

Abstract
Myocardial fibrosis (MF) plays an important part in cardiovascular diseases. The main cytological characteristics of MF is the increased number of myofibroblasts, which have multiple sources such as EMT, EndMT, myeloid progenitors, monocytes, and fibrocytes. Recent data showed that pericytes may represent a major source of myofibroblasts in kidney fibrosis. Valproic acid (VPA) is a kind of short-chain fatty acid. It was reported in recent studies that VPA regulates gene expression and influences various signal pathways. HDACs inhibitors can hinder the growth of tumor cells and differentiation of stem cells. And little is known about the effects of HDACs inhibitors on myofibroblasts transdiffererntiaton. This study focused on the role of HDACs in pericyte-myofibroblast trans-differentiation and how HDACs inhibitor VPA influenced proliferation, migration, viability and myofibroblast trans-differentiation of pericytes for the first time. Rat cardiac fibrosis model was induced by Ang II. Immunohistochemistry was employed to examine cardiac fibrosis and flow cytometry was used to analyze whether inflammatory cells involve VPA-induced trans-differentiation. Pericytes proliferation, migration and differentiation to myofibroblasts were performed to examine the role of VPA on pericyte trans-differentiation. Immunoblot and qPCR were applied to identify the signal transduction involving in VPA-induced trans-differentiation. In vivo study showed that HDAC inhibitor VPA blocks cardiac fibrosis, and inflammation inhibition was not involved in this process. VPA treatment inhibited Ang II pericyte proliferation, migration and transdifferentiation to myofibroblast. Furthermore, the inhibition of α-SMA expression by VPA was related to reduce phosphorylation of ERK, and a pharmacological inhibitor of MEK suppressed Ang II-induced α-SMA expression. HDAC4 knockdown resulted in inhibiting Ang II-mediated α-SMA expression as well as the phosphorylation of ERK. Moreover, the inhibitors of protein phosphatase 2A and 1 (PP2A and PP1) restored the Ang II-stimulated α-SMA expression from the inhibitory effect of VPA. Together, the current data indicate that the differentiation of pericytes to myofibroblasts is HDAC4 dependent and requires phosphorylation of ERK.
Keywords: Valproic acid, pericytes, myofibroblasts, fibrosis
Introduction
Myocardial fibrosis (MF) is associated with an increased collagen volume and changed collagen components in heart tissues [1]. MF is an inevitable process in the late stages of multiple cardiac diseases, including hypertension, heart failure, and valvular dysfunction. Higher incidences of arrhythmia, cardiac dysfunction, and even sudden cardiac death are found in patients with MF [2]. MF is mainly characterized by proliferation of fibroblasts and accumulation of extracellular matrix (ECM), which leads to increased cardiac stiffness, affecting the normal systolic and diastolic function of the heart [3]. MF plays an important role in the diagnosis of cardiovascular diseases [4]. Fibroblasts maintain normal structure and function of the heart by preserving homeostasis of ECM. However, under the circumstances of myocardial infarction, overloading stress or activation of neurohumoral factors, fibroblasts pathologically proliferate and transform into myofibroblasts. Myofibroblasts are smooth-muscle-like fibroblasts, serving as a transition state from fibroblasts to smooth muscle cells. The major difference between myofibroblasts and fibroblasts is the α-SMA expression and bundled contractive microfilaments in the cytoplasm of myofibroblasts. The difference of myofibroblasts from smooth muscle cells is the production of type I, III, IV, and V collagen and isomer of fibronectin ED-A [5].
The sources of myofibroblasts in MF mainly include fibroblasts in the heart tissue, epithelial-mesenchymal transition (EMT), endothelial-mesenchymal transition (EndMT), and the myeloid progenitors, monocytes, and fibrocytes in circulation. Lin and colleagues reported that pericytes and perivascular fibroblasts were the main sources of mesenchymal myofibroblasts [6]. Generated from mesenchyma, pericytes can produce collagen and highly branched mesenchymal cells. Pericytes are embedded into basement membrane of microvessels and connected to endothelial cells intercellularly [7]. It has been revealed that the pericyte begins to move away from capillaries, enters cell cycle, and subsequently increases in the population size within 24 h after kidney injuries by UUO surgery as the increase of collagen 1 (α1) (Col1α1) transcript [8].
In eukaryotes, reconstruction of chromatin protein is the essential mechanism of epigenetic regulation of genes [9]. Acetylization of histone increases DNA accessibility by neutralizing positive charge of lysine residues, promoting the separation of DNA and histone, and making nucleosome DNA more accessible to transcriptional factors in order to activate transcription. Acetylization of histone also plays an important role in regulating the interactions of multiple proteins. Acetylization of histone is dependent on histone acetyltransferases (HATs), and its counter-reaction depends on histone deacetylases (HDACs) [10]. Valproic acid (VPA), a short-chain fatty acid, is the first-line drug to treat epilepsy and depression for more than 40 years [11]. Recent studies showed that VPA could inhibit HDACs, leading to high histone acetylation levels [12]. HDACs inhibitors can hinder the growth of tumor cells and differentiation of stem cells [13,14].
The phosphorylation/dephosphorylation of proteins controlled by protein kinases and protein phosphatases (PP) is a critical factor in regulating multiple cellular processes. PP1 and PP2A belong to serine/threonine phosphatases. PP2A is capable of dephosphorylating ERK in vitro, and the protein phosphatase inhibitor okadaic acid (OA) has been shown to rescue ERK phosphorylation from PP2A dephosphorylation in MCF-7 breast cancer cells [15]. HDAC6 associate with PP1 and their inhibitor results in increased PP1-ERK association [16].
Herein we show that the differentiation of cardiac pericytes to myofibroblasts is HDAC4 dependent and requires phosphorylation of ERK. The phosphorylation of ERK is required for α-SMA expression in response to Ang II. Both VPA and HDAC4 knockdown are sufficient to decrease phosphorylation of ERK and block Ang II-stimulated α-SMA expression, and the pharmacological inhibition of PP1 and PP2A rescues the α-SMA expression in response to Ang II.
Material and methods
Experimental animals
Cardiac fibrosis models of rats were established as described in the literature [17]. The Sprague Dawley rat was infused with Ang II [Bachem, 1.5 μg/(min•kg)] for 2 weeks using an osmotic mini-pump (Model 2004, Alza Corp, America). VPA (10 mg/kg) or vehicle (50:50 DMSO: PEG-300) was intraperitoneally injected daily for continuous 2 weeks. The rats were killed at 20 h after the last administration of the compound. The animals were anesthetized with 2% isoflurane, and the hemodynamic data were collected via carotid catheterization (Scisense, UK).
Tissue preparation and histology
Myocardial tissue from the left ventricular transverse section was routinely fixed and embedded. The paraffin sections were stained with picrosirius red and the myocardial collagen volume fraction (CVF) was observed and measured. Method of calculation: Data were recorded by a Nikon camera system, and Image Pro Plus 6.0 software was used to analyze the ratio of the myocardial collagen area and the total area of the visual field. At least 6 fields of each section were analyzed.
Flow cytometry
About 50 mg fresh heart tissues were obtained and washed with PBS to remove the blood remnant. Ophthalmic scissors were used to cut the tissue into pieces of 1 mm3. Pancreatin-EDTA (2.5 g/L) and collagenase (0.25 g/L) were added to digest the heart tissue into single cell suspension, which underwent centrifugation at 1,000 r/min for 5 min. The supernatant was abandoned. The sediment was resuspended into cold PBS and underwent 300 mesh screen filter. Cells were pelleted, followed by resuspending in FACS wash buffer (PBS with 1% BSA and 0.1% sodium azide, 5 × 106/mL) which contained a 1:100 dilution of each fluorescence-conjugated antibody, such as anti-CD11b-APC (eBioscience), anti-CD45-V500 (BD Biosciences), anti-CD34-PE (BD Biosciences), anti-F4/80-PerCp Cy5.5 (eBioscience), and anti-Ly6G-APC Cy7 (BD Biosciences). Surface staining was carried out at 4°C for 20 min. The cells were washed before resuspension for FACS analysis.
Culture of cardiac pericytes
Ventricular myocardial pericytes were isolated based on previously published data [18]. Rats were intraperitoneally injected with 2000 U/kg heparin and anesthetized with 2% pentobarbital solution (45 mg/kg) after 20 min. The heart with a section of the aorta was rapidly removed. Residual blood from the heart was washed with cold Ca2+-free Ringer solution (dissolving the following chemicals in water (in mM): 127 NaCl, 4.6 KCl, 1.1 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 7.6 glucose, 2 pyruvate, 10 creatine, 20 taurine, 5 ribose, 2 aspartic acid, 2 glutamic acid, 1 arginine, and 0.5 uric acid, equilibrated with carbogen). The heart with a section of the aorta was quickly hung on the Langendorff device, with 6 mL/min flow rate. Retrograde perfusion was performed with oxygen saturated digested liquid 110 collagenase B (Roche Diagnostics, Mannheim, Germany), 8 dispase II (Roche), and 200 albumin (Sigma) in mg/100 mL via the aorta for 13-16 min at 37°C. The digestion was stopped when the heart became soft and the color looked pale. The residual digestive liquid was washed by 30 mL Ca2+-free Ringer solution. Left ventricles were cut into 3 × 3 mm pieces in 20 mL Ringer solution, gently homogenized for 30 min at 37°C. After collection of the suspended myocardial microvessels by filtration through a nylon net (mesh size: 200 μm), pericytes were detached selectively from their parent vessels on the net. For this purpose, the net was covered on both sides with further nets of smaller mesh size (50 μm), installed between two perfusable chambers in a custom built apparatus, and flushed continuously with 13 mL freshly prepared protease solution which was pumped back and forth across the net stack in a custom-built apparatus (15 min, 37°C, 6 cycles/min). In order to further purify pericytes from cell mixture which contains two main contaminating cells (< 10%): endothelial cells (ECs) of venular origin and some smooth muscle cells (SMCs). A few small islets of SMCs could be removed by scratching with a sterile glass rod microscopically after 1-week cultivation. In the next 2-3 week, pericyte clumps had formed and were dispersed selectively with dispase II solution. The cultures obtained by reseeding these clumps still contained small numbers of ECs and thus again proliferated undoubtedly rapidly. These contaminating cells could be eliminated finally by maintenance of the culture in starvation medium (medium 199, supplemented with the usual antibiotics, but free of glutamine and serum) for 2 month.
Immunofluorescence
Paraformaldehyde was used to fix CFs. Triton X-100 was employed for penetration of the cell membrane. 1% bovine serum albumin was used to block for 30 min at the room temperature. Rabbit polyclonal anti-α-SMA primary antibody (Abcam, Cambridge, MA), goat polyclonal anti-PDGFRα and anti-PDGFRβ (both from R&D Systems, Minneapolis, MN) were added and incubated at 4°C overnight. After washing with PBS, the cells were incubated with a goat anti-rabbit secondary antibody conjugated to TRITC (Invitrogen, Carlsbad, CA) for 1 h at the room temperature. PBS was used for washing. Finally, DAPI (Sigma-Aldrich) was employed for nuclear re-staining. The cells were observed under a fluorescence microscope.
Cell cycle analysis
100 μL cell suspension containing 5000 cells (with serum medium) was seeded into a 96-well plate. Additionally, one empty well was added 100 μL culture medium without cells, and marginal well was added with PBS. After routine culture for 24 h, culture solution containing Ang II (100 nM) or Ang II (100 nM) +1, 2.5, 5 mM of VPA (P4543, Sigma Aldrich, Sweden), trichostatin A (TSA, Sigma Aldrich) or VPA analogue penta was used for cell culture for another 48 h. DMEM containing 10% FBS was used as a control. Isovolume of media containing CCK-8 (Dojindo, Japan) was added to each well. The cells were incubated at 37°C for 1.5 h. Microplate assay (Thermo, USA) was used to detect the absorbance at 450 nm. Statistical analysis was performed and the experiment was repeated independently for three times.
Pericytes cell viability (%) = [A(dosing)-A(blank)]/[A(0 dosing)-A(blank)] × 100%. A(dosing): Absorbance of pores with cells, CCK solutions, and drugs; A(blank): Concentration of pores with no culture or CCK solution; A(0 dosing): Absorbance of pores with cells and CCK solutions without drugs.
Migration assay
Pericytes were cultured in a 12-well plate coated with fibronectin (10 mg/l) (Sigma Aldrich, St. Louis, MO) until confluence. A 200 μL pipete tip was used to mark two orthogonal straight lines slightly forcibly perpendicular to the cell culture side. PBS was used for washing twice. Intersection of the two orthogonal straight lines was regarded as the center under the inverted microscope, and continuous 5 visual fields were photographed under 5 × objective lens. Cells were starved with serum-free medium for 8 h before treatments. Culture solution containing Ang II (100 nM) or Ang II (100 nM) +1, 2.5, 5 mM of VPA (P4543, Sigma Aldrich, Sweden), trichostatin A (TSA, Sigma Aldrich) or VPA analogue penta was used for cell culture for another 48 h. DMEM containing 10% FBS was used as a control. The pericytes were stained with crystal violet and the migration was recorded. The migration scores were judged according to the below criterion: 0, no migration; 1, migration began along the border but cells did not span the gap/void; 2, migrating cells were spanning the gap/void but did not completely populate the gap/void; 3, migrating cells completely repopulated the gap/void [10]. The average score of each group was calculated and compared.
Immunoblotting and quantitative PCR
Cells were collected, and cell lysis solution was added to extract the proteins and the protein content was determined. The equivalent of 30 μg protein samples were separated by SDS-PAGE, and were transferred onto a PVDF membrane. They were washed with 1 × TBST for three times, 10 min for each time. The membrane was then incubated with primary antibodies including PDGFRα, PDGFRβ (Cell Signaling Technology, Beverly, MA), α-SMA (Sigma Aldrich, St. Louis, MO), β-actin (Abcam) and phospho-p44/42 MAPK (ERK1/2) (Cell Signaling Technology, Beverly, MA) at 4°C overnight. After washing with TBST for three times, HRP-secondary antibody was incubated at the room temperature for 2 h. ECL was used to cover the membrane in the dark room for 5 min. Gel imaging system was used to scan and analyze absorbance.
Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA). Spectrophotometer was used to measure the purity and quantity of RNA. Reverse transcription was performed to produce cDNA using a reverse transcription reagent kit (Promega, Madison, WI). Reverse transcription system: 2 μg total RNA, 0.5 μg oligo-dT primer, 1 μL dNTP mixture, 2 μL RNase inhibitor, and 4 unit reverse transcriptase. Reverse transcription procedures: preserving heat at 42°C for 60 min, and inactivating reverse transcriptase at 95°C for 5 min, 4°C 5 min. Real time-PCR system: cDNA 1 μL, SYBR Green I (Roche Applied Science) 10 μL, forward primer (20 μmol/L) 0.5 μL, reverse primer (20 μmol/L) 0.5 μL, and DEPC-treated water 8 μL. All RT-PCR amplification system was the same. Amplification conditions: Initial denaturation at 94°C for 2 min; template denaturation at 94°C for 30 s; annealing at 57°C for 30 s; extension at 72°C for 30 s. β-actin mRNA was regarded as the internal reference. The following expressing formula was adopted: the expression level of the target gene = 2-ΔΔCt. The primers are listed in Table 1.
Table 1.
Primers used for quantitative real-time PCR
| Gene | 5-3 | Amplified fragment length |
|---|---|---|
| α-SMA | Sense: GGGCTGTCCCCATGAGAATG | 467 bp |
| Antisense: GACTTGCCGCCTACACTGAT | ||
| PDGFRα | Sense: TCAACTCGGCAAGCTCTCAG | 717 bp |
| Antisense: TCCTCAGGAGGCATGTTTGG | ||
| PDGFRβ | Sense: CAGGTGCAAAGGAGTTGTGC | 864 bp |
| Antisense: CAATAAGAACGCTGAGCGGC | ||
| GAPDH | Sense: TGACGGGCACCCTTGATATG | 686 bp |
| Antisense: CCAAGTCACTGTCACACCAGA |
Data analysis
All data were presented as mean ± SEM, and analyzed with SPSS 13.0 statistical software. Means between two groups were compared using t test. Means among multiple groups were compared using one-way ANOVA. Difference among groups was analyzed. P < 0.05 indicated significant difference.
Results
HDAC inhibitor VPA blocks cardiac fibrosis
VPA treatment for two weeks did not notably influence animal body weight (Figure 1A) or Ang II-induced hypertension (Figure 1B), indicating that VPA did not block the initial phase of cardiac remodeling. Moreover, Ang II did not induce significant LV hypertrophy or abnormalities of cardiac systolic and diastolic function. Effects of VPA on cardiac fibrosis were assessed by picrosirus red staining of the LV sections. Two weeks of Ang II treatment led to an evident collagen deposition in cardiac interstitium, which was completely abolished by VPA (Figure 1C, 1D). These data indicated that HDACs inhibitor VPA played an important role in the pathogenesis of myocardial fibrosis.
Figure 1.
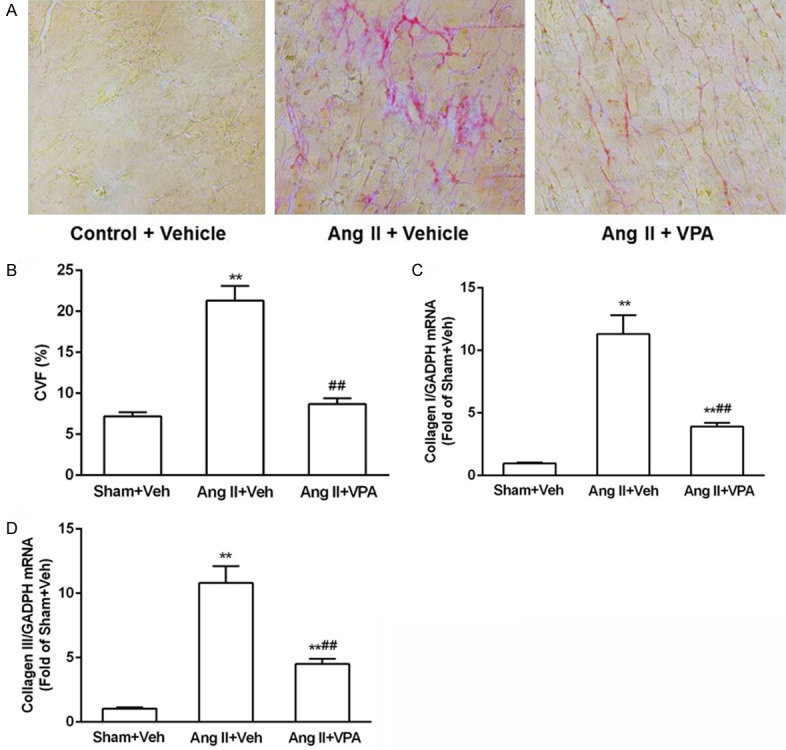
HDACs inhibitor VPA inhibits cardiac fibrosis. A: LV sections stained with picrosirius red for interstitial collagen. LVs were obtained from rats treated with Ang II in the absence or presence of VPA for 2 weeks. Scale bar = 10 μm. B: Quantification of cardiac interstitial collagen area from picrosirius red-stained section with results expressed as (collagen volume fraction, CVF). C and D: Changes in expression levels of collagen I and collagen III mRNA from rats treated with Ang II in the absence or presence of VPA for 2 weeks. Data represent mean ± SEM. *P < 0.05 vs Sham + Vel group, #P < 0.05 vs Ang II + Vel group, **P < 0.01 vs Sham + Vel group, ##P < 0.01 vs Ang II + Vel group.
VPA does not reduce Ang II-mediated inflammation in the LV
Inflammatory cells and factors play an important role in the pathophysiology of cardiac fibrosis, and recent study demonstrated that recruitment of leukocytes to the heart is essential for myocardial fibrosis [19,20]. HDAC inhibitors exert broad anti-inflammatory activity [21]. It is probably that cardiac fibrosis is suppressed by VPA owing to inhibition of Ang II-mediated inflammatory cells recruitment. HE staining of heart section showed that Ang II induced the infiltration of inflammatory cells, but VPA treatment did not reduce the infiltration of inflammatory cells in the heart (Figure 2A). qPCR results indicated that IL-6 level was elevated by Ang II treatment, and VPA did not reduce IL-6 mRNA level (Figure 2B). Flow cytometry analysis indicated that total leukocyte (CD45+ cells) numbers were markedly elevated in the hearts of the rats receiving Ang II (Figure 2C, 2D). Unexpectedly, leukocyte infiltration was not declined in VPA-treated group (Figure 2C and 2D). Furthermore, VPA also failed to decrease Ang II-induced monocytes/macrophages (CD45+CD11b+Ly6G-) and neutrophils (CD45+CD11b+Ly6G+) infiltration to the heart (Figure 2E-G). These data demonstrate that HDAC inhibitor blocks cardiac fibrosis independently of effects on inflammatory responses, there must exist other mechanisms or cells responsible for VPA-induced inhibition of Ang II-mediated myocardial fibrosis.
Figure 2.
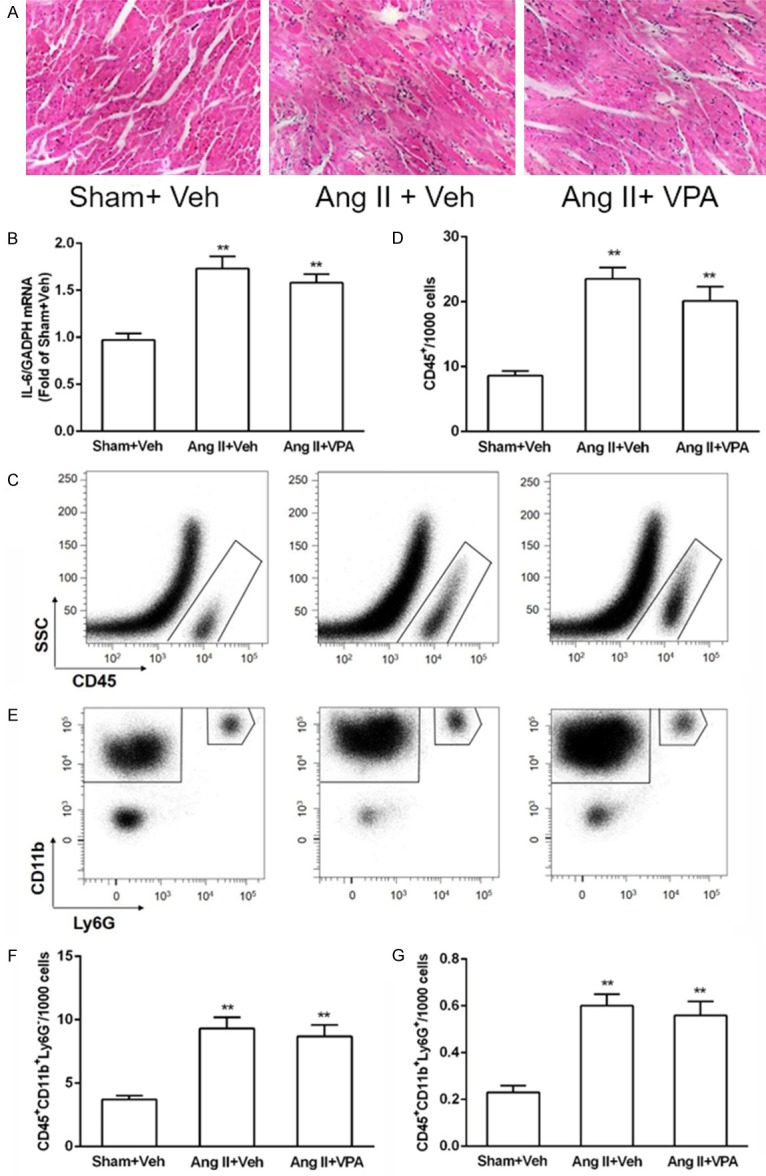
VPA does not reduce Ang II-mediated inflammation in the LV. A: Heart sections of rats treated with Ang II in the absence or presence of VPA were stained by H&E staining, and arrows indicated inflammatory cells. B: IL-6 mRNA expressions was not decreased by VPA treatment. C: Cells falling within the FSC/SSC gate was analyzed for CD45+ positivity against SSC. D: Quantification of CD45+ cells in rat LV, n = 3/group. E: CD45+ cells were then further analyzed for expression of CD11b and Ly6G to identify the monocyte/macrophage and neutrophil populations. F and G: Quantification of indicated populations of monocytes/macrophages (CD45+CD11b+Ly6G-) and neutrophils (CD45+CD11b+Ly6G+) in LV single-cell suspensions, n = 3/group. Data represent mean ± SEM. **P < 0.01 vs Sham + Vel group.
VPA attenuates Ang II-induced pericyte-myofibroblast trans-differentiation in vitro
To examine HDACs inhibition on reversal of pericyte-myofibroblast trans-differentiation, pericytes were induced differentiation by Ang II for 48 h and then treated with VPA at different concentrations in the presence of Ang II for another 48 h. In control groups, cells were incubated with either 10% FBS (pericyte) or Ang II (myofibroblast). Microvascular pericytes are long, branching cells with thin filopodia, whereas Ang II-treated cells adopted spindle-shaped morphology, and treatment with VPA maintained pericytes in a branched phenotype in the presence of Ang II. Immunofluorescence data showed that VPA-treated cells stained less intense for α-SMA, PDGFRα and PDGFRβ, indicating that HDACs inhibition induced a reversal of trans-differentiation (Figure 3A). Consistently, VPA reduced the mRNA levels of the myofibroblast markers α-SMA, PDGFRα and PDGFRβ in a dose-dependent manner that was significant at 5 mM of VPA (Figure 3B). This was verified at the protein level by Western blot analysis (Figure 3C).
Figure 3.
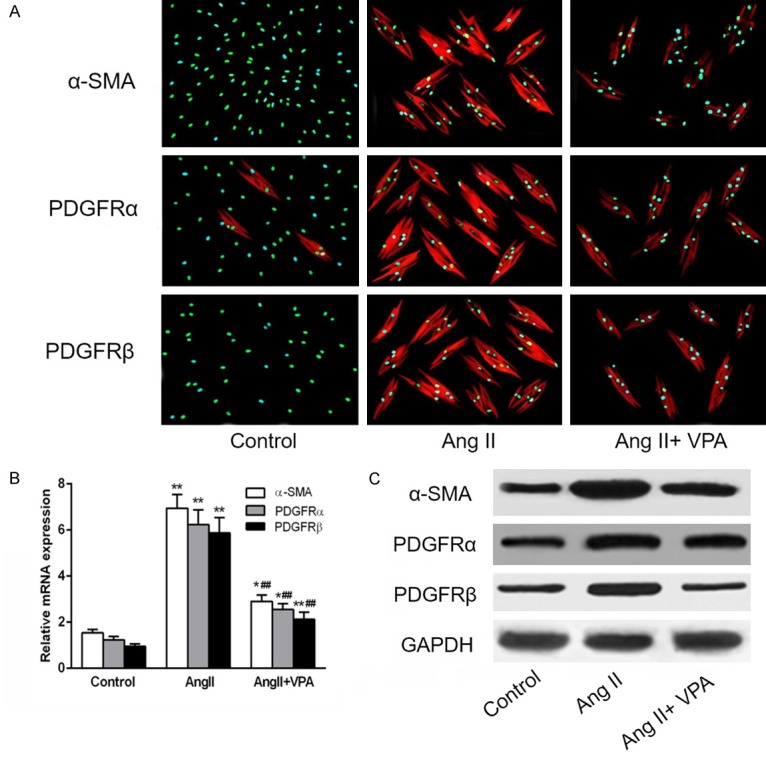
VPA attenuates Ang II-induced pericyte-myofibroblast trans-differentiation in vitro. Pericytes were treated with Ang II in the absence or presence of VPA. (A) Immunofluorescence images showing that VPA inhibited Ang II-stimulated α-SMA, PDGFRα and PDGFRβ expression. (B and C) Quantitative PCR (Q-PCR) (B) and western blot analysis (C) of α-smooth muscle actin (α-SMA), PDGFRα and PDGFRβ of Control, Ang II treated alone and in presence of VPA in pericytes treated with antibody as indicated. Data represent mean ± SEM. *P < 0.05 vs Control group, **P < 0.01 vs Control group, ##P < 0.01 vs Ang II alone treated group.
VPA reduces Ang II-mediated cardiac pericytes proliferation in a dose-dependent manner
To investigate whether VPA affects pericyte cell proliferation, pericytes were treated with Ang II for 48 h. Subsequently, the cells were treated with VPA at different concentration for another 48 h. VPA had a prominent effect on proliferation of pericyte, and 1 mM VPA was sufficient to markedly inhibit the proliferation of pericytes (Figure 4A). To exclude the effect of VPA on the number of viable cells, cell viability was examined. No significant differences in the percentage of PI+ cells were observed when pericytes were exposed to VPA, TSA or Penta for 24 h when compared to control pericytes (Figure 4B).
Figure 4.
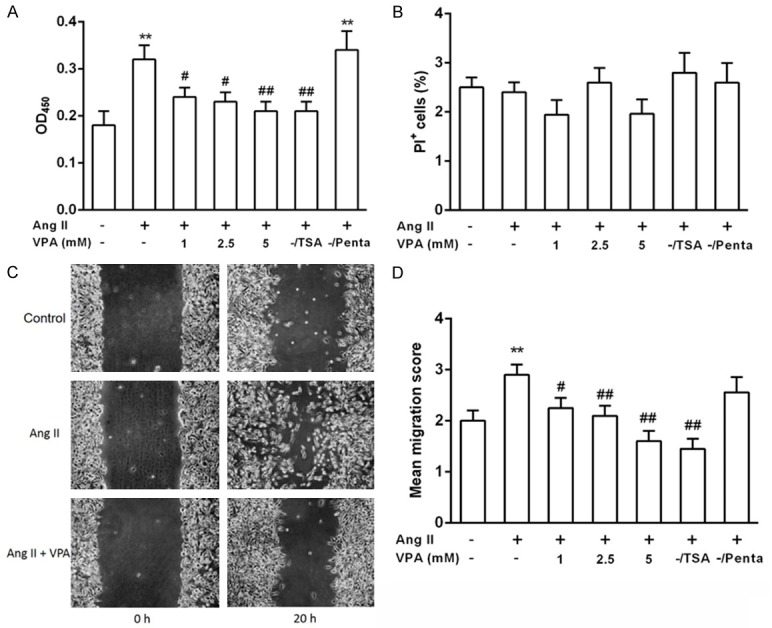
VPA inhibits Ang II-induced proliferation and migration of pericytes. (A) Cultured pericytes were synchronized and then stimulated with Ang II in the absence or presence of VPA, TSA, a pan-HDAC inhibitor or Penta, a VPA analogue. Cell Counting Kit-8 (CCK-8) was used to examine cell proliferation ability. (B) As the indicated treatments in (A), cells were stained with propidium iodide and cell cycle progression analyzed by flow cytometry. (C) Representative images of pericyte cultures in the wound healing assay at 0 and 20 h stimulated with Ang II in the absence or presence of VPA. (D) Migration scores were determined in pericyte exposed to different concentrations of VPA, TSA or Penta. Data represent mean ± SEM. #P < 0.05 vs Ang II +/VPA-group, **P < 0.01 vs Control group, ##P < 0.01 vs Ang II +/VPA-group.
VPA inhibits Ang II-mediated pericytes migration
The effect of VPA on migration of pericytes was examined in a wound healing assay (Figure 4C). The migration score was quantified as described in Materials and Methods. Pericyte migration was greatly reduced when pericytes were exposed to 2.5 mM of VPA. No significant effect on pericyte migration was observed at lower VPA concentration (Figure 4D). The VPA analogue Penta lacking HDAC inhibitory activity had no obvious influence on pericyte migration (Figure 4D). Pericytes exposed to 10 nM TSA, a more potent HDAC inhibitor, showed a markedly reduced migration (Figure 4D). This effect was not seen at 5 nM TSA.
Abrogation of pericytes-to-myofibroblast differentiation is involved in inhibiting the phosphorylation of ERK
To identify the molecular mechanism by which VPA inhibits pericyte-to-myofibroblast differentiation, we first measured the activity of HDACs; The results showed that VPA at 1 mM was sufficient to inhibit the activity of HDACs as indicated by increased acetylation of histone 3 (Figure 5A). ERK/MAPK pathway is significant for mesenchymal cells to differentiate into smooth muscle cells marked by α-SMA expression. We then explored whether the phosphorylation of ERK1/2 was necessary for Ang II-induced myofibroblast differentiation. Ang II alone significantly upregulated the phosphorylation of ERK1/2. In contrast, Ang II and VPA co-treatment decreased the phosphorylation of ERK1/2 (Figure 5B).
Figure 5.
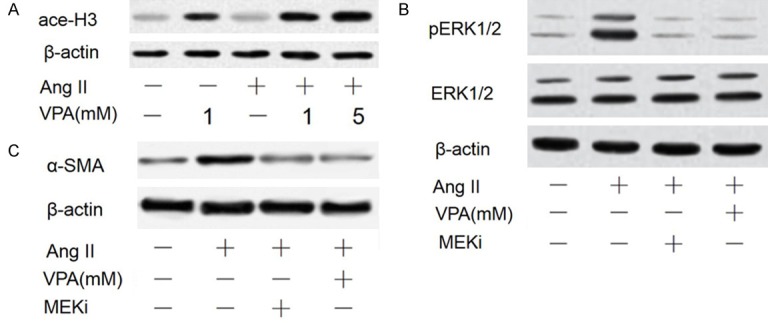
Inhibition of Ang II-mediated ERK activation by HDAC blockade. (A) The acetylation of histone 3 (Lys23) was assessed by Western blot after treatment for 12 h. (B) The abundance of phosphorylated ERK1/2 was determined in pericytes exposed to Ang II with and without VPA for 12 h or pretreated with the MEK inhibitor (MEKi) (50 μM PD98059) for 1 h and then stimulated with Ang II for 12 h. (C) The culture condition was similar to (C), and the expression of α-SMA was measured by western blot in pericytes treated with Ang II with and without VPA and MEKi.
To further clarify that ERK1/2 is involved in regulating pericyte-to-myofibroblast differentiation, a MEK inhibitor, MEKi (50 μM PD98059) was used to pretreat pericytes for 1 h before stimulating the cells with Ang II for 24 h. Western blotting results showed that treatment of cardiac pericytes with MEKi significantly reduced Ang II-induced increase in pERK1/2 and α-SMA expression at protein and mRNA level (Figure 5B, 5C). These results suggest that VPA-mediated reduction in pERK1/2 is enough to inhibit the expression of α-SMA, and the activation of ERK1/2 is necessary for Ang II-mediated α-SMA expression.
HDAC4 is required for the Ang II-induced myofibroblast differentiation
As previously described, HDAC4 is important for TGFβ1-induced α-SMA expression in dermal fibroblasts [22]. We identified whether HDAC4 is also important for α-SMA expression in Ang II-stimulated myofibroblast differentiation in pericytes and whether HDAC4 is involved in the regulation of ERK phosphorylation. We employed siRNA to knock down HDAC4 in our pericytes following Ang II treatment. As shown in Figure 6A, knockdown with siRNA markedly blocked HDAC4 expression at mRNA level. The expression of α-SMA at both the protein and mRNA level was also inhibited by HDAC4 knockdown with or without Ang II (Figure 6B, 6C). HDAC4 knockdown also blocked Ang II-mediated ERK1/2 phosphorylation (Figure 6D), which suggests that ERK1/2 phosphorylation is modulated by HDAC4 in the regulation of Ang II-mediated α-SMA expression (Figure 6).
Figure 6.
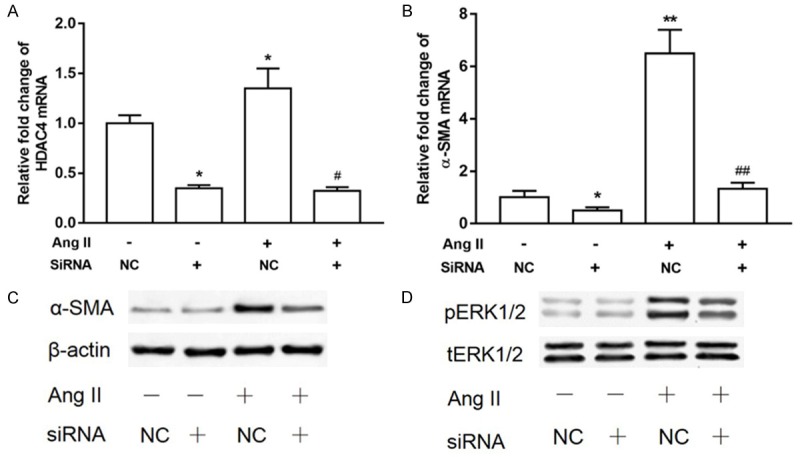
HDAC4 is required for Ang II-mediated α-SMA expression and ERK1/2 activation. (A) SiRNA or negative control (NC) siRNA targeting HDAC4 were transiently transfected into pericytes. The pericytes were subsequently cultivated in the presence or absence of Ang II for 12 h. Quantitative PCR (Q-PCR) was performed to examine HDAC4 (A) and α-SMA (B) mRNA levels (n = 3, means ± SEM). *P < 0.05 vs untreated control; #P < 0.05 vs Ang II alone treated group. (C) HDAC4 knockdown and control pericytes were cultivated in the presence or absence of Ang II for 24 h. Western blot analysis was performed using α-SMA and β-actin antibodies, and then the membranes were stripped and probed for pERK1/2 and total-ERK1/2 (tERK1/2) (D).
Pharmacological inhibitor of PP2A and PP1 restores the expression of α-SMA phosphorylation of ERK1/2
Since both PP1 and PP2A can dephosphorylate ERK1/2 [23], we used two potent PP1 and PP2A inhibitors, Calyculin A and Tautomycin [24], to inhibit PP1 and PP2A in pericytes and examine whether ERK1/2 phosphorylation is necessary for induction of α-SMA in response to Ang II. Western analysis demonstrated that the PP1 and PP2A inhibitors Tautomycin (0.05 nM) and Calyculin A (0.5 nM) significantly rescued α-SMA expression (Figure 7A) from the inhibitory effects of VPA. Moreover, Tautomycin and Calyculin A treatment also rescued ERK1/2 phosphorylation (Figure 7B). Our results suggested that PP1 and PP2A, one or both of them, is/are necessary for the dephosphorization of ERK1/2 and the inhibition of α-SMA expression by VPA.
Figure 7.

PP1 and PP2A inhibitors restore α-SMA expression and ERK1/2 phosphorylation downregulated by VPA. After serum starvation, pericytes were pretreated with the PP1 and PP2A inhibitors T (Tautomycin) and C (Calyculin A) for 1 h followed by treatment with Ang II and VPA. Western blot was performed with α-SMA (A), pERK1/2, tERK1/2 (B), and β-actin antibodies.
Discussion
HDACs and HATs play important roles in the remodeling of chromatin structures [25]. The acetylation and deacetylation of histone is controlled by HATs and HDACs [26,27]. HDACs are increasingly known as targets for the treatment of several diseases, including heart failure and cardiac hypertrophy [28,29]. In the present study, we identified a crucial role for HDAC4 in Ang II-stimulated cardiac pericyte-myofibroblast differentiation via a mechanism regulating the phosphorylation of ERK. Our data showed that VPA, an HDAC inhibitor, attenuated cardiac fibrosis in Ang II-treated rats, which was characterized by reduced type I collagen expression.
We originally hypothesized that repression of fibrosis by class I HDAC inhibitors was associated with a suppression of inflammatory cells or factors, since it has long been recognized that inflammatory cells and factors played an important role in the pathophysiology of cardiac fibrosis [30,31]. In addition, HDACs inhibitors exert broad anti-inflammatory activity [32]. Surprisingly, however, treatment of the mice with VPA failed to block accumulation of monocytes, macrophages and neutrophils in the hearts of Ang II-treated rats.
In response to pressure overload, myocardial infarction or humoral factors stimulation, fibroblasts, fibrocytes and pericytes could proliferate and produce excess amounts of ECM, and transdifferentiate into myofibroblasts, characterized by increased α-SMA and h-collagen I expression. Consistent with this idea, our results showed that VPA had a prominent effect on proliferation of pericyte, and 1 mM VPA was sufficient to markedly inhibit the proliferation of pericytes. The VPA analogue Penta which lacks HDACs inhibitory ability had no effects on pericyte proliferation. These data indicated that the anti-proliferative effect was probably due to the inhibitory ability of VPA on HDACs. That was further supported by experiments using TSA, another more potent HDACs inhibitor, which had similar anti-proliferative effects compared to VPA. In conclusion, these data support a role for VPA in suppressing pericyte proliferation due to its inhibitory effect on HDACs.
The present data show that VPA attenuates pericyte migration. Generally, the influence of cell proliferation and viability on migration must be taken into consideration. In this study, no significant differences in the number of viable cells were observed after exposing pericytes to the different concentrations of VPA in the wound healing assay. Thus, the effect of cell viability should not be considered. Inhibitory effects on proliferation were found in pericytes treated with 1 mM VPA, and the inhibitory effect on migration was first notable when pericytes exposing to 2.5 mM VPA. Thus, a decrease in pericyte proliferation could not influence the observed results. The HDACs inhibitor TSA had a similar effect on pericyte migration. No obvious effect on migration was observed when cells were treated with the VPA analogue Penta, suggesting that inhibitory effect of VPA on pericyte migration was due to its HDACs inhibition ability.
HDACs regulate pathological cardiac conditions such as fibrosis [28] and hypertrophy [33]. Here we show that HDAC4 knockdown not only inhibited α-SMA expression but also suppressed the phosphorylation of ERK1/2. The PP1 and PP2A inhibitors Tautomycin and Calyculin A restored Ang II-mediated α-SMA expression from the inhibitory effects of VPA. HDAC4 forms a complex with the PP2A holoenzyme to regulate its nuclear import and thus, we hypothesize that HDAC4/PP2A complex are parts of the α-SMA transcriptional complex, and HDAC4 is dephosphorylated by PP2A to maintain a state of transcriptional inhibition. When HDAC4 is inhibited by VPA or siRNA knockdown, the PP2A may be released and subsequently dephosphorylate ERK1/2; however, further work is required to confirm this hypothesis.
MAPKs pathways play important role in cell growth, apoptosis and trans-differentiation [34,35]. ERK1/2 is a key growth signaling kinase among the three major MAPKs. ERK phosphorylation is regulated by kinases and protein phosphatases. Once activated, ERK1/2 could active a series of intracellular targets and result in gene expression. Several reports pointed out that Ang-II induced phosphorylation of ERK1/2 in various cell types contributing to remodeling through enhanced proliferation, migration, inflammation and fibrosis [36-38]. In the present study, we notice that phospho-ERK1/2 protein levels were significantly increased under Ang II stimulation, and 1 mM of VPA was sufficient to markedly reduce ERK1/2 phosphorylation, MEK inhibitors significantly block Ang II-induced increase in α-SMA expression at protein and mRNA level. The knockdown or inhibition of HDAC4 dephosphorylates ERK1/2, suggesting that VPA may dephosphorylate ERK by releasing PP2A/1 from HDAC-PP2A/1 complexes leading to increased PP2A/1/ERK association. Moreover, we showed that Tautomycin and Calyculin A could restore the α-SMA expression and phosphorylation of ERK1/2 (Figure 7), however, further work is needed to specify whether PP1 or PP2A alone, or both of them combined, are involved in regulating of VPA on Ang II activity.
In summary, we demonstrated that the activation of ERK played a crucial role in Ang II-stimulated cardiac pericyte-myofibroblast transdifferentiation and that either a HDAC inhibitor or a specific siRNA for HDAC4 can suppress this ERK-dependent Ang II pathway.
Conclusions
This study provides new evidence that VPA inhibits proliferation, migration and differentiation of pericytes into myofibroblasts induced by Ang II and suggests that pericytes could be used as a novel therapeutic target to prevent fibrosis. Future in vivo studies focusing on different types of HDAC inhibitors in pathological conditions will be required to illustrate the effect of HDAC inhibition on pericytes.
Acknowledgements
This research was supported by the funds of Fujian Natural Science Foundation of China (2016J01482) and Fuzhou science and technology project (2016S0008).
Disclosure of conflict of interest
None.
References
- 1.Mendoza-Milla C, Czubryt MP. Tissue inhibitor of metalloproteinases-1 regulation by aldosterone: breaking the balance in cardiac fibrosis. Hypertension. 2016;67:1121–3. doi: 10.1161/HYPERTENSIONAHA.116.06873. [DOI] [PubMed] [Google Scholar]
- 2.van de Schoor FR, Aengevaeren VL, Hopman MT, Oxborough DL, George KP, Thompson PD, Eijsvogels TM. Myocardial fibrosis in athletes. Mayo Clin Proc. 2016;91:1617–1631. doi: 10.1016/j.mayocp.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Meloni M, Izzo V, Vainieri E, Giurato L, Ruotolo V, Uccioli L. Management of negative pressure wound therapy in the treatment of diabetic foot ulcers. World J Orthop. 2015;6:387–393. doi: 10.5312/wjo.v6.i4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Díez J. Diagnosis and treatment of myocardial fibrosis in hypertensive heart disease. Circ J. 2008;72:A8–12. doi: 10.1253/circj.cj-07-1067. [DOI] [PubMed] [Google Scholar]
- 5.Buckley ST, Medina C, Ehrhardt C. Differential susceptibility to epithelial-mesenchymal transition (EMT) of alveolar, bronchial and intestinal epithelial cells in vitro and the effect of angiotensin II receptor inhibition. Cell Tissue Res. 2010;342:39–51. doi: 10.1007/s00441-010-1029-x. [DOI] [PubMed] [Google Scholar]
- 6.Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen YT, Chang FC, Wu CF, Chou YH, Hsu HL, Chiang WC, Shen J, Chen YM, Wu KD, Tsai TJ. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011;80:1170–1181. doi: 10.1038/ki.2011.208. [DOI] [PubMed] [Google Scholar]
- 8.Lin SL, Chang FC, Schrimpf C, Chen YT, Wu CF, Wu VC, Chiang WC, Kuhnert F, Kuo CJ, Chen YM, Wu KD, Tsai TJ, Duffield JS. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol. 2011;178:911–923. doi: 10.1016/j.ajpath.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsai YP, Wu KJ. Epigenetic regulation of hypoxia-responsive gene expression: focusing on chromatin and DNA modifications. Int J Cancer. 2014;134:249–256. doi: 10.1002/ijc.28190. [DOI] [PubMed] [Google Scholar]
- 10.Karen J, Rodriguez A, Friman T, Dencker L, Sundberg C, Scholz B. Effects of the histone deacetylase inhibitor valproic acid on human pericytes in vitro. PLoS One. 2011;6:e24954. doi: 10.1371/journal.pone.0024954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawaoka K, Doi S, Nakashima A, Yamada K, Ueno T, Doi T, Masaki T. Valproic acid attenuates renal fibrosis through the induction of autophagy. Clin Exp Nephrol. 2017;21:771–780. doi: 10.1007/s10157-016-1365-6. [DOI] [PubMed] [Google Scholar]
- 12.Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blaheta RA, Cinatl J Jr. Anti-tumor mechanisms of valproate: a novel role for an old drug. Med Res Rev. 2002;22:492–511. doi: 10.1002/med.10017. [DOI] [PubMed] [Google Scholar]
- 14.Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro Oncol. 2010;12:328–340. doi: 10.1093/neuonc/nop005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miglietta A, Bozzo F, Gabriel L, Bocca C, Canuto RA. Extracellular signal-regulated kinase 1/2 and protein phosphatase 2A are involved in the antiproliferative activity of conjugated linoleic acid in MCF-7 cells. Br J Nutr. 2006;96:22–27. doi: 10.1079/bjn20061619. [DOI] [PubMed] [Google Scholar]
- 16.Chuang MJ, Wu ST, Tang SH, Lai XM, Lai HC, Hsu KH, Sun KH, Sun GH, Chang SY, Yu DS, Hsiao PW, Huang SM, Cha TL. The HDAC inhibitor LBH589 induces ERK-dependent prometaphase arrest in prostate cancer via HDAC6 inactivation and down-regulation. PLoS One. 2013;8:e73401. doi: 10.1371/journal.pone.0073401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams SM, Golden-Mason L, Ferguson BS, Schuetze KB, Cavasin MA, Demos-Davies K, Yeager ME, Stenmark KR, McKinsey TA. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J Mol Cell Cardiol. 2014;67:112–125. doi: 10.1016/j.yjmcc.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nees S, Weiss DR, Senftl A, Knott M, Forch S, Schnurr M, Weyrich P, Juchem G. Isolation, bulk cultivation, and characterization of coronary microvascular pericytes: the second most frequent myocardial cell type in vitro. Am J Physiol Heart Circ Physiol. 2012;302:H69–84. doi: 10.1152/ajpheart.00359.2011. [DOI] [PubMed] [Google Scholar]
- 19.Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial J, Taffet GE, Entman ML. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2010;49:499–507. doi: 10.1016/j.yjmcc.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu J, Lin SC, Chen J, Miao Y, Taffet GE, Entman ML, Wang Y. CCR2 mediates the uptake of bone marrow-derived fibroblast precursors in angiotensin II-induced cardiac fibrosis. Am J Physiol Heart Circ Physiol. 2011;301:H538–547. doi: 10.1152/ajpheart.01114.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011;32:335–343. doi: 10.1016/j.it.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Glenisson W, Castronovo V, Waltregny D. Histone deacetylase 4 is required for TGFbeta1-induced myofibroblastic differentiation. Biochim Biophys Acta. 2007;1773:1572–1582. doi: 10.1016/j.bbamcr.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 23.Sunahori K, Nagpal K, Hedrich CM, Mizui M, Fitzgerald LM, Tsokos GC. The catalytic subunit of protein phosphatase 2A (PP2Ac) promotes DNA hypomethylation by suppressing the phosphorylated mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (MEK)/phosphorylated ERK/DNMT1 protein pathway in T-cells from controls and systemic lupus erythematosus patients. J Biol Chem. 2013;288:21936–21944. doi: 10.1074/jbc.M113.467266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujiki H, Suganuma M. Tumor promotion by inhibitors of protein phosphatases 1 and 2A: the okadaic acid class of compounds. Adv Cancer Res. 1993;61:143–194. doi: 10.1016/s0065-230x(08)60958-6. [DOI] [PubMed] [Google Scholar]
- 25.Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics. 2012;4:5. doi: 10.1186/1868-7083-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 27.Patel J, Pathak RR, Mujtaba S. The biology of lysine acetylation integrates transcriptional programming and metabolism. Nutr Metab (Lond) 2011;8:12. doi: 10.1186/1743-7075-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, Yin Z, Ahn Y, Jeong MH, Bang YJ, Kim N, Kim JK, Kim KK, Epstein JA, Kook H. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation. 2006;113:51–59. doi: 10.1161/CIRCULATIONAHA.105.559724. [DOI] [PubMed] [Google Scholar]
- 29.Cho YK, Eom GH, Kee HJ, Kim HS, Choi WY, Nam KI, Ma JS, Kook H. Sodium valproate, a histone deacetylase inhibitor, but not captopril, prevents right ventricular hypertrophy in rats. Circ J. 2010;74:760–770. doi: 10.1253/circj.cj-09-0580. [DOI] [PubMed] [Google Scholar]
- 30.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKinsey TA. Targeting inflammation in heart failure with histone deacetylase inhibitors. Mol Med. 2011;17:434–441. doi: 10.2119/molmed.2011.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, Rindt H, Gorczynski RJ, Olson EN. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem. 2003;278:28930–28937. doi: 10.1074/jbc.M303113200. [DOI] [PubMed] [Google Scholar]
- 34.Pan BS, Wang YK, Lai MS, Mu YF, Huang BM. Cordycepin induced MA-10 mouse leydig tumor cell apoptosis by regulating p38 MAPKs and PI3K/AKT signaling pathways. Sci Rep. 2015;5:13372. doi: 10.1038/srep13372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu C, Chen F, Han X, Xu H, Wang Y. Role of TGF-β1/p38 MAPK pathway in hepatitis B virus-induced tubular epithelial-myofibroblast transdifferentiation. Int J Clin Exp Pathol. 2014;7:7923–30. [PMC free article] [PubMed] [Google Scholar]
- 36.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- 37.Touyz RM, Yao G, Viel E, Amiri F, Schiffrin EL. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J Hypertens. 2004;22:1141–1149. doi: 10.1097/00004872-200406000-00015. [DOI] [PubMed] [Google Scholar]
- 38.Karén J, Rodriguez A, Friman T, Dencker L, Sundberg C, Scholz B. Effects of the histone deacetylase inhibitor valproic acid on human pericytes in vitro. PLoS One. 2011;6:e24954. doi: 10.1371/journal.pone.0024954. [DOI] [PMC free article] [PubMed] [Google Scholar]