rating scale intended to measure the severity of depression symptoms. The Hamilton Depression Rating Scale (HAM-D) was based on the most prevalent symptoms of patients hospitalized for depression. It is important to note that at that time a list of clinical criteria used to diagnose depression did not exist. Instead, a depression diagnosis was simply given if the clinician felt that the label applied.
The practice of giving physicians’ samples of new compounds to try out on their patients came to an abrupt halt in 1962 with the thalidomide tragedy. Thalidomide, given to pregnant women primarily for nausea, resulted in severe congenital anomalies. The US Food and Drug Administration (FDA) responded strongly to this tragedy and began thorough scrutiny of drug development programs. Governmental regulators worldwide responded in kind.
Although the initial charge of the FDA was only to establish the safety of new drugs before they are distributed to patients, the role of regulatory agencies has expanded greatly in the past half a century. As of now, investigational pharmacological agents cannot be tested for efficacy unless pharmaceutical companies and others sponsoring the trial obtain written approval for the design and conduct of the proposed research. As a result, all efficacy trials for ADs have been approved by the US FDA and similar agencies prior to their conduct.
The standards regulatory agencies developed for these trials were primarily informed by one notable trial of AD efficacy. In 1965, the British Medical Research Council (1965) conducted a large (or so considered at the time), double blind, randomized, placebo-controlled trial of imipramine, phenelzine, electroconvulsive therapy (ECT), and placebo. The BMRC trial was intended not only to establish the advantage of these treatments over placebo, but it was also designed to evaluate whether subgroups of these depressed patients had different responses to the various treatments.
Results of this trial showed superior symptom reduction with imipramine and ECT compared with placebo while phenelzine did not show such superiority. Better response patterns were seen among men treated with imipramine; however, there was a slower onset of action overall with imipramine compared with placebo. It is clear in hindsight that many of the conclusions of the study suffered from preconceived bias. In addition, the statistical power of the trial had not been evaluated since it was the first of its kind and so the results were uncritically accepted. Despite these limitations that we can see in hindsight, this trial set the tone for the future testing of ADs in the clinical trial setting.
Not surprisingly, this has had a chilling effect on the development of alternate models for testing new investigational psychopharmacological agents. Getting new ADs to market meant clearing the bar set by the FDA by conducting a trial that would be accepted as proof of efficacy. It seems that this bar was set such that only clinical trials following the model of the BMRC trial would be accepted for regulatory review. With only minor tweaks, this model has persisted for over half a century; inertia has reigned as sponsoring pharmaceutical companies have not risked investing in clinical trial models that may not be accepted by the FDA and regulators have been content with the current model without regard for its actual utility or scientific validity.
Red herrings in the search for the cause of antidepressant trial failure
In the early 2000s it became clear that AD clinical trials were plagued by a high rate of failure. The 50% failure rate of AD trials was assumed to occur as a result of an increasing and variable placebo response (Walsh et al., 2002; Khan et al., 2003a). Analysis of AD trial data up to 2000 revealed that a high placebo response almost certainly predicted trial failure, and this made sense on intuitive grounds.
This finding set off a cascade of retrospective investigations into the cause of such high placebo responses. The assumption was that features of patient selection and trial design and execution were contributing to a higher magnitude of placebo response. Retrospective analyses aimed to discover which factors might be culprits. Dosing schedule (Khan et al., 2003b), duration (Khan et al., 2001), rating scales (Khan et al., 2004b), interview techniques (Demitrack et al., 1998; Kobak and Thase, 2007), and number of treatment arms (Khan et al., 2004a; Papakostas and Fava, 2009) emerged as potential features that could be controlled to contain a rising placebo response.
However, prospective implementation of trial designs and techniques that appeared promising for containing the placebo response have not been shown to be effective, and recent reports (Khin et al., 2011; Khan et al., 2017) failed to identify any association of these design features with the increase in placebo response. Previous associations between placebo response, trial efficacy outcomes, and overall rate of success have not held up in recent analyses. Moreover, despite efforts to contain it, the placebo response in AD trials has increased by ~6% over the past 30 years while the success rate has actually increased by 15% (Khan et al., 2017).
The mean drug–placebo differences in treatment response and their corresponding effect sizes have remained the same over the decades (with an effect size of about 0.3). The only notable change in trial design has been in the sample sizes of treatment arms, which have increased from under a hundred patients between drug and placebo to several hundred in many cases. These nonintuitive findings were puzzling, and it seemed that somehow the increase in sample size over the years must have been related. We wondered whether it was possible that the change in sample size has played a role in AD trial efficacy outcomes.
Gibertini et al. (2012) plotted effect sizes along with the treatment arm sample sizes from AD clinical trials and it became evident that the AD efficacy data showed the exact pattern that would be expected when studies are underpowered. What the authors found was that this plot quite elegantly depicted the funneling effect of sample size, wherein small studies yield variable results and wide scatter, and larger studies converge around the mean like the shape of a pyramid. Sample sizes were clearly related to the variability of effect size.
However, it was still unclear how this related to the previous findings suggesting the importance of the magnitude of placebo response. To address this, we plotted the estimates of placebo response (from the dataset in Khan et al., 2017) along with placebo arm sample size in the same fashion as Gibertini et al. (2012) and found a similar effect (Fig. 1). It appears that underpowering has led to a wide variation in placebo responses in smaller trials and that larger trials show more stable, less variable estimates of the response to placebo around 34%.
Fig. 1.
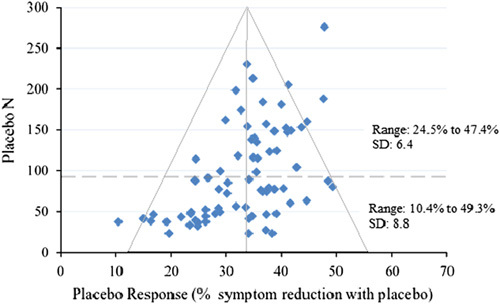
Funnel plot of placebo response plotted with placebo arm sample size. Dotted line represents the median split of placebo arm N (87 N: an unbiased threshold used to examine the differential effects of smaller and larger sample sizes).
Interestingly, very low placebo responses, some showing less than 20% improvement with placebo, only appear in trials with less than 50 patients assigned to placebo. These same small trials that scatter widely at the bottom of the pyramid are just as likely to have a very low placebo response as they are to have one that approaches 50%. Ironically, this high variability in the estimates of placebo response and effect size in these smaller trials coincides with a less than 40% rate of success. Thus, underpowering appears to account for a substantial proportion of AD clinical trial failure.
Why size matters
What is known about studies with low statistical power is that their findings are unreliable – they find statistical significance for the experimental condition at a rate that mirrors chance. Even more importantly, the estimates of the treatment effect size gleaned from such studies are likely to be overestimates in the case of positive studies and underestimates of the true effect if the study was negative (Ioannidis, 2005; Chmura Kraemer et al., 2006; Gibertini et al., 2012; Reinhart, 2015; Blackford, 2017).
Considering this, the estimates of less than 25% symptom reduction with placebo were likely to be underestimated. In other words, the abnormally high effect sizes seen in smaller trials historically were very likely inflated, false positives. It is also likely that these lucky results are disproportionately available for analysis because of selection or publication bias (Turner et al., 2008). This is conspicuous in the missing lower right section of the funnel in Fig. 1 representing larger placebo responses in small samples.
This also provides a logical explanation as to why the association of the magnitude of placebo response and trial success has not held up over time as sample sizes have increased. A low magnitude of placebo response is only necessary to find a significant drug–placebo difference and thus a successful outcome when small sample sizes are involved. Fletcher (2008) put it eloquently: “…small studies are ‘imprecise’ and have wide confidence intervals, it is only the ones with abnormally large effects that manage to achieve statistical significance.”
While it was necessary for a small trial to have a low placebo response and an unusually high effect size to find statistical success, more recent larger trials have been able to achieve statistical significance with more modest (and likely truer) effect sizes of ADs. In these larger studies, despite having larger placebo responses than the ‘lucky’ trials with smaller sample sizes, the drug response maintained a very consistent superiority of about 10%. So, it is no surprise that the historical 50% success rate of AD trials has increased along with sample sizes.
And it is also no surprise that associations found to be predictive of the magnitude of placebo response have not been replicated prospectively in larger sample sizes. Looking at the collective data in relation to sample size, it is likely that the previous associations of trial design variables with lower placebo responses were statistical artifacts of underpowering. The suggestion that certain trial design factors may control placebo response has perhaps been the greatest red herring of all – placebo responses much lower than the mean were likely the result of statistical error.
The true flaws in AD clinical trials may have evaded us all these years owing to the distraction of the rising placebo response. Although a partial solution has been found. Currently, assuming a modest effect size of about 0.3, if one powers adequately at ~150 patients per treatment condition, then the success rate of the trial will be around 90%.

In retrospect, the evidence is strong that earlier studies were likely too small and therefore underpowered. Given this, these smaller AD trials (particularly those with <100 patients per treatment condition) should have been treated with the same caution as pilot studies. As Leon et al. (2011) stated: ‘A pilot study is not a hypothesis testing study…[because] a pilot study does not provide a meaningful effect size estimate for planning subsequent studies due to the imprecision inherent in data from small samples.’
But why were these small trials not considered small? Why were the positive trials treated with the certainty and afforded the confidence that is usually reserved for very large confirmatory studies? Perhaps the imprecision of inadequate powering is assumed to only impact failed studies. Perhaps it is the ‘belief in the law of small numbers’ that we are all susceptible to, where the rules of large samples are incorrectly applied to small samples (Tversky and Kahneman, 1971). Perhaps we were not heeding the risks of low statistical power well enough, as has been the case in many scientific fields (Cohen, 1962; Freiman et al., 1978; Sedlmeier and Gigerenzer, 1989; Halpern et al., 2002; Maxwell, 2004; Button et al., 2013).
Perhaps it was because the history of ADs in clinical trials led us to have unrealistic expectations for their effect sizes? Next, we explore how this came to be.
The origin of unrealistic expectations
Early trials evaluating older generation ADs generally showed sizeable response rates, with upwards of 60% of patients showing satisfactory improvement. For other complex conditions like pain, response rates with drug was about three in four, while the rule for placebo response continued to be informed by Beecher’s assertion that one-third of patients were likely to be responders (Beecher, 1955). Expectations for wide gaps between drug and placebo response gained further traction when the findings from the British MRC depression trial followed Beecher’s pattern of a one-third response rate for placebo and high response rates near 60–70% for imipramine. Such easily observable AD superiority was borne out by Roland Kuhn and the discovery of other tricyclic ADs. Resting on clinical judgments, these agents all appeared to easily clear the placebo hurdle. But, results of this kind would prove difficult to replicate in early regulatory trials for ADs.
Throughout the 1970s, after the thalidomide tragedy brought regulatory forces (US FDA) into the fray, clinical trial methodology became regulated and standardized. In parallel, the face of American psychiatry underwent significant changes resulting in the Diagnostic and Statistical Manual, 3rd ed. (DSM-III, published in 1978). With these new guidelines came the introduction of empirical diagnosis instead of intuitive, clinical judgment-based diagnosis. These two major historical events completely changed the landscape and it was in this emerging landscape that the methodology for assessing AD efficacy took form (Paul Leber, 27 October 2016, personal communication, former head of the psychiatric drug division of the US FDA).
Specifically, the US FDA designed clinical trial methods on the basis of certain assumptions. For patients with chronic (‘syndromal’) illnesses, a drug that simply made a patient feel better would not be approved for efficacy. A commonly cited example is that morphine may make a patient with cancer feel better, but it is not an effective treatment for the actual illness (Thomas Laughren, May 2007, personal communication, former head of psychiatric products for the US FDA). In other words, the potential drug is required to improve the whole syndrome. Oddly enough, the DSM-III fits into this mould perfectly with its pragmatic syndromal approach to depression.
However, there was no easy way to measure such syndromal improvement. Thus, the introduction of the Hamilton Depression Rating Scale in the 1960s was a welcomed event. There were, however, peculiarities of the scale that upon closer look might have made it an ill fit for use in depression trials.
First, Hamilton (1960) had developed what was essentially a clinical scale to assess his own patients. Therefore, the scale’s psychometric properties were not evaluated in relation to healthy controls. The stability of the measures and their ability to distinguish depressed patients from normal groups were not considered during its creation. Second, there were no data as to its sensitivity when used repeatedly (practice effect, normalizing effect, effect of active intervention, etc.). Even more important was the fact that the symptoms listed in the HAM-D did not exactly match the criteria developed for the DSM-III.
Not paying heed to all the vagaries of this new measurement tool, the physicians and scientists at the US FDA, pharmaceutical companies, and academics with interest in mood disorders assumed that treatment effects in future depression trials would remain the same with complex measurement scales such as the HAM-D as with the simple, singular clinical assessments of the past. The measurement tool had changed and the expected effect sizes were grossly overestimated using data from trials with different endpoints, but the statistical probability formulae, including the power calculations, were not changed. Furthermore, older generation ADs such as monoamine oxidase inhibitors and tricyclics were evaluated in small trials that could have been severely underpowered, resulting in selective publication of overestimated effect sizes.
As a result, the AD regulatory trials of the 1970s and 1980s and later were designed with the expectation of a response pattern like that seen in the earlier studies. Specifically, investigators and regulatory agencies were expecting strong and consistent superiority of ADs over placebo. Although in the beginning power calculations were inconsistently carried out, the number of patients enrolled in previous nonregulatory trials informed the enrollment goals of the early regulatory trials. Now that power calculations have become ubiquitous in the trial design process, we can see that these smaller trials, designed with the expectation of high drug and low placebo responses, were underpowered for their demonstrated effect sizes. We now understand that this led to the AD showing superiority over placebo at a disappointing rate of about 50%. A lot of handwringing followed this disappointing performance of ADs in the first couple decades of regulatory AD trials.
Some investigators have suggested that ADs were not effective (Kirsch et al., 2008; Fournier et al., 2010). Others claimed that clinical investigators were manipulating the data. Others have argued that inter-rater and intrarater variabilities contributed to this phenomenon (Demitrack et al., 1998; Kobak et al., 2005; Kobak and Thase, 2007). This particular suggestion has not been borne out in prospective implementation of rater monitoring, which has led to potentially higher reliability, but less sensitivity and accuracy (Khan et al., 2014a, 2014b).
In the midst of this controversy, some of the biostatisticians at pharmaceutical companies such as Forest, Eli Lilly, and Pfizer came to doubt the reliability of findings like those from the British MRC trials. It seems that they had come to accept that the modern design of clinical trials afforded much lower treatment effect sizes for ADs than was expected. In response, it seems that they lowered their expectations and significantly increased the sample size in more recent trials. Clearly, they were right as much better success rates are seen in these larger trials.
Following this logic, if one were to assume that the AD-placebo differences were more in the range of 10% (or a modest effect size of 0.3) and power for this effect size (by including around 150 patients per treatment) then the success rate of an AD trial would likely continue to be over 90%. This is demonstrated clearly in the table. By accepting that AD treatment effect sizes are generally modest in modern clinical trials, one is no longer designing and conducting trials that operate like a coin flip, at the mercy of chance. We can stop wasting resources trying to manipulate the coin or the person flipping it, as has been our wont for the past several decades.
However, this solution does not silence the critics who are concerned about the utility of ADs or the clinical trial methodologists who advocate for inclusion of ‘purer samples of depressed patients’ (implying that such ‘purer samples’ would reveal the truth that ADs are very effective and silence the critics). How can it be that these medications that appear to have strong effects when used in practice (Kramer, 2016) can perform so unimpressively in clinical trials? Next, we explain a major flaw in AD clinical trials that has perhaps gone unnoticed for too long.
The perils of measuring complex syndromes with composite scale
It seems that we are overlooking and taking for granted the very thing a clinical trial relies on the most – the chosen measurement. Little attention has been paid to the fact that the historical shift from a stable, single-factor outcome measure to a very complex, multidimensional scale is likely to have changed how easily we can see treatment effects. Again, this issue is firmly rooted in statistical principles.
The seductive and relatively simple statistical concepts proposed by Ronald Fisher (1925) to test null hypotheses in scientific trials have done well by most measures. It is easy to count the number of bacilli that die with a specific antibiotic versus a control as an example of acute and simple models of pathology. Surprisingly, Fisher’s statistical formulae have also done well in trials for complex diseases such as hypertension and diabetes mellitus type 2. The chances of failure for a new antidiabetic treatment with a known mechanism of action is 0% (Khan et al., 2018b). The failure rate of new antihypertensive agents is about 6% (Khan et al., 2018a). Furthermore, hypertension and diabetes trials are subjected to significant placebo responses and there is evidence suggesting that response to placebo in trials for these conditions has been increasing substantially over the past few decades (Khan et al., 2018a, 2018b).
So, what is the difference between these successful trials in other conditions and depression trials? It seems that there are two main factors operating here. First, trials for hypertension and diabetes drugs are powered quite adequately for their typical effect sizes. Such trials usually contain more than five times the number of patients needed to show the mean difference between drug and placebo. Second, and even more importantly, is the fact that they have a single outcome measure that is uniform in presentation (either the change in diastolic blood pressure or level of HbA1c). These measures are not determined by a collection of symptoms or a series of sequenced questions – they are discrete, surrogate indices for the entire condition of hypertension and diabetes. There are very few moving parts in these measurements.
This is in strong contrast to the dependent measures used in depression trials. Such measures are typically composite scores (such as the HAM-D or Montgomery-Asberg Depression Rating Scale total score or a similar composite score) that result in a total value by combining subparts of a scale. These subparts of the total score aim to capture elements of any and all potential signs and symptoms typical of the depressed patients studied thus far. In such multidimensional measures, moving parts abound.
However, the assumption is that depression rating scales measure one underlying depression construct and therefore patients who score similarly on the composite have a similar ‘severity’ of depression and are comparable to one another. This assumption is unfounded, as composite scale scores for depression are neither uniform in what they are measuring or how they are measuring it. They are, in fact, highly diverse in the signs and symptoms of depression that they measure.
The lack of uniformity and reliability in depression measurements has been extensively explored by Eiko Fried of the Netherlands. In one publication (Fried and Nesse, 2015) he states: ‘three symptoms – sleep problems, weight/appetite problems, and psychomotor problems – encompass opposite features (insomnia vs. hypersomnia; weight/appetite gain vs. loss; psychomotor retardation vs. agitation)’. In fact, patients with similar total scores can represent ‘roughly 1000 unique combinations of symptoms that all qualify for a diagnosis of MDD, some of which do not share a single symptom’ (Fried and Nesse, 2015). In addition, different symptoms carry varying degrees of functionality and impairment (Fried et al., 2014).
These nuances are not apparent when summarized. Fried (2015) put it directly: ‘…sum-scores obfuscate important differences between symptoms on the one hand and between individuals on the other hand’. This overlap (or lack thereof) between symptoms on scales is illustrated elegantly in Fig. 2 as synthesized in the article by Fried (2017).
Fig. 2.
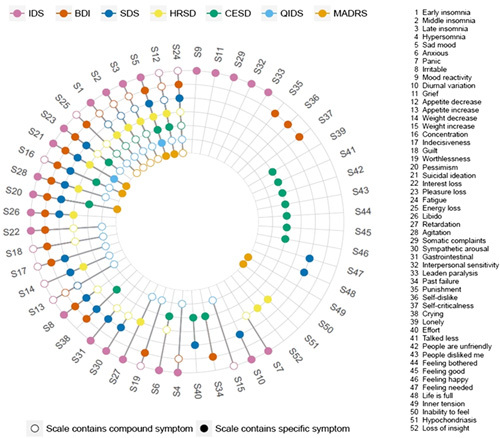
Co-occurrence of 52 depression symptoms across seven depression rating scales. Colored circles for a symptom indicate that a scale directly assesses that symptom, while empty circles indicate that a scale only measures a symptom indirectly. For instance, the IDS assesses item 4 hypersomnia directly; the BDI measures item 4 indirectly by a general question on sleep problems; and the SDS does not capture item 4 at all. Note that the nine QIDS items analyzed correspond exactly to the DSM-5 criterion symptoms for MDD. BDI, 21-item Beck Depression Inventory-II; CESD, Center for Epidemiologic Studies Depression Scale; DSM-5, Diagnostic and Statistical Manual, 5th ed.; HRSD, Hamilton Rating Scale for Depression; IDS, Inventory of Depressive Symptoms; MADRS, Montgomery-Asberg Depression Rating Scale; SDS, Zung Self-Rating Depression Scale; QIDS, Quick Inventory of Depressive Symptoms. Reprinted from: Fried (2017). Copyright [Elsevier], [Amsterdam, Netherlands]. All permission requests for this image should be made to the copyright holder.
Furthermore, ADs may not have the same effect on all symptoms uniformly (Ballard et al., 2018). For example, a specific AD may be sedating and thus make the patient with hypersomnia worse or it may have a sleep loss effect for a patient with insomnia. As to be expected, it is difficult, if not impossible to evaluate such detailed effects in a clinical trial where everything is designed to mute any noise rather than augment it.
In addition, repeated measures of the signs and symptoms of depression are likely to change unpredictably – some getting better, others remaining the same, and a few getting worse. For example, even though a patient with insomnia may be getting better sleep, his mood may still be low and his psychomotor activity not affected at all. Such pattern of response has been well documented by early clinical investigators (Derogatis et al., 1972). The way that these symptoms can interact with one another in a system (Fried, 2015; Ballard et al., 2018) has not been duly considered.
All of these effects, going on just below the surface, are glossed over in a composite scale score. When a patient receives a HAM-D total score of 25 at the start of trial and later drops to 15, we have absolutely no idea what happened and in what ways that patient improved. With all of the underlying elements pulling on one another and moving in opposite ways, the overall readout from the total score is generally that little movement has occurred at all.
What we are discussing is the lack of precision of these measurement tools. It is important to remember that the statistical principles of clinical trials conceptualized by Ronald Fisher were designed for measurements that operate with high precision and are direct and self-contained, as in the case of HbA1c or diastolic blood pressure. In multidimensional composite measurements like the scales and techniques used in depression, schizophrenia, epilepsy, and chronic pain studies, where by the end of the trial patient symptom scores have spread in all directions, a much larger sample size is needed to make sense out of the mess. Ironically, clinical trials for these conditions also show modest effect sizes and high failure rates because of underpowering.
Aside from the dulling of effects that occurs when these scales are used, there are perhaps more impactful, nonstatistical issues with the measurement techniques we use in depression trials. Because psychological constructs cannot be measured or observed directly, the measures used in depression are reliant on patient recall and self-report (Fried, 2015). Interview questions from depression scales are highly complicated and usually require much explanation. Even nondepressed individuals have trouble with the level of detailed recall required for these scales (the wide range of question topics shown in Fig. 2). We administer these complicated, indirect questions to people who are already cognitively impaired as a result of the depressive condition. Repeat questioning of such impaired patients undoubtedly leads to masking of potential differences.
Besides these impediments, expectancy may further confound patient recall and reporting. In other words, patients recruited into AD clinical trials typically hold the impression that ADs work. Such expectancy has been shown to increase the magnitude of both placebo response and AD response (Papakostas and Fava, 2009; Sinyor et al., 2010; Rutherford and Roose, 2013; Berna et al., 2017). This expectancy may play a particularly large role among Americans participating in AD trials as there is considerable direct marketing to consumers through public media in the USA (Shiv et al., 2005). Such exposure is likely to condition potential patients to have high expectations for ADs as marketing exposure is involuntary. Incredibly, US pharmaceutical companies spent over $5 billion on consumer advertising in the last year.
Moreover, major depression and other psychiatric disorders are ‘likely to encompass a group of disorders that are heterogenous with respect to etiology and pathophysiology’ (Hasler, 2006). Evidence has been mounting that several biological and psychopathological endophenotypes may be candidate variables associated with depressive subtypes (Hasler et al., 2004). Therefore, a group of patients meeting the DSM criteria for major depression are very unlikely to share the same pathological processes and phenotypic characteristics. This undoubtedly leads to differential sensitivity to ADs and therefore differing response rates. This heterogenous presentation of depression may not influence patients’ response to placebo. Such diversity in the disease process of major depression is not compatible with symptom-based composite scale measurement techniques.
In essence, antidepressant clinical trials have been plagued by the task of measuring a heterogenous syndrome with diverse patterns of symptom change using a multifaceted measurement tool that may not be measuring the same thing across patients or over time (Fried and Nesse, 2014; Fried et al., 2016). In this context, it is not surprising that the effect sizes for a trial measuring a simple factor such as Clinical Global Impression score or just one item on a scale (such as depressed mood) are much larger than ones obtained from complex composite scores such as the HAM-D or the Montgomery-Asberg Depression Rating Scale. Added to the fact that these measures rely on the potentially faulty memory and self-report of depression-impaired patients with high expectations, it is easy to see how treatment effects could be obscured.
But, what about finding treatments for depression that have much higher effect sizes? We will now examine these possibilities and explain why they might not solve the problem related to antidepressant clinical trials.
Chasing the rainbow
The search for the cause of depression (and thus, its magic bullet) has continued for more than a century. Brain scanning (Schmaal et al., 2017) and minute examination of blood products and endocrine functions (Nemeroff and Vale, 2005) have been the avenues for discovery in this pursuit. Yet the search for depression biomarkers has not been fruitful (Carroll et al., 1981; Hasler, 2010).
In addition, it is evident that drugs such as alcohol, cocaine, amphetamines, and marijuana, to name a few, have profound actions on mood and related behaviors – one does not need a complicated composite score to measure this. Their dramatic effects would be evident on any scale, psychometric or otherwise. Of course, these effects may not be positive, but it is a truism that many drugs can drastically affect mood and behavior in obvious ways.
Similarly, more aggressive therapies such as ECT, ketamine infusions, and vagus nerve and trans-magnetic stimulation also have noticeable, widespread effects on mood and behavior. However, these treatments are highly invasive, mostly short-lived, and carry significant risk of unwanted side effects due to severe disruption of the central nervous system (CNS). However, contemporary antidepressants such as selective serotonin and serotonin and norepinephrine reuptake inhibitors are long-lasting with effects that appear over time. They have fairly minor side effects and act modestly as acute relief for symptoms without major disruptions of the CNS. They are also likely to have better prophylactic effects (Geddes et al., 2003).
These points simply suggest that relatively benign treatments, with effects that are subtle and that occur over time, are likely to show modest efficacy in clinical trials. This is particularly true given the conundrum of measurement. To find a highly effective, quick-acting, long-lasting, noninvasive antidepressant with a specific mechanism of effect that does not act by severely disrupting CNS functioning is like chasing the rainbow to find the pot of gold – it seems that as we get closer, the end of the rainbow simply moves further away.
So where does that leave us?
It is clear that there are many historically-rooted flaws in the way we are measuring antidepressant treatment effects in clinical trials. Essentially, over the last 60 years we have assumed that all treatments will function the same and the clinical trial models of penicillin and the BMRC trials will work well to determine their efficacy. However, we cannot expect antidepressants to perform like penicillin where we can count the bacilli and know if it is working. We cannot expect the modern antidepressants to perform like the early tricyclics, which were prone to unblinding because of side effects and were measured with clinical impressions.
Unrealistic expectations based on the early history of antidepressant efficacy trials put modern clinical trials for depression on a course destined for high levels of failure. Expecting the high effect sizes seen in previous trials has paved the way for underpowering and unreliable results, which is only now beginning to be remedied with the inclusion of larger sample sizes.
In addition, depression measurements involving complex, multifaceted scales with multiple underlying moving parts may not have the accuracy or the sensitivity to measure antidepressant treatment effects, particularly when they are based on patient self-report and are subject to expectation bias. These limitations of depression measurements have gone underappreciated as criticisms of antidepressants themselves have taken center stage.
Furthermore, the elements of psychotherapy also muddy the waters when it comes to the outcomes of antidepressant clinical trials. Specifically, all patients regardless of drug or placebo assignment, receive all components of treatment (i.e. thorough evaluation, explanation of and an opportunity to vocalize distress, an expert healer with enthusiasm and positive regard, a plausible treatment) in addition to the influences described earlier (Frank and Frank, 1991). In addition, the role of the pill itself seems critical in relieving depressive symptoms as relief is much higher in depression trial participants receiving either active or placebo pill compared with those receiving all other elements of treatment in the trial except for the assignment of a pill (Leuchter et al., 2014).
Where do we go from here?
First, we must acknowledge the limitations of clinical trials for drugs treating complex syndromes. Syndromes like these that cannot be captured well in a univariate, singular, consistent measure are going to have limited effect sizes. Trials using such complicated scales to measure what is already a complex condition are going to continue to require large sample sizes to be able to parse through the noise and receive the signal. Accordingly, depression trials will continue to need to be powered adequately with several hundreds of patients to get reliable results.
Next, we must appreciate how various treatments for certain conditions will fundamentally perform in clinical trials – we need to appreciate these differences when conceptualizing and conducting clinical trials as well as when interpreting their data. The goal has always been to find effective, long-lasting, and innocuous treatments for depression. However, such treatments that are not too toxic, not too habituating, and not too disruptive of the CNS will inevitably be subtle and show more modest effect sizes in clinical trials as they are currently designed.
It is worth repeating 18 years later that ‘clinical trials are not primarily designed to identify the optimal effect of antidepressants, but rather to rapidly assess their efficacy [over placebo] …accordingly, clinical trials may identify the lower bound of the effect size’ (Khan et al., 2000). The assessment of efficacy in clinical trials is not designed to inform the real-life effectiveness of these antidepressants in practice; it is simply a hurdle to clear.
Importantly, we need to realize that the placebo response is not the mystical and all-powerful phenomenon that we once thought. It is very much related to the inherent issues of measuring complex syndromes for which direct observation is elusive, such as depression. The placebo response has received perhaps too much focus and credit for determining the fate of an antidepressant trial, when it has in fact become a predictable and surmountable factor in adequately powered trials. It is essential to remember that red herrings abound in this messy world of clinical trials.
Finally, we need to remain critical of the constructs and premises that rationalize our assumptions when it comes to clinical trials. Historical precedent may not apply as medical treatments and diagnosis evolve. Just as well, innovation in scientific and statistical methods need to be thoroughly scrutinized before we can justify their widespread adoption.
So what has the 60-year history of antidepressant clinical trials taught us? When it comes to assessing the efficacy of various treatments and conditions, one size does not fit all – future clinical trials are going to require skilled tailoring.
Acknowledgements
This analysis was funded by the Northwest Clinical Research Center (Bellevue, Washington, USA).
Conflicts of interest
There are no conflicts of interest.
References
- Affidavit of William Thomas Beaver, M.D. in the case of Pharmaceutical Manufacturers Association v. Robert H. Finch and Herbert Ley, Civil Action No. 3797, United States District Court for the District of Delaware. Dr. Beaver was the clinical pharmacologist at Georgetown University who is credited with drafting the initial regulations defining ‘adequate and controlled’ clinical studies. (Personal correspondence, Peter Barton Hutt Esq. and Dr. Robert Temple, FDA, December, 2007, FDA History Office Files).
- Ballard ED, Yarrington JS, Farmer CA, Lener MS, Kadriu B, Lally N, et al. (2018). Parsing the heterogeneity of depression: an exploratory factor analysis across commonly used depression rating scales. J Affective Disord 231:51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beecher HK. (1955). The powerful placebo. JAMA 159:1602–1606. [DOI] [PubMed] [Google Scholar]
- Berna C, Kirsch I, Sr Zion, Lee YC, Jensen KB, Sadler P, et al. (2017). Side effects can enhance treatment response through expectancy effects: an experimental analgesic randomized controlled trial. Pain 158:1014–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford JU. (2017). Leveraging statistical methods to improve validity and reproducibility of research findings. JAMA Psychiatry 74:119–120. [DOI] [PubMed] [Google Scholar]
- British Medical Research Council (1965). Clinical trial of the treatment of depressive illness. BMJ 1:881–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WA, Rosdolsky M. (2015). The clinical discovery of imipramine. Am J Psychiatry 172:426–429. [DOI] [PubMed] [Google Scholar]
- Button KS, Ioannidis JPA, Mokrysz C, Nosek BA, Flint J, Robinson ESJ, et al. (2013). Power failure: why small sample size undermines the reliability of neuroscience. Nature Reviews 14:365–376. [DOI] [PubMed] [Google Scholar]
- Carroll BJ, Feinberg M, Greden JF, Tarika J, Albala AA, Haskett RF, et al. (1981). A specific laboratory test for the diagnosis of melancholia. Standardization, validation, and clinical utility. Arch Gen Psychiatry 38:15–22. [DOI] [PubMed] [Google Scholar]
- Chalmers I. (2010). Why the 1948 MRC trial of streptomycin used treatment allocation based on random numbers. J R Soc Med 104:383–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmura Kraemer H, Mintz J, Noda A, Tinklenberg J, Yesavage JA. (2006). Caution regarding use of pilot studies to guide power calculations for study proposals. Arch Gen Psychiatry 63:484–489. [DOI] [PubMed] [Google Scholar]
- Cohen J. (1962). The statistical power of abnormal-social psychological research: A review. J Abnorm Soc Psychol 65:145–153. [DOI] [PubMed] [Google Scholar]
- Demitrack MA, Faries D, Herrera JM, Potter WZ. (1998). The problem of measurement error in multisite clinical trials. Psychopharmacol Bull 34:19–24. [PubMed] [Google Scholar]
- Derogatis LR, Lipman RS, Covi L, Rickels K. (1972). Factorial invariance of symptoms dimensions in anxious and depressive neuroses. Arch Gen Psychiary 27:659–665. [DOI] [PubMed] [Google Scholar]
- Fisher RA. (1925). Statistical methods experimental design and scientific inference. Reprint. Oxford, NY: Oxford University Press, 2003. [Google Scholar]
- Fletcher J. (2008). Interpreting an underpowered trial. BMJ 337:a2957. [DOI] [PubMed] [Google Scholar]
- Fournier JC, DeRubeis RJ, Hollon SD, Dimidijan S, Amsterdam JD, Shelton RC, et al. (2010). Antidepressant drug effects and depression severity: a patient-level meta-analysis. JAMA 303:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank JD, Frank JB. (1991). Persuasion and healing: a comparative study of psychotherapy. Baltimore, MD: The Johns Hopkins University Press. [Google Scholar]
- Freiman JA, Chalmers TC, Smith H, Kuebler RR. (1978). The importance of Beta, the Type II error and sample size in the design and interpretation of the randomized controlled trials- survey of 71 negative trials. N Engl J Med 299:690–694. [DOI] [PubMed] [Google Scholar]
- Fried E. (2015). Problematic assumptions have slowed down depression research: why symptoms, not syndromes are the way forward. Front Psychol 6:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried E. (2017). The 52 symptoms of major depression: lack of content overlap among seven common depression scales. J Affective Disord 208:191–197. [DOI] [PubMed] [Google Scholar]
- Fried EI, Nesse RM. (2014). Depression is not a consistent syndrome: an investigation of unique symptom patterns in the STAR*D study. J Affective Disord 172:96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried EI, Nesse RM. (2015). Depression sum-scores don’t add up: why analyzing specific depression symptoms is essential. BMC Medicine 13:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried EI, Nesse RM, Zivin K, Guille C, Sen S. (2014). Depression is more than the sum score of its parts: individual DSM symptoms have different risk factors. Psychol Med 44:2067–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried EI, van Borkulo CD, Epskamp S, Schoevers RA, Tuerlunckx F, Borsboom D. (2016). Measuring depression over time… or not? Lack of unidimensionality and longitudinal measurement invariance in four common rating scales of depression. Psychol Assess 28:1354–1367. [DOI] [PubMed] [Google Scholar]
- Geddes JR, Carney SM, Davies C, Furukawa TA, Kupfer DJ, Frank E, et al. (2003). Relapse prevention with antidepressant drug treatment in depressive disorders: a systematic review. Lancet 361:653–661. [DOI] [PubMed] [Google Scholar]
- Gibertini M, Nations KR, Whitaker JA. (2012). Obtained effect size as a function of sample size in approved antidepressants: a real-world illustration in support of better trial design. Int Clin Psychopharmacol 27:100–106. [DOI] [PubMed] [Google Scholar]
- Halpern SD, Karlawish JHT, Berlin JA. (2002). The continuing unethical conduct of underpowered clinical trials. JAMA 288:358–362. [DOI] [PubMed] [Google Scholar]
- Hamilton M. (1960). A rating scale for depression. J Neurol Neurosurg Psychiatry 23:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler G. (2006). Evaluating endophenotypes for psychiatric disorders. Rev Bras Psiquiatr 28:91–92. [DOI] [PubMed] [Google Scholar]
- Hasler G. (2010). Pathophysiology of depression: do we have any solid evidence of interest to clinicians? World Psychiatry 9:155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler G, Drevets WC, Manji HK, Charney DS. (2004). Discovering endophenotypes for major depression. Neuropsychopharmacology 29:1765–1781. [DOI] [PubMed] [Google Scholar]
- Ioannidis JP. (2005). Why most published research findings are false. PLoS Med 2:e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Warner HA, Brown WA. (2000). Symptom reduction and suicide risk in patients treated with placebo in antidepressant clinical trials. Arch Gen Psychiatry 57:311–317. [DOI] [PubMed] [Google Scholar]
- Khan A, Khan SR, Leventhal RM, Brown WA. (2001). Symptom reduction and suicide risk in patients treated with placebo in antidepressant clinical trials: a replication analysis of the Food and Drug Administration database. Int J Neuropsychopharmacol 4:113–118. [DOI] [PubMed] [Google Scholar]
- Khan A, Detke M, Khan S, Mallinckrodt C. (2003a). Placebo response and antidepressant clinical trial outcome. J Nerv Ment Dis 191:211–218. [DOI] [PubMed] [Google Scholar]
- Khan A, Khan SR, Walens G, Kolts R, Giller EL. (2003b). Frequency of positive studies among fixed and flexible dose antidepressant clinical trials: an analysis of the Food and Drug Administration Summary Basis of Approval reports. Neuropsychopharmacology 28:552–557. [DOI] [PubMed] [Google Scholar]
- Khan A, Kolts RL, Thase ME, Krishnan K, Brown WA. (2004a). Research design features and patient characteristics associated with the outcome of antidepressant clinical trials. Am J Psychiatry 161:2045–2049. [DOI] [PubMed] [Google Scholar]
- Khan A, Brodhead AE, Kolts RL. (2004b). Relative sensitivity of the Montgomery-Asberg depression rating scale, the Hamilton depression rating scale and the Clinical Global Impressions rating scale in antidepressant clinical trials: a replication analysis. Int Clin Psychopharmacol 19:1–4. [DOI] [PubMed] [Google Scholar]
- Khan A, Faucett J, Brown W. (2014a). Magnitude of change with antidepressants and placebo in antidepressant clinical trials using structured, taped and appraised rater interviews compared to traditional semi-structured interviews. Psychopharmacology 231:4301–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Faucett J, Brown W. (2014b). Magnitude of placebo response and response variance in antidepressant clinical trials using structured, taped and appraised rater interviews compared to traditional rating interviews. J Psychiatr Res 51:88–92. [DOI] [PubMed] [Google Scholar]
- Khan A, Fahl Mar K, Faucett J, Khan Schilling S, Brown WA. (2017). Has the rising placebo response impacted antidepressant clinical trial outcome? Data from the US Food and Drug Administration 1987-2013. World Psychiatry 16:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Fahl Mar K, Schilling J, Brown WA. (2018a). Does the rising placebo response impact antihypertensive clinical trial outcomes? An analysis of Food and Drug Administration data 1990–2016. PLoS One 13:e0193043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Fahl Mar K, Schilling J, Brown WA. (2018b). Magnitude and pattern of placebo response in clinical trials of oral antihyperglycemic agents: data from the Food and Drug Administration 1999–2015. Diabetes Care 41:994–1000. [DOI] [PubMed] [Google Scholar]
- Khin NA, Chen Y, Yang Y, Yang P, Laughren TP. (2011). Exploratory analyses of efficacy data from major depressive disorder trials submitted to the US Food and Drug Administration in support of New Drug Applications. J Clin Psychiatry 72:464–472. [DOI] [PubMed] [Google Scholar]
- Kirsch I, Deacon BJ, Huedo-Medina TB, Scoboria A, Moore TJ, Johnson BT. (2008). Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med 5:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobak K, Thase ME. (2007). Why do clinical trials fail?: The problem of measurement error in clinical trials: Time to test new paradigms? J Clin Psychopharmacol 27:1–5. [DOI] [PubMed] [Google Scholar]
- Kobak KA, Feiger AD, Lipsitz JD. (2005). Interview quality and signal detection in clinical trials. Am J Psychiatry 162:628. [DOI] [PubMed] [Google Scholar]
- Kramer PD. (2016). Ordinarily well: the case for antidepressants. New York, NY: Farrar, Straus, and Giroux. [Google Scholar]
- Leon AC, Davis LL, Kraemer HC. (2011). The role and interpretation of pilot studies in clinical research. J Psychiatr Res 45:626–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuchter AF, Hunter AM, Tartter M, Cook IA. (2014). Role of pill-taking, expectation and therapeutic alliance in the placebo response in clinical trials for major depression. Br J Psychiatry 205:443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell SE. (2004). The persistence of underpowered studies in psychological research: causes, consequences, and remedies. Psychol Methods 9:147–163. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, Vale WW. (2005). The neurobiology of depression: inroads to treatment and new drug discovery. J Clin Psychiatry 66:5–13. [PubMed] [Google Scholar]
- Papakostas GI, Fava M. (2009). Does the probability of receiving placebo influence clinical trial outcome? A meta-regression of double-blind, randomized clinical trials in MDD. Eur Neuropsychopharmacology 19:34–40. [DOI] [PubMed] [Google Scholar]
- Reinhart A. (2015). Statistics done wrong: a woefully complete guide. San Francisco, CA: No Starch Press, Inc. [Google Scholar]
- Rutherford BR, Roose SP. (2013). A model of placebo response in antidepressant clinical trials. Am J Psychiatry 170:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmaal L, Hibar DP, Samann PG, Hall GB, Baune BT, Jahanshad N, et al. (2017). Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the ENIGMA Major Depressive Disorder Working Group. Mol Psychiatry 22:900–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlmeier P, Gigerenzer G. (1989). Do studies of statistical power have an effect on the power of studies? Psychol Bull 105:309–316. [Google Scholar]
- Shiv B, Carmon Z, Ariely D. (2005). Placebo effects of marketing actions: consumers may get what they pay for. J Mark Res 42:383–393. [Google Scholar]
- Sinyor M, Levitt AJ, Cheung AH, Schaffer A, Kiss A, Dowlati Y, et al. (2010). Does inclusion of a placebo arm influence response to active antidepressant treatment in randomized controlled trials? results from pooled and meta-analyses. J Clin Psychiatry 71:270–279. [DOI] [PubMed] [Google Scholar]
- Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. (2008). Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med 358:252–260. [DOI] [PubMed] [Google Scholar]
- Tversky A, Kahneman D. (1971). Belief in the law of small numbers. Psychol Bull 76:105–110. [Google Scholar]
- Walsh BT, Seidman SN, Sysko R, Gould M. (2002). Placebo response in studies of major depression: variable, substantial, and growing. JAMA 287:1840–1847. [DOI] [PubMed] [Google Scholar]