

Abstract
Where convenient phenotypic readouts are available, saturation mutagenesis coupled to deep sequencing provides a rapid and facile method to infer sequence determinants of protein structure, stability and function. We provide brief descriptions and currently available options for the various steps involved, and mention limitations of current implementations. We also highlight recent applications such as estimating relative stabilities and affinities of protein variants, mapping epitopes, protein model discrimination and prediction of mutant phenotypes. Most mutational scans have so far been applied to single genes and proteins. Additional methodological improvements are required to expand the scope to study intergenic epistasis and intermolecular interactions in macromolecular complexes.
Introduction
Mutant phenotypes are a key resource for obtaining insights into protein function. Many such phenotypes, studied together, provide a rich repository for understanding determinants of protein structure, stability and folding.
When a convenient phenotypic or ligand binding screen or selection is available, the saturation mutagenesis or deep mutational scanning approach, where every amino acid is individually mutated to every other amino acid, is of great value. Using next-generation sequencing, one can then link genotype to phenotype without the need for laborious processes involving protein purification and characterization. These studies have the capability of examining all possible single-site mutations, most of which have not been sampled in nature by evolution.
The intent of this review is to highlight recent advances and limitations in methods for generation and screening of saturation mutagenesis libraries, and particularly to highlight novel applications of the basic approach. An outline of the methodology and some of the initial applications have been detailed in earlier comprehensive reviews [1–3].
Generation and sequencing of saturation mutagenesis libraries
The expanding list of proteins subjected to saturation mutagenesis for various applications largely consist of bacterial antibiotic resistance proteins [4,5], enzymes [6,7], proteins that are essential for host survival, and small protein domains [8–10]. Such experiments differ in the type and size of the library and the method used for screening or selection.
Amongst the methods used for library generation, PFunkel is one of the popular early methods [11]. The PFunkel method requires a bacteriophage preparation of a Uracil containing ssDNA template. This is followed by PCR cycling using kinased mutagenic oligos as primers, with subsequent degradation of the Uracil containing template with Uracil DNA glycosylase and ExoIII. To abrogate the need of bacteriophage propagation, a nicking mutagenesis method [12] was developed that requires the template to instead have a 7-bp BbvCI restriction site. These sites allow successive creation and degradation of a wild-type ssDNA template through nicking, with a pair of endonucleases (Nt.BbvCI and Nb.BbvCI) that each recognize the same site but nick only one strand. Programmed allelic series (PALS) mutagenesis is another variant that utilizes a microarray based synthesis of mutagenic primers targeting a sequence of interest [13]. Programmed mutations are introduced by primer extension. While convenient, with the above ‘one pot’ reactions it is not possible to know the relative efficiency of mutagenesis at each position until the deep sequencing is complete. Another convenient method is the inverse-PCR based methodology for creating mutagenic libraries on templates of virtually any size in a 96 well plate format, which allows for rapid monitoring of individual reactions. Following PCR, all reactions can be pooled for the remaining steps [14].
Alternatively, large numbers of double-stranded DNA sequences can now be rapidly synthesized in a pooled fashion (upto 12,000 sequences per pool) and are commercially available at modest cost [15] (currently marketed by Agilent Technologies, Twist Biosciences, and Custom Array Inc). The synthesis is currently limited to a maximum of 200 bases. An earlier methodology, called MITE (Mutagenesis by Integrated TilEs) made use of six synthetic oligo pools (tiles) for generating a site-saturation library of a 264 aa long protein [16]. Each tile differed in a central variable region and shared homology with the vector at both ends. These technologies bypass the need for carrying out mutagenesis and multiple mutations can be encoded in a given sequence. However, they also appear to result in additional mutations. The errors are reported to be as high as 60% (Agilent and Custom Array) to 40% (Twist Biosciences) [17]. Although the origins of these errors have not been rigorously tested, non-matching reads are usually caused by a combination of oligo library synthesis errors, post-synthesis amplification errors, as well as sequencing errors. These errors often complicate the downstream single mutant analysis because of the limited read lengths conveniently accessible by current deep sequencing technologies. For many applications it is desirable to make use of a saturation mutagenesis method that results in generation of primarily single mutants. This ensures that majority of the reads will have a single mutation, even in cases where one is unable to sequence the whole gene in a single read.
The dominant platform of choice for deep sequencing is Illumina. The read length routinely achievable on most Illumina platforms is currently about 2*250bp. Certain applications may require longer read lengths. Sequencing platforms such as PacBio (Pacific Biosciences) and Nanopore sequencing have long read lengths [18], but these platforms have both lower accuracy and throughput with error rates typically greater than 10%. These platforms are therefore currently not routinely used [19]. To overcome this read length limitation, there are innovative methods that allow assembly of short-barcoded reads to form full length sequence that are beyond the read length limits of Illumina. These methods rely on barcoding single DNA molecules in such a way so as to allow reconstruction the full- length sequence based on barcode reading, with accuracies as high as 99.97% [20]. Brief descriptions of these methods are indicated in Table 1. While potentially powerful, most of the above methods are not widely used at present, presumably because of the additional steps involved, relative to normal amplicon sequencing.
Table 1.
Methods for reconstruction of longer reads from short amplicon sequencing
| Method | Steps involved | Ref |
|---|---|---|
| Tag- directed | 1. DNA fragments are ligated to tagged adaptors and amplified | [52] |
| 2. PCR products are concatemerized, sonicated and ligated to a ‘breakpoint’ adaptor | ||
| 3. Breakpoint reads are grouped based on tag sequence to facilitate local assembly | ||
|
| ||
| Droplet- capture based | 1. Isolation of single DNA molecules in droplets | [53,54] |
| 2. Amplification and fragmentation | ||
| 3. Barcoding of the fragments | ||
| 4. Sequencing and full length assembly using barcodes | ||
|
| ||
| Circularization based | 1. Barcoding with two distinct barcodes via PCR amplification of single molecules | [20] |
| 2. Random fragmentation and circularization of fragments | ||
| 3. Amplicon size selection and sequencing | ||
| 4. Assembly of reads by barcode pairing | ||
|
| ||
| DMS-BarSeq | 1. Barcoded strains are transformed with a specific variant | [49] |
| 2. Plate position-specific indexing is done for each strain | ||
| 3. Growth curves of individual strains are reconstructed from the deep sequencing data | ||
Choice of screening method
An efficient screen is critical and should be, robust, parallelizable and sensitive. The resolution of the screen depends crucially on the methodology adopted as well as on the phenotype assayed. Very small changes in activity/stability/binding affinity, especially the ones that are beneficial, are often missed in many screens. Yeast surface display has become a popular method of choice for phenotype screening. Theoretical and technical considerations for performing FACS based mutational screens of full length proteins have been extensively discussed [21]. For some applications involving fitness estimates or non-binary interactions, growth based screens may be more physiologically relevant for assays of protein function. Some of the recent studies that used different methods of screening have been summarized in Table 2. Most saturation mutagenesis studies till date have focussed on obtaining the enrichment ratios of mutants relative to WT, pre and post selection. For FACS based studies, this will depend on the choice of sorting and gating parameters as well as ligand concentrations and incubation time used [22]. For growth based selections, the enrichment ratios will depend on the protein expression levels and applied selection pressure. Hence, obtaining the frequency distribution of mutants across different bins (that comprise either different binding affinities or expression levels) may be more informative and useful than enrichment ratios, since the amount of protein for each variant displayed or produced may vary considerably [23].
Table 2.
Methodological details for diverse saturation mutagenesis studies of protein function
| Cell Type | Protein/Gene name | Library size | Mutagenesis and screening methodologies | Ref |
|---|---|---|---|---|
| Bacterial | TEM-1 β-lactamase | 287 positions, 3 libraries in single mutant backgrounds with 5434 mutants in each | Pfunkel mutagenesis, selection on various antibiotic concentrations | [19] |
| Bacterial | APH kinase | 264 positions, 4234 mutants | MITE (Mutagenesis by Integrated TilEs), growth selection on various aminoglycosides | [16] |
| Bacterial | Ras | 165 residues, 2 libraries in WT and single mutant backgrounds | Mutagenesis using partially overlapping primers, screening by bacterial two-hybrid system | [31] |
| Yeast | Gal4 | 64 positions, 1196 mutants | PALS (Programmed allelic series) mutagenesis, screening based on yeast two-hybrid system | [13] |
| Yeast | Hsp82 ATPase domain | 219 positions, 4021 mutants | EMPIRIC methodology, growth rate screening | [9] |
| Mammalian | BCR-ABL1 kinase domain | 20 positions, 380 mutants | CRISPR-Cas9-based genome editing approach, fluorescence- based screening by bulk competition of murine BalF3 cells | [55] |
| Bacterial | GFP | 51715 protein variants Upto 15 mutations | Random mutagenesis, FACS based screening | [34] |
| Yeast | BH3 peptides | 1026 variants | Synthetic peptides, screened by SORTCERY (FACS screening and gating strategy that rank orders variants based on their relative counts in bins sorted based on affinities) | [56,57] |
| Mammalian | Ubiquitin fused to EGFP | N- terminal residues | Landing pad cell line was developed to transfer libraries of mammalian genes into the mammalian genome | [58] |
A recent study [24] employed two different screens for two proteins to screen for mutants with increased solubility and then screened positive hits for activity to address the trade-off between enzyme solubility and activity. This also allowed the authors to assess the merits and demerits of both growth-based vs. display-based screens. Although the phenotypes assigned by the two complementary methods for both the proteins correlated well, the number of false positives obtained with yeast surface display based methodology was marginally higher than the growth-based selection. Since the readout of surface display is the per-cell number of epitope tags labelled by a fluorescently conjugated antibody, the relationship between fluorescence and fraction of protein surface displayed breaks down when destabilization can increase accessibility of the epitope tag, resulting in false positives.
Development of dedicated servers and web tools that allow rapid analysis of the deep sequencing data have further aided in extracting useful and reliable data, although given the diversity of approaches, applications and sequencing platforms, typically some computational expertise is essential to analyze data from mutational scanning experiments. ENRICH is a python based software that transforms raw sequencing reads from the pre and post selection populations into enrichment ratios [25,26]. Alternatively the enrichments can be expressed as likelihood based calculations [27] rather than ratios. Other tools that allow pre-processing of the Illumina NGS data include TRIMMER [28], FLASH (fast length adjustment of short reads) [29] and PEAR (Illumina Pair-End read merger for DNA fragments) [30], which are now routinely used to merge overlapping reads in paired end libraries, with a fragment size shorter than twice the read length.
Studies with multisite mutant libraries
Studies involving creation of multisite mutant libraries by random mutagenesis or by saturation mutagenesis in the background of a single mutation to test which alleles interact with the primary mutation, find diverse applications such as studying protein-ligand interactions [22,31], redesigning interfaces [32] and studying the contribution of epistasis in the fitness landscape of proteins and RNA[10,33–35].
One such study involved assessment of more than 100,000 mutants including 40,000 double mutants in an RNA recognition motif in S.cerevisiae that yielded a systematic picture of intragenic epistasis [36]. Another study screened a library containing all 160,000 variants of PhoQ at four key interface positions and used a two-step selection coupled to next-generation sequencing to identify 1659 functional variants [32]. Both positive and negative selection was combined to map the sequence space underlying the interface formed by bacterial two-component signalling proteins, PhoQ-PhoP in vivo. Another recent study examined 1000 mutants at 9 residues at the active site of hsp90 in the background of seven individual mutations that had a wild type like phenotype [33]. This allowed the authors to study slightly deleterious mutations that have the potential to become fixed in the background of other permissive mutations.
Evolutionary pathways of protein function are complex and moulded by the strength and duration of the selection pressure. The protein fitness landscape for Amp resistance was probed along the evolutionary pathway from TEM-1 (resistant to ampicillin) to TEM-15 (resistant to cefotaxime) [19]. Both pairwise and tertiary epistasis was studied by constructing and analysing single mutant libraries of the full length protein in the background of known single mutations that switch specificities to cefotaxime.
Recent applications of deep mutational scanning methodology
Protein model discrimination
As interactions between pairs of residues tend to remain conserved throughout evolution, compensatory mutations in these pairs can be used to infer residue proximity in the corresponding three-dimensional protein structures. This residue-residue contact information can be experimentally obtained from double mutant saturation mutagenesis libraries (Figure 1A). Using E. coli CcdB toxin as a test protein, an experimental method termed as saturation suppressor mutagenesis coupled to deep sequencing (SAS-seq) was developed to acquire reliable residue contact information [37]. This was used to determine the functional conformation adopted by the membrane protein DgkA in vivo. In principle, large double-mutant libraries to identify suppressors of multiple individual inactive mutations can be subjected to deep sequencing to identify large number of suppressor pairs which can be subsequently used for structure prediction as described above. A similar rationale is used in complementary methods that infer residue contacts from correlated substitution patterns from a multiple sequence alignment; however, these methods typically need a large number of sequences in the MSA for accurate contact prediction and little contact information is available for highly conserved positions. These methods usually yield large numbers of false positives and false negatives and do not necessarily predict residues in physical contact. Hence saturation suppressor methodology can be used to augment information from MSA based methods and also identify globally stabilizing mutations [37].
Figure 1. Select applications of the site-saturation mutagenesis coupled to deep sequencing approach.
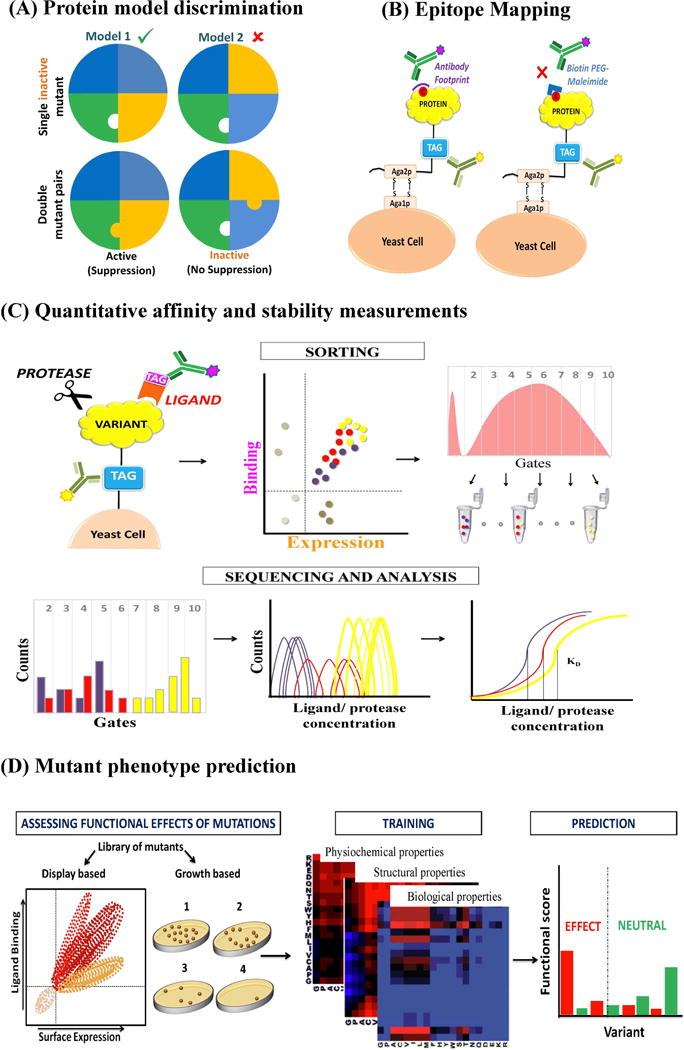
(A) Strategy to discriminate between possible models of a given protein using SAS-seq. A loss of function mutation (‘X’) in a given residue in the protein is identified (shown in white). This mutation is introduced into an existing saturation mutagenesis library, which is screened to identify suppressors based on activity, through deep sequencing. Mutations in the protein form a suppressor pair if the two residues interact. These suppressor pairs can be subsequently used to discriminate correct models (Model 1) from incorrect ones (Model 2). (B) The gene encoding the protein with a cysteine mutation (shown as a red dot) is displayed as a fusion protein on the surface of yeast cell. Ligand (here, antibody) binding to the displayed protein is monitored by FACS. Cysteine on the displayed protein is labeled with Biotin-PEG2-maleimide (shown in blue). If this cysteine is a part of the ligand/protein binding site, then the label will prevent binding of protein/ligand to the displayed protein leading to loss in fluorescence signal. (C) Yeast surface display is used to express proteins of interest which can be detected by labelled antibody against a tag. The ligand is fluorescently labelled and the cells displaying a given variant of the target are sorted into multiple bins based on their affinities. Alternatively, the displayed protein can be subjected to protease digestion to assess stability. Since each variant is sorted multiple times, it is associated with a histogram of counts spread over one or multiple bins. The experiment is repeated using various ligand/protease concentrations spanning a range of dissociation constants to infer sigmoidal titration curves which are used to infer relative affinity or stability. (D) The functional effect of single mutants present in the library can be assessed through growth based or display based methods. These data can then be used to understand the molecular bases of observed phenotypes. Known structural and physiochemical properties such as those derived from measurements of average solvent accessibilities in a database of known structures or free energies of transfer from neutral to aqueous solution of cyclohexane, as well as determinants of phenotypes obtained from the mutational scanning experiments can then be used for the training models for mutant phenotype prediction. The parameters obtained from training dataset are applied to a test dataset and results are usually converted into discrete functional scores to predict if a mutation has a deleterious effect or is neutral.
Epitope mapping
Mutagenesis coupled to deep sequencing has been extensively exploited for screening protein binder libraries [2]. However there are fewer studies that use this to delineate ligand binding sites including antibody epitopes [38,39]. One complication is that mutations at both buried and exposed, ligand binding sites can affect protein function. Distinguishing between the two is non-trivial in the absence of an accurate structural model. Another potential complication is that surface residues distal from a ligand binding site can exert allosteric effects, though a recent study [40] suggests this may not be a major concern. An ideal substitution for detecting protein-ligand interfaces should exhibit a large difference in mutational effect between interface and non-interface positions. A recent study analyzed large mutational datasets of fourteen proteins [41]. Most of the data sets reported mutational effect scores as the log-transformed ratio of mutant frequency before and after selection, divided by wild type frequency before and after selection. A systematic analysis of the datasets revealed that alanine substitutions were amongst the worst discriminators for interface and non-interface positions. Although deep mutational scanning can reveal the functional consequences of all possible single amino acid substitutions in a protein, these experiments can sometimes be expensive or unwieldy, depending upon the application. In many cases, scanning mutagenesis with one or a few amino acids is useful for determining functionally important positions, probing protein-ligand interactions and answering other specific questions. A recent approach for mapping protein: ligand binding sites and conformational epitopes makes use of single cysteine variants coupled to chemical labelling [42] (Figure 1B). The method relies on masking the epitopes residues by label rather than mutation for disrupting binding. An added advantage of the approach is that it can aid in distinguishing between buried and exposed residues and unlike Ala scanning, the method identifies most residues at the interface, instead of just hot-spot residues.
Quantitative affinity and stability measurements from deep sequencing data
With advances in technology and reduction in sequencing costs, there has been a considerable increase in the use of saturation mutagenesis to obtain qualitative information (binders and non-binders; active vs. inactive etc.) but fewer attempts to obtain semi-quantitative and quantitative estimates of parameters such as binding affinity (KD) and stability (Cm, Tm). Employing variant sorting similar to the SORTCERY method, deep sequencing data was used to infer dissociation constants for 1000 variants of scFv’s to fluorescent antigen through analysis of their titration curves [43]. The target molecule (here ‘antigen’) is fluorescently labelled and the cells displaying a given variant of the antibody are sorted into multiple bins based on their affinities. Since each variant antibody is sorted multiple times, it is associated with a histogram of counts spread over one or multiple bins. The study is repeated using various antigen concentrations spanning a range of KDs to construct sigmoidal titration curves (Figure 1C). However differences in antibody expression in different variants can complicate interpretation of such data.
In a recent comprehensive study, computational protein design, next-generation gene synthesis, and a high-throughput protease susceptibility were combined to measure folding and stability for more than 15,000 de novo designed miniproteins, 1000 natural proteins, 10,000 point mutants, and 30,000 negative control sequences [17]. Analysis of the data was used to systematically examine how sequence determines folding, stability and guide successive iterations between design and experiment to increase the design success rate. Using deep sequencing data on proteins displayed on the yeast surface that were subjected to various concentrations of protease, the authors could obtain relative stability estimates for a large number of variants without purification. It is noteworthy, that the proteins studied in the above paper have small loop lengths, thereby proteolysis under native conditions due to local fluctuations, was not observed. This will likely be a confounding factor for larger proteins [44]. Moreover, the approach is currently limited by the length of the oligonucleotide synthesis to very small proteins. This methodology has been extended to design more than 20000 mini proteins that target influenza haemaglutinin and botulinium neurotoxin B. The high affinity binders, selected through yeast surface display, provided potent prophylactic and therapeutic protection against influenza [45].
Prediction of mutant phenotypes
Unlike with antibiotic resistance, for most human diseases the genotype-phenotype relationship is often not straightforward. This is due to factors such as multiple interactions of the proteins at the cellular level, heterozygosity, protein threshold and protein level effects and in many cases, unknown inheritance patterns. While studies attempt to infer relative effects of mutations on fitness, their application to understand natural evolution or predict clinical significance of mutations in disease is subject to several limitations. In natural evolution, the selection pressures are unknown and variable, and possibly very different from those observed in the laboratory. In addition, over long timescales, small differences in fitness that are undetectable in the laboratory can lead to substantial differences in fixation probability [46]. Similar considerations apply to correlating fitness measurements from cellular screens with clinical data. Another concern is the use of heterologous promoters to drive expression of the gene of interest, typically at levels that are non-physiological. With respect to disease causing mutations, it is important to have validated clinical data on the functional effects of multiple point mutants in the system of interest and negative control data from healthy individuals, as the phenotypes would likely depend on factors specific to each protein. Such validated control data can be used to calibrate the output of experimental mutational scans to aid in improved prediction of mutant phenotypes [47] (Figure 1D).
Many proteins exhibit a threshold effect in phenotype that necessitates saturation mutagenesis scans at different expression levels of the protein, to rank order mutants in terms of their activity. In a recent study, deep mutational scanning data for ~1700 single-site mutants of the 100 residue protein, CcdB were collected at multiple different expression levels. The data were analyzed to provide possible explanations for the patterns of mutational tolerance observed and then validated by purifying and studying individual mutants in terms of stability, solubility and folding kinetics. While these studies are laborious, they provide crucial information. The data suggested that mutational effects on folding kinetics rather than stability are important determinants of in vivo phenotypes and add to efforts in predicting fitness effects of mutations [48].
Another comprehensive effort to understand and obtain mutational phenotypes for single-site mutants for human proteins, combined random codon-mutagenesis and multiplexed functional variation assays with computational imputation and refinement to produce exhaustive maps for human missense variants [49]. The framework was applied to four proteins: UBE2I, SUMO1, TPK1, and CALM1/2/3. The functional impact of ~16,000 missense variants was experimentally characterized and several pathogenic variants were identified. These functional complementation assays test the variant gene’s ability to rescue the phenotype caused by reduced activity of the wild type gene.
Future directions
Over the past five years, several macromolecular systems have been probed by saturation mutagenesis, coupled to deep sequencing. While proteins subjected to such mutational scans till date are fairly diverse in structure and function, important categories of proteins that are underrepresented in these analyses are membrane proteins [50,51], nucleic acid: protein complexes[36] and natively unfolded proteins for which mutational effects are not as well understood as for globular proteins. The lack of deep sequencing technology that combines long read lengths with high accuracy is a significant limitation that complicates quantitative analyses of multisite mutations, epistatic interactions and macromolecular complexes. Other limitations include efficient transfer of mutational libraries into mammalian cells, understanding and accurately predicting how mutations affect protein structure and stability, how best to correlate mutational data from laboratory selections/screens to clinical data, and to predict phenotypic effects of mutations at the organismal level. Despite these challenges, with the explosion of information-rich saturation mutagenesis datasets, a vast amount of information is now available for understanding determinants of protein function and stability, to delineate evolutionary trajectories, guide protein design and importantly, predicting and understanding the effects of mutations on mutant phenotypes.
Acknowledgments
Funding information
This work was funded in part by a grant to RV from the Department of Biotechnology (grant number NO.BT/COE/34/SP15219/2015, DT.20/11/2015), Government of India. We also acknowledge funding for infrastructural support from the following programs of the Government of India: DST FIST, UGC Centre for Advanced study, Ministry of Human Resource Development (MHRD), and the DBT IISc Partnership Program. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest: None
References
- 1.Tripathi A, Varadarajan R. Residue specific contributions to stability and activity inferred from saturation mutagenesis and deep sequencing. Curr Opin Struct Biol. 2014;24:63–71. doi: 10.1016/j.sbi.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Wrenbeck EE, Faber MS, Whitehead TA. Deep sequencing methods for protein engineering and design. Curr Opin Struct Biol. 2017;45:36–44. doi: 10.1016/j.sbi.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Araya CL, Fowler DM. Deep mutational scanning: assessing protein function on a massive scale. Trends Biotechnol. 2011;29:435–442. doi: 10.1016/j.tibtech.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Firnberg E, Labonte JW, Gray JJ, Ostermeier M. A Comprehensive, High-Resolution Map of a Gene's Fitness Landscape. Mol Biol Evol. 2016;33:1378. doi: 10.1093/molbev/msw021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stiffler MA, Hekstra DR, Ranganathan R. Evolvability as a function of purifying selection in TEM-1 beta-lactamase. Cell. 2015;160:882–892. doi: 10.1016/j.cell.2015.01.035. [DOI] [PubMed] [Google Scholar]
- 6.Klesmith JR, Bacik JP, Michalczyk R, Whitehead TA. Comprehensive Sequence-Flux Mapping of a Levoglucosan Utilization Pathway in E. coli. ACS Synth Biol. 2015;4:1235–1243. doi: 10.1021/acssynbio.5b00131. [DOI] [PubMed] [Google Scholar]
- 7.Wrenbeck EE, Azouz LR, Whitehead TA. Single-mutation fitness landscapes for an enzyme on multiple substrates reveal specificity is globally encoded. Nat Commun. 2017;8:15695. doi: 10.1038/ncomms15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Starita LM, Young DL, Islam M, Kitzman JO, Gullingsrud J, Hause RJ, Fowler DM, Parvin JD, Shendure J, Fields S. Massively Parallel Functional Analysis of BRCA1 RING Domain Variants. Genetics. 2015;200:413–422. doi: 10.1534/genetics.115.175802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mishra P, Flynn JM, Starr TN, Bolon DNA. Systematic Mutant Analyses Elucidate General and Client-Specific Aspects of Hsp90 Function. Cell Rep. 2016;15:588–598. doi: 10.1016/j.celrep.2016.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olson CA, Wu NC, Sun R. A comprehensive biophysical description of pairwise epistasis throughout an entire protein domain. Curr Biol. 2014;24:2643–2651. doi: 10.1016/j.cub.2014.09.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Firnberg E, Ostermeier M. PFunkel: efficient, expansive, user-defined mutagenesis. PLoS One. 2012;7:e52031. doi: 10.1371/journal.pone.0052031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wrenbeck EE, Klesmith JR, Stapleton JA, Adeniran A, Tyo KE, Whitehead TA. Plasmid-based one-pot saturation mutagenesis. Nat Methods. 2016;13:928–930. doi: 10.1038/nmeth.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitzman JO, Starita LM, Lo RS, Fields S, Shendure J. Massively parallel single-amino-acid mutagenesis. Nat Methods. 2015;12:203–206. doi: 10.1038/nmeth.3223. 204 p following 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain PC, Varadarajan R. A rapid, efficient, and economical inverse polymerase chain reaction-based method for generating a site saturation mutant library. Anal Biochem. 2014;449:90–98. doi: 10.1016/j.ab.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Kosuri S, Church GM. Large-scale de novo DNA synthesis: technologies and applications. Nat Methods. 2014;11:499–507. doi: 10.1038/nmeth.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Melnikov A, Rogov P, Wang L, Gnirke A, Mikkelsen TS. Comprehensive mutational scanning of a kinase in vivo reveals substrate-dependent fitness landscapes. Nucleic Acids Res. 2014;42:e112. doi: 10.1093/nar/gku511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17•.Rocklin GJ, Chidyausiku TM, Goreshnik I, Ford A, Houliston S, Lemak A, Carter L, Ravichandran R, Mulligan VK, Chevalier A, et al. Global analysis of protein folding using massively parallel design, synthesis, and testing. Science. 2017;357:168–175. doi: 10.1126/science.aan0693. High throughput protease suspecibility assay to measure stability for thousands of variants for small proteins, that allows iteriative protein design and characterization to further understand determinants of protein stability and design stabilized variants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinberg B, Ostermeier M. Shifting Fitness and Epistatic Landscapes Reflect Trade-offs along an Evolutionary Pathway. J Mol Biol. 2016;428:2730–2743. doi: 10.1016/j.jmb.2016.04.033. [DOI] [PubMed] [Google Scholar]
- 20.Stapleton JA, Kim J, Hamilton JP, Wu M, Irber LC, Maddamsetti R, Briney B, Newton L, Burton DR, Brown CT, et al. Haplotype-Phased Synthetic Long Reads from Short-Read Sequencing. PLoS One. 2016;11:e0147229. doi: 10.1371/journal.pone.0147229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21•.Kowalsky CA, Klesmith JR, Stapleton JA, Kelly V, Reichkitzer N, Whitehead TA. High-resolution sequence-function mapping of full-length proteins. PLoS One. 2015;10:e0118193. doi: 10.1371/journal.pone.0118193. Theoretical and technical considerations for performing growth and FACS based mutational screens of full length proteins have been provided and best practices to simplify the experimental design are discussed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen-Khait R, Schreiber G. Low-stringency selection of TEM1 for BLIP shows interface plasticity and selection for faster binders. Proc Natl Acad Sci U S A. 2016;113:14982–14987. doi: 10.1073/pnas.1613122113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1:755–768. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 24.Klesmith JR, Bacik JP, Wrenbeck EE, Michalczyk R, Whitehead TA. Trade-offs between enzyme fitness and solubility illuminated by deep mutational scanning. Proc Natl Acad Sci U S A. 2017;114:2265–2270. doi: 10.1073/pnas.1614437114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fowler DM, Araya CL, Gerard W, Fields S. Enrich: software for analysis of protein function by enrichment and depletion of variants. Bioinformatics. 2011;27:3430–3431. doi: 10.1093/bioinformatics/btr577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26•.Rubin AF, Gelman H, Lucas N, Bajjalieh SM, Papenfuss AT, Speed TP, Fowler DM. A statistical framework for analyzing deep mutational scanning data. Genome Biol. 2017;18:150. doi: 10.1186/s13059-017-1272-5. A statistical model for analyzing deep sequencing data that uses the frequency of each variant before and after selection obtained from deep sequencing to calculate enrichment ratios and estimate fitness. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bloom JD. Software for the analysis and visualization of deep mutational scanning data. BMC Bioinformatics. 2015;16:168. doi: 10.1186/s12859-015-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30:614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bandaru P, Shah NH, Bhattacharyya M, Barton JP, Kondo Y, Cofsky JC, Gee CL, Chakraborty AK, Kortemme T, Ranganathan R, et al. Deconstruction of the Ras switching cycle through saturation mutagenesis. Elife. 2017;6 doi: 10.7554/eLife.27810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Podgornaia AI, Laub MT. Protein evolution. Pervasive degeneracy and epistasis in a protein-protein interface. Science. 2015;347:673–677. doi: 10.1126/science.1257360. [DOI] [PubMed] [Google Scholar]
- 33.Bank C, Hietpas RT, Jensen JD, Bolon DN. A systematic survey of an intragenic epistatic landscape. Mol Biol Evol. 2015;32:229–238. doi: 10.1093/molbev/msu301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sarkisyan KS, Bolotin DA, Meer MV, Usmanova DR, Mishin AS, Sharonov GV, Ivankov DN, Bozhanova NG, Baranov MS, Soylemez O, et al. Local fitness landscape of the green fluorescent protein. Nature. 2016;533:397–401. doi: 10.1038/nature17995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puchta O, Cseke B, Czaja H, Tollervey D, Sanguinetti G, Kudla G. Network of epistatic interactions within a yeast snoRNA. Science. 2016;352:840–844. doi: 10.1126/science.aaf0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Melamed D, Young DL, Gamble CE, Miller CR, Fields S. Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)-binding protein. RNA. 2013;19:1537–1551. doi: 10.1261/rna.040709.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahoo A, Khare S, Devanarayanan S, Jain PC, Varadarajan R. Residue proximity information and protein model discrimination using saturation-suppressor mutagenesis. Elife. 2015;4 doi: 10.7554/eLife.09532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kowalsky CA, Faber MS, Nath A, Dann HE, Kelly VW, Liu L, Shanker P, Wagner EK, Maynard JA, Chan C, et al. Rapid fine conformational epitope mapping using comprehensive mutagenesis and deep sequencing. J Biol Chem. 2015;290:26457–26470. doi: 10.1074/jbc.M115.676635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Blarcom T, Rossi A, Foletti D, Sundar P, Pitts S, Bee C, Melton Witt J, Melton Z, Hasa-Moreno A, Shaughnessy L, et al. Precise and efficient antibody epitope determination through library design, yeast display and next-generation sequencing. J Mol Biol. 2015;427:1513–1534. doi: 10.1016/j.jmb.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 40.Anishchenko I, Ovchinnikov S, Kamisetty H, Baker D. Origins of coevolution between residues distant in protein 3D structures. Proc Natl Acad Sci U S A. 2017;114:9122–9127. doi: 10.1073/pnas.1702664114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gray VE, Hause RJ, Fowler DM. Analysis of Large-Scale Mutagenesis Data To Assess the Impact of Single Amino Acid Substitutions. Genetics. 2017;207:53–61. doi: 10.1534/genetics.117.300064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42•.Najar TA, Khare S, Pandey R, Gupta SK, Varadarajan R. Mapping Protein Binding Sites and Conformational Epitopes Using Cysteine Labeling and Yeast Surface Display. Structure. 2017;25:395–406. doi: 10.1016/j.str.2016.12.016. Method for mapping protein:ligand binding sites and conformational epitopes by combining cysteine scanning mutagemesis with chemical labelling followed by deep sequencing to discriminate binders vs non-binders, as well as distinguish buried and exposed residues. [DOI] [PubMed] [Google Scholar]
- 43•.Adams RM, Mora T, Walczak AM, Kinney JB. Measuring the sequence-affinity landscape of antibodies with massively parallel titration curves. Elife. 2016:5. doi: 10.7554/eLife.23156. An experimental approach that can be used to measure the binding affinities for thousands of variants in parallel, by constructing titration curves from the relative reads obtained through deep sequencing for a given variant sorted at various ligand concentrations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park C, Marqusee S. Quantitative determination of protein stability and ligand binding by pulse proteolysis. Curr Protoc Protein Sci. 2006 doi: 10.1002/0471140864.ps2011s46. Chapter 20: Unit 20 11. [DOI] [PubMed] [Google Scholar]
- 45.Chevalier A, Silva DA, Rocklin GJ, Hicks DR, Vergara R, Murapa P, Bernard SM, Zhang L, Lam KH, Yao G, et al. Massively parallel de novo protein design for targeted therapeutics. Nature. 2017;550:74–79. doi: 10.1038/nature23912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boucher JI, Bolon DN, Tawfik DS. Quantifying and understanding the fitness effects of protein mutations: Laboratory versus nature. Protein Sci. 2016;25:1219–1226. doi: 10.1002/pro.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47•.Miosge LA, Field MA, Sontani Y, Cho V, Johnson S, Palkova A, Balakishnan B, Liang R, Zhang Y, Lyon S, et al. Comparison of predicted and actual consequences of missense mutations. Proc Natl Acad Sci U S A. 2015;112:E5189–5198. doi: 10.1073/pnas.1511585112. Predicted and actual effects of clinically relevant mutations are compared using mice models that revealved a high false postive rate and gap in our ability to relate genotype to phenotype in clinical cases. Factors that may lead to overprediction of deleterious phenotypes are discussed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48•.Tripathi A, Gupta K, Khare S, Jain PC, Patel S, Kumar P, Pulianmackal AJ, Aghera N, Varadarajan R. Molecular Determinants of Mutant Phenotypes, Inferred from Saturation Mutagenesis Data. Mol Biol Evol. 2016;33:2960–2975. doi: 10.1093/molbev/msw182. Deep mutational scanning data for single-site mutants were collected at multiple different expression levels and the data were analyzed and validated by purifying and studying individual mutants to understand determinants of stability, solubility and folding kinetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weile J, Sun S, Cote AG, Knapp J, Verby M, Mellor JC, Wu Y, Pons C, Wong C, van Lieshout N, et al. Expanding the Atlas of Functional Missense Variation for Human Genes. bioRxiv. 2017 doi: 10.1101/166595. [DOI] [Google Scholar]
- 50.Fujii S, Matsuura T, Sunami T, Nishikawa T, Kazuta Y, Yomo T. Liposome display for in vitro selection and evolution of membrane proteins. Nat Protoc. 2014;9:1578–1591. doi: 10.1038/nprot.2014.107. [DOI] [PubMed] [Google Scholar]
- 51.Schutz M, Schoppe J, Sedlak E, Hillenbrand M, Nagy-Davidescu G, Ehrenmann J, Klenk C, Egloff P, Kummer L, Pluckthun A. Directed evolution of G protein-coupled receptors in yeast for higher functional production in eukaryotic expression hosts. Sci Rep. 2016;6:21508. doi: 10.1038/srep21508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hiatt JB, Patwardhan RP, Turner EH, Lee C, Shendure J. Parallel, tag-directed assembly of locally derived short sequence reads. Nat Methods. 2010;7:119–122. doi: 10.1038/nmeth.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53•.Lan F, Haliburton JR, Yuan A, Abate AR. Droplet barcoding for massively parallel single-molecule deep sequencing. Nat Commun. 2016;7:11784. doi: 10.1038/ncomms11784. Method to obtain long reads from short-read sequencing of single DNA molecules that are encapsulated, fragmented and barcoded in picolitre droplets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Redin D, Borgstrom E, He M, Aghelpasand H, Kaller M, Ahmadian A. Droplet Barcode Sequencing for targeted linked-read haplotyping of single DNA molecules. Nucleic Acids Res. 2017;45:e125. doi: 10.1093/nar/gkx436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma L, Boucher JI, Paulsen J, Matuszewski S, Eide CA, Ou J, Eickelberg G, Press RD, Zhu LJ, Druker BJ, et al. CRISPR-Cas9-mediated saturated mutagenesis screen predicts clinical drug resistance with improved accuracy. Proc Natl Acad Sci U S A. 2017;114:11751–11756. doi: 10.1073/pnas.1708268114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56•.Reich LL, Dutta S, Keating AE. SORTCERY-A High-Throughput Method to Affinity Rank Peptide Ligands. J Mol Biol. 2015;427:2135–2150. doi: 10.1016/j.jmb.2014.09.025. A FACS-based sorting procedure that ranks the affinities of library members for a given target using yeast surface display followed by deep sequencing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reich LL, Dutta S, Keating AE. Generating High-Accuracy Peptide-Binding Data in High Throughput with Yeast Surface Display and SORTCERY. Methods Mol Biol. 2016;1414:233–247. doi: 10.1007/978-1-4939-3569-7_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matreyek KA, Stephany JJ, Fowler DM. A platform for functional assessment of large variant libraries in mammalian cells. Nucleic Acids Res. 2017;45:e102. doi: 10.1093/nar/gkx183. [DOI] [PMC free article] [PubMed] [Google Scholar]