

Abstract
Dysregulation of Fused in Sarcoma (FUS) gene expression is associated with fronto-temporal lobar degeneration (FTLD), and missense mutations in the FUS gene have been identified in patients affected by amyotrophic lateral sclerosis (ALS). However, molecular and cellular defects underlying FUS proteinopathy remain to be elucidated. Here, we examined whether genes important for mitochondrial quality control play a role in FUS proteinopathy. In our genetic screening, Pink1 and Park genes were identified as modifiers of neurodegeneration phenotypes induced by wild type (Wt) or ALS-associated P525L-mutant human FUS. Down-regulating expression of either Pink1 or Parkin genes ameliorated FUS-induced neurodegeneration phenotypes. The protein levels of PINK1 and Parkin were elevated in cells overexpressing FUS. Remarkably, ubiquitinylation of Miro1 protein, a downstream target of the E3 ligase activity of Parkin, was also increased in cells overexpressing FUS protein. In fly motor neurons expressing FUS, both motility and processivity of mitochondrial axonal transport were reduced by expression of either Wt- or P525L-mutant FUS. Finally, down-regulating PINK1 or Parkin partially rescued the locomotive defects and enhanced the survival rate in transgenic flies expressing FUS. Our data indicate that PINK1 and Parkin play an important role in FUS-induced neurodegeneration. This study has uncovered a previously unknown link between FUS proteinopathy and PINK1/Parkin genes, providing new insights into the pathogenesis of FUS proteinopathy.
Introduction
Since the discovery of Fused in Sarcoma, also known as Translocated in Liposarcoma (FUS/TLS) as a characteristic component of protein inclusion bodies in FUS proteinopathy (1,2), great efforts have been made to understand this group of devastating neurodegenerative disorders (3–5). A number of cellular and animal models have been developed to model FUS proteinopathy (6–18). Accumulating evidence supports that FUS gene dysregulation may play an important role in the pathogenesis of FUS proteinopathy. In FUS-ALS patients, mutations in both the protein coding region and the untranslated region have been identified (19). However, in the majority of cases of fronto-temporal lobar degeneration with FUS pathology (FTLD-FUS), no FUS mutations have been identified; instead, increased FUS expression and FUS protein aggregation have been reported (17,19). Nonetheless, little is known about how mutations in or dysregulation of the FUS gene cause neuronal death in FUS proteinopathy.
To investigate molecular pathogenesis of FUS proteinopathy, we previously generated transgenic flies expressing human FUS protein, either the wild type (Wt) or ALS-associated mutants, R524S or P525L (6). Our characterization of phenotypes in these transgenic flies and cellular models of FUS proteinopathy has revealed that increased FUS expression induces mitochondrial damage (20). This prompted us to examine the genetic interaction between FUS and genes involved in mitochondrial dynamics and quality control and to search for potential modifier genes.
Recent studies have uncovered several mitochondrial quality control mechanisms, including selective degradation of mitochondrial proteins or removal of the entire organelle by mitophagy (21,22). In particular, PINK1 (PTEN induced putative kinase 1) and Parkin act in a common pathway that targets damaged mitochondria towards autophagic removal to control mitochondrial quality (22). They also have anti-apoptotic function and protect cells against various stress factors. PINK1 is a key kinase to phosphorylate ubiquitin and Parkin, both of which are required for Parkin activation (23,24). In Hela cells, damaged mitochondria recruit Parkin to their outer membrane in a PINK1-dependent manner (25–29). PINK1 and Parkin proteins have anti-apoptotic function and protect cells against various stress factors. Other phosphorylation substrates of PINK1 include TRAP1 and Miro (30,31). Parkin is an E3 ligase acting downstream of PINK1 and targeting multiple mitochondrial proteins (32–34). In addition to regulating p53 function (35), Parkin protects cells against apoptosis by ubiquitinating AIMP2, PARIS or BAX proteins (36–38). PINK1 and PARK2 (Parkinson protein 2) are mutated in a fraction of patients with autosomal recessive Parkinson’s disease. Although extensively studied, functional roles of PINK1 and Parkin in neurons remain controversial.
Unexpectedly, specific down-regulation of PINK1 and Park in FUS transgenic flies partially rescued neurodegeneration phenotypes induced by FUS. Consistent with these results, our biochemical and cell biology data show that FUS expression increases the levels of PINK1 and Parkin proteins and affects the subcellular distribution of Parkin. Ubiquitinylation of a Parkin substrate, Miro1, is increased by FUS expression. Expression of either Wt- or ALS-mutant FUS in fly motor neurons induces significant defects in mitochondrial axonal transport. Finally, down-regulation of Pink1 or Parkin partially rescues locomotive deficits in larvae expressing Wt- or ALS-mutant FUS in motor neurons. These data provide new insights into molecular mechanisms underlying FUS proteinopathy and suggest potential targets for developing therapeutic approaches to FUS proteinopathy associated with FUS mutation or dysregulation.
Results
Down-regulation of expression of PINK1 or Parkin in FUS-transgenic flies partially rescues FUS-induced retinal degeneration
To understand molecular pathogenesis of FUS proteinopathy, we have systematically characterized our transgenic fly model for FUS proteinopathy. Our recent work showed that expression of either Wt- or an ALS-associated P525Lmutant FUS induced mitochondrial fragmentation and dysfunction, with the P525L-mutant showing more severe effects (20). These data indicate that FUS-induced mitochondrial damage may contribute to the pathogenic processes. These observations led us to test genes important for mitochondrial quality control for their potential effects in modifying neurodegeneration phenotype in FUS transgenic flies. Taking advantage of the retinal degeneration phenotype induced by FUS expression in our transgenic FUS flies (6), we carried out genetic interaction screen experiments, focusing on genes involved in controlling mitochondrial fission/fusion and mitochondrial quality control. As shown in Figure 1, expression of the FUS protein, either the Wt- or P525L-mutant, led to prominent retinal degeneration (see control panels in Fig. 1A and B), consistent with our previous study (6). Specific down-regulation of the fly Pink1 or Parkin genes in fly retinae by RNA interference (RNAi; Fig. 1A–C) remarkably attenuated the retinal degeneration phenotype induced by FUS expression (red arrows in Fig. 1A), although neither siPink1 nor siParkin by themselves affected retinal morphology in the control flies (Supplementary Material, Fig. S1). More potent rescuing effects were achieved when both Pink1 and Parkin genes were down-regulated (Fig. 1A). Consistently, overexpression of Drosophila Pink1 exacerbated the neurodegeneration phenotype in flies expressing the P525L-mutant FUS (as marked by the white arrow in the Pink1 panel of Fig. 1A), although Parkin overexpression did not seem to affect the retinal phenotype (Fig. 1A). Scanning electron microscopy (SEM) revealed that FUS expression disrupted the normal ommatidial organization and induced ectopic bristles, whereas knock-down PINK1 or Parkin partially restored ommatidial organization (Fig. 1B). It should be noted that in these flies, the alteration of either PINK1 or Parkin expression did not affect FUS expression, either at the mRNA or protein levels (see Supplementary Material, Fig. S2).
Figure 1.
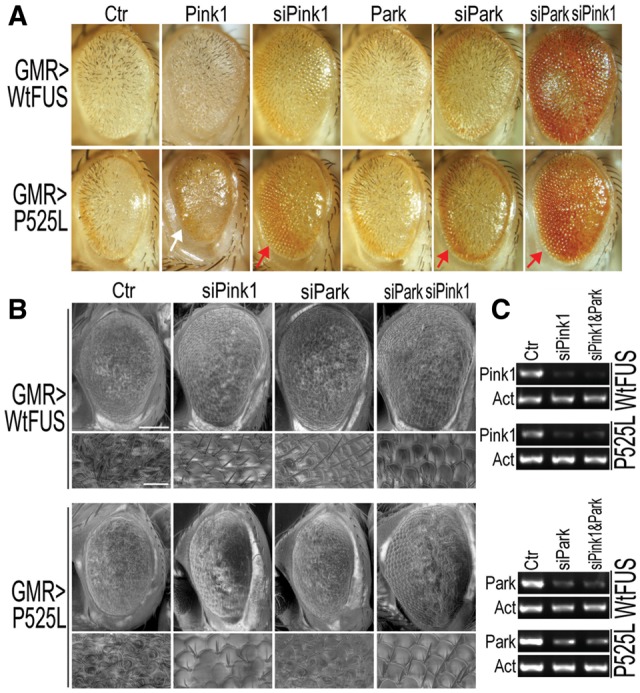
Down-regulating Pink1 or Parkin in Drosophila partially rescues retinal degeneration in flies expressing Wt- or P525L-mutant FUS. (A) Bright field microscopy (A) of fly eyes show that RNAi against Pink1 or Parkin (siPink1 or siPark, respectively) mitigates retinal degeneration caused by Wt- or P525L-mutant FUS, whereas overexpression of Pink1 exacerbates the eye phenotype in flies expressing the P525L-mutant FUS. (B) Scanning EM (SEM) images showing areas of the fly eyes with restored ommatidial organization. Scale bars in panel B: 100 μm (upper panels) or 20 μm (lower panels at higher magnification of SEM). (C) The down-regulation of Pink1 and Park gene expression was verified by RT-PCR.
Increased FUS expression leads to accumulation of PINK1 and Parkin proteins
The observation that down-regulation of PINK1 and/or Parkin suppresses FUS-induced neurodegeneration in vivo prompted us to examine the underlying mechanisms. Following co-transfection of HEK293 cells with PINK1-HA together with GFP-tagged Wt- or P525L-mutant FUS or with the vector control expressing a nuclear localized GFP (NLS-GFP), we examined the subcellular distribution and steady-state levels of PINK1 and Parkin (Fig. 2A–D and E–G, respectively). Immunofluorescent staining showed that FUS expression increased PINK1 protein level (compare PINK1 immunostaining signals in Wt- or P525L-mutant FUS panels with that in the control group, Fig. 2A). In addition, the percentage of cells showing cytoplasmic punctate pattern of PINK1 expression was significantly increased in the cells expressing Wt- or P525L-FUS than that in the control group (Fig. 2B). It has been previously reported that the PINK1 protein undergoes proteolysis when associated with the outer mitochondrial membrane to produce a 52-kDa form, leading to stabilization of PINK1 on the mitochondrial outer membrane (39). Consistent with the previous data, both the full-length and the cleaved species of PINK1 were detected (marked by arrowhead and arrow, respectively, in the PINK1 panel of Fig. 2C) by anti-HA antibody, and the level of both species of PINK1-HA increased significantly in cells expressing either Wt- or P525L-mutant FUS as compared with the control cells (Fig. 2C and D).
Figure 2.

Expression of Wt- or P525L-mutant FUS in mammalian cells led to accumulation of PINK1 and Parkin proteins. (A) Immunofluorescent staining reveals the punctate distribution of PINK1 in cells expressing either Wt- or P525L-mutant FUS. Following co-transfection of HEK293 cells with different plasmids [PINK1-HA together with the nuclear localized GFP control (NLS-GFP), or Wt-FUS or P525L-mutant FUS tagged with GFP], immunostaining was performed using anti-TOM20 and anti-HA antibodies 20 h post-transfection. (B) Less than 25% of the control cells exhibited punctate PINK1 staining, whereas cells expressing either Wt- or P525L-mutant FUS show a significant increase in the percentages of cells with the punctate pattern of PINK1 distribution (puncta are marked by the white arrows). Data represent three independent experiments with at least 50 cells in each group in each experiment. (C, D) PINK1 accumulated in Wt-or P525L-FUS expressing cells. (E) Immunostaining images of cells co-transfected with different plasmid combinations (Parkin-Flag together with NLS-GFP, or with Wt- or P525L-mutant FUS tagged with GFP). Cells were stained with anti-TOM20 and anti-Flag antibodies 20h after transfection. (F, G) The level of Parkin protein was determined by Western blotting analyses of cell lysates prepared from inducible HEK293 cells expressing either Wt- or P525L-mutant FUS upon induction using tetracycline (0.5 μg/mL; 20 h). Data represent independent experiments. Scale bars: 20 μm. Data were analyzed using one-way ANOVA (*: P < 0.05; **: P < 0.01; ***: P < 0.001).
Immunofluorescent staining experiments revealed that the Parkin protein signals were also increased in cells expressing the Wt- or P525L-mutant FUS as compared with the cells expressing the NLS-GFP control or neighboring non-transfected cells (Fig. 2E). To confirm this observation, we prepared inducible stable HEK293 cells that expressed myc-tagged Wt- or P525L-mutant FUS, or the vector control upon induction using tetracycline (Tet). The expression of FUS was induced by tetracycline (0.5 μg/ml, 20 h), and cells were lysed for Western blotting analyses. The endogenous Parkin was detected by a specific monoclonal antibody against Parkin (40). In addition to the full-length Parkin band (marked by the asterisk in Fig. 2F), a higher molecular weight band was detected (Fig. 2F, marked by the arrow). This Parkin species was significantly increased following induction of Wt- or P525L-mutant FUS expression (Fig. 2G). This band was previously found predominantly in a detergent-insoluble fraction, and regarded as a ubiquitinated species (38,41–43). Further analyses of these cells following biochemical fractionation to separate the Triton X-soluble versus Triton X-insoluble/SDS-soluble fractionations demonstrate that expression of Wt- or P525L-mutant FUS led to an increase in the level of the detergent Triton X-insoluble/SDS-soluble species of the Parkin protein (compare lanes 5–6 with lane 3 in the Parkin panels of Supplementary Material, Fig. S3), whereas in the control cells, most Parkin protein was detected in the Triton-X soluble fraction (lane 1 in Supplementary Material, Fig. S3).
A common sign of mitochondrial damage is the change in mitochondrial membrane potential, leading to mitochondrial depolarization. An early response to mitochondrial depolarization is the accumulation of PINK1 on the mitochondrial surface (24,39). PINK1 stabilization and Parkin relocalization to mitochondria are believed to initiate mitophagy (32–34). We examined the subcellular distribution of PINK1 in response to increased expression of FUS. Upon expression of either Wt- or P525L-mutant FUS, PINK1 accumulated in a punctate pattern to partially colocalize with mitochondria (marked by the white arrows), in contrast to the diffuse pattern of PINK1 in the control cells (Fig. 2A). In cells expressing either Wt- or P525L-mutant FUS, the percentage of cells with punctate PINK1 distribution pattern was 51 or 63%, respectively, as compared with 25% in the control cells (Fig. 2B). We then asked if FUS expression affected Parkin, which is recruited to damaged mitochondrial outer membrane in a PINK1-dependent manner (32–36). A low level of Parkin immunostaining signals distributed diffusely was detected in the cytoplasm in the control cells. The Parkin staining signals were significantly increased, appearing as multiple foci when either Wt- or P525L-mutant FUS was expressed (Fig. 2E). Such Parkin foci did not seem to colocalize with the mitochondrial marker, TOMM20. Together, these results indicate that expression of the Wt- or P525L-mutant FUS induces accumulation of PINK1 and Parkin proteins.
FUS promotes ubiquitination of Miro1 protein
Miro1 is a mitochondrial Rho GTPase and a downstream target of the PINK1/Parkin activation. Phosphorylation and/or ubiquitination of Miro1 protein have been associated with its degradation (44,45). We tested if FUS expression affected Miro1 protein level. Expression of either Wt- or P525L-mutant FUS did not seem to affect the total level of Miro1 (Fig. 3A, B). To test if ubiquitinylation of Miro1 was altered, we co-transfected HEK293 cells with HA-tagged ubiquitin and myc-tagged Miro1 together with either the GFP vector control, or Wt- or P525L-mutant FUS tagged with GFP. Following immunoprecipitation using the myc antibody to pull down Miro1, ubiquitinated Miro1 (Ub-Miro1) was detected in Western blotting using the anti-HA antibody. The level of Ub-Miro1 was increased in cells expressing either Wt- or P525L-mutant FUS, although the steady-state protein level of neither transfected nor the endogenous Miro1 protein was affected by FUS expression (Fig. 3A–C). We also examined the ubiquitination of Miro2. In contrast to Miro1, ubiquitination of Miro2 was not changed when Wt- or P525L-mutant FUS was expressed (Supplementary Material, Fig. S4). This is consistent with the previous observation that the level of ubiquitinated Miro2 protein did not change upon mitochondrial depolarization (44).
Figure 3.

Expression of Wt- or P525L-mutant FUS enhances MIRO1 ubiquitination. (A, B) Endogenous MIRO1 protein level is not altered in Wt- or P525L-mutant FUS expressing cells. Cells were co-transfected with different plasmids (NLS-GFP control or Wt- or P525L-mutant FUS tagged with GFP). Protein levels were determined using Western blotting with corresponding antibodies. No significant changes were detected in the MIRO1 protein level among cells expressing either GFP control or Wt-FUS or P525L-mutant FUS. Data represent three independent experiments and are analyzed using one-way ANOVA. (C) Expression of Wt-FUS or P525L-mutant FUS promotes MIRO1 ubiquitination. Cells were co-transfected with different combinations of plasmids as shown above the gel: HA-ubiquitin (HA-Ub) together with myc-MIRO1 and GFP, or Wt- or P525L-mutant FUS. Following immunoprecipitation using anti-myc, the ubiquitinyted MIRO1 was detected by HA antibody. Poly-ubiquitinated MIRO1 was increased in cells expressing Wt- or P525L-mutant FUS as compared with the control cells. Data represent three independent experiments.
Expression of Wt- or ALS mutant-FUS disrupts axonal transport of mitochondria
The Miro/Milton/KHC motor protein complex is required for axonal transport of mitochondria (46,47). Although the total protein level of Miro1 did not seem to be reduced by FUS overexpression, Miro1 ubiquitination may lead to removal of Miro1 from the mitochondrial outer membrane, and subsequently detachment of the motors from mitochondria (44,45). We examined whether mitochondrial trafficking in motor neurons (MNs) was impaired in our transgenic flies expressing FUS. Time-lapse confocal microscopy was carried out to monitor mitochondria in the MN axons of the ventral nerve cords (VNC) of 3rd instar larvae expressing either Wt- or P525L-mutant FUS with the mitochondria visualized by mito-GFP fluorescence. Individual mitochondria were tracked over time, and their duty cycles were quantified (Fig. 4A). Moving mitochondria were grouped into net anterograde-moving (AM) or net retrograde-moving (RM), with the duty cycles and velocity of moving mitochondria measured. The percentages of time spent by mitochondria in anterograde-running (AR), retrograde-running (RR) and stop (ST) were also calculated. Expression of either Wt- or P525L-mutant FUS in MNs led to disrupted duty cycles of mitochondrial movement in both directions. AM-mitochondria in FUS expressing MNs showed a moderate reduction in the time spent in the anterograde movement (Fig. 4B). RM-mitochondria in FUS expressing MNs exhibited shortened motile phase and prolonged stationary phase, with mutant-FUS larvae exhibiting significantly more severe disruption in mitochondrial transport (Fig. 4C). The percentage of time spent on pausing was 16.7 ± 8.8% in the control mitochondria, but increased to 22.2 ± 11.6% and 26.2 ± 12.5%, respectively, in the mitochondria of the Wt- or P525L-mutant FUS expressing MNs. We then measured the velocity of moving mitochondria in both anterograde and retrograde directions. Retrograde moving mitochondria were15% or 23% slower in Wt- or P525L-mutant FUS expressing MNs than those in the control group, whereas the average velocity of anterograde movement remained unchanged (Fig. 4D). We next took a closer look at the kinetic properties of mitochondrial transport, specifically individual run or pause events. Expression of the P525L-mutant FUS significantly reduced the duration of mitochondrial anterograde runs without affecting the duration of pauses (Fig. 4E). Expression of either Wt- or P525L-mutant FUS led to shorter runs in RM-mitochondria, again with mitochondria in the P525L-mutant expressing neurons showing more severe disruption in the retrograde running, leading to a 35% increase in the average duration of pauses (Fig. 4F). Expression of P525L-mutant FUS resulted in more frequent pause and reversal events in the retrograde direction, whereas expression of Wt-FUS did not affect frequency of pauses or reversals (Fig. 4G and H). Thus, expression of Wt- or P525L-mutant FUS reduced the motility and processivity of mitochondrial transport by increasing the frequency and duration of interruptions and decreasing the proportion of motile phases, with P525L-mutant FUS exhibiting more severe effects on mitochondria transport. Although the defects were observed in both directions, the retrograde mitochondrial transport was preferentially affected by the expression of either Wt- or P525L-mutant FUS. These data suggest that mitochondrial damage and possibly defects in mitochondrial repair may contribute to FUS-induced neurotoxicity.
Figure 4.
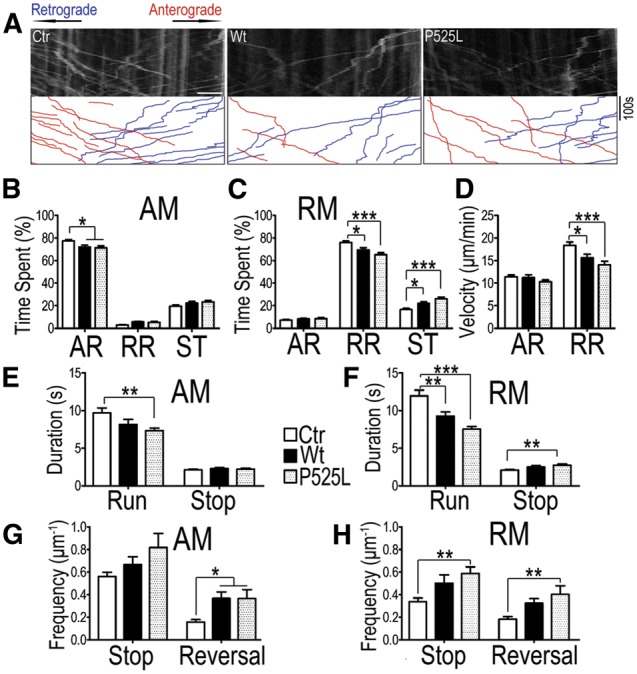
Axonal mitochondrial transport defects in MNs of FUS-transgenic flies as detected by the mitochondrial imaging in MN axons in the ventral nerve cord (VNC). (A) Representative kymographs of axonal mitochondria labeled by mito-GFP in MNs of control (Ctr) or transgenic flies expressing either Wt- or P525L-mutant FUS. A 40-μm axonal region was photo-bleached before image acquisition and video recorded for 6 min with VNC placed on the left of the acquired images. The anterograde (plus end-directed) and retrograde (minus end-directed) trafficking of mitochondria are marked in red and blue respectively. Scale bar: 5 μm. (B–H) Quantification of mitochondrial transport in MN axons of flies expressing Wt- or P525L-mutant FUS as compared with the control flies. Images were processed and quantified using ImageJ. (B, C) show duty cycles of the mitochondria that exhibit net anterograde movement (AM) and retrograde movement (RM). Duty cycles of individual mitochondria are divided into anterograde run (AR), retrograde run (RR), and stop (ST). A significant decrease in the time spent on moving (AR in B and RR in C, respectively) together with a significant increase in time spent pausing is detected in flies expressing either Wt- or P525L-mutant FUS as compared with the control flies. (D) The velocity of moving mitochondria in the anterograde and retrograde runs (AR and RR respectively). Flies expressing either Wt- or P525L-mutant FUS showed significantly reduced velocity of RR mitochondria, as compared with the control flies. (E, F) Duration of individual mitochondrial running or pausing events in different fly groups. Duration of mitochondrial anterograde movement (AM) in flies expressing the P525L-mutant FUS is reduced (E); whereas the duration of retrograde movement (RM) was decreased in flies expressing either Wt- or P525L-mutant FUS as compared with the control group. (G, H). The frequency of reversal of mitochondrial movement in AM or RM. The reversal of AM mitochondria was increased in flies expressing either Wt- or P525L-mutant FUS; whereas the P525L-mutant FUS group showed a significant increase in the frequencies of both stop and reversal of mitochondrial RM. At least 120 mitochondria from > 10 fly larvae were analyzed for each group. Data were analyzed using one-way ANOVA (*, P < 0.05; ***, P < 0.001). Fly genotypes, Ctr: D42-Gal4/UAS-mitoGFP/UAS-RFP; Wt: D42-Gal4/UAS-mitoGFP/UAS-Wt-hFUS-RFP; P525L: D42-Gal4/UAS-mitoGFP/UAS-P525L-hFUS-RFP.
We also examined mitochondrial axonal trafficking in primary cultures of rat cortical neurons electroporated with GFP, Wt- or P525L-mutant FUS, together with mitoRed. Increased stationary phase was observed in the RM-mitochondria in rat cortical axons expressing Wt-FUS; and the velocity of RM-mitochondria decreased in neurons expressing either Wt- or P525L-mutant FUS (Supplementary Material, Fig. S5A–D). Consistent with the observation in fly MN axons, the retrograde mitochondrial transport was more severely affected by FUS expression, demonstrating conserved features of FUS-induced mitochondrial axonal transport defects in mammalian neurons. Remarkably, the total level of the endogenous Pink1 protein (the full-length and cleaved form as marked by arrowhead and arrow, respectively; Supplementary Material, Fig. S5E) was increased in cortical neurons transfected with the P525L-mutant FUS and to a lesser extent in the Wt- FUS group (Supplementary Material, Fig. S5F). The level of the full-length PINK1 was significantly increased following expression of either Wt- or P525L-mutant FUS in these neurons (Supplementary Material, Fig. S5G). We also attempted to examine changes in Parkin protein levels in these neurons. However, the high basal level of Parkin together with relatively low transfection efficiency in these neurons made it difficult to detect significant increases in Parkin protein levels.
Down-regulation of PINK1 and Parkin expression restores locomotive function and eclosion ability of FUS-transgenic flies
Most of the FUS mutations have been identified in ALS patients. Our published work shows that expression of Wt- or P525L-mutant FUS in fly motor neurons results in locomotive defects, as well as abnormal morphology at neuromuscular junctions in 3rd instar larvae (6). Denervation and impaired neurotransmission may be caused by deficient energy supply as a result of disrupted mitochondrial transport observed in flies expressing either the Wt- or P525L-mutant FUS protein. We then asked whether PINK1 and Parkin also modulate FUS-induced motor neuron degeneration. Knockdown of PINK1 and Parkin in fly motor neurons led to improved larval locomotive function in larvae expressing the P525L-mutant FUS together with RNAi against Pink1 or Parkin, without altering the protein level of mutant FUS (Fig. 5A–C). Interestingly, down-regulation of Pink1 in motor neurons suppressed the pupal eclosion failure caused by expression of the P525L-mutant FUS (Fig. 5D). In contrast to 0% eclosion rate in the larvae expressing the P525L-mutant FUS protein, 89% of the pupae co-expressing P525L-mutant FUS and RNAi against Pink1 exhibited complete or incomplete eclosion. Together, these data supports that the PINK1/Parkin pathway contributes to FUS-induced neurodegeneration and functional impairment.
Figure 5.

Knockdown of Pink1 or Parkin in fly MNs ameliorates functional defects in larval movement and eclosion induced by FUS expression. (A, B) The impaired locomotion of larvae expressing P525L-mutant FUS was partially restored by knockdown of Pink1 or Parkin in MNs (A), although down-regulation of Pink1 or Parkin did not affect locomotive function in the control flies (B). At least 30 larvae from each group were analyzed, and data were analyzed using one-way ANOVA (*: P < 0.05; ***: P < 0.001). (C) The FUS protein level was not affected by knockdown of Pink1 or Parkin, as determined by Western blotting. (D) Knockdown of Pink1 largely restored the eclosion ability of pupae expressing the P525L-mutant FUS. At least 250 pupae were examined for each group.
Discussion
Our data presented in this study demonstrate that excessive PINK1 or Parkin may play pathogenic roles in FUS proteinopathy, and that their down-regulation could ameliorate FUS-induced neurodegeneration phenotypes. Loss-of-function mutations in PINK1 and PARK2 genes are associated with autosomal recessive Parkinson’s disease (PD) (48). Previous studies report that both PINK1 and Parkin proteins play anti-apoptotic roles (30,35–38). Loss-of-function mutations in Drosophila Pink1 or Parkin lead to locomotion defects and male sterility as a result of muscle cell death and defective spermatid individualization (49,50). Mitochondrial dysfunction is an early manifestation in these PD models and patients. PINK1 deficiency leads to reduced complex I activity (51); and some PD patients exhibit Complex I specific impairment, suggesting the role of mitochondrial dysfunction in the pathogenesis of PINK1-related PD (52). In mammalian cells, PARK2 mRNA increases in response to mitochondrial stress, and the level of Parkin protein is crucial for viability under stress conditions (41). Moreover, overexpression of Parkin suppresses cell loss in a model of TDP-43 proteinopathy (53).
Our data show that expression of Wt- or P525L-mutant FUS increases the levels of PINK1 and Parkin proteins and that FUS-induced neurotoxicity can be suppressed by reducing the level of PINK1 or Parkin in vivo. Knockdown of both PINK1 and Parkin seems to more efficiently rescue the retinal degeneration phenotype compared to knockdown of either alone, suggesting that PINK1 and Parkin may act synergistically in this setting. In contrast to the neuroprotective activities of PINK1 and Parkin demonstrated in previous studies (30,37), our results suggest that increased PINK1 or Parkin expression may contribute to neurotoxicity and that maintaining appropriate levels of PINK1 and Parkin may be essential for neuronal health.
The mitochondria and ER appeared damaged in the spinal cord MNs of a juvenile ALS patient carrying the P525L-mutation in the FUS gene (54). The expression of FUS mutants R521G and R521H in cultured mouse DRG neurons led to mitochondrial shortening (55). Our recent work has demonstrated that mitochondrial damage is a prominent feature in FTLD-FUS patients (20). Expression of either Wt- or ALS-mutant FUS in cultured cells and transgenic animals results in mitochondrial damage (55), supporting the notion that mitochondrial impairment may be a common feature shared by Amyotrophic lateral sclerosis with FUS-positive pathology and FTLD-FUS. FUS overexpression leads to an increase in the levels of PINK1 and Parkin, suggesting activation of this pathway as a consequence of mitochondrial damage. It should be noted that the increase in the Parkin level is accompanied by a loss of protein solubility and redistribution into inclusion-like structures reminiscent of the aggregation and inclusion formation of Parkin induced by PD-related mutations or cellular stresses (56,57). It has been shown that the level of Parkin is regulated by FUS, whose knockdown reduced the level of Park2 mRNA in the mouse brain (58). The elevation in the Parkin protein level induced by FUS expression may result from inefficient degradation of insoluble forms or other mechanisms to be discovered. The abnormal accumulation of Parkin in addition to its functional activation by PINK1 may play a role in FUS-induced neurodegeneration.
The fission and fusion processes are essential for maintaining mitochondrial quality (59–61). PINK1/Parkin pathway promotes mitochondrial fission, and their overexpression results in retinal degeneration (62,63). It is possible that activated PINK1/Parkin pathway exacerbates the mitochondrial fragmentation induced by Wt- or P525L-mutant FUS (20). Therefore, reducing PINK1 and Parkin expression in this case may play a protective role in neurons in transgenic FUS flies.
It has been documented previously that mitochondria were removed by autophagy when A53T mutant α-synuclein was expressed in cultured neurons and that the autophagic removal was not restricted to depolarized or damaged mitochondria. Mitochondrial removal and neuronal death could be blocked by shRNA against Parkin or Beclin1 (24,64). One possibility raised by our observations is that excessive mitophagy might play a role in the pathogenesis of FUS proteinopathy. However, in flies expressing Wt- or P525L-mutant FUS, knockdown of Beclin1 or other autophagy-related genes did not show obvious protective effect against retinal degeneration (Supplementary Material, Fig. S6).
Another possibility is that increased Pink1 and Parkin levels in response to mitochondrial damage affect mitochondrial trafficking. Miro1 has been identified as an essential component for axonal mitochondrial transport. Miro1 mutations results in defective mitochondrial transport in both directions (46,47). We have demonstrated that expression of Wt- or P525L-mutant FUS induces mitochondrial damage, including mitochondrial membrane potential loss (20). Our data presented here indicate that expression of Wt- or P525L-mutant FUS leads to changes in PINK1/Parkin proteins and results in defects in mitochondrial axonal transport.
Together, these observations led us to propose a working model for FUS-induced mitochondrial transport defects. Increased expression of Wt-FUS or expression of ALS-mutant FUS leads to elevated FUS mitochondrial localization that results in mitochondrial damage and dysfunction, such as reduced mitochondrial membrane potential (20). This may lead to increased levels of PINK1 and Parkin, and subsequently increased Miro1 ubiquitination (possibly by Parkin). It is conceivable that Miro1 ubiquitination makes it less active in mitochondrial transport machinery, possibly leading to dissociation of Miro1 from moving mitochondria and thereby reducing mitochondrial transport. Consistent with this model, our data show that elevated Miro1 ubiquitination is accompanied by accumulation of PINK1 and Parkin proteins. It suggests that insufficient Miro1 function may cause defective mitochondrial transport. Because FUS expression did not seem to affect the overall level of the Miro1 protein, it remains to be tested whether a reduction in the functionally active Miro1 protein directly causes mitochondrial transport defects. On the other hand, it has been reported that the level of Miro1/2 protein remarkably decreased in cell lines deficient in PINK1; and this reduction is independent of mitochondrial depolarization (45). If this is the case in transgenic FUS flies, it is possible that Miro1-independent mechanism(s) may operate to mediate FUS induced defects in mitochondrial axonal transport. Clearly, further studies are necessary to elucidate the mechanisms by which down-regulating Pink1 or Parkin protects against FUS-induced neurodegeneration.
Axonal transport of organelles, vesicles, RNA-protein particles and translation machineries is essential for the function and survival of neurons (64). Dysregulation of this process or defects in the transport machinery have been associated with neurodegenerative diseases, including Alzheimer disease (AD), Parkinson disease (PD), Huntington disease (HD) and amyotrophic lateral sclerosis (ALS) (65–70). Specifically, mutations in genes involved in axonal transport have been identified in these diseases, such as p150Glued, Rab7 and KIF1B (69,71).
In particular, anterograde transport of mitochondria from the soma to distal neurites meets the local energy requirement. Denervation and impaired neurotransmission observed in flies expressing either the Wt- or P525L-mutant FUS protein (6) may be caused by deficient energy supply as a result of disrupted mitochondrial transport. On the other hand, retrograde transport is important for mitochondrial quality control. Damaged mitochondria are transported back to the soma for repair or clearance, probably depending on the severity of mitochondrial damage (72). A variety of genetic mutations affect mitochondrial retrograde transport (69). As is shown in a Drosophila model of Friedreich ataxia, abnormal retrograde transport of mitochondria is correlated to accumulation of depolarized mitochondria in the distal segmental nerves and NMJs (73). Supporting this notion, mitochondrial membrane potential is reduced in cells expressing Wt- or P525L-mutant FUS (20), and the retrograde transport is preferentially disrupted in Wt- or P525L-mutant FUS expressing flies. This disruption of the retrograde mitochondrial transport may further increase the stress load in affected neurons. Mitochondrial transport is crucial for maintaining neuronal function and survival. Alterations or defects in axonal transport of mitochondria have been associated with a range of neurodegenerative diseases, including AD (66), PD (70) and ALS (74). In these models, the mutated genes encode either a component of axonal transport machinery, a cytoplasmic protein or a mitochondrially localized protein. For example, TFAM is a mitochondrial resident protein whose depletion leads to mitochondrial transport defects (70). Our data indicate that mutations in or dysregulation of a nuclear RNA binding protein, FUS, lead to defects in axonal transport of mitochondria.
In this study, we have demonstrated that locomotive impairment in the fly model for FUS proteinopathy is accompanied by disruption of axonal mitochondrial transport in motor neurons. PINK1 and Parkin have been identified as key players that promote FUS-induced neurodegeneration. Although the accumulation of PINK1 and Parkin proteins is likely a consequence of mitochondrial damage, it remains to be investigated whether PINK1 and Parkin exert pro-cell death effect by activating mitophagy. Nonetheless, the fact that down-regulating Pink1 and Parkin expression protects against FUS-induced neurodegeneration in vivo highlights the requirement of appropriate levels and activity of PINK1/Parkin pathway in maintaining neuronal viability and function. Thus, the possible shared molecular machinery involved in the pathogenesis of Parkinson’s disease and of ALS or FTLD warrants further investigation. Protective effects of suppressing PINK1/Parkin is also observed in flies expressing Wt-FUS, which may help to explain the pathogenesis in FUS-proteinopathy patients with elevated FUS levels rather than with missense mutations (19). Our study suggests mitochondrial damage and transport defects as potential targets in future development of treatment for FUS proteinopathy.
Materials and Methods
Drosophila strains and antibodies
FUS expressing transgenic flies were described previously (6). The RNAi fly lines were obtained from Vienna Drosophila Resource Center (VDRC). The Gal4 lines were obtained from Bloomington Stock Center. Fly lines overexpressing Miro1 and Miro1 RNAi lines were previously reported (45).
The following antibodies were used in this study: mouse anti-Miro1 (Sigma), rabbit anti-FUS (ProteinTech Group, PTG), mouse anti-FUS (PTG), rabbit anti-TOMM20 (PTG), mouse anti-actin (PTG), mouse anti-HA (CWBIO), mouse anti-Flag (CWBIO), mouse anti-myc (CWBIO) and mouse anti-GFP (PTG), and secondary antibodies were: Alexa Flour 488-conjugated goat anti-mouse IgG (Molecular Probes), Alexa Flour594-conjugated goat anti-rabbit IgG (Molecular Probes), Alexa Flour 647-conjugated donkey anti-mouse IgG (Molecular Probes), HRP-conjugated anti-mouse IgG (GE) and HRP-conjugated anti-rabbit IgG (GE).
Live imaging of mitochondrial transport in fly motor neurons
Image acquisition and analyses of mitochondrial transport in fly motor neurons were performed according to published protocols with slight modifications (75). Briefly, D42-Gal4 was used to drive the expression of FUS and mitochondrial localized GFP (mitoGFP) in a subset of motor neurons (76,77). Wandering stage larva expressing control vector, FUS (wild type or ALS-mutant) together with mitoGFP were used for live imaging under a 60X oil lens. A 40-μm region in the third abdominal segment (A3) was photo-bleached before imaging acquisition. Time-lapse images were acquired using Olympus FV1000 confocal, at max frame rate for 6 min. Individual moving mitochondria were tracked in ImageJ.
Immunofluorescent staining and immunoprecipitation assays
Cells were fixed with 3.7% PFA containing 5% sucrose and 0.1% TritonX-100 in 1XPBS. Cells were washed using 1XPBS three times and then blocked with blocking buffer (5% normal goat serum, 0.05% Tween20 in 1XPBS) for 1 h at RT. Antibodies were diluted with blocking buffer. After staining with antibodies, cells were counterstained with Hoechst 33342 before mounting. Images were acquired using Olympus FV1000 system.
HEK293 cells were used for transfection and analysis of Miro1/2 ubiquitination. NLS-GFP, Wt or P525L FUS-GFP with HA-Ub and myc-hMiro1/2 were co-transfected into HEK293T cells. Cells were treated with 10 μM MG132 at 23 h post-transfection. After MG132 treatment for 3 h, cells were washed with PBS and lysed for 30 min on ice in the lysis buffer. Cell lysates were spun down at 13 000 rpm at 4°C for 15 min and supernatant was immunoprecipitated 4 h with anti-myc and protein A agarose (Roche) at 4°C. The immunoprecipitates were washed three times with lysis buffer. The beads were then directly boiled in 1XSDS loading buffer.
For western blotting, cells were lysed in SDS-PAGE buffer and heated to 99 °C for 10 min, and samples were then loaded onto SDS-PAGE gel.
Scanning electron microscopy (SEM)
SEM was performed as described previously (6). Briefly, intact flies were progressively dehydrated in ethanol (70%, 85%, 95%, 100%; 60 min each step) and then in 100% ethanol overnight. Dehydrated flies were applied to a CPD 030 critical point dryer, with CO2 as the purge agent. SEM images were acquired under the FEI Quanta 200 FEG electron microscope in the low vacuum mode.
Larval movement assay
The larval movement assay was carried out as previously described (20). Briefly, the larval mobility index was measured as the number of peristaltic waves during a period of 2 min in the late third instar larvae. The fly larvae were expressing various genes and/or RNAi under the OK371-Gal4 driver in a controlled environment (25 °C, humidity 50% ± 5%, illumination 2800 ±100 lux).
Statistical analysis
Statistical analyses were performed using one-way ANOVA, followed by Bonferroni’s correction for post-hoc analysis when comparing individual groups. The bar graphs with error bars represent mean ± standard error of the mean (SEM). Significance is indicated by asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We thank Dr Xinnan Wang (Stanford University), Dr Ming Guo (University of California, Los Angeles), Drs Yongqing Zhang, Aiyu Yao and Li Liu (CAS) for generously providing reagents and valuable suggestions in live imaging studies. We thank members of the Wu lab for stimulating discussions and helpful suggestions. We thank Dr Eileen Bigio, Warren McGee and other members of the Wu group for critical reading of the manuscript. We are grateful for generous help from Drs C Wang, D Han and J Xie at National Center for Nanoscience and Technology in our SEM work.
Conflict of Interest Statement. None declared.
Funding
National Major Basic Research Program of China (2013CB917803 to M.C., L.Z. and J.L.; 2013CB531301 to Q.X.); the National Natural Science Foundation of China (91132710 to M.C., L.Z. and J.L.; 31430048 to Q.X.); CAMS-I2M (2006I2M1004 to Q.X.); NIH (R01NS084412 and R01MH080378 to B.L.; R56NS074763 and RO1AG033004 to J.Y.W.); ALS Therapy Alliance (R01AG054008 to J.Y.W.).
References
- 1. Vance C., Rogelj B., Hortobagyi T., De Vos K.J., Nishimura A.L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P. et al. (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science, 323, 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kwiatkowski T.J., Jr., Bosco D.A., Leclerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T. et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science, 323, 1205–1208. [DOI] [PubMed] [Google Scholar]
- 3. Lattante S., Rouleau G.A., Kabashi E. (2013) TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum. Mutat., 34, 812–826. [DOI] [PubMed] [Google Scholar]
- 4. Ling S.C., Polymenidou M., Cleveland D.W. (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron, 79, 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Da Cruz S., Cleveland D.W. (2011) Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol., 21, 904–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen Y., Yang M., Deng J., Chen X., Ye Y., Zhu L., Liu J., Ye H., Shen Y., Li Y. et al. (2011) Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein Cell, 2, 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fushimi K., Long C., Jayaram N., Chen X., Li L., Wu J.Y. (2011) Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy. Protein Cell, 2, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lanson N.A., Jr., Maltare A., King H., Smith R., Kim J.H., Taylor J.P., Lloyd T.E., Pandey U.B. (2011) A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum. Mol. Genet., 20, 2510–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miguel L., Avequin T., Delarue M., Feuillette S., Frebourg T., Campion D., Lecourtois M. (2012) Accumulation of insoluble forms of FUS protein correlates with toxicity in Drosophila. Neurobiol. Aging, 33, 1008.e1001–1008.e 1015. [DOI] [PubMed] [Google Scholar]
- 10. Wang J.W., Brent J.R., Tomlinson A., Shneider N.A., McCabe B.D. (2011) The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J. Clin. Invest., 121, 4118–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murakami T., Yang S.P., Xie L., Kawano T., Fu D., Mukai A., Bohm C., Chen F., Robertson J., Suzuki H. et al. (2012) ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum. Mol. Genet., 21, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kryndushkin D., Wickner R.B., Shewmaker F. (2011) FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast model of amyotrophic lateral sclerosis. Protein Cell, 2, 223–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ju S., Tardiff D.F., Han H., Divya K., Zhong Q., Maquat L.E., Bosco D.A., Hayward L.J., Brown R.H., Jr., Lindquist S. et al. (2011) A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol., 9, e1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang C., Zhou H., Tong J., Chen H., Liu Y.J., Wang D., Wei X., Xia X.G. (2011) FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet., 7, e1002011.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verbeeck C., Deng Q., Dejesus-Hernandez M., Taylor G., Ceballos-Diaz C., Kocerha J., Golde T., Das P., Rademakers R., Dickson D.W. et al. (2012) Expression of Fused in sarcoma mutations in mice recapitulates the neuropathology of FUS proteinopathy and provides insight into disease pathogenesis. Mol. Neurodegener., 7, 53.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun Z., Diaz Z., Fang X., Hart M.P., Chesi A., Shorter J., Gitler A.D. (2011) Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol., 9, e1000614.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mitchell J.C., McGoldrick P., Vance C., Hortobagyi T., Sreedharan J., Rogelj B., Tudor E.L., Smith B.N., Klasen C., Miller C.C. et al. (2013) Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol., 125, 273–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shelkovnikova T.A., Peters O.M., Deykin A.V., Connor-Robson N., Robinson H., Ustyugov A.A., Bachurin S.O., Ermolkevich T.G., Goldman I.L., Sadchikova E.R. et al. (2013) Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J. Biol. Chem., 288, 25266–25274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sabatelli M., Moncada A., Conte A., Lattante S., Marangi G., Luigetti M., Lucchini M., Mirabella M., Romano A., Del Grande A. et al. (2013) Mutations in the 3' untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet., 22, 4748–4755. [DOI] [PubMed] [Google Scholar]
- 20. Deng J., Yang M., Chen Y., Chen X., Liu J., Sun S., Cheng H., Li Y., Bigio E.H., Mesulam M. et al. (2015) FUS Interacts with HSP60 to promote mitochondrial damage. PLoS Genet., 11, e1005357.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu S., Peng G., Wang Y., Fang S., Karbowski M. (2011) The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell, 22, 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Youle R.J., Narendra D.P. (2011) Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol., 12, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koyano F., Okatsu K., Kosako H., Tamura Y., Go E., Kimura M., Kimura Y., Tsuchiya H., Yoshihara H., Hirokawa T. et al. (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature, 510, 162–166. [DOI] [PubMed] [Google Scholar]
- 24. Lazarou M., Narendra D.P., Jin S.M., Tekle E., Banerjee S., Youle R.J. (2013) PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J. Cell Biol., 200, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C.A., Sou Y.S., Saiki S., Kawajiri S., Sato F. et al. (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol., 189, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Narendra D.P., Jin S.M., Tanaka A., Suen D.F., Gautier C.A., Shen J., Cookson M.R., Youle R.J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol., 8, e1000298.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vives-Bauza C., Zhou C., Huang Y., Cui M., de Vries R.L., Kim J., May J., Tocilescu M.A., Liu W., Ko H.S. et al. (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA, 107, 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Narendra D., Tanaka A., Suen D.F., Youle R.J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol., 183, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen Y., Dorn G.W., 2nd (2013) PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science, 340, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pridgeon J.W., Olzmann J.A., Chin L.S., Li L. (2007) PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol., 5, e172.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kondapalli C., Kazlauskaite A., Zhang N., Woodroof H.I., Campbell D.G., Gourlay R., Burchell L., Walden H., Macartney T.J., Deak M. et al. (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol., 2, 120080.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sarraf S.A., Raman M., Guarani-Pereira V., Sowa M.E., Huttlin E.L., Gygi S.P., Harper J.W. (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature, 496, 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park J., Lee S.B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J.M. et al. (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature, 441, 1157–1161. [DOI] [PubMed] [Google Scholar]
- 34. Yang Y., Gehrke S., Imai Y., Huang Z., Ouyang Y., Wang J.W., Yang L., Beal M.F., Vogel H., Lu B. (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. USA, 103, 10793–10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. da Costa C.A., Sunyach C., Giaime E., West A., Corti O., Brice A., Safe S., Abou-Sleiman P.M., Wood N.W., Takahashi H. et al. (2009) Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson's disease. Nat. Cell Biol., 11, 1370–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee Y., Karuppagounder S.S., Shin J.H., Lee Y.I., Ko H.S., Swing D., Jiang H., Kang S.U., Lee B.D., Kang H.C. et al. (2013) Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci., 16, 1392–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shin J.H., Ko H.S., Kang H., Lee Y., Lee Y.I., Pletinkova O., Troconso J.C., Dawson V.L., Dawson T.M. (2011) PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell, 144, 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johnson B.N., Berger A.K., Cortese G.P., Lavoie M.J. (2012) The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proc. Natl. Acad. Sci. USA, 109, 6283–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jin S.M., Lazarou M., Wang C., Kane L.A., Narendra D.P., Youle R.J. (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol., 191, 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Burns M.P., Zhang L., Rebeck G.W., Querfurth H.W., Moussa C.E. (2009) Parkin promotes intracellular Abeta1-42 clearance. Hum. Mol. Genet., 18, 3206–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun X., Liu J., Crary J.F., Malagelada C., Sulzer D., Greene L.A., Levy O.A. (2013) ATF4 protects against neuronal death in cellular Parkinson's disease models by maintaining levels of parkin. J. Neurosci., 33, 2398–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Um J.W., Park H.J., Song J., Jeon I., Lee G., Lee P.H., Chung K.C. (2010) Formation of parkin aggregates and enhanced PINK1 accumulation during the pathogenesis of Parkinson's disease. Biochem. Biophys. Res. Commun., 393, 824–828. [DOI] [PubMed] [Google Scholar]
- 43. Sriram S.R., Li X., Ko H.S., Chung K.K., Wong E., Lim K.L., Dawson V.L., Dawson T.M. (2005) Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum. Mol. Genet., 14, 2571–2586. [DOI] [PubMed] [Google Scholar]
- 44. Wang X., Winter D., Ashrafi G., Schlehe J., Wong Y.L., Selkoe D., Rice S., Steen J., LaVoie M.J., Schwarz T.L. (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell, 147, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu S., Sawada T., Lee S., Yu W., Silverio G., Alapatt P., Millan I., Shen A., Saxton W., Kanao T. et al. (2012) Parkinson's disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet., 8, e1002537.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guo X., Macleod G.T., Wellington A., Hu F., Panchumarthi S., Schoenfield M., Marin L., Charlton M.P., Atwood H.L., Zinsmaier K.E. (2005) The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron, 47, 379–393. [DOI] [PubMed] [Google Scholar]
- 47. Russo G.J., Louie K., Wellington A., Macleod G.T., Hu F., Panchumarthi S., Zinsmaier K.E. (2009) Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J. Neurosci., 29, 5443–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martin I., Dawson V.L., Dawson T.M. (2011) Recent advances in the genetics of Parkinson's disease. Annu. Rev. Genom. Hum. Genet., 12, 301–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Clark I.E., Dodson M.W., Jiang C., Cao J.H., Huh J.R., Seol J.H., Yoo S.J., Hay B.A., Guo M. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature, 441, 1162–1166. [DOI] [PubMed] [Google Scholar]
- 50. Greene J.C., Whitworth A.J., Kuo I., Andrews L.A., Feany M.B., Pallanck L.J. (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA, 100, 4078–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Morais V.A., Verstreken P., Roethig A., Smet J., Snellinx A., Vanbrabant M., Haddad D., Frezza C., Mandemakers W., Vogt-Weisenhorn D. et al. (2009) Parkinson's disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med., 1, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gu M., Gash M.T., Cooper J.M., Wenning G.K., Daniel S.E., Quinn N.P., Marsden C.D., Schapira A.H. (1997) Mitochondrial respiratory chain function in multiple system atrophy. Mov. Disord., 12, 418–422. [DOI] [PubMed] [Google Scholar]
- 53. Hebron M., Chen W., Miessau M.J., Lonskaya I., Moussa C.E. (2014) Parkin reverses TDP-43-induced cell death and failure of amino acid homeostasis. J. Neurochem., 129, 350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huang E.J., Zhang J., Geser F., Trojanowski J.Q., Strober J.B., Dickson D.W., Brown R.H., Jr., Shapiro B.E., Lomen-Hoerth C. (2010) Extensive FUS-immunoreactive pathology in juvenile amyotrophic lateral sclerosis with basophilic inclusions. Brain Pathol., 20, 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tradewell M.L., Yu Z., Tibshirani M., Boulanger M.C., Durham H.D., Richard S. (2012) Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum. Mol. Genet., 21, 136–149. [DOI] [PubMed] [Google Scholar]
- 56. Wang C., Ko H.S., Thomas B., Tsang F., Chew K.C., Tay S.P., Ho M.W., Lim T.M., Soong T.W., Pletnikova O. et al. (2005) Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin's protective function. Hum. Mol. Genet., 14, 3885–3897. [DOI] [PubMed] [Google Scholar]
- 57. Wang C., Tan J.M., Ho M.W., Zaiden N., Wong S.H., Chew C.L., Eng P.W., Lim T.M., Dawson T.M., Lim K.L. (2005) Alterations in the solubility and intracellular localization of parkin by several familial Parkinson's disease-linked point mutations. J. Neurochem., 93, 422–431. [DOI] [PubMed] [Google Scholar]
- 58. Lagier-Tourenne C., Polymenidou M., Hutt K.R., Vu A.Q., Baughn M., Huelga S.C., Clutario K.M., Ling S.C., Liang T.Y., Mazur C. et al. (2012) Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci., 15, 1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen H., Chomyn A., Chan D.C. (2005) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem., 280, 26185–26192. [DOI] [PubMed] [Google Scholar]
- 60. Chen H., Detmer S.A., Ewald A.J., Griffin E.E., Fraser S.E., Chan D.C. (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol., 160, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Twig G., Elorza A., Molina A.J., Mohamed H., Wikstrom J.D., Walzer G., Stiles L., Haigh S.E., Katz S., Las G. et al. (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J., 27, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Poole A.C., Thomas R.E., Andrews L.A., McBride H.M., Whitworth A.J., Pallanck L.J. (2008) The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA, 105, 1638–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yu W., Sun Y., Guo S., Lu B. (2011) The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum. Mol. Genet., 20, 3227–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Choubey V., Safiulina D., Vaarmann A., Cagalinec M., Wareski P., Kuum M., Zharkovsky A., Kaasik A. (2011) Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem., 286, 10814–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sheng Z.H., Cai Q. (2012) Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci., 13, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Calkins M.J., Reddy P.H. (2011) Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer's disease neurons. Biochim. Biophys. Acta, 1812, 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gunawardena S., Her L.S., Brusch R.G., Laymon R.A., Niesman I.R., Gordesky-Gold B., Sintasath L., Bonini N.M., Goldstein L.S. (2003) Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron, 40, 25–40. [DOI] [PubMed] [Google Scholar]
- 68. Magrane J., Hervias I., Henning M.S., Damiano M., Kawamata H., Manfredi G. (2009) Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum. Mol. Genet., 18, 4552–4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Perlson E., Maday S., Fu M.M., Moughamian A.J., Holzbaur E.L. (2010) Retrograde axonal transport: pathways to cell death? Trends Neurosci., 33, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sterky F.H., Lee S., Wibom R., Olson L., Larsson N.G. (2011) Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc. Natl. Acad. Sci. USA, 108, 12937–12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chevalier-Larsen E., Holzbaur E.L. (2006) Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta, 1762, 1094–1108. [DOI] [PubMed] [Google Scholar]
- 72. Miller K.E., Sheetz M.P. (2004) Axonal mitochondrial transport and potential are correlated. J. Cell Sci., 117, 2791–2804. [DOI] [PubMed] [Google Scholar]
- 73. Shidara Y., Hollenbeck P.J. (2010) Defects in mitochondrial axonal transport and membrane potential without increased reactive oxygen species production in a Drosophila model of Friedreich ataxia. J. Neurosci., 30, 11369–11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. De Vos K.J., Chapman A.L., Tennant M.E., Manser C., Tudor E.L., Lau K.F., Brownlees J., Ackerley S., Shaw P.J., McLoughlin D.M. et al. (2007) Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet., 16, 2720–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sanyal S. (2009) Genomic mapping and expression patterns of C380, OK6 and D42 enhancer trap lines in the larval nervous system of Drosophila. Gene Expr. Patterns, 9, 371–380. [DOI] [PubMed] [Google Scholar]
- 76. Yao A., Jin S., Li X., Liu Z., Ma X., Tang J., Zhang Y.Q. (2011) Drosophila FMRP regulates microtubule network formation and axonal transport of mitochondria. Hum. Mol. Genet., 20, 51–63. [DOI] [PubMed] [Google Scholar]
- 77. Li Y., Ray P., Rao E.J., Shi C., Guo W., Chen X., Woodruff E.A., 3rd, Fushimi K., Wu J.Y. (2010) A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA, 107, 3169–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.