

Abstract
Introduction:
Paraganglioma (PGL) is a rare neuroendocrine tumor. Currently, the malignancy is defined as the presence of metastatic spread at presentation or during follow-up. Several gene mutations are listed in the pathogenesis of PGL, among which succinate dehydrogenase (SDHX), particularly the SDHB isoform, is the main gene involved in malignancy. A 55-year-old male without evidence of catecholamine secretion had surgery for PGL of the urinary bladder. After 1 year, he showed a relapse of disease and demonstrated malignant PGL without evidence of catecholamine secretion with a germline heterozygous mutation of succinate dehydrogenase B (SDHB). After failure of a second surgery for relapse, he started medical treatment with sunitinib daily but discontinued due to serious side effects. Cyclophosphamide, vincristine, and dacarbazine (CVD) chemotherapeutic regimen stopped the disease progression for 7 months.
Conclusion:
Malignant PGL is a very rare tumor, and SDHB mutations must be always considered in molecular diagnosis because they represent a critical event in the progression of the oncological disease. Currently, there are few therapeutic protocols, and it is often difficult, as this case demonstrates, to decide on a treatment option according to a reasoned set of choices.
Keywords: cyclophosphamide-vincristine-dacarbazine, paraganglioma, SDHB, sunitinib
1. Introduction
Paraganglioma (PGL) or extra-adrenal pheochromocytoma is a rare neuroendocrine tumor arising from chromaffin cells in many different paraganglia in the body. The PGL affects approximately 2 to 5 patients per million per year and can occur between 30 and 50 years of age, with a similar gender affinity. PGLs are classified as sympathetic or parasympathetic, depending on their origin. Thoraco-abdomino-pelvic PGLs are of sympathetic origin and produce catecholamines, their metabolites and other peptides; in contrast, PGLs of the head and neck are of parasympathetic origin and do not usually produce catecholamines.[1] A proper histological recognition of the potential malignancy of these tumors is not yet available, although a scoring system based on microscopic morphology and clinical features has been proposed.[2] Malignancy is involved in approximately 10% of PGLs and is unfortunately established by metastatic spread. Currently, genetic studies attribute an etiopathogenic role to a mutation of succinate dehydrogenase complex subunits (SDHs) in an increasing number of PGLs. Familial PGL syndromes are autosomal dominant inherited diseases that are caused by mutations in SDHX genes and have been classified as PGL1, PGL2, PGL3, PGL4, and PGL5 based on mutations in SDHD, SDHAF2, SDHC, SDHB, and SDHA, respectively.[3] The SDHB mutation in PGL4 is associated with an increased risk of malignancy. The morbidity associated with these tumors is related mainly to their metastatic potential.[4] Here, we report a case of a male patient who was affected by sporadic nonsecreting malignant paraganglioma of the urinary bladder with a PGL4 syndrome carrier of a SDHB heterozygous germline mutation. The patient was treated with 2 therapeutic regimen options, which had different results. Preliminary evidence indicates that PGL carrying SDHB mutations have a potential role in the prediction of response to antiangiogenic therapy. In a retrospective study,[5] most of the patients who exhibited a positive response to sunitinib therapy were SDHB mutation carriers, and some of them exhibited a lasting response up to 2 years later. However, we obtained in this case, a significant result with the cyclophosphamide-vincristine-dacarbazine (CVD) chemotherapy scheme. It shows that genetics plays a key role in diagnosis and follow up program but cannot still drive a therapeutic choice.
2. Case report and methods
The patient's family history included tireopathy, dyslipidaemia, hypertension, and oncological disease, such as bladder cancer (father), ovarian cancer (mother), and gastric cancer (maternal grandfather). Physiological history was negative. The patient, 55-year-old male of Caucasian ethnicity coming from Central Italy, suffered from chronic obstructive bronchopneumopathy, reflux oesophagitis, and colon diverticulosis. Furthermore, many years ago, he had a left breast adenoma and colon poliposis removed endoscopically.
He went to an endocrinology outpatient clinic because a year previously, following a hematuria and abdominal pain, he had surgery for a PGL of the urinary bladder, 7 cm of maximum diameter, Ki67 8% to 10%, with positive immunohistochemical staining for chromogranin A, synaptophysin, neuron specific enolase and S100 protein.
The objective examination was negative, and he had a normal blood pressure and heart beat.
The assays of a urinary 24-hour sample of metanephrines and normetanephrines were negative (35.5 mcg/24 h and 8.1 mcg/24 h, respectively). Chromogranin A level was negative (32 mcg/L). Molecular analysis of the SDHB gene identified a germline heterozygous variant mutation in exon 7, c.689G > A p.Arg230His, which has been described as being associated with PGL4 syndrome and considered pathogenetic.[6] The first 18FDG PET/TC performed showed suspicious lymph nodes (SUV 8.13) in the right iliac region (Fig. 1), and surgical removal was positive for cromaffin tissue. Follow-up at 3 months with 18FDG PET/TC showed a new uptake in a large lymph node in the right iliac region (30 mm) and in the aorto-caval region (9 mm). MIBG scan was negative. The patient was successfully treated with a second surgical excision of the iliac and aorto-caval lymph nodes, resulting from metastasis. A 18FDG PET/TC after 4 months demonstrated a worsening of metastatic spread, with a positive uptake of lymph nodes in mediastinum, in the aorto-caval region, the right iliac and inguinal region and the 7/8 liver segment.
Figure 1.
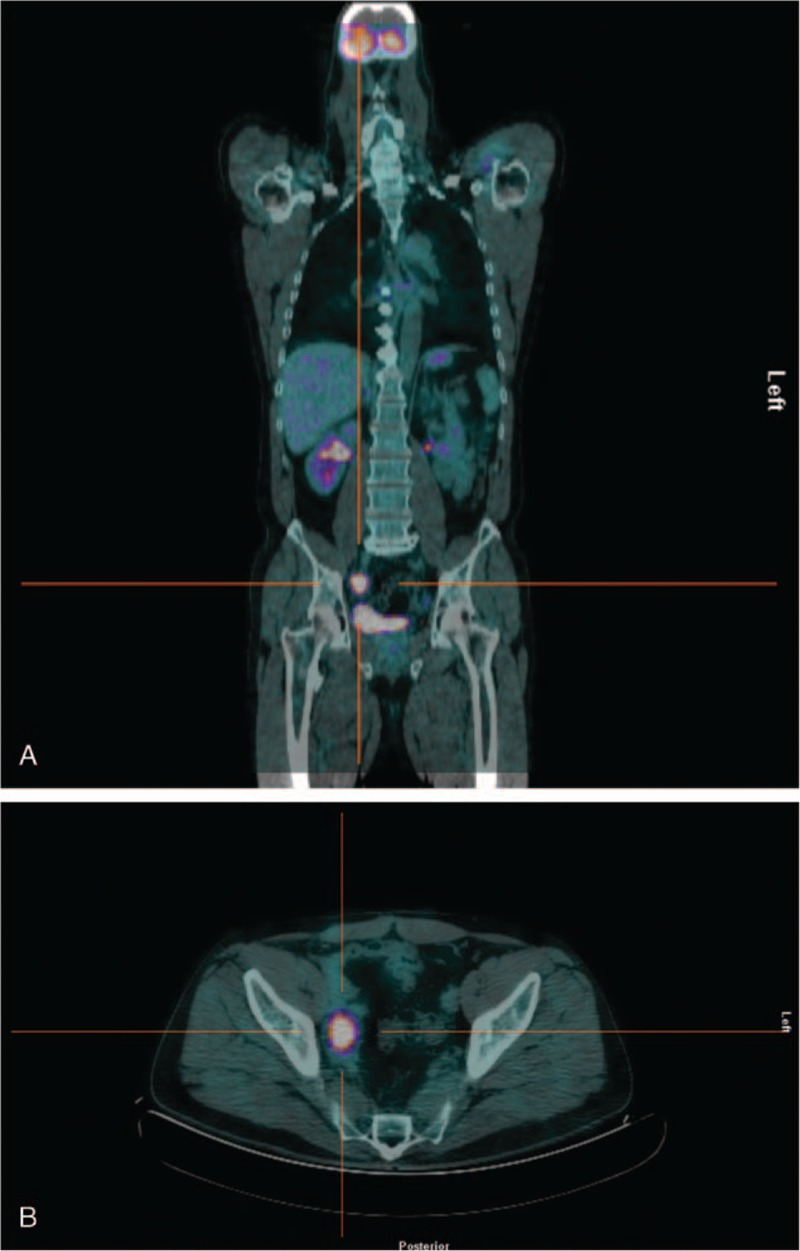
18FDG PET/CT whole body: pathological uptake in lymph nodes of right iliac region (A), particular of right iliac region (B).
The patient started antiangiogenic therapy with sunitinib orally, 50 mg flat per day. Treatment was associated with severe gastrointestinal side effects, which led to reducing the dose to 25 mg after 4 months of treatment (Table 1). TC whole body performed 6 months after starting of the sunitinib treatment documented disease progression, which consisted of a volumetric and numeric increase of over and under diaphragmatic lymph nodes (Fig. 2), with evidence of lung, liver and bone metastatic lesions, which were also documented with scintigraphy. Therefore, it was decided to start a chemotherapeutic treatment consisting of a combination of cyclophosphamide 750 mg/m2, vincristine 1.4 mg/m2, and dacarbazine 600 mg/m2 (CVD) for the first 4 months and reduced by 25% in the following 5 months for a total of 9 months (Table 1). This treatment schedule led to stabilizing the disease and subjective clinical benefit. Following a close follow-up, after 9 months of consecutive cycles of chemotherapy, the patient did not have disease progression for 7 months stopping treatment, when a TC whole body showed progression of the disease with the appearance of lumbar aortic lymphadenopathies. The clinical and radiological pictures indicated that the chemotherapy treatment should be resumed according to the previous scheme. At 3 months after the onset of treatment, TC reported a further progression of the disease due to para-aortic and right iliac lymph nodes. Currently, the patient is following the previous CVD schedule treatment with a relative benefit in terms of disease progression. Written consent for publishing this case report was obtained from the patient.
Table 1.
First treatment: sunitinib 50 mg administered orally for 4 months, then reduced to 25 mg for other 2 months.

Figure 2.
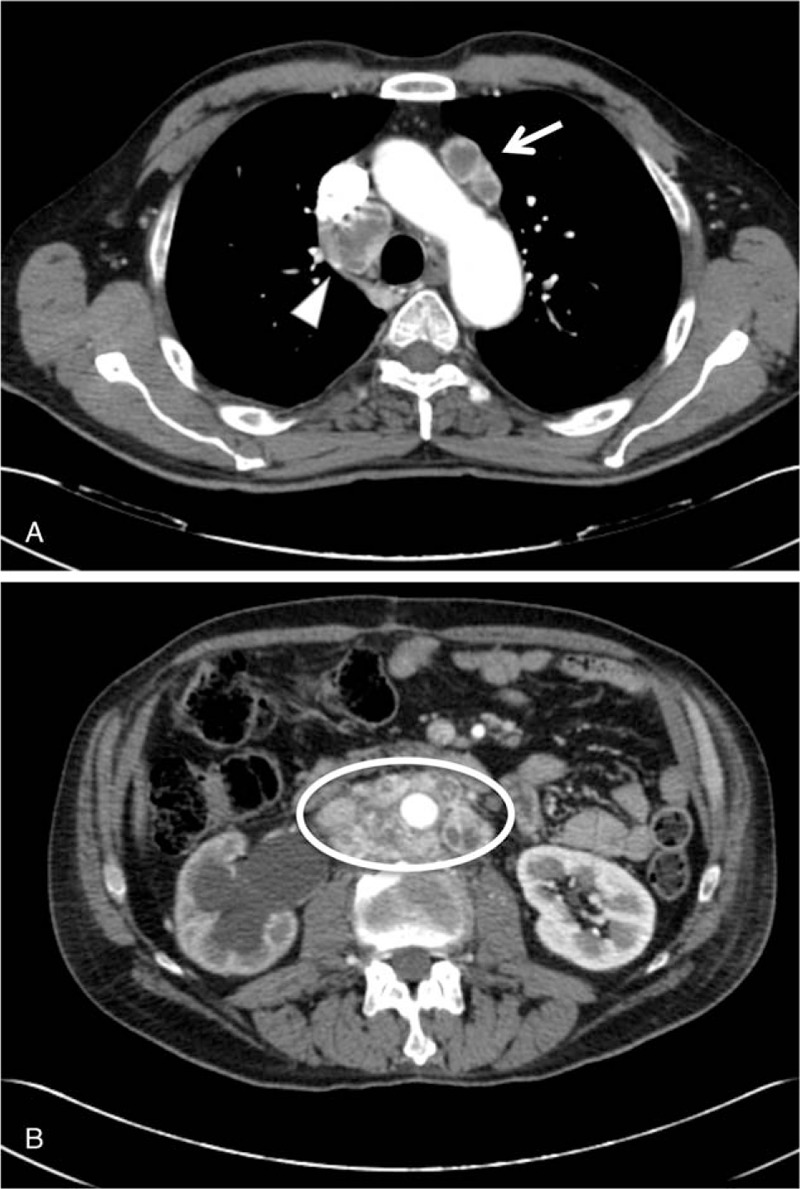
TC whole body: radiological evidence of disease progression. (A) Pathological over diaphragmatic lymph nodes, arrow indicates prevascular lymph nodes, triangle indicates lymph nodes in the Barety region. (B) Pathological under diaphragmatic lymph nodes, circle indicates para-aortic, paracaval and intercavo-aortic lymph nodes that include cava vein, aorta and, right ureter.
3. Discussion
Nonsecreting paragangliomas are typically associated with head and neck parasympathetic PGLs.[1] The incidence of malignant PGLs is <1 million people/year. The malignancy of phaeochromocytoma and PGL is estimated to be approximately 10% to 20%, and it is more common in sympathetic PGLs than in phaeochromocytomas.[7] PGL carriers with a mutation involving the SDHB gene are considered to potentially be at risk of malignancy, and a close biochemical and imaging follow-up must be reserved for them. SDHX inactivation is considered to be a cause of a pseudo-hypoxic response due to an accumulation of metabolites that are involved in the activation of the Hypoxia Inducible Factor 1 alpha (HIF-1α) pathway, resulting in a marked increase in neoangiogenesis and the expression of both vasoendothelial growth factor (VEGF) and its receptors in the neoplastic tissues.[8] The close follow-up program and molecular analysis that patients affected by PGL must undergo are crucial, as this case demonstrates, especially because of its unusual presentation that is not associated with catecholamine secretion. The treatment of choice typically consists of the complete surgical removal of the tumor. Therapeutic strategies for malignant PGLs are lacking. Most of them are derived from retrospective studies with few patients included or anecdotal case reports. In this case, treatment posed further difficulties due to the lack of hormonal secretion. Indeed, after surgery failure, the only therapeutic chance was placed on medical treatment because the use of a radiopharmaceutical targeted agent was not indicated. Therapy options have been discussed in the setting of expert multidisciplinary meetings including endocrinologists, oncologists, radiologists and nuclear medicine physicians. SDHB-mutated tumors promote the angiogenic pathway and activate genes that can promote the epithelial-mesenchymal transition associated with metastatic invasiveness.[9] The encouraging response related to sunitinib with manageable toxicity in some patients affected by metastatic PGL[10] encouraged us to start treatment with this antiangiogenic drug (Table 1). As described, we noticed the same side effect, especially in the gastrointestinal tract, even when the dose was reduced significantly. Unfortunately, this treatment in this case did not induce tumor growth arrest. Therefore, for the progression of the disease, the patient started the CVD chemotherapy regimen (Table 1). CVD was well tolerated by the patient, without any evidence of side effects. This combination was able to induce tumor growth arrest without evidence of disease progression and with subjective clinical benefit. These results are in accordance with a recent study, which revealed that 47% of the treated patients showed a radiological tumor response and disease stabilization with a gain in progression-free survival.[11]
In conclusion, sympathetic PGL tumors are associated with a higher rate of malignancy and lower overall survival rates. They often are characterized by high secretory activity. This very rare case of urinary bladder PGL4 syndrome in a patient with a germline SDHB mutation highlights the need to classify these tumors both genetically and histologically to differentiate benign from malignant PGLs. Currently, the experience coming from this case report suggests that all PGLs should be screened by molecular analysis and that the follow up must be very close in germline SDHB-mutated tumors. The lack of knowledge about the histological behavior and natural history of the disease makes clinical management of PGL a great challenge for endocrinologists. Prospective studies are needed to develop a flowchart and therapeutic strategies.
Author contributions
Conceptualization: Antonio Stigliano, Vincenzo Toscano.
Data curation: Antonio Stigliano, Maria Rosaria Nardone.
Formal analysis: Elisa Petrangeli.
Investigation: Antonio Stigliano, Pina Lardo, Anna Maria Aschelter, Iolanda Matarazzo, Gabriela Capriotti, Francesca Schiavi.
Methodology: Antonio Stigliano, Lidia Cerquetti.
Supervision: Paolo Marchetti.
Footnotes
Abbreviations: CVD = cyclophosphamide, vincristine and dacarbazine, HIF-1α = hypoxia inducible factor 1 alpha, PGL = paraganglioma, SDH = succinate dehydrogenase, VEGF = vasoendothelial growth factor.
The authors have no conflicts of interest to disclose.
References
- [1].Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2014;99:1915–42. [DOI] [PubMed] [Google Scholar]
- [2].Kimura N, Takayanagi R, Takizawa N, et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endoc Relat Cancer 2014;21:405–14. [DOI] [PubMed] [Google Scholar]
- [3].Santos P, Pimenta T, Taveira-Gomes A. Hereditary phaeochromocytoma. Int J Surg Pathol 2014;22:393–400. [DOI] [PubMed] [Google Scholar]
- [4].Fishbein L, Nathanson KL. Pheochromocytoma and paraganglioma: understanding the complexities of the genetic background. Cancer Genet 2012;205:1–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ayala-Ramirez M, Chougnet CN, Habra MA, et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic para- gangliomas. J Clin Endocrinol Metab 2012;97:4040–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000;15:7–12. [DOI] [PubMed] [Google Scholar]
- [7].Ayala-Ramirez M, Feng L, Johnson MM, et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab 2011;96:717–25. [DOI] [PubMed] [Google Scholar]
- [8].Favier J, Gimenez-Roqueplo AP. Genetics of paragangliomas and pheochromocytomas. Med Sci (Paris) 2012;28:625–32. [DOI] [PubMed] [Google Scholar]
- [9].Loriot C, Burnichon N, Gadessaud N, et al. Epithelial to mesenchymal transition is activated in metastatic pheochromocytomas and paragangliomas caused by SDHB gene mutations. J Clin Endocrinol Metab 2012;97:E954–62. [DOI] [PubMed] [Google Scholar]
- [10].Joshua AM, Ezzat S, Asa SL, et al. Rationale and evidence for sunitinib in the treatment of malignant paraganglioma/pheochromocytoma. J Clin Endocrinol Metab 2009;94:5–9. [DOI] [PubMed] [Google Scholar]
- [11].Tanabe A, Naruse M, Nomura K, et al. Combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine in patients with malignant pheochromocytoma and paraganglioma. Horm Cancer 2013;4:103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]