

Abstract
Absence of the protein dystrophin causes Duchenne muscular dystrophy. Dystrophin directly binds to microtubules in vitro, and its absence in vivo correlates with disorganization of the subsarcolemmal microtubule lattice, increased detyrosination of α-tubulin, and altered redox signaling. We previously demonstrated that the dystrophin homologue utrophin neither binds microtubules in vitro nor rescues microtubule lattice organization when overexpressed in muscles of dystrophin-deficient mdx mice. Here, we fine-mapped the dystrophin domain necessary for microtubule binding to spectrin-like repeats 20–22. We show that transgenic mdx mice expressing a full-length dystrophin/utrophin chimera completely lacking microtubule binding activity are surprisingly rescued for all measured dystrophic phenotypes, including full restoration of microtubule lattice organization. Conversely, despite the presence of dystrophin at the sarcolemma, β-sarcoglycan-deficient skeletal muscle presents with a disorganized and densified microtubule lattice. Finally, we show that the levels of α-tubulin detyrosination remain significantly elevated to that of mdx levels in transgenic mdx mice expressing nearly full-length dystrophin. Our results demonstrate that the microtubule-associated perturbations of mdx muscle are distinct, separable, and can vary independently from other parameters previously ascribed to dystrophin deficiency.
Introduction
Dystrophin is a 427-kDa cytoplasmic protein predominantly expressed in striated muscle (1). Mutations in dystrophin which abolish expression or reduce its functionality lead to Duchenne muscular dystrophy (DMD) or Becker muscular dystrophy, a milder form of the disease (2). Approximately one in every 4000 boys is born with DMD (3), and each of them will inevitably become wheelchair-bound and succumb to fatal cardiac arrest or respiratory failure mainly during their twenties, with an increasing percentage reaching their thirties (4). Current treatment is limited to ventilatory support, which prolongs life, and corticosteroids, which also provide benefit, but can cause serious side effects (5,6).
In healthy skeletal muscle, dystrophin is enriched at subsarcolemmal protein assemblies known as costameres, where it couples actin and intermediate filaments to a membrane-associated glycoprotein complex (7). In addition, dystrophin directly binds microtubules (8,9), suggesting it may play a role in organizing them into a rectilinear lattice beneath the sarcolemma. The microtubule lattice becomes disorganized when dystrophin expression is ablated as in the mdx mouse (8–10) or when dystrophin protein is absent from costameres due to perturbations in other proteins that are required for proper costameric localization of dystrophin, such as ankyrin-B, ankyrin-G, β2-spectrin, dynactin-4 and obscurin (11–13).
Available studies reveal that microtubules are essential in skeletal muscle for myotube formation (14–16) whereas microtubule organization is important for proper Golgi localization in adult skeletal muscle fibers (17,18). In addition, it has been shown that post-translational modification of the microtubule network via detyrosination of α-tubulin in mdx cardiac and skeletal muscle contributes to increased production of reactive oxygen species and aberrant calcium regulation (19–21). Previously, we showed that disorganization and/or densification of the microtubule lattice correlates with a decrease in skeletal muscle torque production following eccentric contraction-induced injury (9). However, the relative contributions of microtubule post-translational modification, lattice disorganization and densification to the dystrophic phenotype remain to be determined for both mdx mice and human DMD patients.
To further understand the physiologic role of the dystrophin/microtubule interaction and its importance for the molecular pathogenesis of dystrophinopathy, we biochemically mapped the minimal domain within dystrophin necessary for microtubule binding. We previously showed that the dystrophin homologue utrophin neither binds microtubules in vitro, nor does it restore microtubule lattice organization when overexpressed in the dystrophin-deficient mdx mouse (9). Here, we show that in vivo expression of a dystrophin/utrophin chimera lacking in vitro microtubule binding activity ameliorates all measured aspects of the mdx mouse phenotype, including microtubule lattice organization. Our results demonstrate that the in vitro microtubule binding and in vivo microtubule organizing functions of dystrophin are independent and separable activities. Furthermore, several microtubule-associated abnormalities of mdx muscle including organization, densification and detyrosination vary independently from each other and do not consistently correlate with other well established dystrophy phenotypes.
Results
Based on our previous study (9), we hypothesized that the in vitro microtubule binding domain of dystrophin lies within spectrin-like repeats 20–24. Since utrophin lacks microtubule binding activity (9), we attempted to further define the minimal microtubule binding domain in dystrophin by analyzing dystrophin/utrophin hybrid constructs in which varying numbers of homologous utrophin repeats were substituted into dystrophin (Supplementary Material, Fig. S1A). Given the highly similar microtubule binding activities of dystrophin and Dp260 (9), Dp260 was chosen as a template over full-length dystrophin due to its smaller size, ease of cloning and higher protein expression. The exchange of dystrophin repeats 23 and/or 24 for utrophin repeats 21 and/or 22 (Dp260 R24Utr and R23-24Utr) did not affect microtubule binding activity (Supplementary Material, Fig. S1A). However, substitution of dystrophin repeat 20 for utrophin repeat 18 (Dp260 R20Utr) resulted in a measureable decrease in microtubule binding affinity, similar to that of the substitution of repeats 22–24 with utrophin repeats 20–22 (Dp260 R22–24Utr). Moreover, the substitution of two or all repeats within the region of dystrophin repeats 20–22 with utrophin repeats 18–20 (Dp260 R20–21Utr, R20–22Utr, R20–23Utr, R21–24Utr and R20–24Utr) resulted in complete loss of specific in vitro microtubule binding activity (Supplementary Material, Fig. S1A and Table S1).
To verify that no single repeat within repeats 20–22 is individually responsible for microtubule binding, we analyzed multiple Dp260 constructs deleted for individual spectrin-like repeats 20–24, or hinges 3 or 4 exchanged for the homologous utrophin hinges (Supplementary Material, Table S1). Deletion of each individual repeat from repeat 20 to repeat 24 or swapping of hinge 3 or 4 for the homologous hinge from utrophin did not appreciably impair the microtubule binding activity of Dp260 (Supplementary Material, Table S1). These data indicate that no single repeat within repeats 20–24 is individually responsible for the microtubule binding activity of dystrophin and that neither hinge 3 nor hinge 4 contributes to microtubule binding activity. Taken together, these data indicate that dystrophin repeats 20–22 comprise the minimal region of dystrophin necessary for binding microtubules in vitro.
To confirm the results observed with internal deletion constructs (9) and in Dp260/utrophin hybrids (Supplementary Material, Fig. S1A), we analyzed a full-length dystrophin construct in which repeats 20–24 were replaced with the homologous repeats 18–22 of utrophin, as well as a utrophin construct where repeats 18–22 were substituted with dystrophin repeats 20–24 (Fig. 1A–C). As expected, insertion of utrophin repeats 18–22 into Dys R20–24Utr conferred a loss of microtubule binding function, corroborating the lack of microtubule binding we observed for Dp260 R20–24Utr (Fig. 1). Interestingly, constructs encoding only dystrophin repeats 20–22 or 20–24 did not bind microtubules (Supplementary Material, Fig. S1B and C) while insertion of dystrophin repeats 20–24 failed to confer a gain of microtubule binding function to Utr R18–22Dys (Fig. 1A–C). Collectively, our data suggest that dystrophin repeats 20–22 are necessary for direct microtubule binding by dystrophin but are not sufficient to directly bind microtubules in isolation or even within the context of utrophin.
Figure 1.
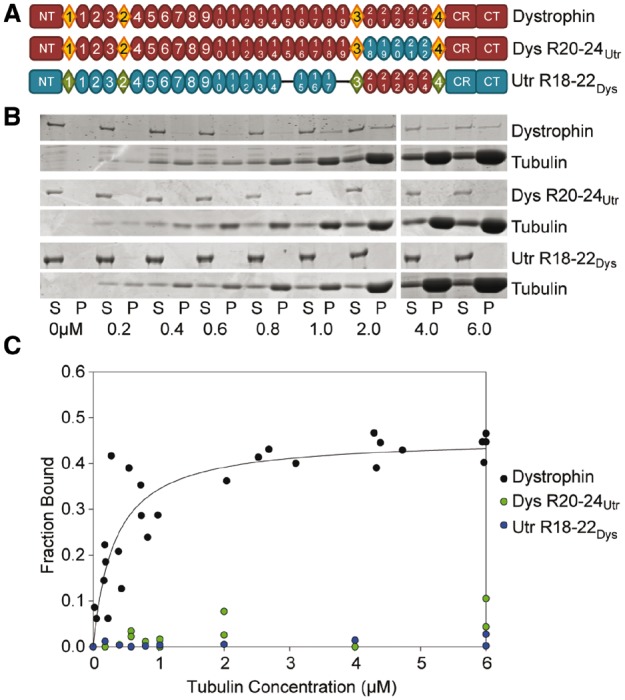
Domain structure and microtubule binding properties of dystrophin/utrophin hybrid constructs. (A) Schematic representation of full-length dystrophin and utrophin hybrid constructs used in this study. Ovals represent spectrin-like repeats, diamonds represent hinge regions. NT, N-terminus; CR, cysteine-rich domain; CT, C-terminus. Blue ovals represent portions of utrophin swapped into dystrophin. Red ovals represent dystrophin repeats swapped into utrophin. (B) Representative coomassie blue stained SDS-PAGE microtubule cosedimentation gels for dystrophin, Dys 20–24Utr, and Utr 18–22Dys. S, supernatant (fraction not bound to microtubules); P, pellet (fraction bound to microtubules). Tubulin dimer concentration is listed below each pair of lanes. (C) Microtubule binding curves. Substituting dystrophin repeats 20–24 for utrophin repeats 18–22 ablates-specific microtubule binding. However, substituting utrophin repeats 18–22 for dystrophin repeats 20–24 does not confer microtubule binding to utrophin. Full-length dystrophin data was previously published and shown here for reference (9).
To determine the functional importance of microtubule binding by dystrophin repeats 20–22, we utilized the full-length dystrophin/utrophin hybrid Dys 20–24Utr to create a transgene under the control of the human skeletal α-actin (HSA) promoter, which is expressed solely in skeletal muscle (Fig. 2A). Transgenic founder mice were crossed onto the mdx background and named DysΔMTB-mdx mice. In DysΔMTB-mdx mice, Dys 20–24Utr is expressed at ∼2-fold above the endogenous dystrophin levels observed in WT mice (Fig. 2B and C). DysΔMTB-mdx mice display WT levels of utrophin, and α-tubulin (Fig. 2B and C), both of which are significantly elevated in mdx mice (8,19,20,22). In addition, DysΔMTB-mdx mice display incorporation of Dys 20–24Utr into a biochemically-stable dystrophin–glycoprotein complex, indicating that it is forming the proper structure within skeletal muscle (Fig. 2D).
Figure 2.

Generation of a transgenic mouse model expressing a dystrophin incapable of directly binding microtubules. (A) Constructs used to generate transgenic mice expressing a muscle-specific dystrophin incapable of binding microtubules. Ovals and diamonds are as in Figure 1. HSA, human skeletal α-actin promoter; NT, N-terminus; CR, cysteine rich domain; CT, C-terminus. (B–C) Dystrophin protein levels in DysΔMTB-mdx mice are ∼170% of WT levels. Utrophin and α-tubulin levels are elevated in mdx mice as compared with WT levels, but are restored to WT levels in the presence of transgenic dystrophin. nNOS and β-dystroglycan levels are reduced in mdx mice, but restored to WT levels in DysΔMTB-mdx mice. DM1A and B512 both recognize α-tubulin (see Materials and Methods), although DM1A shows a greater difference in immunoreactivity when probing mdx lysates than B512, as we previously reported (8). Sixty micrograms of quadriceps muscle lysate were run in each lane. Quantitation of protein levels is relative to pan-actin levels for all proteins. n = 3 mice per genotype. Statistics were carried out using one-way ANOVA with post-hoc t-test analyses with P < 0.05 considered significant *Statistically different from WT and DysΔMTB-mdx. **Statistically different from WT and mdx. (D) Transgenic dystrophin incorporates into the dystrophin–glycoprotein complex of DysΔMTB-mdx mice as evidenced by its presence in the elution fraction of the WGA enrichment. The dystroglycans and syntrophins are also present in the elution fraction. S, starting lysate; V, void fraction not bound to WGA beads; E, elution fraction from WGA beads.
Immunofluorescence analyses on isolated quadriceps muscles from WT, mdx and DysΔMTB-mdx mice verify that Dys 20–24Utr is correctly localized to the sarcolemma and restores localization of the dystroglycans, sarcoglycans, dystrobrevin and syntrophin, all of which are either reduced or absent at the mdx sarcolemma (Fig. 3). In addition, the increased localization of utrophin at the sarcolemma of the mdx mouse is reduced to WT levels in the DysΔΜΤΒ-mdx mouse, corroborating our western blot findings (Fig. 2B). Moreover, histologic assessments of DysΔMTB-mdx muscle were similar to that of WT muscle (Supplementary Material, Fig. S2A and B). DysΔMTB-mdx muscle also retains the contiguous neuromuscular junction morphology of WT muscle rather than the fragmented morphology typically observed in the mdx mouse (Supplementary Material, Fig. S2C). Taken together, these data indicate that transgenically expressed Dys 20–24Utr correctly localizes within DysΔMTB-mdx skeletal muscle tissue and properly associates into the dystrophin–glycoprotein complex.
Figure 3.
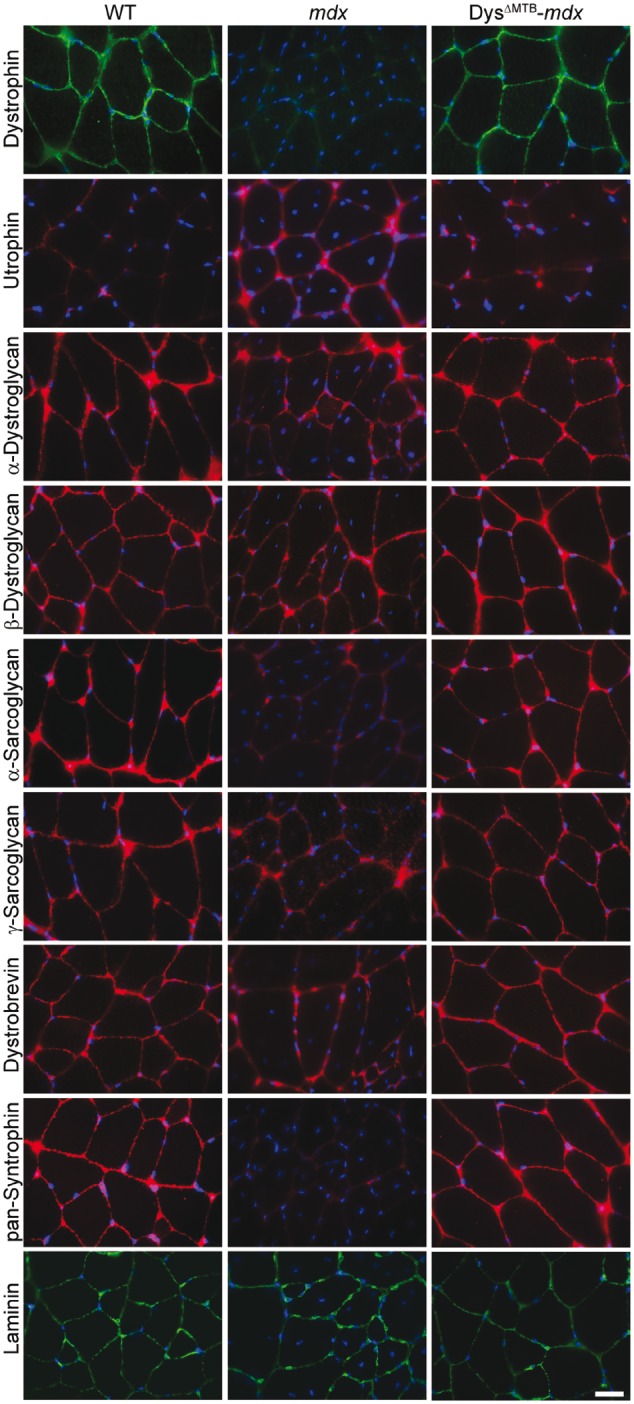
Restoration of the sarcolemmal dystrophin–glycoprotein complex in DysΔMTB-mdx mice. Dystrophin is absent in mdx mice. Utrophin levels are upregulated in mdx mice as compared with WT and DysΔMTB-mdx mice. DysΔMTB-mdx mice show presence of all dystrophin–glycoprotein complex components assayed at the sarcolemma. Bar, 20 μm.
After establishing restoration of the dystrophin–glycoprotein complex in muscle from DysΔMTB-mdx mice, we quantitatively assessed the morphology of the subsarcolemmal microtubule lattice. Intriguingly, the microtubule lattice of the DysΔMTB-mdx mouse appeared to be completely rescued from the disorganized and densified microtubule morphology observed in the mdx mouse (Fig. 4A). Quantitative analysis of the microtubule lattice revealed that DysΔMTB-mdx exhibited microtubule density (Fig. 4B) and directionality (Fig. 4C) parameters that were not different from WT muscle, but were significantly different from muscle of non-transgenic mdx littermates.
Figure 4.
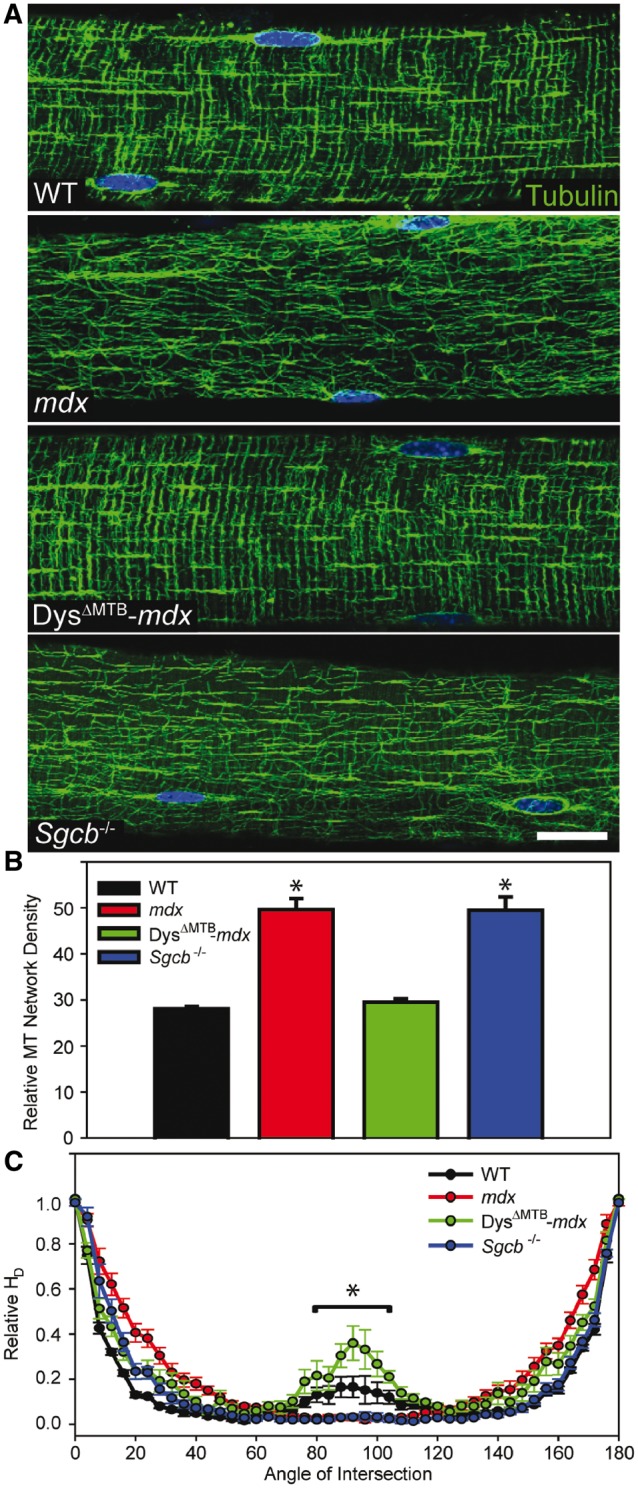
Rescue of subsarcolemmal microtubule lattice organization in DysΔMTB-mdx mice. (A) Microtubules are organized into a rectilinear lattice beneath the sarcolemma in WT and DysΔMTB-mdx mice in contrast to the perturbed microtubule lattice in mdx and Sgcb-/- mice. Images are representative of those obtained for n ≥ 10 fibers from each of n ≥ 3 mice per genotype. Bar, 20 μm. (B) WT and DysΔMTB-mdx mouse EDL fibers display the least dense microtubule network whereas mdx and Sgcb-/- mouse fibers display higher microtubule network density. *Statistically different from WT and DysΔMTB-mdx. (C) Transversely-oriented microtubules (centered around 90°) were significantly greater in WT and DysΔMTB-mdx mice compared with mdx and Sgcb-/- mice. Longitudinally oriented microtubules (0° and 180°) were not different between the mouse lines. Statistics were performed using one-way (density) or two-way (orientation) ANOVA with post-hoc t-test analyses with P < 0.05 considered significant. *WT and DysΔMTB-mdx mice are significantly different from mdx and Sgcb-/- between 80° and 100°.
Because our microtubule imaging data of DysΔMTB-mdx muscle demonstrated a clear separation between the in vitro microtubule binding and in vivo organizing functions of dystrophin, we investigated whether sarcolemmal dystrophin expression is sufficient for normal microtubule lattice organization in a different model of dystrophy. The mouse lacking β-sarcoglycan (Sgcb-/-) has a perturbed dystrophin–glycoprotein complex yet expresses WT levels of dystrophin (23,24). Sgcb-/- muscle retains the dystroglycans and dystrophin at the sarcolemma, but is lacking the entire sarcoglycan complex due to the absence of the beta subunit. In addition, utrophin is retained at WT levels in Sgcb-/- muscle (23,24). Thus, loss of β-sarcoglycan models muscular dystrophy caused by defects in the dystrophin–glycoprotein complex in a manner distinct from the mdx mouse. Surprisingly, Sgcb-/- muscle displayed a highly densified and disorganized microtubule lattice quantitatively similar to mdx muscle (Fig. 4A–C) despite the retention of sarcolemmal dystrophin (23,24). These data indicate that subsarcolemmal microtubule lattice organization is not solely regulated by dystrophin. While it is possible that the sarcoglycan complex may serve as a second important microtubule binding site in the dystrophin–glycoprotein complex, we find that equivalent amounts of α-tubulin co-purify in WGA-enriched fractions from WT and Sgcb-/- muscle (Supplementary Material, Fig. S3).
Transgenic utrophin expression in mdx muscle fails to fully correct all behavioral and physiological parameters of dystrophy that have been measured (9). Therefore, we measured each of these same parameters in the DysΔMTB-mdx and Sgcb-/- mice. Cage activity after mild exercise (25) revealed that DysΔMTB-mdx mice performed like WT animals while their non-transgenic littermates and Sgcb-/- mice displayed mdx levels of inactivity (Fig. 5A). To assess the effects of eccentric contraction on force production (26), we measured both the loss of torque in the anterior crural muscles in vivo (Fig. 5B) and loss of force in isolated EDL muscles ex vivo (Fig. 5C) in response to a series of eccentric contractions. In both assays, DysΔMTB-mdx mice showed minimal torque and force losses not different from WT mice. In addition, they performed significantly better than their non-transgenic littermates and Sgcb-/- mice, which in turn performed significantly better than mdx mice in the ex vivo isolated EDL assays. All other physiological measures of isolated EDL muscle function in the DysΔMTB-mdx mouse were not different from WT (Table 1) while the Sgcb-/- muscle performed most similarly to mdx muscle. These data collectively indicate that the DysΔMTB-mdx mouse is fully corrected for all measured mdx phenotypes.
Figure 5.
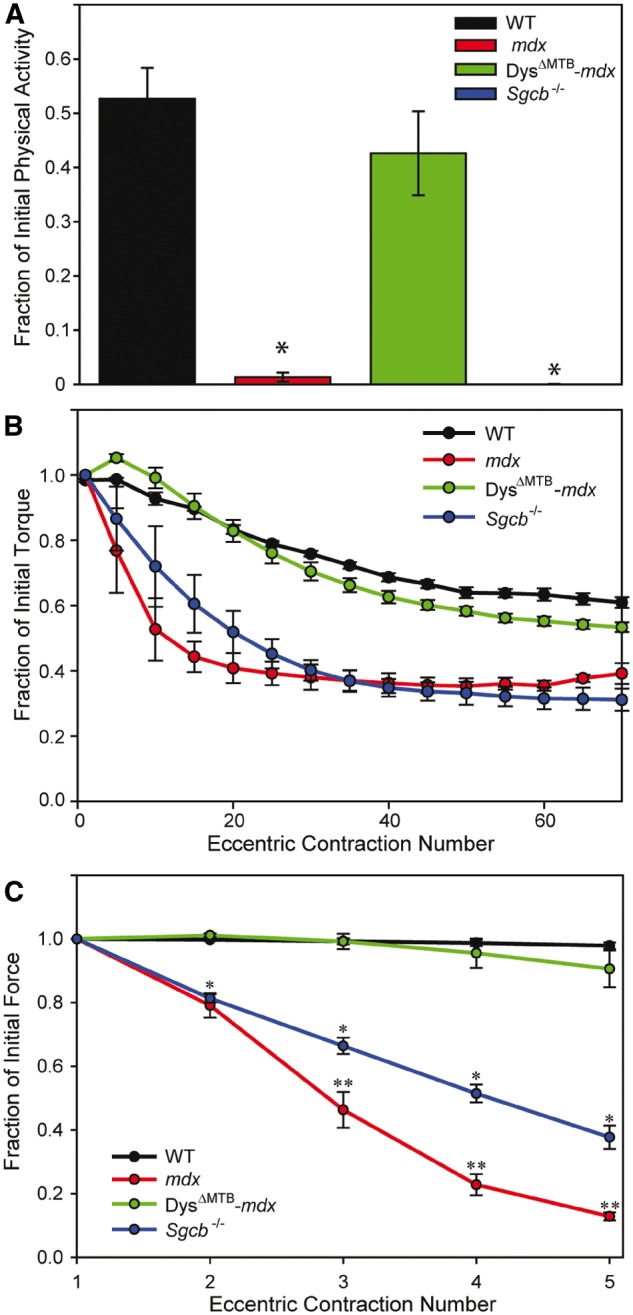
Normal skeletal muscle physiology in DysΔMTB-mdx mice. (A) Physical activity of mice immediately following mild treadmill exercise. WT and DysΔMTB-mdx mice exhibited ∼40–50% of their initial cage activity after mild exercise whereas post-exercise activity in mdx and Sgcb-/- mice dropped >99% of their initial activity. n ≥ 4 for each line. (B) As compared with WT and DysΔMTB-mdx mice, mdx and Sgcb-/- mice showed drastic loss of in vivo torque production of anterior crural muscles during a series of eccentric contractions. n ≥ 4 for each line. (C) As compared with EDL muscles of WT and DysΔMTB-mdx mice, those of mdx mice show significant loss of ex vivo force after eccentric contraction. Isolated Sgcb-/- muscles also displayed significant loss of force after eccentric contraction, but not to the extent seen in mdx mice. n ≥ 4 for each line. Statistics were performed using two-way ANOVA with post-hoc t-test analyses with P < 0.05 considered significant. *Statistically different from WT and DysΔMTB-mdx mice. **Statistically different from WT, DysΔMTB-mdx and Sgcb-/- mice. WT data were previously reported and shown here for comparison (9).
Table 1.
Physiological parameters of isolated EDL muscles used in ex vivo force measurements
| WT | mdx | DysΔMTB-mdx | Sgcb-/- | P value | |
|---|---|---|---|---|---|
| EDL mass (mg) | 12.8 ± 0.4a | 18.1 ± 0.7b | 11.7 ± 0.2c | 22.7 ± 0.4b,c,d | P < 0.001 |
| L0 (mm) | 13.3 ± 0.2a | 13.1 ± 0.2 | 12.2 ± 0.2b,c | 13.6 ± 0.1d | P = 0.003 |
| CSA (cm2) | 0.021 ± 0.001a | 0.030 ± 0.001b | 0.021 ± 0.001c | 0.036 ± 0.001b,c,d | P < 0.001 |
| Passive stiffness (N/m) | 13.9 ± 0.9a | 19.9 ± 0.8b | 13.1 ± 1.3c | 16.2 ± 1.2b,c,d | P < 0.001 |
| P0 (mN) | 449 ± 8.8a | 415 ± 12.8 | 424 ± 21.3 | 347 ± 25.0b,c,d | P < 0.001 |
| Specific P0 (N/cm2) | 21.8 ± 0.5a | 14.1 ± 0.3b | 20.6 ± 0.9c | 9.7 ± 0.6b,c,d | P < 0.001 |
| ΔP0 (%) | 11.1 ± 3.1a | 86.7 ± 2.6b | 11.0 ± 5.1c | 80.9 ± 2.5b,d | P < 0.001 |
| Force drop (%) | 9.4 ± 5.7a | 87.1 ± 1.3b | 8.5 ± 2.3c | 62.3 ± 3.0b,c,d | P < 0.001 |
Values are means ± SE.
L0 = optimal muscle length, CSA = physiological cross sectional area, P0 = maximal isometric tetanic force. ΔP0 = change in P0 after eccentric contractions. Force drop =change in eccentric force from the first to the fifth eccentric contraction. n ≥ 4 for each line. Statistics were performed using two-way ANOVA with post-hoc t-test analyses with P < 0.05 considered significant.
aWT data were previously reported and shown here for comparison (9)
bSignificantly different than WT.
cSignificantly different than mdx.
dSignificantly different than DysΔMTB-mdx.
Recent studies support a causal relationship between α-tubulin detyrosination and increased expression of the intermediate filament protein desmin with increased production of reactive oxygen species and aberrant calcium regulation in mdx skeletal and cardiac muscle (19–21,27). Therefore, we performed quantitative western blot analysis for α-tubulin detyrosination and desmin in skeletal muscle from WT, mdx, DysΔMTB-mdx, Fiona-mdx, DysΔ71-78-mdx and Sgcb-/- mice (Fig. 6A and B). The Fiona-mdx mouse model expresses full-length utrophin (28), whereas the DysΔ71-78-mdx mouse model expresses nearly full-length dystrophin lacking only a portion of the unstructured C-terminus encoded by exons 71–78 (29), both under control of the HSA promoter. We have used the DysΔ71-78-mdx mouse model in previous studies (9) because it stood as the most phenotypically rescued transgenic mdx model available. Of the six mouse lines analyzed, mdx, DysΔ71-78-mdx, and Sgcb-/- all showed significantly increased levels of detyrosinated α-tubulin and desmin immunoreactivity (Fig. 6A and B). The high levels of detyrosinated α-tubulin and desmin immunoreactivity in DysΔ71–78-mdx muscle is rather counterintuitive since this line is corrected for all measured phenotypes of dystrophy with the exception of passive muscle stiffness (9,29). In contrast, the Fiona-mdx mouse shows abnormal microtubule lattice organization and density (9), but WT levels of detyrosinated α-tubulin and desmin immunoreactivity (Fig. 6A and B), as well as WT passive muscle stiffness (Table 1).
Figure 6.

Analysis of cytoskeletal composition. (A–B). deTyr-tubulin (detyrosination of α-tubulin) and desmin levels are increased in mdx, DysΔ71-78-mdx and Sgcb-/- mice, but remain at WT levels in DysΔMTB-mdx and Fiona-mdx mice. n ≥ 3 for each genotype. *P < 0.05, **P < 0.01, ***P < 0.001 as compared with WT. (C) Each tubulin fraction was blotted for total α-tubulin (DM1A) and deTyr-tubulin. n ≥ 3 for each genotype. Proportions of each isoform in each fraction were plotted for each mouse strain and analyzed within each strain and between strains using ANOVA with post-hoc t-test analyses with P < 0.05 considered significant. No statistically significant differences were seen for any measured parameter.
Finally, we performed a microtubule fractionation analysis (30,31) to measure the proportion of detyrosinated α-tubulin immunoreactivity in free tubulin, free microtubules and membrane-associated microtubules (Fig. 6C). For all six mouse strains analyzed, the proportion of detyrosinated α-tubulin in each fraction was not different from the distribution of total α-tubulin. In addition, the distributions of detyrosinated α-tubulin were not significantly different among the different mouse strains. Our data demonstrate that mdx, DysΔ71-78-mdx and Sgcb-/- muscles all show increased levels of detyrosinated α-tubulin immunoreactivity that retains WT distribution within the different states of polymerization and membrane association.
Discussion
Here, we quantitatively compared a number of skeletal muscle microtubule perturbations associated with dystrophinopathy in mice across several transgenically rescued lines and with mice ablated for β-sarcoglycan (Table 2). Our principal findings are (1) that the microtubule-binding and microtubule-organizing functions of dystrophin are distinct and separable, (2) that sarcolemmal dystrophin expression is insufficient to maintain microtubule lattice organization in the face of at least one other perturbation to the dystrophin-glycoprotein complex (β-sarcoglycan deficiency) and (3) that elevated post-translational α-tubulin detyrosination does not directly correlate with subsarcolemmal microtubule density, lattice organization or rescue of nearly all skeletal muscle pathology in mdx mice by transgenic expression of nearly full-length dystrophin.
Table 2.
Summary of measured parameters across all mouse lines analyzed
| WT | mdx | DysΔMTB-mdx | Fiona-mdx | DysΔ71-78-mdx | Sgcb-/- | |
|---|---|---|---|---|---|---|
| MT density | WTa | ↑a | WT | ↑a | WTa | ↑ |
| MT organization | WTa | ↓a | WT | ↓a | WTa | ↓ |
| In vivo ECC torque loss | WTa | ↑a | WT | ↑a | WTa | ↑ |
| Passive stiffness | WTa | ↑a | WT | WTa | ↑a | ↑ |
| deTyr-Tubulin | WT | ↑ | WT | WT | ↑ | ↑ |
| Desmin | WT | ↑ | WT | WT | ↑ | ↑ |
| Dystrophic pathology | No | Yes | No | Nob | Noc | Yesd |
Up arrows indicate a significant increase above WT values, down arrows indicate a significant decrease below WT values.
aData previously measured using the exact methods employed in this study and shown for comparison (9).
bPreviously reported (28).
cPreviously reported (29).
dPreviously reported (24).
Our recent demonstration that utrophin lacks both microtubule binding and organizing activities (9) followed previous work showing that utrophin is also incapable of localizing neuronal nitric oxide synthase (nNOS) to the sarcolemma (32,33). Our new results separating the in vitro microtubule binding and in vivo organizing activities of dystrophin parallel a study in which in vitro experiments identified the α1 helix of spectrin-like repeat 17 in sarcolemmal nNOS localization, but failed to detect critical in vivo contributions by the α2 and α3 helices of spectrin-like repeats 16 and 17, respectively (33). While the dystrophin N-terminal (ABD1) and middle rod (ABD2) actin binding domains first identified using in vitro assays (34,35) have been verified in vivo (36,37), subsequent in vitro experiments ascribing greater functional importance to the first calponin homology domain (CH1) of ABD1 (38) are less certain in light of the high in vivo functionality recently demonstrated for a dystrophin deleted for CH1 (39). Collectively, these results underscore the necessity to validate with in vivo experiments, dystrophin-related functions such as phospholipid binding that have so far been studied using only in vitro assays (e.g., 40). The possibility of species-dependent differences in dystrophic phenotype or phenotypic rescue (41) also reinforce the need to assess microtubule perturbations and associated pathological consequences in human DMD patients.
We also demonstrate that sarcolemmal dystrophin expression is not sufficient to organize the microtubule lattice, as evidenced by imaging data from skeletal muscle of the Sgcb-/- dystrophic mouse model. Clearly, dystrophin makes some unique contribution since transgenic expression of DysΔ71–78, but not utrophin, restores microtubule lattice organization to mdx muscle (9). Because the DysΔMTB-mdx mouse displays corrected microtubule lattice morphology and density in the absence of microtubule binding activity by Dys 20–24Utr, we hypothesize that dystrophin may indirectly control subsarcolemmal microtubule lattice organization through one or more intermediary proteins. In support of this hypothesis, dystrophin is trafficked to the sarcolemma via interactions with the microtubule associated proteins ankyrin-B, β2-spectrin and dynactin-4, and its retention at the sarcolemma requires ankyrin-G (11,12). Moreover, proper localization of ankyrin-B to the sarcolemma relies on the giant scaffolding protein obscurin (13). Because the ankyrin-B/G binding site in dystrophin lies in close proximity to the binding site for β-dystroglycan (11), a challenge for future studies to map the microtubule organizing function of dystrophin will be to design in vivo screens that can perturb ankyrin binding without disrupting the critical interaction with β-dystroglycan binding.
While the exact pathophysiological consequences of microtubule derangement in skeletal muscle remain to be fully elucidated, recent work suggests that subsarcolemmal microtubule densification, disorganization and/or post-translational detyrosination contribute to pathology in the dystrophin-deficient mdx mouse (9,20,21,27). Specifically, eccentric contraction-induced force loss in dystrophin-deficient mdx muscle was ameliorated by microtubule depolymerization with colchicine (20), or by inhibition of α-tubulin detyrosination with the parthenolide prodrug dimethylamino-parthenolide (21). Our new data show that detyrosination of α-tubulin remains significantly elevated to mdx levels in DysΔ71–78-mdx mice that are rescued for virtually all other dystrophy phenotypes including eccentric contraction-induced force drop (Table 2 and ref. 9). The elevated α-tubulin detyrosination (Fig. 6A), desmin immunoreactivity (Fig. 6B), and passive stiffness values (Table 1 and ref. 9) shared by mdx, DysΔ71–78-mdx and Sgcb-/- muscle tempt speculation that these three parameters may be functionally linked. Such a link was recently demonstrated for the former two with cytoskeletal stiffness in cardiac muscle (27). Finally, our new data show that elevated α-tubulin detyrosination is separable and independent from the microtubule densification and disorganization that also manifest in dystrophin-deficient mdx muscle. Microtubule lattice derangement in mdx muscle correlates with reduced in vivo torque production after eccentric contraction in both mdx (9) and Sgcb-/- muscle (Fig. 5B). Thus, elucidating the causes of microtubule lattice density versus microtubule lattice disorganization and their contributions to the dystrophic phenotype will require new in vivo structure/function approaches.
Materials and Methods
Reagents
Anti-α-tubulin (DM1A, WB: 1:1000, IF: 1:200, epitope: amino acids 426-450), anti-α-tubulin (B512, 1:1000, epitope: C-terminal third), anti-pan-actin (C4, 1:5000), anti-Desmin (DE-U-10, 1:1000), anti-GAPDH (71.1, 1:10,000) and anti-laminin (L9393, 1:1000) antibodies were purchased from Sigma Aldrich. Anti-dystrophin (Dys2, 1:50), anti-β-dystroglycan (NCL-b-DG, 1:100), anti-α-sarcoglycan (NCL-a-SG, 1:100) and anti-γ-sarcoglycan (NCL-g-SG, 1:100) antibodies were purchased from Leica. Anti-utrophin (8A4, 1:50) antibody was purchased from Santa Cruz Biotechnology. Anti-α-dystroglycan (IIH6C4, WB: 1:500, IF: 1:100), anti-pan-syntrophin (1351, WB: 1:1000, IF: 1:100) and anti-deTyr-tubulin (AB3201, 1:500) antibodies were purchased from Millipore. An additional anti-deTyr-tubulin (1:10,000) antibody was previously described (42). Anti-dystrobrevin (23, 1:100) antibody was purchased from BD Biosciences. Anti-nNOS (Z-RNN3, 1:50) antibody, Alexa Fluor secondary antibodies (1:250) and Alexa Fluor α-bungarotoxin (1:500) were purchased from Invitrogen. DyLight 680 and 800 secondary antibodies (1:10,000) were purchased from Pierce. Wheat germ agglutinin agarose beads were purchased from Vector Laboratories. Pre-formed microtubules and paclitaxel were purchased from Cytoskeleton. DNA primers were purchased from Integrated DNA Technologies.
Cloning
All dystrophin and utrophin plasmid constructs were PCR-amplified from existing N-terminally FLAG-tagged pFastBac1 constructs generated in our previous studies (8,9,43–46), inserted into the Gateway entry vector pENTR/D-TOPO (Invitrogen) and sequence verified. All PCRs were performed using PfuII Ultra HS polymerase (Stratagene). Once verified, entry vectors were recombined into the Gateway insect cell destination vector, pDEST8, using LR Clonase II (Invitrogen) and subsequently expressed in Sf9 insect cells using the Bac-to-Bac system (Invitrogen). Deletion constructs were built as previously described (47). In brief, PCR primers were designed such that they amplified the entire plasmid except the deleted region. The linear PCR products were circularized via the addition of T4 polynucleotide kinase and T4 DNA ligase (New England Biolabs) and sequence verified. To generate dystrophin/utrophin hybrid constructs, PCR primers were designed to amplify the entire dystrophin plasmid except the repeats to be exchanged with primers containing 15 nucleotide overhangs homologous to the ends of the utrophin repeats chosen for substitution. In addition, the homologous utrophin repeats for substitution were PCR amplified with primers containing 15 nucleotide overhangs homologous to the dystrophin repeat sequences flanking the hybrid region. The two resulting PCR products were mixed in equimolar ratios and assembled into a single vector using the Gibson Assembly Master Mix (New England Biolabs).
Protein expression and purification
FLAG-tagged dystrophin and utrophin proteins were expressed and purified in Sf9 insect cells using the Bac-to-Bac protocol (Invitrogen). In brief, recombinant baculoviral DNA was transfected into a small culture of Sf9 insect cells using CellFectin II (Invitrogen). Four days later, the media containing the recombinant baculovirus was harvested and the transfected cells were analyzed for protein expression by anti-FLAG western blot. Once expression was verified, large cultures were incubated for 3 days with amplified baculovirus before being harvested for protein purification. For purification, cells were lysed using 1% Triton in phosphate buffered saline (PBS: 8 mM NaH2PO4, 42 mM Na2HPO4, 150 mM NaCl, pH 7.5) containing protease inhibitors (100 nM aprotinin, 1 mM benzamidine, 10 μM E-64, 10 μM leupeptin, 1 mM pepstatin A and 1 mM phenylmethanesulfonylfluoride) and protein purified using M2 FLAG affinity agarose (Sigma) as previously described (8,45,46). Proteins were dialyzed into microtubule buffer (50 mM HEPES, 50 mM KCl, 1 mM MgCl2, 1 mM EGTA, pH 7.5) overnight at 4°C before being concentrated and used in in vitro microtubule cosedimentation assays.
Mice
All animals were housed and treated in accordance with the standards set by the University of Minnesota Institutional Animal Care and Use Committee. WT mice used in this study were C57BL/10 obtained from Jackson Laboratories. Congenic Fiona-mdx, DysΔ71–78-mdx and DysΔMTB-mdx were bred for at least ten generations onto the mdx (C57BL/10ScSn-Dmdmdx/J) mouse strain obtained from Jackson Laboratories. Sgcb-/- mice were originally obtained from Jackson Laboratories (B6.129-Sgcbtm1Kcam/1J) and backcrossed onto C57BL/6 mice. All mice used in this study were male.
Generation of transgenic mice
The HSA promoter was cloned in place of the cytomegalovirus promoter in the Gateway version of the mammalian expression vector pcDNA, pDEST40. This new plasmid (pDEST-HSA) was used to create a transgene with the Dys R20–24Utr construct, built in the pENTR/D-TOPO vector for in vitro binding studies as described above. pDEST-HSA-Dys R20–24Utr DNA was restriction digested to linearize the transgene, gel extracted, and sent to the Mouse Genetics Core at The Scripps Research Institute for pronuclear injection into C57BL/6J mice. Transgenic founder mice were identified by PCR using HSA-specific primers (Forward: 5′-GTC AGG AGG GGC AAA CCC GC-3′, Reverse: 5′-GTC GCT GCC CTT CTC GAG CC-3′, Product Size: 187 bp) and crossed with C57BL/10 mice to check for transgene transmission. Transgenic dystrophin protein expression was assessed in several different muscles from each transgenic line crossed onto the mdx (C57BL/10ScSn-Dmdmdx/J) background using quantitative western blotting. All DysΔMTB-mdx mice used in this study were compared with non-transgenic littermate mdx mice as controls.
Biochemical characterization of the dystrophin-glycoprotein complex
Quadriceps muscle tissue was harvested from each mouse line, snap frozen in liquid nitrogen, ground with a mortar and pestle and lysed for 30 min in 1% Triton in PBS containing protease inhibitors. Lysates were clarified by centrifugation, an aliquot collected as a starting lysate sample, and the remaining lysate incubated with wheat germ agglutinin conjugated agarose beads overnight at 4°C while rotating. The next day, the lysate was removed from the beads and kept as the void fraction. The beads were washed three times with 0.1% Triton in PBS. After washing, the beads were boiled in 2× Laemmli sample buffer (5% sodium dodecyl sulfate, 192 mM sucrose, 108 mM Tris, 710 mM β-mercaptoethanol, 0.0007% bromophenol blue) for 5 min. This fraction was taken as the elution. Equivalent volumes from each fraction were run side-by-side.
Immunofluorescence
Quadriceps muscles from each mouse line were cryopreserved in OCT and 10 μm transverse sections cut. Sections were fixed in 4% paraformaldehyde in PBS for 10 min and subsequently washed three times in PBS with 0.1% Triton for 5 min each. Primary antibodies were incubated overnight at 4°C in a humidified chamber. The next day, slides were washed three times in PBS with 0.1% Triton for 5 min each and goat anti-mouse or anti-rabbit secondary antibody coupled to Alexa Fluor 488 or 568 was incubated on the sections for 30 min at 37°C. Sections were washed three times in PBS with 0.1% Triton for 5 min each and slides sealed using SlowFade Gold antifade reagent with DAPI to visualize nuclei. Images were acquired on a Deltavision PersonalDV deconvolution microscope equipped with a 40× 1.35 NA oil objective.
Neuromuscular junction imaging
Gastrocnemius muscles from each mouse line were cryopreserved in OCT and 20 μm longitudinal sections cut. Sections were fixed in 4% paraformaldehyde in PBS for 10 min and subsequently washed three times in PBS for 5 min each. Alexa Fluor 488-conjugated α-bungarotoxin was incubated on the sections for 30 min at 37°C. Sections were washed three times in PBS for 5 min each and slides sealed using SlowFade Gold antifade reagent with DAPI to visualize nuclei. Images were acquired on a Deltavision PersonalDV deconvolution microscope equipped with a 100× 1.40 NA oil objective.
Ex vivo EDL force measurements
EDL muscles of 6-month-old mice were passively shortened to 95% resting length and then stimulated for 200 ms while the muscle was simultaneously lengthened to 105% resting length at 0.5 length/s. To prevent fatigue, each eccentric contraction was separated by 3 min of rest. Force production was plotted as a percentage of initial force.
In vivo muscle torque measurements
In brief, torque about the ankle joint of 12-week-old mice was measured for each eccentric contraction and plotted as a fraction of the first eccentric contraction. To prevent fatigue, each eccentric contraction was separated by 3 min of rest.
Exercise-induced inactivity
In brief, prior to activity cage assessments, 10-week-old mice were acclimated to the treadmill for three consecutive days. On the fourth day, baseline voluntary activity was assessed for 30 min prior to exercise using activity cages (AccuScan Instruments Inc.). Mice were then placed on the treadmill set at 15° decline. Without the use of electrical shock, mice were encouraged to walk for 5 min at 5 m/min followed by 10 min at 15 m/min. After exercise, mice were placed back in the same activity cages and activity monitored for 30 min after exercise. Data were plotted as to represent the percentage of pre-exercise activity the mice retained after exercise.
Tubulin fractionation
Tubulin fractionation was performed as previously described (30,31). In brief, quadriceps muscle from each mouse strain analyzed was snap frozen in liquid nitrogen and ground into a fine powder using a mortar and pestle. The tissue was then lysed in MT stabilization buffer (10 mM sodium phosphate, 0.5 mM EGTA, 0.5 mM GTP, 0.5 mM MgCl2, 50% glycerol (v/v), 5% DMSO (v/v) and protease inhibitors, pH 6.95). The solution was centrifuged at 100,000 g for 20 min at room temperature and the supernatant taken as Free Tubulin. The pellet was dissolved in MT destabilization buffer (250 mM sucrose, 10 mM sodium phosphate, 0.5 mM GTP, 0.5 mM MgCl2 and protease inhibitors, pH 6.95) for 2 h on ice. After centrifugation for 20 min at 4°C, the supernatant was taken as Free MTs. The pellet was dissolved on ice for 10 min in Triton lysis buffer (20 mM Tris–HCL, 150 mM NaCl, 1 mM EDTA, 10% glycerol (v/v), 1% Triton-X100 (v/v) and protease inhibitors, pH 7.5), centrifuged at 14,000 g for 10 min at 4°C and the supernatant taken as Membrane MTs. The last steps to isolate the Membrane MTs were repeated once more to increase the yield. The remaining pellet contained insoluble cytoskeletal proteins and was discarded after verifying the absence of tubulin in the fraction.
Additional methods
EDL muscle fiber imaging and quantitation were all performed exactly as previously described (9,48). In vitro microtubule cosedimentation assays were performed as previously described (9). Additional information on ex vivo EDL force measurements, in vivo anterior crural muscle torque measurements, and mouse activity cage measurements was previously described (9).
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
The authors would like to thank Drs. Wenhua Liu and Evelyn Ralston (National Institute of Arthritis and Musculoskeletal and Skin Diseases) for providing the directionality analysis program and Dr. DeWayne Townsend (University of Minnesota) for a breeding pair of Sgcb-/- mice.
Conflict of Interest statement. None declared
Funding
The study was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases grant to J.M.E. [RO1 AR042423]. J.J.B. and J.T.O. were supported by the National Institutes of Health Training Program in Muscle Research [AR007612]. J.J.B. was also supported by a University of Minnesota Doctoral Dissertation Fellowship. J.T.O., T.L.M. and D.M.N. were each supported by fellowships from the National Institute on Aging Training Program for Functional Proteomics of Aging [T32 AG029796]. D.M.T. was supported by an American Heart Association Predoctoral Fellowship [12PRE12040402].
References
- 1. Hoffman E.P., Brown R.H., Kunkel L.M. (1992) Dystrophin: the protein product of the Duchene muscular dystrophy locus. Biotechnology, 24, 457–466. [PubMed] [Google Scholar]
- 2. Worton R.G., Thompson M.W. (1988) Genetics of Duchenne muscular dystrophy. Annu. Rev. Genet., 22, 601–629. [DOI] [PubMed] [Google Scholar]
- 3. Mendell J.R., Shilling C., Leslie N.D., Flanigan K.M., Al-Dahhak R., Gastier-Foster J., Kneile K., Dunn D.M., Duval B., Aoyagi A., et al. (2012) Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol., 71, 304–313. [DOI] [PubMed] [Google Scholar]
- 4. Rall S., Grimm T. (2012) Survival in Duchenne muscular dystrophy. Acta Myol., 31, 117–120. [PMC free article] [PubMed] [Google Scholar]
- 5. Angelini C. (2007) The role of corticosteroids in muscular dystrophy: a critical appraisal. Muscle Nerve, 36, 424–435. [DOI] [PubMed] [Google Scholar]
- 6. Ricotti V., Ridout D.A., Scott E., Quinlivan R., Robb S.A., Manzur A.Y., Muntoni F. (2013) Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J. Neurol. Neurosurg. Psychiatry, 84, 698–705. [DOI] [PubMed] [Google Scholar]
- 7. Ervasti J.M. (2003) Costameres: The Achilles’ heel of Herculean muscle. J. Biol. Chem., 278, 13591–13594. [DOI] [PubMed] [Google Scholar]
- 8. Prins K.W., Humston J.L., Mehta A., Tate V., Ralston E., Ervasti J.M. (2009) Dystrophin is a microtubule-associated protein. J. Cell Biol., 186, 363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Belanto J.J., Mader T.L., Eckhoff M.D., Strandjord D.M., Banks G.B., Gardner M.K., Lowe D.A., Ervasti J.M. (2014) Microtubule binding distinguishes dystrophin from utrophin. Proc. Natl. Acad. Sci. U. S. A., 111, 5723–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Percival J.M., Gregorevic P., Odom G.L., Banks G.B., Chamberlain J.S., Froehner S.C. (2007) rAAV6-Microdystrophin rescues aberrant Golgi complex organization in mdx skeletal muscles. Traffic, 8, 1424–1439. [DOI] [PubMed] [Google Scholar]
- 11. Ayalon G., Davis J.Q., Scotland P.B., Bennett V. (2008) An Ankyrin-based mechanism for functional organization of dystrophin and dystroglycan. Cell, 135, 1189–1200. [DOI] [PubMed] [Google Scholar]
- 12. Ayalon G., Hostettler J.D., Hoffman J., Kizhatil K., Davis J.Q., Bennett V. (2011) Ankyrin-B interactions with spectrin and dynactin-4 are required for dystrophin-based protection of skeletal muscle from exercise injury. J. Biol. Chem., 286, 7370–7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Randazzo D., Giacomello E., Lorenzini S., Rossi D., Pierantozzi E., Blaauw B., Reggiani C., Lange S., Peter A.K., Chen J., et al. (2013) Obscurin is required for ankyrinB-dependent dystrophin localization and sarcolemma integrity. J. Cell Biol., 200, 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saitoh O., Arai T., Obinata T. (1988) Distribution of microtubules and other cytoskeletal filaments during myotube elongation as revealed by fluorescence microscopy. Cell Tissue Res., 252, 263–273. [DOI] [PubMed] [Google Scholar]
- 15. Chang W., Webster D.R., Salam A.A., Gruber D., Prasad A., Eiserich J.P., Bulinski J.C. (2002) Alteration of the C-terminal amino acid of tubulin specifically inhibits myogenic differentiation. J. Biol. Chem., 277, 30690–30698. [DOI] [PubMed] [Google Scholar]
- 16. Perez O.D., Chang Y.T., Rosania G., Sutherlin D., Schultz P.G. (2002) Inhibition and reversal of myogenic differentiation by purine-based microtubule assembly inhibitors. Chem. Biol., 9, 475–483. [DOI] [PubMed] [Google Scholar]
- 17. Ralston E., Lu Z., Ploug T. (1999) The organization of the Golgi complex and microtubules in skeletal muscle is fiber type-dependent. J. Neurosci., 19, 10694–10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ralston E., Ploug T., Kalhovde J., Lomo T. (2001) Golgi complex, endoplasmic reticulum exit sites, and microtubules in skeletal muscle fibers are organized by patterned activity. J. Neurosci., 21, 875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prosser B.L., Ward C.W., Lederer W.J. (2011) X-ROS signaling: rapid mechano-chemo transduction in heart. Science, 333, 1440–1445. [DOI] [PubMed] [Google Scholar]
- 20. Khairallah R.J., Shi G., Sbrana F., Prosser B.L., Borroto C., Mazaitis M.J., Hoffman E.P., Mahurkar A., Sachs F., Sun Y., et al. (2012) Microtubules underlie dysfunction in Duchenne muscular dystrophy. Sci. Signal, 5, ra56.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kerr J.P., Robison P., Shi G., Bogush A.I., Kempema A.M., Hexum J.K., Becerra N., Harki D.A., Martin S.S., Raiteri R., et al. (2015) Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat. Commun., 6, 8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matsumura K., Ervasti J.M., Ohlendieck K., Kahl S.D., Campbell K.P. (1992) Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature, 360, 588–591. [DOI] [PubMed] [Google Scholar]
- 23. Araishi K., Sasaoka T., Imamura M., Noguchi S., Hama H., Wakabayashi E., Yoshida M., Hori T., Ozawa E. (1999) Loss of the sarcoglycan complex and sarcospan leads to muscular dystrophy in β-sarcoglycan-deficient mice. Hum. Mol. Genet., 8, 1589–1598. [DOI] [PubMed] [Google Scholar]
- 24. Durbeej M., Cohn R.D., Hrstka R.F., Moore S.A., Allamand V., Davidson B.L., Williamson R.A., Campbell K.P. (2000) Disruption of the β-Sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol. Cell, 5, 141–151. [DOI] [PubMed] [Google Scholar]
- 25. Kobayashi Y.M., Rader E.P., Crawford R.W., Iyengar N.K., Thedens D.R., Faulkner J. A., Parikh S.V., Weiss R.M., Chamberlain J.S., Moore S. a., et al. (2008) Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature, 456, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petrof B.J., Shrager J.B., Stedman H.H., Kelly A. M., Sweeney H.L. (1993) Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl Acad. Sci. U. S. A., 90, 3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Robison P., Caporizzo M.A., Ahmadzadeh H., Bogush A.I., Chen C.Y., Margulies K.B., Shenoy V.B., Prosser B.L. (2016) Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science, 352, aaf0659.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tinsley J., Deconinck N., Fisher R., Kahn D., Phelps S., Gillis J.M., Davies K. (1998) Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med., 4, 1441–1444. [DOI] [PubMed] [Google Scholar]
- 29. Crawford G.E., Faulkner J.A., Crosbie R.H., Campbell K.P., Froehner S.C., Chamberlain J.S. (2000) Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J. Cell Biol., 150, 1399–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ostlund R.E., Leung J.T., Hajek S.V. (1979) Biochemical determination of tubulin-microtubule equilibrium in cultured cells. Anal. Biochem., 96, 155–164. [DOI] [PubMed] [Google Scholar]
- 31. Fassett J.T., Xu X., Kwak D., Wang H., Liu X., Hu X., Bache R.J., Chen Y. (2013) Microtubule actin cross-linking factor 1 regulates cardiomyocyte microtubule distribution and adaptation to hemodynamic overload. PLoS One, 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li D., Bareja A., Judge L., Yue Y., Lai Y., Fairclough R., Davies K.E., Chamberlain J.S., Duan D. (2010) Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J. Cell Sci., 123, 2008–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lai Y., Zhao J., Yue Y., Duan D. (2013) α2 and α3 helices of dystrophin R16 and R17 frame a microdomain in the α1 helix of dystrophin R17 for neuronal NOS binding. Proc. Natl Acad. Sci. U. S. A., 110, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Way M., Pope B., Cross R.A., Kendrick-Jones J., Weeds A.G. (1992) Expression of the N-terminal domain of dystrophin in E. coli and demonstration of binding to F-actin. FEBS Lett., 301, 243–245. [DOI] [PubMed] [Google Scholar]
- 35. Rybakova I.N., Amann K.J., Ervasti J.M. (1996) A new model for the interaction of dystrophin with F-actin. J. Cell Biol., 135, 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Warner L.E., DelloRusso C., Crawford R.W., Rybakova I.N., Patel J.R., Ervasti J.M., Chamberlain J.S. (2002) Expression of Dp260 in muscle tethers the actin cytoskeleton to the dystrophin-glycoprotein complex and partially prevents dystrophy. Hum. Mol. Genet., 11, 1095–1105. [DOI] [PubMed] [Google Scholar]
- 37. Hanft L.M., Rybakova I.N., Patel J.R., Rafael-Fortney J.A., Ervasti J.M. (2006) Cytoplasmic gamma-actin contributes to a compensatory remodeling response in dystrophin-deficient muscle. Proc. Natl Acad. Sci. U. S. A., 103, 5385–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Singh S.M., Bandi S., Winder S.J., Mallela K.M.G. (2014) The actin binding affinity of the utrophin tandem calponin-homology domain is primarily determined by its N-terminal domain. Biochemistry, 53, 1801–1809. [DOI] [PubMed] [Google Scholar]
- 39. Wein N., Vulin A., Falzarano M.S., Szigyarto C.A.K., Maiti B., Findlay A., Heller K.N., Uhlén M., Bakthavachalu B., Messina S., et al. (2014) Translation from a DMD exon 5 IRES results in a functional dystrophin isoform that attenuates dystrophinopathy in humans and mice. Nat. Med., 20, 992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sarkis J., Vie V., Winder S.J., Renault A., Le Rumeur E., Hubert J.F. (2013) Resisting sarcolemmal rupture: dystrophin repeats increase membrane-actin stiffness. FASEB J, 27, 359–367. [DOI] [PubMed] [Google Scholar]
- 41. Wang B., Li J., Qiao C., Chen C., Hu P., Zhu X., Zhou L., Bogan J., Kornegay J., Xiao X. (2008) A canine minidystrophin is functional and therapeutic in mdx mice. Gene Ther., 15, 1099–1106. [DOI] [PubMed] [Google Scholar]
- 42. Gundersen G.G., Kalnoski M.H., Bulinski J.C. (1984) Distinct populations of microtubules: tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell, 38, 779–789. [DOI] [PubMed] [Google Scholar]
- 43. Poirier M.G., Eroglu S., Marko J.F. (2002) The bending rigidity of mitotic chromosomes. Mol. Biol. Cell, 13, 2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rybakova I.N., Humston J.L., Sonnemann K.J., Ervasti J.M. (2006) Dystrophin and utrophin bind actin through distinct modes of contact. J. Biol. Chem., 281, 9996–10001. [DOI] [PubMed] [Google Scholar]
- 45. Henderson D.M., Lee A., Ervasti J.M. (2010) Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc. Natl Acad. Sci. U. S. A., 107, 9632–9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Henderson D.M., Belanto J.J., Li B., Heun-Johnson H., Ervasti J.M. (2011) Internal deletion compromises the stability of dystrophin. Hum. Mol. Genet., 20, 2955–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Imai Y., Matsushima Y., Sugimura T., Terada M. (1991) A simple and rapid method for generating a deletion by PCR. Nucleic Acids Res., 19, 2785.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu W., Ralston E. (2014) A new directionality tool for assessing microtubule pattern alterations. Cytoskeleton, 71, 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.