

Abstract
Molybdenum cofactor deficiency is an extremely rare and fatal metabolic disorder that should be considered in the differential diagnosis of hypoxic-ischemic encephalopathy. Magnetic resonance imaging findings are useful in diagnosis. The short-echo-time magnetic resonance spectrum was characterized by a total loss of signal and lipid and lactate peaks. In this case, conventional magnetic resonance imaging and magnetic resonance spectroscopy findings of this extremely rare disease whose pathophysiology was not known were presented.
Keywords: Molybdenum cofactor deficiency, MRI, Spectroscopy
Introduction
Molybdenum cofactor deficiency (MCD) is an extremely rare and fatal metabolic disorder that is characterized by severe and progressive neurologic deterioration in early infancy. Less than 150 cases have been reported in the literature [1]. MCD is an autosomal recessively inherited disorder and reveals findings within the first few days after birth. The major clinical findings are severe neonatal seizures, feeding difficulties, and a progressive neurologic deficit. Diagnosis is late in many cases, and these cases usually pass away in early infancy. Diagnosis can be made by typical magnetic resonance imaging (MRI) findings in the early period. Progressive neurodegeneration is unavoidable in cases with early diagnosis. We presented the clinical presentation and characteristic conventional and spectroscopic MRI findings on this under-recognized disease.
Case
An 11-month-old female patient came to the pediatric neurology clinic with an increase in seizure frequency and serious eating difficulty. She was the first child of second-degree consanguineous parents. There was no history of neurologic diseases in the family. There was a history of three previous abortus of the mother before this delivery. The patient was born to a 41-year-old mother at normal gestational week by vaginal delivery, weighing 3050 g. The patient was diagnosed with infantile spasm-like seizures 1 week after birth, which were controlled with phenobarbital. Physical examination revealed microcephaly, hypotonia, and hyper-reflexia. The head circumference was 42.6 cm (between the 3rd and the 10th percentiles), the weight was 7250 g (between the 3rd and the 10th percentiles), and the height was 71 cm (between the 3rd and the 10th percentiles). The measurements were consistent with developmental retardation. Electroencephalographic recordings manifested diffuse, bilateral, and hemispheric epileptiform discharges. There was no detectable uric acid in the urinary sample, indicated as 0 mg/dL. Serum uric acid levels revealed a progressive fall. Serum uric acid measurements obtained intermittently at different times were less than 2 mg/dL on the first and the second measurements, was less than 1 mg/dL on the third measurement, and was 0 mg/dL on the last measurement, which were lower than the normal limits (N: 2.6-6.0 mg/dL). On posterior-anterior chest x-ray, a shunt catheter, which was implanted for hydrocephalus in an external center, was seen, and rotatory scoliosis of the thoracal vertebrae was noted (Fig. 1).
Fig. 1.
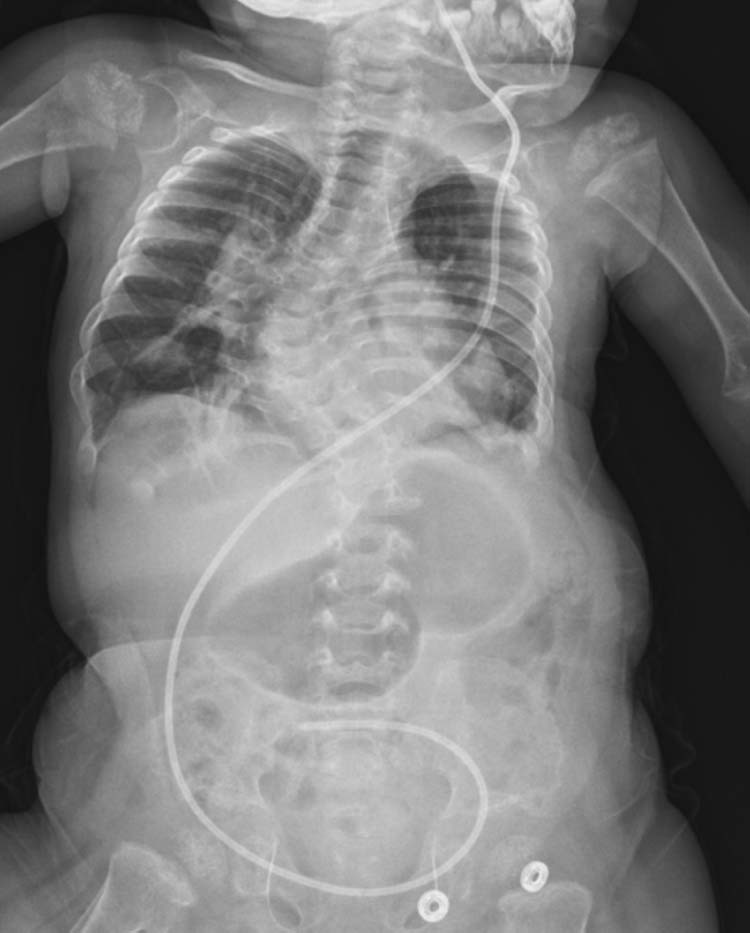
On x-ray involving the chest and the abdomen, rotatory scoliosis of the thoracal vertebrae was noted in addition to the ventriculoperitoneal shunt catheter that extended along the cranium and the abdomen.
MRI was performed for diagnosis (1.5-T Siemens Magnetom Aera, Germany). MRI of the brain revealed extensive subcortical and periventricular white matter loss, cystic encephalomalacia and hyperintensity in white matter, dysgenesis of the corpus callosum, and ventriculomegaly with generalized volume loss (Fig. 2). In addition, focal polymicrogyria and focal agyria in the cerebral hemisphere at the cortical location, brainstem-cerebellar atrophy, and enlargement of the retrocerebellar cyst were noted on brain MRI images (Fig. 3). The short-echo-time single-voxel proton magnetic resonance (MR) spectroscopy was performed. The volume of interest was located in the parieto-occipital and frontal areas and included both white and gray matter. There were significant lipid-lactate peaks at 1.3 ppm, and all other metabolites were reduced on spectroscopy (Fig. 4). MCD was considered at the prediagnosis with laboratory and imaging findings. Our diagnosis was confirmed by genetic analysis.
Fig. 2.

T1-weighted axial (A), T2-weighted coronal (B), T2-weighted axial (C), and axial fluid-attenuated inversion recovery sequence (D) of magnetic resonance images show ventriculomegaly, cystic encephalomalacia, and extensive subcortical and periventricular white matter loss and hyperintensity in white matter with atrophy.
Fig. 3.

Axial fluid-attenuated inversion recovery (A-C), T2-weighted coronal (D) and axial (E), and T1-weighted sagittal (F) sequence of magnetic resonance images show polymicrogyria (arrows), agyri (curved arrows), brainstem-cerebellar atrophy (arrowheads), and enlargement of the retrocerebellar cyst (stars).
Fig. 4.

The short-echo-time single-voxel proton MR spectroscopy shows significant lipid-lactate peaks at 1.3 ppm and reduced all other metabolites in frontal (A) and parieto-occipital (B) areas, including both white and gray matter. NAA, N-acetylaspartate.
Discussion
MCD predominantly affects the central nervous system [2]. Molybdenum is a cofactor required for the functions of sulfite oxidase, xanthine dehydrogenase, and aldehyde oxidase enzymes for humans. In molybdenum deficiency, activities of these 3 enzymes decrease. Four genes (MOCS1, MOCS2, MOCS3, and GEPH) are needed for the production pathway of molybdenum cofactor. Mutation in any of these genes causes deterioration in molybdenum formation. The most common mutation observed is in the MOCS1 gene, followed by the MOCS2 gene [3]. Neuronal damage is rapidly progressive and severe as a result of the accumulation of toxic concentrations of sulfite in the brain [4]. MRI studies reveal diffuse brain atrophy with arrested development of myelination, gliosis, and cystic necrosis of white matter. Microcephaly is common [4].
More than 90% of the cases present early, in the first few days of life with intractable seizures, hypertonia, and feeding difficulties. The mean age at diagnosis was 12.5 months [1]. The median survival is 36 months [4]. The most common clinical findings were intractable seizures, feeding difficulties, dysmorphic facies such as puffy cheeks, long face micrognathia, large ears, and periferic hypertonicity [5]. Seizure is the most prominent initial sign and usually leads to a metabolic workup [4]. There are elevated levels of sulfite, thiosulfate, S-sulfocysteine, taurine, xanthine, and hypoxanthine and low amounts of uric acid in the serum [5].
Less than 150 cases have been reported worldwide [1]. Actual incidence might be greater than known because patients might have passed away without diagnosis [2]. The global incidence of MCD is likely to be higher as a result of failure to diagnose and under-reporting [2]. The neurologic injury caused by MCD is not fully understood [3]. It is considered that elevated levels of neurotoxic metabolites (sulfite and taurine) or the deficit of the product sulfate are cause of neurologic injury [6]. There is no effective treatment for the disease now, but research on animal models anticipates hope for future availability of effective treatments in humans [6]. Current treatment for individuals with this disorder aims to provide relief of symptoms; for seizures, in particular, anticonvulsant drugs are used. Dietary restriction of sulfur-containing amino acids may decrease sulfite excretion but is not able to stop the neurologic progression of the disease [4]. Efficient uptake of cyclic pyranopterin monophosphate (can be produced in Escherichia coli) and restoration of molybdenum cofactor-dependent enzyme activities were reported [4].
Isolated sulfite oxidase deficiency reveals autosomal recessive transition, and its clinic is similar to MCD. These 2 disorders can be differentiated by biochemical analysis. There are limited data on long-term outcome or brain MRI features [2]. The MRI findings of MCD are global cerebral edema, cystic encephalomalacia, cortical and white matter atrophy, focal or bilateral changes within the globi pallidi, thalamic and subthalamic regions, dysgenesis of the corpus callosum, brainstem-cerebellar atrophy, and ventriculomegaly [2], [7]. These imaging findings may be confused with severe hypoxic-ischemic encephalopathy (HIE), and most of the patients are misdiagnosed as having HIE. The degree of spectral abnormality corresponds to the severity of HIE. In severe HIE cases, in particular, spectral findings may be similar to spectral findings of our patient. Diagnosis can be made easily by laboratory findings in suspected cases, which distinguish it from HIE. A significant reduction in N-acetylaspartate is an indication of neuronal destruction, and lipid peak is an indication of white matter destruction on MR spectroscopy [7]. Accumulation of lactate is in agreement with the presence of cavitations seen on MR images, as cerebrospinal fluid is rich in lactate. Decreases in choline and inositol exclude demyelinization (increase in choline) and gliosis (increase in inositol) [7].
In conclusion, MCD is an extremely rare metabolic disorder. Conventional MRI and MR spectroscopy are quite useful in diagnosis. Having good knowledge of spectroscopic findings, in particular, is very useful in the differential diagnosis of MCD.
Footnotes
Competing Interests: The authors declare that there are no financial or other relations that could lead to a conflict of interest.
References
- 1.Fathalla W.M., Mohamed K.A., Ahmed E. Molybdenum cofactor deficiency: report of a new case and literature review. Ibnosina J Med Biomed Sci. 2010;2(3):133–138. http://www.ijmbs.org [Google Scholar]
- 2.Vijayakumar K., Gunny R., Grunewald S., Carr L., Chong K.W., DeVile C. Clinical neuroimaging features and outcome in molybdenum cofactor deficiency. Pediatr Neurol. 2011;45(4):246–252. doi: 10.1016/j.pediatrneurol.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Reiss J. Molybdenum cofactor and sulfite oxidase deficiency. Metabolomics (Los Angel) 2016;6(3):184. [Google Scholar]
- 4.Mechler K., Mountford W.K., Hoffmann G.F., Ries M. Ultra-orphan diseases: a quantitative analysis of the natural history of molybdenum cofactor deficiency. Genet Med. 2015;17(12):965–970. doi: 10.1038/gim.2015.12. 25764214 Epub 2015 Mar 12; PubMed PMID. [DOI] [PubMed] [Google Scholar]
- 5.Johnson J.L., Waud W.R., Rajagopalan K.V., Duran M., Beemer F.A., Wadman S.K. Inborn errors of molybdenum metabolism: combined deficiencies of sulfite oxidase and xanthine dehydrogenase in a patient lacking the molybdenum cofactor. Proc Natl Acad Sci U S A. 1980;77(6):3715–3719. doi: 10.1073/pnas.77.6.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayram E., Topçu Y., Karakaya P., Yis U., Çakmakçı H., Ichida K. Molybdenum cofactor deficiency: review of 12 cases (MoCD and review) Eur J Paediatr Neurol. 2013;17(1):1–6. doi: 10.1016/j.ejpn.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 7.Salvan A.M., Chabrol B., Lamoureux S., Gouny S.C., Cozzone P.J., Dur J.V. In vivo brain proton MR spectroscopy in a case of molybdenum cofactor deficiency. Pediatr Radiol. 1999;29(11):846–848. doi: 10.1007/s002470050710. [DOI] [PubMed] [Google Scholar]