

Translational control of gene expression is generally exerted in the initiation phase of protein synthesis. The reactions of the initiation pathway bring the ribosome to the mRNA and place the initiation codon in the peptidyl (P) site of the ribosome, base-paired with the anticodon of methionyl initiator tRNA (Met-tRNA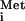 ). The aminoacyl (A) site in the ribosome is left open to accept the elongator tRNA that decodes the second codon in the ORF. Formation of this 80S initiation complex in eukaryotic cells depends on 11 or more soluble initiation factors (eIFs), identified primarily through biochemical studies of translation in rabbit reticulocyte extracts. Most of these eIFs are essential for normal cell growth in budding yeast, confirming their importance in vivo (1). Nevertheless, a number of mRNAs contain specialized regulatory elements that allow them to dispense with one or more eIFs during translation initiation. These elements, called internal ribosome entry sites (IRESs), recruit the ribosome directly to the initiation region of the mRNA. Their independence of certain eIFs allows viral mRNAs containing IRESs to be translated in infected cells where one or more eIFs have been impaired, either by the virus to provide a selective advantage for its mRNAs or by the host to impede virus gene expression. The most striking instance of this strategy is provided by cricket paralysis virus (CrPV) mRNA, which seems to be translated without any eIFs or even Met-tRNA
). The aminoacyl (A) site in the ribosome is left open to accept the elongator tRNA that decodes the second codon in the ORF. Formation of this 80S initiation complex in eukaryotic cells depends on 11 or more soluble initiation factors (eIFs), identified primarily through biochemical studies of translation in rabbit reticulocyte extracts. Most of these eIFs are essential for normal cell growth in budding yeast, confirming their importance in vivo (1). Nevertheless, a number of mRNAs contain specialized regulatory elements that allow them to dispense with one or more eIFs during translation initiation. These elements, called internal ribosome entry sites (IRESs), recruit the ribosome directly to the initiation region of the mRNA. Their independence of certain eIFs allows viral mRNAs containing IRESs to be translated in infected cells where one or more eIFs have been impaired, either by the virus to provide a selective advantage for its mRNAs or by the host to impede virus gene expression. The most striking instance of this strategy is provided by cricket paralysis virus (CrPV) mRNA, which seems to be translated without any eIFs or even Met-tRNA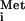 (2). The work reported by Sarnow and colleagues (3) in this issue of PNAS shows that the unique CrPV initiation mechanism can operate in budding yeast cells, so long as the canonical initiation pathway involving initiation factor 2 (eIF2) has been impaired. These findings open up the possibility of using powerful yeast genetics to dissect the details of this remarkable mechanism and to define the factors that modulate its efficiency in living cells.
(2). The work reported by Sarnow and colleagues (3) in this issue of PNAS shows that the unique CrPV initiation mechanism can operate in budding yeast cells, so long as the canonical initiation pathway involving initiation factor 2 (eIF2) has been impaired. These findings open up the possibility of using powerful yeast genetics to dissect the details of this remarkable mechanism and to define the factors that modulate its efficiency in living cells.
For most eukaryotic mRNAs, it is thought that translation initiation occurs by the scanning mechanism. In the current model for this complex process, a small (40S) ribosomal subunit carrying eIF3 and eIF1A interacts with a ternary complex (TC) consisting of eIF2 bound to GTP and Met-tRNA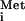 . The resulting 43S complex binds to the 5′ terminal m7GpppN cap structure (forming the 48S complex) and migrates into the 5′ untranslated region (UTR) of the mRNA until encountering an AUG start codon in a suitable sequence context. Binding of the 43S complex to the mRNA is stimulated by the eIF4F complex, consisting of the cap-binding protein eIF4E, a DEAD-box RNA helicase (eIF4A), and a scaffolding polypeptide (eIF4G) with binding sites for the other two eIFs in the 4F complex. The ATP-dependent helicase activity of eIF4A is thought to help unwind secondary structure in the 5′ UTR to facilitate movement of the ribosome along the mRNA during scanning (1, 4). The poly(A)-binding protein (PABP), bound to the poly(A) tail, also interacts with eIF4G, so that both ends of the mRNA are tethered to eIF4G (5). It is thought that eIF3 bridges the interaction between this messenger ribonucleoprotein (mRNP) complex and the ribosome by interacting simultaneously with eIF4G and the 40S subunit (6). Formation of a 48S complex with Met-tRNA
. The resulting 43S complex binds to the 5′ terminal m7GpppN cap structure (forming the 48S complex) and migrates into the 5′ untranslated region (UTR) of the mRNA until encountering an AUG start codon in a suitable sequence context. Binding of the 43S complex to the mRNA is stimulated by the eIF4F complex, consisting of the cap-binding protein eIF4E, a DEAD-box RNA helicase (eIF4A), and a scaffolding polypeptide (eIF4G) with binding sites for the other two eIFs in the 4F complex. The ATP-dependent helicase activity of eIF4A is thought to help unwind secondary structure in the 5′ UTR to facilitate movement of the ribosome along the mRNA during scanning (1, 4). The poly(A)-binding protein (PABP), bound to the poly(A) tail, also interacts with eIF4G, so that both ends of the mRNA are tethered to eIF4G (5). It is thought that eIF3 bridges the interaction between this messenger ribonucleoprotein (mRNP) complex and the ribosome by interacting simultaneously with eIF4G and the 40S subunit (6). Formation of a 48S complex with Met-tRNA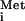 base-paired to the start codon also requires eIF1 and eIF1A (7). This assembly is then recognized by eIF5, which stimulates hydrolysis of GTP in the TC, leading to release of eIF2-GDP but leaving behind Met-tRNA
base-paired to the start codon also requires eIF1 and eIF1A (7). This assembly is then recognized by eIF5, which stimulates hydrolysis of GTP in the TC, leading to release of eIF2-GDP but leaving behind Met-tRNA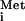 in the P site (1, 4). Finally, eIF5B catalyzes joining of the 60S subunit to form the 80S initiation complex, with hydrolysis of a second molecule of GTP (8).
in the P site (1, 4). Finally, eIF5B catalyzes joining of the 60S subunit to form the 80S initiation complex, with hydrolysis of a second molecule of GTP (8).
An increasing number of mRNAs have been identified that are translated by alternative mechanisms, known collectively as internal initiation, in which the 40S ribosome bypasses the cap and binds to an IRES located in the 5′ UTR. The best-studied IRESs, which occur in viral mRNAs, are large highly structured elements, often stabilized by RNA binding proteins. In bypassing the cap, IRESs dispense with the requirement for eIF4E, the cap-binding subunit of eIF4G (2). This fact underlies the ability of picornaviruses, including poliovirus and foot and mouth disease virus, to inhibit host cell protein synthesis by expressing proteases that cleave the PABP- and eIF4E-binding domain from the N terminus of eIF4G (6, 9). It also explains how these mRNAs can be efficiently translated despite their highly structured 5′ UTRs and the presence of multiple AUG codons (10) that should waylay scanning ribosomes and prevent them from reaching the authentic start site downstream (11, 12). Insertion of an IRES between the two cistrons of a dicistronic mRNA allows the downstream cistron to be translated even when cap-dependent translation of the first cistron is impaired, as in poliovirus-infected cells (13).
Except for eIF4E, PABP, and the N terminus of eIF4G, all other canonical eIFs are required to form a 48S complex on the IRES of encephalomyocarditis virus (EMCV) (14), another picornavirus. The essential core domain of eIF4G, containing the binding sites for eIF3 and eIF4A, interacts directly with a portion of the IRES in a manner facilitated by eIF4A. This direct eIF4G–IRES interaction supplements the mRNA binding functions of eIF4E and PABP (15). Hepatitis C virus (HCV) mRNA presents a more extreme case of an IRES that functions independently of the canonical eIFs. In this case, a naked 40S ribosome can bind directly to the IRES, placing the start codon in proximity to the P site, without assistance from any other eIFs or ATP hydrolysis. Binding of Met-tRNA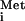 to the start codon positioned in the P site, mediated by the TC, is still required to form an 80S initiation complex on the HCV IRES (2, 16). Recent exciting results using cryoelectron microscopy show that the 40S subunit makes numerous contacts with the IRES and undergoes a conformational change that might clamp the IRES onto the ribosome (17).
to the start codon positioned in the P site, mediated by the TC, is still required to form an 80S initiation complex on the HCV IRES (2, 16). Recent exciting results using cryoelectron microscopy show that the 40S subunit makes numerous contacts with the IRES and undergoes a conformational change that might clamp the IRES onto the ribosome (17).
The ultimate factor-independent IRESs occur in a group of insect viruses with picornavirus-like RNA genomes. These IRESs direct translation of the second ORF in the viral genome, encoding the structural protein precursor, and are often referred to as intergenic region (IGR) IRESs. The predicted base-pairing interactions of the CrPV IGR IRES is shown in Fig. 1. Remarkably, the ORF under the control of the IGR IRES initiates with an alanine codon (GCU) in CrPV (Fig. 1) or with a glutamine codon (CAA) in Plautia stali intestine virus (PSIV), rather than with AUG, and the N-terminal residue of the PSIV protein was shown to be glutamine rather than methionine (18, 19). The preceding triplet is not AUG, and it can be replaced with a stop codon without altering translation (18, 20). These findings eliminated the possibility that a non-AUG codon was misread by Met-tRNA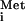 followed by removal of the methionine by methionine aminopeptidase. Rather, it seems that the eIF2/GTP/Met-tRNA
followed by removal of the methionine by methionine aminopeptidase. Rather, it seems that the eIF2/GTP/Met-tRNA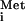 TC is simply not required for initiation by these IRESs.
TC is simply not required for initiation by these IRESs.
Figure 1.

Predicted structure of the CrPV IGR-IRES. Base pairs conserved among the IGR IRESs of cricket paralysis-like viruses and unpaired residues are shown in bold. The underlined GCU triplet is decoded ribosomal A site into the N-terminal alanine of the protein encoded by the second ORF of the viral mRNA. Arrows mark nucleotides where the leading edge of the ribosome was mapped by the technique of “ribosomal toeprinting” in 40S/IGR IRES complexes. Only the toeprints at residues 6226 and 6227 correlated with the ability of the IRES to function in internal initiation (21). The drawing was provided courtesy of C. Hellen (State University of New York, Brooklyn).
In vitro experiments showed that a naked 40S ribosome can bind directly to the IGR IRES in CrPV, with the first decoded triplet (GCU) in the A site. It is believed that a pseudoknot structure in the IRES (Fig. 1) takes the place of the TC in the P site, perhaps mimicking Met-tRNA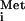 . A 60S subunit can join this complex to form an 80S complex in the absence of eIF5, eIF5B, and GTP hydrolysis. Thus, it seems that none of the canonical eIFs or Met-tRNA
. A 60S subunit can join this complex to form an 80S complex in the absence of eIF5, eIF5B, and GTP hydrolysis. Thus, it seems that none of the canonical eIFs or Met-tRNA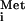 are required for initiation by the IGR IRES. The A site codon is decoded by the cognate aminoacyl-tRNA delivered to the ribosome by elongation factor 1A (EF-1A), and a pseudotranslocation event occurs (i.e., without prior peptide bond formation) to move this tRNA into the P site, after which normal elongation ensues (21).
are required for initiation by the IGR IRES. The A site codon is decoded by the cognate aminoacyl-tRNA delivered to the ribosome by elongation factor 1A (EF-1A), and a pseudotranslocation event occurs (i.e., without prior peptide bond formation) to move this tRNA into the P site, after which normal elongation ensues (21).
Interestingly, the activity of the IGR IRES was enhanced in cultured mammalian cells when the unfolded protein response (UPR) was induced (21). This condition is known to activate the protein kinase PERK that phosphorylates the α-subunit of eIF2 (22). The phosphorylated factor (eIF2[αP]) inhibits the guanine nucleotide exchange factor eIF2B, preventing recycling of eIF2-GDP to eIF2-GTP and thereby reducing TC formation (4). It is believed that this mechanism provides a means of reducing the rate of protein synthesis when processing or folding of secreted proteins in the endoplasmic reticulum (ER) is impeded. As expected, cap-dependent translation, or translation directed by the EMCV IRES, was reduced when the UPR was induced (21). Thus, the factor-independent mechanism of the IGR IRES was specifically enhanced by a reduction in TC levels, although it could be argued that some other event evoked by unfolded proteins could be responsible for activation of the IRES.
In this issue of PNAS, Sarnow and colleagues (3) provide direct evidence that IGR IRES function is indeed stimulated when TC levels are diminished in vivo. They introduced dicistronic mRNAs into Saccharomyces cerevisiae containing the IGR IRES upstream of the URA3 ORF, encoding an enzyme of uracil biosynthesis. The dicistronic construct was expressed in a strain lacking the endogenous URA3 gene, such that growth on medium lacking uracil required URA3 expression from the dicistronic construct. As in many other attempts to detect IRES activity in budding yeast, the IGR IRES did not function and the strain failed to produce the URA3 protein or to grow on medium lacking uracil. However, URA3 expression directed by the IGR IRES was activated by two independent genetic manipulations known to reduce TC levels in vivo.
In one case, constitutively activated forms of the yeast eIF2α kinase GCN2 were expressed in the strain, leading to high-level phosphorylation of eIF2α. A large amount of genetic and biochemical evidence indicates that this degree of eIF2α phosphorylation evokes a reduction in eIF2B activity and consequent decline in TC levels, which reduces protein synthesis and cell growth (12). Importantly, the induction of URA3 expression by the activated GCN2c kinase was eliminated by a mutation in the IRES that destroys the critical pseudoknot structure. Additionally, there was no evidence of monocistronic mRNAs containing URA3, which could potentially translate URA3 by the scanning mechanism, in either the wild-type or GCN2c strains. Hence, the evidence is quite strong that IGR IRES function was induced in the GCN2c mutant. The second manipulation that activated URA3 translation from the dicistronic construct was a deletion of 2 of the 4 IMT genes encoding Met-tRNA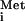 in yeast. Again, there is strong genetic evidence that ternary complex levels are reduced in this mutant below the level needed to sustain wild-type rates of protein synthesis (23). As expected, the effect of deleting IMT genes in stimulating URA3 expression was independent of GCN2 (3).
in yeast. Again, there is strong genetic evidence that ternary complex levels are reduced in this mutant below the level needed to sustain wild-type rates of protein synthesis (23). As expected, the effect of deleting IMT genes in stimulating URA3 expression was independent of GCN2 (3).
The IGR IRES also functioned in translationally competent yeast extracts, and mutational analysis confirmed the expected dependence on the ability to form the pseudoknot in the IRES. Moreover, translation directed by the IGR IRES was relatively resistant to edeine, an inhibitor of start codon recognition by canonical 48S initiation complexes containing the TC bound to the P site. At a concentration of edeine where cap-dependent translation of the first cistron in a dicistronic mRNA was greatly impaired, translation of the second cistron under the control of the IGR IRES was completely unaffected (3). Thus, whether examined in yeast cells or yeast extracts, translation directed by the IGR IRES does not involve TC binding to the P site, just as shown previously in the mammalian system. Based on the genetic data, one would predict that the TC was present at low levels in the cell-free extracts, allowing the IGR IRES to function. Moreover, the IGR IRES should be functional in wild-type yeast cells when cultured under conditions of amino acid limitation in which GCN2 kinase function is activated (12).
Why would a reduction in TC levels stimulate the IGR IRES? One possibility is that the IRES-directed mechanism cannot compete with the standard initiation pathway for free 40S subunits unless the latter mechanism is impaired. In this event, it might be found that interfering with eIF3 or eIF1A, two other factors required in addition to eIF2 for 43S complex formation (4), would also activate the IGR IRES in yeast cells. The same outcome could be predicted for mutations that reduce the abundance of 60S ribosomal subunits, as this leads to an excess of free 40S subunits by a different mechanism. Alternatively, it could be imagined that the TC competes with the IRES pseudoknot for binding to the P site of the ribosome, perturbing IRES function by a more active mechanism. This latter hypothesis is supported by the fact that IGR IRES activity was inhibited by adding eIF2 to an extract where this factor was not limiting for cap-dependent translation (21).
A requirement for reduced TC levels to activate the IGR IRES might serve an important biological function for the virus. In mammalian cells, many viruses activate an eIF2α kinase known as PKR, contributing to a shut-off of protein synthesis in infected cells as part of an antiviral defense mechanism (24). If eIF2α becomes phosphorylated in insect cells infected with CrPV, then the IGR IRES and the translation of viral structural proteins under its control would be activated at a time during infection when host-cell protein synthesis is impaired. In this way, the virus could subvert the host's ribosomes for production of viral proteins.
It is interesting that a number of IRES elements identified in mammalian cellular mRNAs are active under conditions where one or more canonical eIFs is impaired. A subset of IRESs can function in the G2/M phase of the cell cycle where the dephosphorylation of eIF4E is thought to reduce cap-dependent translation (2, 25, 26). An IRES in the mRNA encoding the cationic amino acid transporter Cat-1 is activated in amino acid-starved cells where eIF2α is transiently phosphorylated and eIF4E activity is reduced (27). In yeast, a segment of Escherichia coli lacI sequence seems to have IRES activity, but only in stationary phase or carbon-starved cells where protein synthesis is reduced (28). Thus far, it is not clear in these cases whether IRES activity depends on a reduction in eIF function. Even if it is, this could serve primarily to induce the translation of a trans-acting factor required for IRES activity. For example, eIF2α phosphorylation by GCN2 in amino acid-starved yeast cells stimulates translation of GCN4 mRNA through a specialized scanning-reinitiation mechanism involving short upstream ORFs (uORFs) in the mRNA leader (12). Similarly, translation of ATF4 mRNA is stimulated in mammalian cells in response to eIF2α phosphorylation by GCN2 or PERK through a mechanism involving uORFs (29). In any case, it seems likely that many cellular mRNAs containing IRESs will encode proteins that function under starvation or stress conditions because they can be translated efficiently under conditions where cap-dependent translation is reduced. Indeed, many cellular mRNAs in the growing list of transcripts thought to contain IRESs encode regulators of transcription, translation, or cell growth (2).
In addition to the finding that the CrPV IRES functions in yeast cells, it was reported that two naturally occurring yeast mRNAs, YAP1 and TIF4631 (encoding eIF4G), contain IRESs that function in actively growing cells (30). With these numerous developments, the power of yeast genetics now can be enlisted for the molecular dissection of IRES function in vivo. The dicistronic constructs described above, where URA3 translation is directed by the IRES, should be particularly useful in such efforts because proven strategies exist for selecting mutants with increased or decreased URA3 expression. For IRESs that require a subset of canonical eIFs, the factor requirements can be defined in vivo by using the large number of yeast strains available containing mutations in different eIFs, and the precise activities of the required eIFs in internal initiation can be probed in great detail. Novel proteins or RNAs that are required specifically for IRES function may also be identified. Finally, genetically engineered yeast strains have been produced in which the ribosomal RNA is encoded by a single episomal rDNA repeat (31). Such strains permit mutational analysis of rRNA and should be invaluable for studying base pair interactions between portions of the IRES and RNA segments in the ribosome.
Acknowledgments
I thank Tom Dever for helpful comments on the manuscript and Bobbie Felix for assistance in its preparation.
Footnotes
See companion article on page 12972.
References
- 1.Hershey J W B, Merrick W C. In: Translational Control of Gene Expression. Sonenberg N, Hershey J W B, Mathews M B, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 2000. pp. 33–88. [Google Scholar]
- 2.Hellen C U, Sarnow P. Genes Dev. 2001;15:1593–1612. doi: 10.1101/gad.891101. [DOI] [PubMed] [Google Scholar]
- 3.Thompson S R, Gulyas K D, Sarnow P. Proc Natl Acad Sci USA. 2001;98:12972–12977. doi: 10.1073/pnas.241286698. . (First Published October 30, 2001; 10.1073/pnas.241286698) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hinnebusch A G. In: Translational Control of Gene Expression. Sonenberg N, Hershey J W B, Mathews M B, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 2000. pp. 185–243. [Google Scholar]
- 5.Sachs A B, Varani G. Nat Struct Biol. 2000;7:356–361. doi: 10.1038/75120. [DOI] [PubMed] [Google Scholar]
- 6.Lamphear B J, Kirchweger R, Skern T, Rhoads R E. J Biol Chem. 1995;270:21975–21983. doi: 10.1074/jbc.270.37.21975. [DOI] [PubMed] [Google Scholar]
- 7.Pestova T V, Borukhov S I, Hellen C U T. Nature (London) 1998;394:854–859. doi: 10.1038/29703. [DOI] [PubMed] [Google Scholar]
- 8.Pestova T V, Lomakin I B, Lee J H, Choi S K, Dever T E, Hellen C U T. Nature (London) 2000;403:332–335. doi: 10.1038/35002118. [DOI] [PubMed] [Google Scholar]
- 9.Gradi A, Svitkin Y V, Imataka H, Sonenberg N. Proc Natl Acad Sci USA. 1998;95:11089–11094. doi: 10.1073/pnas.95.19.11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pelletier J, Flynn M E, Kaplan G, Racaniello V, Sonenberg N. J Virol. 1988;62:4486–4492. doi: 10.1128/jvi.62.12.4486-4492.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kozak M. J Cell Biol. 1989;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hinnebusch A G. In: Translational Control. Hershey J W B, Mathews M B, Sonenberg N, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1996. pp. 199–244. [Google Scholar]
- 13.Pelletier J, Sonenberg N. Nature (London) 1988;334:320–325. doi: 10.1038/334320a0. [DOI] [PubMed] [Google Scholar]
- 14.Pestova T V, Hellen C U T, Shatsky I V. Mol Cell Biol. 1996;16:6859–6869. doi: 10.1128/mcb.16.12.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pestova T V, Shatsky I N, Hellen C U T. Mol Cell Biol. 1996;16:6870–6878. doi: 10.1128/mcb.16.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pestova T V, Shatsky I N, Fletcher S P, Jackson R J, Hellen C U T. Genes Dev. 1998;12:67–83. doi: 10.1101/gad.12.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spahn C M, Kieft J S, Grassucci R A, Penczek P A, Zhou K, Doudna J A, Frank J. Science. 2001;291:1959–1962. doi: 10.1126/science.1058409. [DOI] [PubMed] [Google Scholar]
- 18.Sasaki J, Nakashima N. J Virol. 1999;73:1219–1226. doi: 10.1128/jvi.73.2.1219-1226.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasaki J, Nakashima N. Proc Natl Acad Sci USA. 2000;97:1512–1515. doi: 10.1073/pnas.010426997. . (First Published January 31, 2000; 10.1073/pnas.10.1073/pnas.010426997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson J E, Powell M J, Hoover S E, Sarnow P. Mol Cell Biol. 2000;20:4990–4999. doi: 10.1128/mcb.20.14.4990-4999.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson J E, Pestova T V, Hellen C U T, Sarnow P. Cell. 2000;102:511–520. doi: 10.1016/s0092-8674(00)00055-6. [DOI] [PubMed] [Google Scholar]
- 22.Ron D, Harding H P. In: Translational Control of Gene Expression. Sonenberg N, Hershey J W B, Mathews M B, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 2000. pp. 547–560. [Google Scholar]
- 23.Dever T E, Yang W, Åström S, Byström A S, Hinnebusch A G. Mol Cell Biol. 1995;15:6351–6363. doi: 10.1128/mcb.15.11.6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaufman R. In: Translational Control of Gene Expression. Sonenberg N, Hershey J W B, Mathews M B, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 2000. pp. 503–527. [Google Scholar]
- 25.Cornelis S, Bruynooghe Y, Denecker G, Van Huffel S, Tinton S, Beyaert R. Mol Cell. 2000;5:597–605. doi: 10.1016/s1097-2765(00)80239-7. [DOI] [PubMed] [Google Scholar]
- 26.Pyronnet S, Pradayrol L, Sonenberg N. Mol Cell. 2000;5:607–616. doi: 10.1016/s1097-2765(00)80240-3. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez J, Yaman I, Mishra R, Merrick W C, Snider M D, Lamers W H, Hatzoglou M. J Biol Chem. 2001;276:12285–12291. doi: 10.1074/jbc.M009714200. [DOI] [PubMed] [Google Scholar]
- 28.Paz I, Abramovitz L, Choder M. J Biol Chem. 1999;274:21741–21745. doi: 10.1074/jbc.274.31.21741. [DOI] [PubMed] [Google Scholar]
- 29.Harding H P, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 30.Zhou W, Edelman G M, Mauro V P. Proc Natl Acad Sci USA. 2001;98:1531–1536. doi: 10.1073/pnas.98.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wai H H, Vu L, Oakes M, Nomura M. Nucleic Acids Res. 2000;28:3524–3534. doi: 10.1093/nar/28.18.3524. [DOI] [PMC free article] [PubMed] [Google Scholar]