

Abstract
To cope with DNA damage, mitochondria developed a pathway by which severely damaged or unrepairable mtDNA molecules are abandoned and degraded, and new molecules are resynthesized using intact templates, if available. In this unit, we describe a method that harnesses this pathway to completely eliminate mtDNA from mammalian cells by transiently overexpressing the Y147A mutant of human uracil-N-glycosylase (mUNG1). We also provide an alternate protocol for mtDNA depletion using combined treatment with ethidium bromide (EtBr) and dideoxycytidine (ddC). Support protocols detail approaches for 1) genotyping ρ0 cells of human, mouse and rat origin by PCR; 2) quantitation of mtDNA by qPCR; and 3) preparation of calibrator plasmids for mtDNA quantitation.
Keywords: mtDNA, ρ0 cells, mtDNA damage, cybrids, mtDNA copy number
INTRODUCTION
Mitochondria possess limited ability to repair damage inflicted to their DNA (mtDNA) since the only complete DNA repair pathway available in these organelles is base excision repair (BER). Mitochondria compensate for this limitation by taking advantage of the high redundancy of mtDNA, which ranges from 5–10 copies per cell in sperm to as many as 500,000 copies per cell in oocytes (I. N. Shokolenko & Alexeyev, 2015). To cope with DNA damage, mitochondria developed a pathway whereby severely damaged or unrepairable mtDNA molecules are abandoned and degraded, and new molecules are resynthesized using intact templates, if available (I. Shokolenko, Venediktova, Bochkareva, Wilson, & Alexeyev, 2009; I. N. Shokolenko & Alexeyev, 2015; I. N. Shokolenko, Wilson, & Alexeyev, 2013, 2016; Spadafora, Kozhukhar, Chouljenko, Kousoulas, & Alexeyev, 2016). In this unit, we describe a method that harnesses this pathway to completely eliminate mtDNA from mammalian cells by transiently overexpressing a Y147A mutant of human uracil-N-glycosylase (mUNG1). We also provide an alternate protocol for mtDNA depletion using combined treatment with ethidium bromide (EtBr) and dideoxycytidine (ddC). Support protocols detail approaches for 1) genotyping ρ0 cells of human, mouse and rat origin by PCR; 2) quantitation of mtDNA by qPCR; and 3) preparation of calibrator plasmids for mtDNA quantitation.
BASIC PROTOCOL 1. INDUCING mtDNA LOSS WITH mUNG1
Introductory paragraph
This method employs mtDNA damage induced by mUNG1 to destroy mtDNA and thereby induce the loss of mtDNA (ρ0 phenotype). Cells are first transfected with a plasmid that encodes a mitochondrially-targeted mUNG1 and a fluorescent marker. Transfected cells are sorted using FACS and plated in a medium permissive for growth of ρ0 cells. Colonies are picked into wells of a 24-well plate, expanded, and genotyped by PCR. The resulting ρ0 cells become auxotrophic for uridine and pyruvate and therefore must be grown in media supplemented with these two compounds.
Materials
A CO2 incubator.
An appropriate growth medium.
A cell line for the derivation of the ρ0 phenotype. The protocol involves cell cloning after transfection, and therefore primary and other cells with limited proliferative lifespan are not suitable. In our hands, rapidly proliferating cell lines typically produce better results.
50 mg/ml uridine.
100 mM pyruvate.
A plasmid encoding both a fluorescence marker (e.g., EGFP, mCherry, etc.) and mUNG1 (e.g. pMA3790, Addgene #70110). While endotoxin-free plasmid preparation may be required for particularly sensitive cell lines, regular Qiagen miniprep purity is adequate in many cases.
A transfection reagent. There is no transfection reagent that works well for all cell lines. If a suitable transfection reagent has not been identified, a good place to start is to screen several test samples from Mirus Bio https://www.mirusbio.com/
Cell culture treated 35-mm dishes or 6-well plates.
Cell culture treated 150-mm dishes and 24-well plates.
A fluorescence-activated cell sorter (FACS).
3-mm cloning disks (Andwin Scientific, Fisher cat# NC9417109).
Protocol steps
-
In 35-mm cell culture dishes or 6-well plates, set up transfections of the cell line of interest with either pMA3790 or mock (control) according to recommendations of the manufacturer of the transfection reagent. In our hands, Xfect (Clontech cat# 631317) works well with many cell lines.
While endotoxin-free plasmid preparation may be required for particularly sensitive cell lines, regular Qiagen miniprep purity is adequate in many cases. -
72 hours after transfection, conduct FACS for EGFP-positive cells using mock-transfected cells as negative control to set up sorting gates.
Depending on the cell line, a shorter (48h) or longer (96h) growth time prior to sorting may be used. Gate first on viable cells using FSC vs SSC plot, and then on top 5–10% brightest transfected cells. For optimal results, transfection efficiency should be greater than 10%. -
Collect 2,000 positive cells in 2 ml of sterile growth medium and plate 100, 300 and 900 cells into 150-mm dishes using growth medium supplemented with UP (50 μg/ml uridine and 1 mM pyruvate).
Collecting 2,000 positive cells works well with all cell lines tested so far. However, for cell lines that attach poorly, or for those in which mtDNA loss negatively affects attachment, a greater number of positive cells may need to be collected/plated. Also, if mtDNA-depleted cells attach poorly, the growth period prior to FACS may be shortened to avoid platingρ0 cells.If a flow cytometer is not available, cells transfected with pMA3790 can be plated for selection with G418 (plate 50%, 10% and 1% of all transfected cells into 150-mm cell culture dishes using medium supplemented with UP and appropriate concentration of G418). -
Place dishes into a CO2 incubator, and observe colony formation.
Depending on growth rate, colonies usually become visible within 1–2 weeks. -
In 2–3 weeks, pick small colonies into 24-well plates using cloning disks.
Typically, the protocol results in a population of colonies that are heterogeneous in size. One should resist temptation of picking colonies as soon as they become visible because cells that escape mtDNA elimination grow faster than ρ0 cells and form colonies first. It is useful to circle smaller colonies on the bottom of the dish with a fine-point marker prior to picking them with cloning disks. Alternatively, cloning cylinders (Fisher 14-512-78) may be used. -
When cells in 24-well plates reach >20% confluence, trypsinize them with 80 μl of 0.05% trypsin/0.5mM EDTA in PBS.
Depending on cell line, a more concentrated trypsin solution (e.g. 0.25%) may be needed to efficiently dissociate cells. Disperse cells by vigorous pipetting using sterile 200 μl aerosol barrier tips. Remove 10 μl of cell suspension for analysis by DirectPCR (see SUPPORT PROTOCOL 1). Add 1 ml of fresh medium supplemented with uridine and pyruvate to remaining cells and return 24-well plate to CO2 incubator for growth.
After identification of wells with ρ0 cells by DirectPCR, expand putative ρ0 cells and confirm the ρ0 phenotype by repeating DirectPCR and testing them for inability to grow in the absence of uridine and pyruvate, inability to utilize galactose as carbon source, and/or by testing for the loss of respiratory chain function using Seahorse XF-24 extracellular flux analyzer.
ALTERNATE PROTOCOL 1 (optional). GENERATION OF ρ0 CELLS BY mtDNA DEPLETION WITH EtBr AND ddC
Introductory paragraph
This is a modification of a traditional method for mtDNA depletion using EtBr. This method employs both EtBr and dideoxycytidine (ddC) to reduce toxicity. Cells are treated over a period of up to 4 weeks with a mix of EtBr and ddC, and samples are collected during splitting for DNA isolation and mtDNA copy number determination by duplex qPCR (see SUPPORT PROTOCOL 2). When the average mtDNA copy number in the population approaches 1 molecule per cell, serial dilutions of cells are plated in medium supplemented with 50 μg/ml uridine and 1 mM pyruvate. When colonies grow ≥3mm in diameter, they are picked into 24-well plates, expanded and genotyped by DirectPCR as in BASIC PROTOCOL 1.
Materials
A CO2 incubator.
An appropriate growth medium.
50 mg/ml uridine.
100 mM pyruvate.
EtBr 1 mg/ml.
ddC 200 mM in water.
Cell culture treated 100-mm and 150-mm dishes.
24-well tissue culture treated plates.
3-mm cloning disks (Andwin Scientific, Fisher cat# NC9417109).
Protocol steps
Plate cells into 100 mm dishes in growth medium supplemented with uridine (50 μg/ml) and pyruvate (1 mM) and allow to attach overnight.
-
Replace the medium with the same medium supplemented with 1 μg/ml EtBr and 200 μM ddC and return dish to CO2 incubator.
Concentrations of EtBr and ddC should be optimized for each cell line so that cells continue to grow actively after the first 10 days of depletion. With some concentrations, cells initially respond to treatment but eventually develop resistance (see Figure 1). Since mtDNA depletion is a lengthy process, it is advisable to test several concentrations of EtBr and ddC in parallel. Recommended starting ranges are 0.1 to 2 ug/ml EtBr and 50–200 μM ddC. As depletion progresses, some concentrations can be eliminated as inferior based on results of qPCR. -
Continue to grow cells until 80–90% confluent, changing medium every other day.
When cells are nearly confluent, split them by trypsinization. Split ratios are cell line- and depletion time-dependent. Split ratios of 1:4 to 1:10 can be used for faster growing cells like mouse fibroblasts and many human cell lines within the first 10 days of depletion. For slower growing cells, and for faster growing cells after the first 10 days of depletion when their growth slows down, lower split ratios are recommended. When splitting, collect aliquots for DNA isolation and determination of mtDNA copy number by duplex qPCR(SUPPORT PROTOCOL 2). -
When mtDNA copy number approaches 1 per cell, trypsinize cells and plate 100, 500 and 2,500 cells into separate 150-mm cell culture dishes for cloning using growth medium supplemented with UP, but devoid of EtBr and ddC.
At this point, cells may cease proliferating and appear unhealthy. Plating different cell numbers is meant to compensate for any attachment defects that cells may exhibit at this point. When clones appear, pick them into 24-well plates, expand, and genotype as in BASIC PROTOCOL 1.
Figure 1.

A representative time course of mtDNA depletion using BASIC PROTOCOL 2. 4B6 cells (I. N. Shokolenko, Fayzulin, et al., 2013) were treated either with EtBr alone, or with a mix of 0.5 μg/ml EtBr and 50 μM ddC. Note that an EtBr 0.5/ddC50 mix is almost as effective at mtDNA depletion as 2 μg/ml EtBr, whereas cells eventually develop resistance to 0.5 μg/ml EtBr when it is used alone.
SUPPORT PROTOCOL 1 (optional). GENOTYPING ρ0 CELLS BY DIRECTPCR
This protocol is adapted for use with small quantities of cells. Cells are first lysed in DirectPCR buffer and subjected to proteinase K treatment. Then, proteinase K is inactivated, EDTA and salts are diluted, and the resulting crude DNA solution is used in a duplex PCR to simultaneously amplify nuclear and mitochondrial targets.
Materials
DirectPCR Lysis Reagent (Cell) (Viagen cat# 301-C).
Proteinase K 20 mg/ml.
2x PCR mastermix (e.g. Promega GoTaq Green mastermix M7122).
Thermocycler capable of accommodating 200 μl PCR tubes or 96-well plates.
DNA ladder (e.g. Minnesota Molecular HiLo)
-
Genotyping primer mix (made of 100 μM stocks of individual primers) for each species (25 μM for each primer):
ρ0 genotyping human cells: HVRF AATGTCTGCACAGCCACTTTCCAC, HVRR TCGTAGTGTTCTGGCGAGCAGTTT, hPolRmtmultiR2 CCAGTCTAAATGAGGGCAAGT, hPolRmtdel2 GCGAGTGCTGCCCCGAC. PCR products: mitochondrial 901 bp, nuclear 371 bp.
ρ0 genotyping mouse cells: mMitF AAAGCATCTGGCCTACACCCAGAA, mMitR ACCCTCGTTTAGCCGTTCATGCTA, mNucF CCACGTGCTCTGTATGAGATT, mNucR ATGCTGGCTTATCTGTTCCTT. PCR products: mitochondrial 1,041 bp, nuclear 636 bp.
ρ0 genotyping rat cells: rDloopDiagnF CATCAACACCCAAAGCTGATATTC, rDloopDiagnR TTATAAGGCCAGGACCAAACC, rPrnPmultiR CAGGAAGATGAGGAAGGAGATG, rPrnPcrispr2F CTGGTGGAAGCCGGTAC. PCR products: mitochondrial 1,023bp, nuclear 656bp.
Equipment and reagents for agarose gel electrophoresis.
Gel documentation equipment.
Protocol steps
-
Prepare a cell lysis mastermix.
Mix DirectPCR Lysis Reagent (Cell) with 20 mg/ml proteinase K to achieve final concentration of 1 mg/ml. Make enough mastermix for N+1 samples x 10 μl per sample (e.g., 80 μl for analysis of 7 clones). -
Dispense 10 μl of mastermix into each 200μl PCR tube.
It is often convenient to use 200μl PCR strip tubes or 96-well PCR plates, especially when analyzing large numbers of clones. Add 10 μl of cell suspension (see steps 6 and 7 of BASIC PROTOCOL 1) into each tube (well) and mix by pipetting.
-
Close lids on PCR tubes/seal 96-well PCR plate and place into the block of a thermocycler. Run a program: 55°C for 30 min followed by 95°C for 15 min.
The first segment of this program facilitates cell lysis and protein digestion by proteinase K. The second segment inactivates proteinase K. -
Add 40 μl of distilled water to each tube/well and mix by pipetting.
This step facilitates subsequent PCR by diluting impurities and EDTA, which can be inhibitory. It can be omitted when starting with a small number of cells. -
Assemble and run PCR genotyping reactions. Dispense genotyping mastermix consisting of 10 μl of 2x PCR mastermix +10 μl dH2O+ 0.4 μl of species-specific genotyping primer mix (see MATERIALS) per well. Add 2 μl of DNA solution from step 5 and run the following program: Initial denaturation 1 min 95°C followed by 35 cycles of 95°C for 10 sec, 55°C for 20 sec and 68°C for 30 sec, followed by a 3 min hold at 72°C, followed by infinite hold at 10°C. This program works well with Promega’s GoTaq Green 2x PCR mastermix. Other PCR mastermixes/polymerases may require adjustment of PCR parameters.
To distinguish between a PCR failure and a loss of mtDNA, each genotyping primer mastermix contains two primers for amplification of a nuclear gene and two primers specific for mtDNA. Nuclear primers serve as an amplification control, and should produce a product in ρ0 cells. -
Analyze PCR products by gel electrophoresis.
Use 1% agarose gel in TAE buffer. When using the Horizon 58 gel electrophoresis system (Labrepco SKU: 41060039), the run can be completed in 24 min at 10 V/cm. Adding EtBr (350 ng/ml. Mutagen!) to both buffer and gel allows omission of the DNA staining step. -
Image the agarose gel using a gel documentation system and analyze results.
Typically, in wild type cells, only the mitochondrial target is amplified (Figure 2), because due to the high mtDNA copy number, amplification of the mitochondrial target uses up dNTPs before enough nuclear target is amplified to be visible on an EtBr-stained agarose gel. In ρ0 cells, no amplification of mtDNA is observed and only the product of nuclear DNA (nDNA) amplification is visible. In cells with low mtDNA copy number, amplification of both nuclear and mitochondrial targets can be observed.
Figure 2.

Representative results of PCR genotyping. Cells of A, mouse (4B6); B, human (143B); and C, rat (C6) origin were depleted of mtDNA and genotyped using primers described in the SUPPORT PROTOCOL 1. Lanes 1, DNA size marker (HiLo); 2, WT cells; 3, ρ0 cells; 4, 4B6 cells with reduced mtDNA copy number (Spadafora, Kozhukhar, & Alexeyev, 2016).
SUPPORT PROTOCOL 2 (optional). DETERMINATION OF mtDNA COPY NUMBER BY qPCR
In this protocol, total DNA is isolated from cultured cells, digested with EcoRI restriction endonuclease, and subjected to duplex qPCR using as calibrator a plasmid that contains cloned nuclear and mitochondrial targets (SUPPORT PROTOCOL 3). Starting copy numbers for the nuclear and mitochondrial targets are then determined, and their ratio is used to calculate mtDNA copy number per cell.
Materials
A kit for isolating total cellular DNA (e.g. Qiagen DNeasy Blood and Tissue kit cat#69504).
EcoRI restriction endonuclease and appropriate buffer.
A sensitive UV-spectrophotometer capable of measuring absorbance at 260 and 280 nm (e.g, NanoDrop Lite).
A qPCR instrument (e.g., Bio-Rad CFX-96) capable of resolving at least two fluorophores.
A qPCR mastermix suitable for your qPCR instrument (e.g. Bioline SensiFAST™ Probe No-ROX Kit, cat# Bio-86005 for Bio-Rad CFX-96)
qPCR primers and probes mastermix (See REAGENTS AND SOLUTIONS).
A linearized calibrator plasmid(s) (See REAGENTS AND SOLUTIONS) containing cloned nuclear and mitochondrial targets, e.g. pMA2789 for mouse mtDNA quantitation or pMA4684 for human mtDNA quantitation (Figure 3). Both plasmids are available from the authors.
Figure 3.

Plasmid maps. Abbreviations: ApR, bacterial ampicillin resistance gene; BGH pA, HSVTk pA, and SV40 pA, corresponding polyadenylation signals; CMV, EF1a, and SV40, corresponding promoters; F1 ori, single-stranded origin of replication of the bacteriophage F1; MTS, mitochondrial matrix targeting sequence of human ornithine transcarbamylase; myc, myc tag epitope; mUNG1, a gene encoding Y147A mutant of the human UNG1; NeoR, G418 and kanamycin resistance gene; ColE1ori, bacterial origin of replication; qPCR mNuclear, qPCR mMito, qPCR hNuclear, qPCR hMito, fragments encompassing nuclear and mitochondrial targets in mouse and human genomes, respectively.
Protocol steps
-
Isolate DNA from a sample of cells.
Column-based methods are rapid and produce high-quality DNA. However, methods based on phenol-chloroform extraction work just as well. -
(Optional) Quantitate DNA using a spectrophotometer.
This helps in standardizing the amount of DNA used for qPCR. Digest 20 ng DNA with EcoRI in 20μl reaction.
-
In reaction vessels appropriate for your qPCR instrument (e.g., in 96-well plates or 8-or 12-well tube strips with optically transparent lids for CFX-96), assemble triplicate 10 μl qPCR reactions by mixing 5 μl of the 2x qPCR mix, 2 μl of the EcoRI-digested template DNA, 0.5 μl of the 20x qPCR primer mastermix, and 2.5 μl of H2O. Separately, assemble calibration reactions using as a template serial 10-fold dilutions of linearized calibrator plasmid (102 to 107 copies/μl).
It is useful to prepare a mastermix containing 2x qPCR mix, water, and primers and probes sufficient for the number of intended reactions and to dispense 8 μl of this mix into each well. Typically, 10 μl reactions work well. However, if tube/well sealing/evaporation becomes a problem as evidenced by erratic results, switching to standard 20 μl reactions may prove helpful. -
Run qPCR using conditions recommended by the manufacturer of your 2x qPCR mastermix and analyze results.
At the end of the run, your qPCR instrument’s software should calculate the starting quantities of nuclear and mitochondrial targets. Dividing starting quantity of the mitochondrial target by starting quantity of nuclear target and multiplying the result by 2 (for diploid genome) will provide mtDNA copy number per diploid cell. When it is known that cells are aneuploid and copy number for the nuclear target (hTERT or mTert) is different from 2 per cell, calculations should be adjusted accordingly.
SUPPORT PROTOCOL 3 (optional). PREPARATION OF THE CALIBRATOR PLASMID FOR qPCR
In this protocol, a calibrator plasmid is isolated from E. coli, linearized, extracted with phenol/chloroform followed by chloroform, precipitated with ethanol, re-dissolved in water, and quantitated.
Materials
Terrific Broth
E. coli carrying a calibrator plasmid.
Ampicillin 100 mg/ml.
QiaprepSpin miniprep kit or equivalent.
ScaI restriction endonuclease (New England Biolabs).
Phenol:chloroform:isoamyl alcohol mix (25:24:1).
Chloroform:isoamyl alcohol mix (24:1).
100% ethanol, molecular biology grade.
70% ethanol, molecular biology grade.
TE buffer (10 mM Tris, 1 mM EDTA, pH=8.0)
Sodium acetate 3M (pH=5.2).
NanoDrop Lite or Qubit 3.0.
Protocol steps
Inoculate a single colony of E. coli containing pMA2789 or pMA4684 into 5 ml of TB medium containing 400 μg/ml ampicillin and grow overnight in a shaker-thermostat at 37°C and 250 rpm.
Isolate plasmid DNA using Qiagen’s QuiaprepSpin miniprep kit according to manufacturer’s recommendations. Measure plasmid concentration by A260.
-
Digest 20 μg of pMA2789 or pMA4684 in 100 μl reaction volume for 1h at 37°C in a 1.5 ml microcentrifuge tube.
Each plasmid has a single ScaI site, and digestion results in linearization of the plasmid. -
Extract once with phenol:chloroform:isoamyl alcohol (25:24:1).
Add 100 μl of the phenol:chloroform:isoamyl alcohol mix to 100 μl restriction digest, vortex for 30 sec at maximum speed, and spin down for 2 min at 13,000g. Transfer the upper (aqueous) phase into a new 1.5 ml microcentrifuge tube. -
Extract once with chloroform:isoamyl alcohol (24:1).
Repeat steps above. This step removes traces of phenol, which may inhibit qPCR. -
Precipitate DNA with ethanol.
Transfer the supernatant to a fresh 1.5 ml tube and add 10 μl of 3M sodium acetate pH=5.2 and 250 μl of 100% ethanol, molecular biology grade. Place the tube in a −80°C freezer for 5 min then spin in a microcentrifuge for 2 min at 13,000g. Remove the supernatant with a 200 μl pipettor and wash the pellet with 70% ethanol. Repeat the spin in a microcentrifuge for 2 min at 13,000g. Remove the supernatant with a 200 μl pipettor, spin the tube briefly in a microcentrifuge, and remove traces of supernatant with a 10 μl pipettor. -
Dissolve DNA pellet in 50 μl of TE buffer and measure concentration by A260 using NanoDrop Lite or by fluorescence using Qubit 3.0.
Qubit 3.0 is less sensitive to impurities and provides more accurate measurements of DNA concentration.1 ng/μl of pMA2789 corresponds to 2.14x108 copies/μl and 1 ng/μl of pMA4684 corresponds to 1.98x108 copies/μl.
REAGENTS AND SOLUTIONS
100 μM stock solutions of primers and probes
Use your favorite supplier to synthesize primers and probes (we use Integrated DNA Technologies, Coralville, IA). Spin vials with lyophilized oligonucleotides prior to removing the lid to prevent oligonucleotide loss. Add 10 μl of sterile distilled H2O or buffer (e.g., IDTE=10 mM Tris, pH 7.5 or 8.0, 0.1 mM EDTA) per each nmol of oligonucleotide yield.
Store 2–3 years at −20°C.
PCR genotyping primer mastermixes
Mix equal volumes of all four primers. The resulting mix is 25 μM for each primer.
20x mastermixes of qPCR primers and probes
For human cells, prepare a 20x mastermix containing:
175 μl H2O
10 μl mitochondrial forward CGCTTTCCACACAGACATCA primer
10 μl mitochondrial reverse GGCTGGTGTTAGGGTTCTTT primer
5 μl mitochondrial mitochondrial probe TEX615/ CGCTTCTGGCCACAGCACTTAAAC /IAbRQSp
40 μl nuclear forward GACCAAGCACTTCCTCTACTC primer
40 μl nuclear reverse GGAACCCAGAAAGATGGTCTC primer
20 μl nuclear probe 56-FAM/AGAGAGCTG/ZEN/AGTAGGAAGGAGG GC/3IABkFQ/
Final concentrations of oligonucleotides in this mastermix are: 3.3 μM for mitochondrial forward and reverse primers, 1.67 μM for mitochondrial probe, 13.3 μM for nuclear forward and reverse primers, and 6.67 μM for nuclear probe.
Store up to 3 years at −20°C.
a. For mouse cells, prepare a 20x mastermix containing:
175 μl H2O
10 μl mitochondrial forward ACTTCTAACTAAAAGAATTACAGC primer
10 μl mitochondrial reverse TAGACGAGTTGATTCATAAAATTG primer
5 μl mitochondrial mitochondrial probe TEX615/CCCGAAACCAAACGAGCTACCT/IAbSQ
40 μl nuclear forward CCTCAAGCATTCACCTCTTCTTTG primer
40 μl nuclear reverse CCAAGGACCTGCTCGATGAC primer
20 μl nuclear nuclear probe 56-FAM/ACCACCCTC/ZEN/TCTGACCTCCAGCCA/3IABkFQ
Final concentrations of oligonucleotides in this mastermix are: 3.3 μM for mitochondrial forward and reverse primers, 1.67 μM for mitochondrial probe, 13.3 μM for nuclear forward and reverse primers, and 6.67 μM for nuclear probe.
Store up to 3 years at −20°C.
Terrific Broth
Add the following to 800ml distilled H2O:
12g Tryptone
24g Yeast extract
4ml Glycerol
Adjust to 900ml with distilled H2O
Sterilize by autoclaving
Allow to cool to room temperature
Adjust volume to 1000ml with 100ml of a filter-sterilized solution of 0.17M KH2PO4 and 0.72M K2HPO4
Store up to 6 months at room temperature.
Solution of 0.17M KH2PO4 and 0.72M K2HPO4
23.1g KH2PO4
164.3g K2HPO4
H2O to 1L
Filter sterilize.
Store up to 2 years at room temperature.
Ampicillin 100 mg/ml
1g ampicillin
10 ml H2O
Filter sterilize.
Store up to 6 months at +4°C.
200 mM ddC
422.436 mg ddC
10 ml H2O
Filter sterilize.
Store up to 1 year at −20°C.
EtBr 10 mg/ml
100 mg EtBr
10 ml H2O
Filter sterilize.
Store up to 1 year at +4°C.
Uridine 50 mg/ml
5g uridine
100 ml H2O
Filter sterilize.
Store up to 1 year at +4°C.
Pyruvate 100 mM
1.1g sodium pyruvate
100 ml H2O
Filter sterilize.
Store up to 1 year at +4°C.
Proteinase K 20 mg/ml
100 mg Proteinase K
2.5 ml glycerol, ultrapure
2.5 μl 3M CaCl2
250 μl 1M Tris, pH=8.0
2.25 ml H2O
Store up to 1 year at +4°C.
COMMENTARY
Background Information
Most mammalian cells produce the bulk of their ATP through oxidative phosphorylation (OxPhos), which occurs in mitochondria. Mitochondria are unique among mammalian cell organelles in that they house their own genetic information in the form of mitochondrial DNA (mtDNA). The mitochondrial genome is represented by a covalently closed circular double-stranded molecule, with a typical length of 16,569 base pairs in humans. mtDNA encodes 37 genes, including 13 polypeptide components of OxPhos, 2 rRNAs, and 22 tRNAs (Anderson et al., 1981; Bibb, Van Etten, Wright, Walberg, & Clayton, 1981). Since the discovery of the role of mtDNA mutations in human disease, aging, and cellular bioenergetics, there has been ongoing interest in the study of interactions between mitochondrial and nuclear genomes and epigenomes. Studies of mitochondrial disease in humans are confounded by the limited availability of patient material and by the diversity of the genetic (nuclear) background, which can profoundly modulate the expression of a mitochondrial defect (Giordano et al., 2014). Fortunately, the cytoplasmic hybrid (cybrid) technology introduced by King and Attardi (King & Attardi, 1989) greatly facilitates the study of mitochondrial disease. This technology takes advantage of so-called ρ0 cells, which lack mtDNA and therefore can be used as recipients of mitochondria in fusions with patient platelets or with cytoplasts derived from fibroblasts by extrusion or chemical inactivation of their nuclei (Barrientos, Kenyon, & Moraes, 1998; Bayona-Bafaluy, Manfredi, & Moraes, 2003; Kenyon & Moraes, 1997). The resulting cytoplasmic hybrids (cybrids) have a relatively uniform genetic background, thus facilitating biochemical analyses.
The isolation of ρ0 cells for these studies typically involves prolonged [as long as 16 weeks (Vaillant & Nagley, 1995)] treatment with permissive concentrations of DNA-intercalating agents such as ethidium bromide (EtBr) or ditercalinium chloride (DC). Therefore, techniques that can achieve elimination of mtDNA within shorter time frames are of considerable interest. Successful elimination of mtDNA has been reported with the help of mitochondrially targeted fusion between EcoRI restriction endonuclease and Enhanced Green Fluorescent Protein (EGFP). When expressed in recipient cells, this fusion construct enters mitochondria and destroys mitochondrial DNA (Kukat et al., 2008). While this technique represents a considerable advancement over treatment with EtBr, it has limitations. First, overexpression of this fusion may result in mistargeting to the nucleus, where it can induce mutagenic and cytotoxic double-strand breaks. Also, the utility of this method is limited to elimination of mitochondrial genomes that harbor EcoRI recognition sites.
In this unit, we describe an alternate approach that relies on the use of the Y147A mutant of human uracil-N-glycosylase (mUNG1). Uracil-N-glycosylase (UNG) catalyzes the first step of the BER of uracil, which is a premutagenic DNA lesion that can arise in DNA through deamination of cytosine. This enzyme is found in both the mitochondria (UNG1) and the nucleus (UNG2), and its activity results in the scission of a glycosidic bond between uracil and deoxyribose, thus creating an abasic (AP) site. It has been reported that mUNG1 has expanded substrate specificity and can also hydrolyze glycosidic bonds with thymidine in DNA, thus creating AP sites at the locations of T residues (Kavli et al., 1996). These AP sites have three possible outcomes: 1) they can be faithfully repaired by BER machinery (Krokan & Bjoras, 2013), 2) they can be bypassed through translesion synthesis by Pol γ (Liu & Demple, 2010) or PRIMPOL (Garcia-Gomez et al., 2013), or 3) the mtDNA molecule containing an overwhelming number of lesions can be degraded (I. Shokolenko et al., 2009; I. N. Shokolenko & Alexeyev, 2015; I. N. Shokolenko, Wilson, et al., 2013; I. N. Shokolenko et al., 2016; Spadafora, Kozhukhar, Chouljenko, et al., 2016). Our basic protocol takes advantage of the third pathway to generate ρ0 cells. It should be noted that while our protocol is not free of some of the shortcomings of the EcoRI-based protocol, such as potential mistargeting to the nucleus and subsequent nDNA damage, mUNG1 induces less severe damage (AP sites) as compared to EcoRI (double-strand breaks), which is repaired more readily and efficiently.
Critical Parameters and Troubleshooting
mUNG1 drives mtDNA degradation by introducing enough abasic sites to overwhelm mitochondrial BER machinery. Therefore, it is critical to achieve high levels of expression and efficient mitochondrial targeting of mUNG1. In pMA3790, expression of mUNG1 is driven by a CMV promoter (Figure 3, which is notorious both for its propensity to be silenced (Teschendorf, Warrington, Siemann, & Muzyczka, 2002) and for the variability of its strength among different cell lines (Qin et al., 2010). Therefore, if pMA3790 fails to deliver the expected results, mUNG1 expression can be optimized by changing the promoter that drives mUNG1. Alternatively, expression of UL12.5M185 instead of or together with mUNG1 may resolve the problem. UL12.5M185 is a herpesvirus protein that has also been shown to efficiently eliminate mtDNA (Spadafora, Kozhukhar, Chouljenko, et al., 2016). A construct encoding UL12.5M185 is available from AddGene (#70109).
All ρ0 cell lines that we isolated grow slower than their parents. Both the BASIC PROTOCOL 1 and the ALTERNATE PROTOCOL 1 usually result in a mixed population of wild type and ρ0 cells. Therefore, one should allow ample time for ρ0 cells to grow and form colonies prior to colony picking. By this time, wild type colonies may exceed 5 mm in diameter. When picking colonies, focus on smaller ones, keeping in mind that ρ0 cells grow slower.
In the ALTERNATE PROTOCOL 1, regular media changes (every 2–3 days) are critical, because without them, cells tend to develop resistance to treatment. In this protocol, mtDNA elimination is achieved through the inhibition of mtDNA replication by EtBr and ddC followed by the “dilution” of remaining mtDNA molecules through proliferation and mtDNA turnover. The procedure is successful only when mtDNA replication is more sensitive to the treatment than proliferation. In some cell lines, mtDNA replication is only blocked at cytostatic EtBr concentrations. Therefore, the alternate protocol employs a combination of EtBr and ddC. ddC has a much milder effect on proliferation, but by itself is unable to induce mtDNA loss in most cell lines. Combining EtBr and ddC allows us to minimize the necessary EtBr concentration, thus relieving a proliferation block while still maintaining an efficient block to mtDNA replication (Figure 1). Therefore, if the protocol fails due to a proliferative arrest, one should attempt to lower the EtBr concentration with a concomitant increase in ddC concentration, if necessary. On the other hand, while reaching mtDNA copy number of 1 or lower before plating cells for cloning is optimal, cells may cease proliferating before reaching this point. If this is the case, clone them anyway. With some cell lines, we were able to isolate ρ0 cells even when plating at 15 copies of mtDNA per cell. If no ρ0 clones are observed, repeat the experiment with a different EtBr:ddC ratio.
mtDNA is present in cells in a much higher copy number than a typical single-copy gene. As a result, mtDNA amplification can suppress amplification of nDNA and therefore skew the results of copy number determination by qPCR. Therefore, the recommended 20x mastermixes of qPCR primers and probes contain low concentrations of mitochondrial primers, so that they can be used up before the dNTP pool of the qPCR mastermix becomes exhausted (Figure 4).
Figure 4.
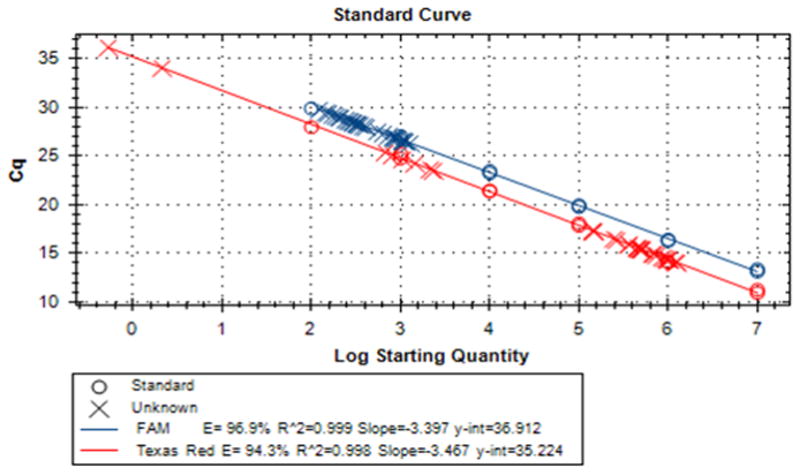
A representative standard curve for mtDNA copy number determination by duplex qPCR.
Anticipated Results
When using either of the BASIC PROTOCOLS, after plating for cloning, one should expect a size-heterogenous mix of colonies, of which the majority of large colonies are going to be those with retained mtDNA (WT), whereas many (but not all) small colonies will be ρ0. When genotyping by PCR, one should expect that only one band, either mtDNA or nDNA, will be observed on an agarose gel. The presence of both bands may indicate either the generation of a cell line with low mtDNA copy number (Figure 2) or the contamination of a ρ0 clone with WT cells.
Time Considerations
When using the BASIC PROTOCOL 2, one should expect a steady gradual decrease of mtDNA copy number down to about 1 copy per cell within 2–4 weeks (Figure 1). After plating for cloning, with either of the BASIC PROTOCOLS, one can expect that colonies on the plate will become visible within 10 days. Small (presumably ρ0) colonies can be picked within 17–21 days. After picking colonies into 24-well plates, wells containing WT clones should be ready for analysis within 1 week, whereas wells containing ρ0 clones will be ready to analyze within 10–14 days.
Significance Statement.
Mitochondrial DNA (mtDNA) is dispensable for the viability of most studied mammalian cell lines. However, its loss is associated with the loss of oxidative phosphorylation (OxPhos) capacity and with auxotrophy for uridine and pyruvate. Therefore, cells devoid of mtDNA (ρ0 or rho-0 cells) represent a convenient experimental system to study OxPhos-related phenomena. Furthermore, ρ0 cells can be repopulated with mtDNA from various sources, including fibroblasts and platelets from patients with mitochondrial disease, thus enabling studies of the effects of different mtDNA mutations in a relatively uniform genetic background.
Acknowledgments
These studies were supported by National Institutes of Health grants OD010944 and HL66299 and by Department of Defense CDMR award PR150220.
Footnotes
KEY REFERENCE Spadafora, D., Kozhukhar, N., Chouljenko, V. N., Kousoulas, K. G., & Alexeyev, M. F. (2016). Methods for Efficient Elimination of Mitochondrial DNA from Cultured Cells. PloS One, 11(5), e0154684. doi: 10.1371/journal.pone.0154684
Contributor Information
Natalya Khozhukhar, University of South Alabama, Department of Physiology and Cell Biology, Tel: (251) 460-6726, Fax: (251) 460-6386.
Domenico Spadafora, University of South Alabama, Flow Cytometry Core Lab, Tel: (251) 460-7989, Fax: (251) 460-6386.
Yelitza Rodriguez, University of South Alabama, Department of Physiology and Cell Biology, Tel: (251) 460-6726, Fax: (251) 460-6386.
Mikhail Alexeyev, University of South Alabama, Department of Physiology and Cell Biology, Tel: (251) 460-6789, Fax: (251) 460-6386.
LITERATURE CITED
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, … Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Barrientos A, Kenyon L, Moraes CT. Human xenomitochondrial cybrids. Cellular models of mitochondrial complex I deficiency. Journal of Biological Chemistry. 1998;273(23):14210–14217. doi: 10.1074/jbc.273.23.14210. [DOI] [PubMed] [Google Scholar]
- Bayona-Bafaluy MP, Manfredi G, Moraes CT. A chemical enucleation method for the transfer of mitochondrial DNA to rho(o) cells. Nucleic Acids Research. 2003;31(16):e98. doi: 10.1093/nar/gng100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA. Sequence and gene organization of mouse mitochondrial DNA. Cell. 1981;26(2 Pt 2):167–180. doi: 10.1016/0092-8674(81)90300-7. [DOI] [PubMed] [Google Scholar]
- Garcia-Gomez S, Reyes A, Martinez-Jimenez MI, Chocron ES, Mouron S, Terrados G, … Blanco L. PrimPol, an archaic primase/polymerase operating in human cells. Molecular Cell. 2013;52(4):541–553. doi: 10.1016/j.molcel.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano C, Iommarini L, Giordano L, Maresca A, Pisano A, Valentino ML, … Carelli V. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain. 2014;137(Pt 2):335–353. doi: 10.1093/brain/awt343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavli B, Slupphaug G, Mol CD, Arvai AS, Peterson SB, Tainer JA, Krokan HE. Excision of cytosine and thymine from DNA by mutants of human uracil-DNA glycosylase. EMBO Journal. 1996;15(13):3442–3447. [PMC free article] [PubMed] [Google Scholar]
- Kenyon L, Moraes CT. Expanding the functional human mitochondrial DNA database by the establishment of primate xenomitochondrial cybrids. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(17):9131–9135. doi: 10.1073/pnas.94.17.9131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246(4929):500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- Krokan HE, Bjoras M. Base excision repair. Cold Spring Harbor Perspectives in Biology. 2013;5(4):a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukat A, Kukat C, Brocher J, Schafer I, Krohne G, Trounce IA, … Seibel P. Generation of rho0 cells utilizing a mitochondrially targeted restriction endonuclease and comparative analyses. Nucleic Acids Research. 2008;36(7):e44. doi: 10.1093/nar/gkn124. gkn124 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Demple B. DNA repair in mammalian mitochondria: Much more than we thought? Environmental and Molecular Mutagenesis. 2010;51(5):417–426. doi: 10.1002/em.20576. [DOI] [PubMed] [Google Scholar]
- Qin JY, Zhang L, Clift KL, Hulur I, Xiang AP, Ren BZ, Lahn BT. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PloS One. 2010;5(5):e10611. doi: 10.1371/journal.pone.0010611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokolenko I, Venediktova N, Bochkareva A, Wilson GL, Alexeyev MF. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Research. 2009;37(8):2539–2548. doi: 10.1093/nar/gkp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokolenko IN, Alexeyev MF. Mitochondrial DNA: A disposable genome? Biochimica et Biophysica Acta. 2015;1852(9):1805–1809. doi: 10.1016/j.bbadis.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokolenko IN, Fayzulin RZ, Katyal S, McKinnon PJ, Wilson GL, Alexeyev MF. Mitochondrial DNA ligase is dispensable for the viability of cultured cells but essential for mtDNA maintenance. Journal of Biological Chemistry. 2013;288(37):26594–26605. doi: 10.1074/jbc.M113.472977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokolenko IN, Wilson GL, Alexeyev MF. Persistent damage induces mitochondrial DNA degradation. DNA Repair (Amst) 2013;12(7):488–499. doi: 10.1016/j.dnarep.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokolenko IN, Wilson GL, Alexeyev MF. The “fast” and the “slow” modes of mitochondrial DNA degradation. Mitochondrial DNA A DNA Mapp Seq Anal. 2016;27(1):490–498. doi: 10.3109/19401736.2014.905829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spadafora D, Kozhukhar N, Alexeyev MF. Presequence-Independent Mitochondrial Import of DNA Ligase Facilitates Establishment of Cell Lines with Reduced mtDNA Copy Number. PloS One. 2016;11(3):e0152705. doi: 10.1371/journal.pone.0152705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spadafora D, Kozhukhar N, Chouljenko VN, Kousoulas KG, Alexeyev MF. Methods for Efficient Elimination of Mitochondrial DNA from Cultured Cells. PloS One. 2016;11(5):e0154684. doi: 10.1371/journal.pone.0154684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorf C, Warrington KH, Jr, Siemann DW, Muzyczka N. Comparison of the EF-1 alpha and the CMV promoter for engineering stable tumor cell lines using recombinant adeno-associated virus. Anticancer Research. 2002;22(6A):3325–3330. [PubMed] [Google Scholar]
- Vaillant F, Nagley P. Human cell mutants with very low mitochondrial DNA copy number (rho d) Human Molecular Genetics. 1995;4(5):903–914. doi: 10.1093/hmg/4.5.903. [DOI] [PubMed] [Google Scholar]