

Abstract
The mitochondrial and nuclear genomes coordinate and co-evolve in eukaryotes in order to adapt to environmental changes. Variation in the mitochondrial genome is capable of affecting expression of genes on the nuclear genome. Sex-specific mitochondrial genetic control of gene expression has been demonstrated in Drosophila melanogaster, where males were found to drive most of the total variation in gene expression. This has potential implications for male-related health and disease resulting from variation in mtDNA solely inherited from the mother. We used a family-based study comprised of 47,323 gene expression probes and 78 mitochondrial SNPs (mtSNPs) from n = 846 individuals to examine the extent of mitochondrial genetic control of gene expression in humans. This identified 15 significant probe-mtSNP associations (P) corresponding to 5 unique genes on the mitochondrial and nuclear genomes, with three of these genes corresponding to mitochondrial genetic control of gene expression in the nuclear genome. The associated mtSNPs for three genes (one cis and two trans associations) were replicated (P < 0.05) in an independent dataset of n = 452 unrelated individuals. There was no evidence for sexual dimorphic gene expression in any of these five probes. Sex-specific effects were examined by applying our analysis to males and females separately and testing for differences in effect size. The MEST gene was identified as having the most significantly different effect sizes across the sexes (P). MEST was similarly expressed in males and females with the G allele; however, males with the C allele are highly expressed for MEST, while females show no expression of the gene. This study provides evidence for the mitochondrial genetic control of expression of several genes in humans, with little evidence found for sex-specific effects.
Introduction
Human cells contain DNA from the nuclear and mitochondrial genomes. Mitochondria are responsible for generating most of a cell’s supply of energy (adenosine triphosphate, ATP) for cellular processes via oxidative phosphorylation (OXPHOS). Over 1,000 proteins in the mitochondrial proteome are encoded by the nuclear genome, with the remaining mitochondrial proteins encoded by DNA on a circular, double-stranded haploid mitochondrial genome (mtDNA) that is inherited solely from the mother. In humans, mtDNA is approximately 16 kb in length and contains 37 genes encoding 13 polypeptides, 2 ribosomal RNAs and 22 tRNAs (1).
The mitochondrial and nuclear genomes coordinate and co-evolve in eukaryotes in order to adapt to environmental changes in species and populations (2,3). Studies have shown that variation in mtDNA is capable of affecting expression of genes on the nuclear genome that function to maintain homeostasis of normal cellular function. For example, a study of mitochondrial dysfunction and risk of type 2 diabetes mellitus (T2DM) found that two hybrid cell populations harbouring three mtDNA haplogroups that were previously found to be associated with increased and decreased risk of T2DM were normal for mitochondrial and cellular metabolism (4). An examination of gene expression between the two cell populations found that hybrid cells that increase the risk of T2DM had a significantly lower expression of OXPHOS and were highly expressed for glycolysis when compared to hybrid cells that decrease the risk of T2DM. This indicates that mtDNA is affecting expression of genes on the nuclear genome in order to maintain normal cellular function, and may potentially influence disease status if their coordination is defective.
A study of Drosophila melanogaster found evidence for sex-specific mitochondrial genetic control of gene expression, where polymorphisms in mtDNA were found to affect expression of genes on the nuclear genome (5). A disproportionately large number of these relationships were found in males compared to females, with most genes showing male-biased expression. These results suggest sex-specific effects in the mitochondrial genetic control of nuclear gene expression in Drosophila melanogaster, where males were found to drive most of the total variation in gene expression.
An examination of sex-specific mitochondrial genetic control of gene expression in humans is motivated by the implications for male-related health and disease (6). Because mtDNA is inherited solely from the mother, natural selection favours variants on mtDNA that are neutral or beneficial to females, even if deleterious to males (7). In particular, natural selection can be blind to variants on mtDNA that are harmful to males if these same variants do not have a deleterious effect in females.
This study investigates the extent of mitochondrial genetic control of gene expression in humans, and whether there are sex-specific effects as observed in other species.
Results
Mitochondrial genetic control of gene expression was examined in a family-based study comprising of 47,323 gene expression probes (three probes are located on the mitochondrial genome) and 78 mtSNPs (72 mtSNPs had frequency and 27 had frequency ; see Excel table in the Supplementary Material) in n = 846 individuals from 312 independent families (8,9). Individuals were verified for European ancestry through principal component (PC) analysis and comparison of PCs with those of HapMap3 populations. No individuals were excluded based on ambiguous European ancestry. A total of 3,691,194 probe-mtSNP pairs were tested for association in a linear mixed regression model, which accounts for sample structure with a genetic relatedness matrix (see Materials and Methods). The quantile-quantile plot for expected versus observed P-values is shown in Figure 1. As shown in Table 1, a total of 15 probe-mtSNP associations achieved experiment-wide significance corresponding to five unique probes (), with allele frequencies of the mtSNPs ranging from 0.02 to 0.14. Figure 2 illustrates the relationship between normalised gene expression intensity levels for each of the five probes versus the alleles of the top associated mtSNP. Similar plots using raw gene expression intensities are illustrated in Supplementary Material, Figure S1. The significantly associated mtSNPs for each of the five genes were tested for replication in an independent dataset of n = 452 unrelated individuals from the United Kingdom and The Netherlands in the Fehrmann cohort (10). Three genes (one cis and two trans associations) showed replication with P < 0.05 (Table 1), and the top associated mtSNP for the signal peptidase complex subunit 2 (SPCS2) gene showed a nominal replication with P.
Figure 1.

Quantile-quantile plot for expected versus observed p-values of 3,691,194 probe-mtSNP associations. A strong deviation from the expected is observed for large .
Table 1.
15 probe-mtSNP associations had P < 1.35× corresponding to 5 unique probes. 10 probe-mtSNP associations were significant in a male-specific analysis, and 5 were significant in a female-specific analysis, with no new associations found. For convenience, we present effect sizes from the sex-specific analyses (β, mal and β, fem columns) with further details presented in Supplementary Materials, Tables S1 and S2. Sex-specific effect sizes statistically significant with P < 1.35× are highlighted in bold
| Probe | Mitochondrial SNP | Fehrmann Cohort | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr. | Position | Gene | Probe | mtSNP | MAF | β,full (n = 846) | SE | P | β, mal. (n = 428) | β, fem. (n = 418) | β (n = 452) | SE | P |
| 1 | 28422819:28422868 | SPCS2P4 | ILMN1780382 | MitoA3481G | 0.05 | −1.64 | 0.16 | 1.51× | −1.53 | −1.77 | −0.44 | 0.09 | 1.92× |
| 1 | 28422819:28422868 | SPCS2P4 | ILMN1780382 | MitoA10551G | 0.06 | −1.53 | 0.16 | 1.50× | −1.53 | −1.50 | −0.44 | 0.09 | 1.92× |
| 1 | 28422819:28422868 | SPCS2P4 | ILMN1780382 | MitoT9699C | 0.06 | −1.43 | 0.15 | 1.41× | −1.34 | −1.40 | −0.40 | 0.09 | 1.65× |
| 1 | 28422819:28422868 | SPCS2P4 | ILMN1780382 | MitoT1191C | 0.04 | −1.46 | 0.19 | 3.92× | −1.28 | −1.62 | −0.35 | 0.09 | 1.69× |
| MT | 8249:8287 | MT-CO2 | ILMN1821517 | MitoG8270A | 0.03 | −1.74 | 0.45 | 1.85× | −1.61 | −1.97 | −0.36 | 0.09 | 1.28x |
| 11 | 74660418:74660444 | SPCS2 | ILMN2408645 | MitoA3481G | 0.05 | −1.23 | 0.16 | 3.19× | −1.33 | −1.12 | −0.18 | 0.09 | 0.06 |
| 11 | 74660418:74660444 | SPCS2 | ILMN2408645 | MitoA10551G | 0.06 | −1.19 | 0.16 | 6.35× | −1.33 | −1.01 | −0.18 | 0.09 | 0.06 |
| 5 | 99383348:99383397 | MTND5P12 | ILMN1743078 | MitoC295T | 0.12 | 0.91 | 0.12 | 8.75× | 0.92 | 0.94 | 0.34 | 0.09 | 3.32× |
| 11 | 74660418:74660444 | SPCS2 | ILMN2408645 | MitoT9699C | 0.06 | −1.13 | 0.15 | 1.01× | −1.34 | −0.92 | −0.14 | 0.09 | 0.13 |
| 5 | 99383348:99383397 | MTND5P12 | ILMN1743078 | MitoC464T | 0.10 | 0.99 | 0.13 | 1.13× | 1.00 | 0.95 | 0.37 | 0.09 | 6.58× |
| 5 | 99383348:99383397 | MTND5P12 | ILMN1743078 | MitoT491C | 0.14 | 0.85 | 0.12 | 5.06× | 0.86 | 0.86 | 0.28 | 0.09 | 2.72× |
| MT | 8249:8287 | MT-CO2 | ILMN1821517 | MitoT5005C | 0.02 | −1.76 | 0.60 | 2.91× | −1.62 | −2.06 | −0.20 | 0.09 | 0.03 |
| MT | 8249:8287 | MT-CO2 | ILMN1821517 | MitoA14583G | 0.02 | −1.82 | 0.62 | 6.76× | −1.69 | −2.06 | −0.20 | 0.09 | 0.03 |
| MT | 8249:8287 | MT-CO2 | ILMN1821517 | MitoA4025G | 0.02 | −1.81 | 0.62 | 7.24× | −1.68 | −2.06 | −0.20 | 0.09 | 0.03 |
| 3 | 100414192:100414241 | ADGRG7 | ILMN2125395 | MitoT217C | 0.02 | 2.01 | 0.35 | 1.01× | 2.10 | 1.98 | −0.15 | 0.09 | 0.11 |
Figure 2.
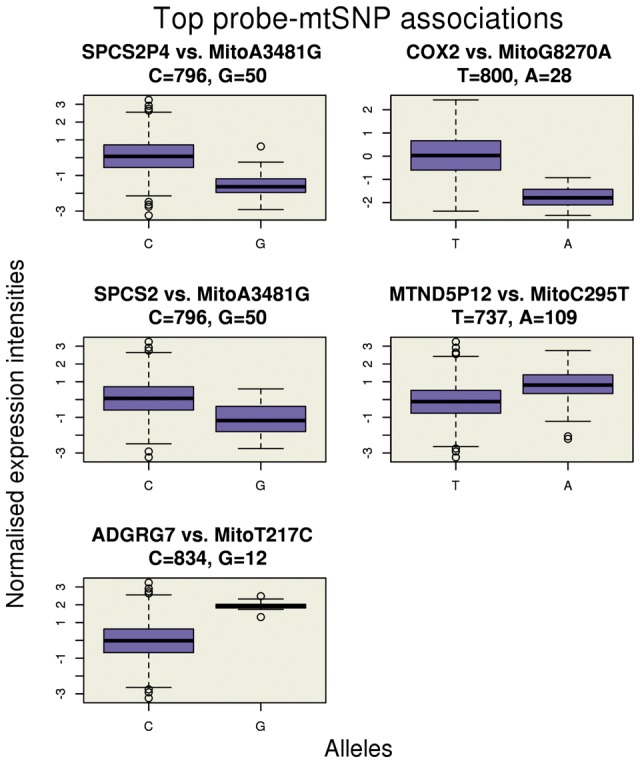
Boxplot of normalised expression intensity levels for each of the top five probes versus the alleles of the top associated mtSNP.
Of the five genes identified as being associated with mtSNPs, only the mitochondrially encoded cytochrome c oxidase II (MT-CO2) gene was located in the mitochondrial genome. A test for cross-hybridization with sequences other than the target transcript (see Methods and Materials) found that the mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 5 pseudogene 12 (MTND5P12) on chromosome 5 showed high similarity with sequences in the MT-ND5 gene on the mitochondrial genome, with 97.6% identity over the aligned region, 42 of 50 matching bps, and no gaps. The potential for cross-hybridisation therefore makes it unclear whether the mtSNP is showing evidence for control of pseudogene MTND5P12 on the nuclear genome or the MT-ND5 gene on the mitochondrial genome. The remaining probe-mtSNP associations correspond to the signal peptidase complex subunit 2 homolog (S. cerevisiae) pseudogene 4 (SPCS2P4) on chromosome 1, the signal peptidase complex subunit 2 (SPCS2) gene on chromosome 11, and the adhesion G protein-coupled receptor G7 (ADGRG7) gene on chromosome 3, which indicate mitochondrial genetic control of gene expression on the nuclear genome. The test for cross-hybridisation showed no evidence that these genes are cross-hybridising with sequences on the mitochondrial genome. In particular, the test for cross-hybridisation revealed a robust relationship between these mtSNPs and gene expression on the nuclear genome even when substantially relaxing the detection criteria. For example, when reducing the matching criteria to only require a match as small as 25 base pairs with 90% identity, no sequence matches to the mitochondrial genome were found for any of the other three genes. Pearson phenotypic correlations of the normalised expression intensities across the probes ranged from to , with correlations between SPCS2P4 and SPCS2 () and MTND5P12 and MT-CO2 () being the largest, and the remaining ranging from to . We ruled out the possibility that each of the detected probe-mtSNPs associations on the nuclear genome were due to cis-eQTLs at the probe sites (results not shown). There was no evidence for sexual dimorphic gene expression in any of these five genes with P > 0.05 (results not shown).
Applying our analysis to n = 428 males and n = 418 females separately identified 10 probe-mtSNP associations with P < 1.35 × in males (Supplementary Material, Table S1) and 5 in females (Supplementary Material, Table S2); however, no new probe-mtSNP associations were identified. The quantile-quantile plot for expected versus observed P-values for each sex is shown in Supplementary Materials, Figures S2 and S3. Figure 3 illustrates the intersections between the sets of significant probe-mtSNP associations from each dataset. As shown, 40% of the total number of associations from the combined analysis were driven by males (i.e. 6 of the 15 associations were not significant in the female-specific analysis), while approximately 7% of the total number of associations from the combined analysis were driven by females (i.e. one of the 15 associations was not significant in the male-specific analysis). Despite this, however, if we observe the effect estimates for the male- and female-specific analyses in Table 1 (β, mal. and β, fem. columns, respectively), we can see that the probe-mtSNP associations have the same direction of effect and are similar in magnitude across sexes. This suggests that these probes-mtSNP associations were not detected in the female-specific analysis at the P < 1.35 × level likely due to the reduced power afforded by the split dataset.
Figure 3.

Intersections between the sets of significant probe-mtSNP associations.
Finally, there was no evidence for differences in effect sizes across all tested genes between the sexes from random-effect inverse-variance-weighted meta-analysis at a significance threshold of P < 1.35 × (Supplementary Material, Fig. S4). The smallest P-value corresponded to the association between the mesoderm specific transcript (MEST) gene on chromosome 7 and mtSNP MitoT4337C with nominal P × . MEST encodes a member of the alpha/beta hydrolase superfamily, and shows a preferential expression from the paternal allele. Females had an effect estimate of −1.13 and corresponding P × , while males had an effect estimate of 1.56 and corresponding P × . That is, on average, males with the C allele had higher gene expression levels than males with the reference allele, while on average, females with the C allele had a lower gene expression levels than females with the reference allele. Due to the low number of minor alleles, MEST showed no evidence for sexually dimorphic gene expression (P = 0.75).
Figure 4 illustrates the raw gene expression intensities of MEST by alleles for each sex, with a similar plot with the normalised gene expression intensities presented in Supplementary Material, Figure S5. As shown, males and females with the G allele had very low expression with some outliers; however, males with the C allele are highly expressed for MEST, whereas females had low expression. MEST had mean raw expression intensity levels of 0.60 (SD: 3.16) across all individuals (n = 846), and mean raw expression intensity levels of 0.73 (SD: 3.34) and 0.46 (SD: 2.95) for males (n = 428) and females (n = 418), respectively. Expression of MEST in males with the G allele (n = 420) and females with the G allele (n = 412) showed similar mean expression intensities of 0.60 (SD: 2.83) and 0.46 (SD: 2.97), respectively; however, males with the C allele (n = 8) are highly expressed for MEST with mean expression intensities of 7.95 (SD: 11.88), whereas females with the C allele (n = 6) show no expression of the gene.
Figure 4.

A boxplot of the raw gene expression intensities of MEST versus the alleles of the associated mtSNP across the sexes.
Discussion
This study examined the extent of mitochondrial genetic control of gene expression in humans. We identified 15 probe-mtSNP associations corresponding to 5 unique genes, and replicated the mtSNP associations for three genes (one cis and two trans associations) in an independent cohort, indicating mitochondrial genetic control of gene expression on both the nuclear and mitochondrial genomes. No evidence was found for sexually dimorphic gene expression in any of these five probes. Sex-specific analyses found a larger number of probe-mtSNP associations in a male-only analysis; however, the effect estimates in the female-specific analysis were similar in direction of effect and magnitude compared to results from the male-specific analysis, suggesting that the disproportionality is likely due to the reduced power afforded by the split dataset. These results are in contrast with results observed in a study of mitochondrial genetic control of gene expression in Drosophila melanogaster, where it was found that 62% of differentially expressed genes were driven by males and had mostly male-biased expression (5).
A comparison of effect sizes between the sexes for each probe-mtSNP association by random-effect inverse-variance-weighted meta-analysis did not reveal sex differences in the mitochondrial genetic control of the expression at the experiment-wide level; however, the top gene, MEST, is of interest because it is imprinted. MEST is a mesoderm-specific transcript that encodes a member of the alpha/beta hydrolase superfamily, where the paternal allele shows preferential expression in fetal tissues, and isoform-specific imprinting in lymphocytes (11). In adult lymphocytes, isoform 1 is expressed only from the paternal allele, and isoform 2 is expressed from both the paternal and maternal alleles. Our results show that the mitochondrial genetic control of the expression of MEST through mtSNP MitoT4337C has sex-specific effects. While the expression of MEST for males and females with the G allele has similar expression levels, males with the C allele are highly expressed for MEST, whereas females with the C allele show no expression of the gene. Because mitochondria are transmitted solely from the mother and isoform 1 is expressed only from the paternal allele, our results indicate that mtSNP MitoT4337C may be involved in the silencing of the maternal allele in isoform 1 (11).
Of particular interest is the evidence for mitochondrial genetic control of nuclear gene expression of SPCS2, a protein coding gene involved in the mitotic cell cycle pathway; pseudogene SPCS2P4, involved in biosynthesis of the N-glycan precursor and transfer to a nascent protein; and ADGRG7, which encodes for G protein-coupled receptor 128, a member of the adhesion G protein-coupled receptor family. Although these associations are quite strong (P), the biological reasonings for their association with polymorphisms on mtDNA is not clear. It should be noted that the mitochondrial genome is a recombination ‘cold-spot’ which restricts the localisation of causal variants through association given the phylogenetic structure of mtDNA (12). A previous study of SNP tagging in whole mitochondrial genome showed that the linkage disequilibrium (LD) between mtSNPs is relatively low; however, the small, positive correlation between LD and distance gave no indication of mitochondrial recombination (13). Thus, these reported associations do not necessarily imply causal relationships between any one particular mtSNP and variation in gene expression. Nevertheless, it is possible that these associations are an example of variation in mtDNA affecting expression of gene on the nuclear genome that function to maintain homeostasis of normal cellular function (4); however, functional genomic studies would need to follow in order to explore this further.
This study provides evidence for the mitochondrial genetic control of gene expression of several genes in humans; however, little evidence was found for sex-specific effects as observed in previous studies in other species. Functional genomic studies may provide insight into the biological motivation for mitochondrial genetic control of genes on the nuclear genome observed in this study.
Materials and Methods
Study participants
The Brisbane Systems Genetics Study (BSGS) is a family-based study comprised of gene expression and genotype data from n = 846 individuals of verified Northern European origin from 312 independent families (8,9). Families consisted of adolescent monozygotic (MZ) (n = 131) and dizygotic (DZ) (n = 301) twins, their siblings (n = 91) and their parents. RNA was collected from whole-blood samples, with expression levels measured in 47,323 genome-wide probes using the Illumina HumanHT-12 v4.0 Beadchip. Individuals were genotyped using the Illumina 610-Quad Beadchip. Following standard QC filtering (including SNPs on the nuclear genome with Hardy-Weinberg equilibrium (HWE) test P, genotyping call rate < 0.95, and minor allele frequency (MAF) < 0.01), 528,509 SNPs and 88 mitochondrial SNPs (mtSNPs) were available for analysis.
Gene expression normalisation and quality control
Variance stabilisation was carried out using the method of Huber et al. (14), where true signal intensities were decoupled from background noise using the Bioconductor vsn package in R. Quantile normalisation was used to map the distribution of signal intensities to a standard distribution to allow for a comparison of signal intensities between probes. To account for known procedural variances (i.e. batch effects), gene expression levels for each probe were regressed on the chip ID, position on chip, and extraction date. Residuals from this analysis were carried forward as the corrected expression levels. Finally, a rank normal transformation was applied to further standardise the gene expression levels. A total of 47,323 gene expression probes were available for analysis.
Mitochondrial genotype and quality control
Quality control of mtSNP data included verifying homozygosity, and checking for errors in maternal inheritance where the child’s genotype did not match the mother’s or where there was variation within siblings. Ten mtSNPs were excluded with individual missingness , leaving a total of 78 mtSNPs available for analysis. Of these mtSNPs, 72 had frequency and 27 had frequency (see Excel table in the Supplementary Material).
Mitochondrial genetic control of gene expression
To investigate mitochondrial genetic control of gene expression, we modelled gene expression levels as a linear function of the presence or absence of the minor allele in a linear mixed regression model using the GCTA software package (15). The model for gene i can be written as,
| (1) |
where, a is the mean expression, and b is the effect estimate for a fixed mitochondrial genotype covariate, X. To accommodate for mtSNPs in GCTA, mtSNPs were coded as 0 or 2 corresponding to the reference allele and minor allele, respectively. G is a random polygenic effect captured by a genetic relatedness matrix (GRM) calculated from 795,723 imputed autosomal HapMap3 SNPs filtered for MAF < 0.01 and HWE test P; and e is the residual. The effect estimate, b, from model 1 can be interpreted as twice the difference in expression levels between the two homozygous alleles. We calculated a likelihood ratio test statistic to assess significance. A P-value was calculated from a -distribution with one degree of freedom. We accounted for multiple testing for both the number of mtSNPs and the number of probes using the Bonferroni method in this and subsequent analyses. Significance was defined as P < 1.35 × (i.e. 0.05/(78 × 47,323)).
Significant probes from the above analysis were tested for cross-hybridisation with sequences other than the target transcript using BLAST (16). In particular, we were interested in probes that were cross-hybridising with sequences on the mitochondrial genome. We considered strong evidence for cross-hybridising if probes sequences had 90% identity over the aligned region, at least 40 of 50 matching bps, and no gaps.
Further, probes showing evidence for mitochondrial genetic control were tested for sexually dimorphic gene expression. Using a model similar to equation 1, we modelled gene expression levels as a linear function of male or female status. Here, b is the effect estimate for a fixed sex covariate, X, where males are coded 0 and females are coded 1, and can be interpreted as the difference in gene expression levels across the sexes. We used the Wald statistic, calculated as the square of the effect estimate divided by the square of the standard error, to assess significance. A P-value was calculated from a -distribution with one degree of freedom.
Sex-specific effects of mtSNP on gene expression were examined by applying model 1 to males and females separately. Differences in effect sizes were compared by random-effect inverse-variance-weighted meta-analysis of each probe using the METAL meta-analysis package (17). The Cochran Q-statistic with corresponding p-value was used to test for heterogeneity between the effect estimates of each mtSNP between the sexes. A significant P-value would suggest that effect estimates is different across the sexes.
mtSNP-probe replication set
We sought replication of the mtSNP associations for each gene identified in the mitochondrial genetic control of gene expression analysis in an independent dataset of n = 452 unrelated individuals from the United Kingdom and The Netherlands in the Fehrmann cohort (10). Some of these individuals are patients, while others are healthy controls. Briefly, RNA was isolated from whole peripheral blood, with gene expression levels quantified using the Illumina HT12v3 platform, described in further detail in (10). Raw expression intensities were normalised using a log2 transformation, and were centred, scaled and corrected for the first 20 principal components. Individuals were genotyped using Illumina 610 Quad platform. QTL mapping was performed as outlined in (18).
Data Access
Gene expression from the BSGS and Fehrmann datasets are available on the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE53195 and GSE20142, respectively.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We gratefully acknowledge the participation of the twins and their families sampled in this work. We would like to thank Lisa Bowdler and Steven Crooks for their technical assistance with the microarray hybridisation, Alison Mackenzie, Marlene Grace and Ann Eldridge for data collection and Dale Nyholt and Scott Gordon for data management.
Conflict of Interest statement. None declared.
Funding
This research was supported by the Australian National Health and Medical Research Council (NHMRC) grants 389892, 496667, 613601, and 1046880. JEP, GWM, PMV and AFM are supported by the NHMRC Fellowship Scheme (1083656, 1107599, 1078037 and 1078399). We acknowledge funding by the Australian Research Council (A7960034, A79906588, A79801419, DP0212016, DP0343921), and the Australian National Health and Medical Research Council (NHMRC) Medical Bioinformatics Genomics Proteomics Program (grant 389891), which funded the genotyping of the adolescent twin family resource. This work is supported by a grant from the European Research Council (ERC Starting Grant agreement number 637640 ImmRisk) to Lude Franke and a VIDI grant (917.14.374) from the Netherlands Organisation for Scientific Research (NWO) to Lude Franke.
Research reported in this publication was supported by the National Institutes of Health under Award Number P01GM099568. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1. Mishra P., Chan D.C. (2014) Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol., 15, 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Greiner S., Bock R. (2013) Tuning a ménage à trois: co-evolution and co-adaptation of nuclear and organellar genomes in plants. Bioessays, 35, 354–365. [DOI] [PubMed] [Google Scholar]
- 3. Woodson J.D., Chory J. (2008) Coordination of gene expression between organellar and nuclear genomes. Nat. Rev. Genet., 9, 383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hwang S., Kwak S.H., Bhak J., Kang H.S., Lee Y.R., Koo B.K., Park K.S., Lee H.K., Cho Y.M. (2011) Gene expression pattern in transmitochondrial cytoplasmic hybrid cells harboring type 2 diabetes-associated mitochondrial DNA haplogroups. PLoS One, 6, e22116.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Innocenti P., Morrow E.H., Dowling D.K. (2011) Experimental evidence supports a sex-specific selective sieve in mitochondrial genome evolution. Science, 332, 845–848. [DOI] [PubMed] [Google Scholar]
- 6. Frank S.A., Hurst L.D. (1996) Mitochondria and male disease. Nature, 383, 224.. [DOI] [PubMed] [Google Scholar]
- 7. Gemmell N.J., Metcalf V.J., Allendorf F.W. (2004) Mother’s curse: the effect of mtDNA on individual fitness and population viability. Trends Ecol. Evol., 19, 238–244. [DOI] [PubMed] [Google Scholar]
- 8. Powell J.E., Henders A.K., McRae A.F., Caracella A., Smith S., Wright M.J., Whitfield J.B., Dermitzakis E.T., Martin N.G., Visscher P.M., et al. (2012) The Brisbane Systems Genetics Study: genetical genomics meets complex trait genetics. PLoS One, 7, e35430.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Powell J.E., Henders A.K., McRae A.F., Kim J., Hemani G., Martin N.G., Dermitzakis E.T., Gibson G., Montgomery G.W., Visscher P.M., et al. (2013) Congruence of additive and non-additive effects on gene expression estimated from pedigree and SNP data. PLoS Genet, 9, e1003502.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fehrmann R.S.N., Jansen R.C., Veldink J.H., Westra H.J., Arends D., Bonder M.J., Fu J., Deelen P., Groen H.J.M., Smolonska A., et al. (2011) Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet., 7, e1002197.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kosaki K., Kosaki R., Craigen W.J., Matsuo N. (2000) Isoform-specific imprinting of the human PEG1/MEST gene. Am. J. Hum. Genet., 66, 309–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eyre-Walker A., Awadalla P. (2001) Does human mtDNA recombine? J. Mol. Evol., 53, 430–435. [DOI] [PubMed] [Google Scholar]
- 13. McRae A.F., Byrne E.M., Zhao Z.Z., Montgomery G.W., Visscher P.M. (2008) Power and SNP tagging in whole mitochondrial genome association studies. Genome Res., 18, 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huber W., von Heydebreck A., Sültmann H., Poustka A., Vingron M. (2002) Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics, 18 Suppl 1, S96–104. [DOI] [PubMed] [Google Scholar]
- 15. Yang J., Lee S.H., Goddard M.E., Visscher P.M. (2011) GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet., 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 17. Willer C.J., Li Y., Abecasis G.R. (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Westra H.J., Peters M.J., Esko T., Yaghootkar H., Schurmann C., Kettunen J., Christiansen M.W., Fairfax B.P., Schramm K., Powell J.E., et al. (2013) Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet., 45, 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.