

Abstract
Sepsis is a major cause of death in intensive care units. The purpose of this study was to investigate the effect of resveratrol (RSV) on sepsis-induced acute lung injury (ALI). The underlying molecular mechanisms were deciphered by both in vitro and in vivo experiments. Polymicrobial sepsis was induced in C57BL/6 mice by cecal ligation and puncture (CLP). RSV pretreatment significantly attenuated CLP-induced acute lung injury, which was associated with enhanced expression of VEGF-B. The protective properties of RSV were assayed in lipopolysaccharide (LPS)-stimulated MH-S cells. We determine that RSV administration inhibited the increased production of TNF-α, IL-6, and IL-1β in LPS-stimulated MH-S cells, which was associated with inhibition of the nuclear factor-κB, P38, and ERK signaling pathways. We also provide evidence that RSV administration reduced LPS-induced apoptosis of MH-S cells by altering the unbalance of Bax/Bcl-2 and inhibiting LPS-induced autophagy. The inhibitory effects of RSV on cytokine levels and apoptosis of alveolar macrophages were both blocked by VEGF-B siRNA. Furthermore, RSV administration regulated LPS-induced C5aR and C5L2 expression, revealing an additional mechanism underlying RSV’s anti-inflammatory and anti-apoptosis effects. Collectively, these results demonstrated that RSV was able to protect against sepsis-induced acute lung injury by activating the VEGF-B signaling pathway.
Keywords: RSV, acute lung injury, alveolar macrophages, anti-apoptosis, anti-inflammation, VEGF-B
Introduction
Despite improvements in supportive treatment for acute lung injury (ALI), ALI is common and devastating clinical disorders with high morbidity and mortality [1]. The pathophysiology of ALI is characterized by complex mechanisms that involve cell inflammation, cytokines, as well as abnormal apoptosis of pulmonary cells, including pulmonary alveolar type II epithelial cells, pulmonary vascular endothelial cells, alveolar macrophages (AMs) [2-5]. AMs account for about 90% of the cells in the bronchoalveolar lavage fluid (BALF) and play an important role in the pathogenesis of ALI [6]. Many current studies have indicated a consistent association between AMs and ALI in humans and animal models [7-9], because AMs apoptosis and dysfunction occur late in sepsis, which is implicated in the increased mortality [10]. However, little is known about the intracellular signaling pathways that regulate the fate of AMs in ALI, and efforts directed towards the accurate mechanisms to improve ALI detection and treatments are indispensable. Discovering new drugs and new strategies therefore remains an urgent therapeutic challenge in the treatment of ALI.
Resveratrol (3, 5, 4’-trans-trihydroxystilbene; RSV), a potent activator of SIRT-1 [11] present in mulberries, peanuts, and grape skins [12,13], is known for its anti-inflammatory, anti-oxidative and anti-apoptotic properties in multiple organs, including the lungs [14,15]. Emerging studies have reported that RSV has protective effect in sepsis. It can significantly extend the life span of septic animals and reduce acute lung injury in a LPS-induced sepsis mouse model [16,17]. RSV could attenuate the apoptosis of pulmonary microvascular endothelial cells (PMVECs) and suppresse ox-LDL-induced macrophage apoptosis [18,19]. However, the molecular mechanisms underlying the protective effects of RSV in septic ALI remain poorly understood.
The VEGF family includes five major factors: VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor [20]. VEGF-B bind to VEGF-receptor 1 (VEGFR-1) and the neuropilin-1 (NRP-1) coreceptor, which are expressed in lung and lung macrophages [21,22]. Recently, VEGF-B was found as a key regulator of endothelial fatty acid uptake and insulin sensitivity [23,24]. Its anti-cancer and neuroprotective effects were reported [25,26]. In addition, VEGF-B expression is reduced in human heart disease, and VEGFB mRNA expression is significantly increased in the hypoxic lung [27,28]. However, unlike VEGF-A, VEGF-B played a potent antiapoptotic effect while lacking a general antigenic activity [29,30]. VEGF-B treatment by intravitreal injection markedly reduced retinal apoptosis in streptozotocin (STZ)-induced diabetes rats [31]. However, the functions of VEGFB in macrophages under septic conditions remain poorly understood.
In the previous study, we have first identified the relationship between RSV treatment and the expression of VEGF-B [32]; we also found that RSV attenuates myocardial ischemia/reperfusion injury through upregulation of VEGF-B [33]. The purpose of this study was to investigate the protective mechanisms of RSV in sepsis-induced acute lung injury, particularly with regard to the VEGF-B signaling.
Materials and methods
Animals and experimental designs
SPF C57BL/6 male mice were purchased from China Laboratory Animal Resource Center of National Institute for Food and Drug Control. The experimental protocol was approved by the Medicine Ethical Committee of Tianjin Nankai Hospital. Mice were acclimatized for at least 1 week before use. Mice were fasted but had free access to water starting 12 h before the surgery. A median incision was made in the lower abdomen after anesthesia, skin was prepared and disinfected, and the cecum was slightly taken out of the incision. A 4-0 braided silk suture was passed through the midpoint between the colon root and cecum terminal, which was used to tighten the cecum while avoiding the cecum artery. After that, a 21 gauge 1 inch needle was used to penetrate the cecum ligation, from which a small drop of intestinal contents was squeezed after the needle was withdrawn, followed by cecum reposition and abdomen closure. For the sham group, the abdomen was opened, cecum exposed and repositioned, and incision closed. After successful modeling, mice in the CLP+RSV group immediately received RSV (40 mg/kg body weight).
Cell culture
MH-S cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in RPMI 1640 medium supplemented with 10 mM HEPES, 2 mM l-glutamine, 100 U/ml streptomycin, 100 U/ml penicillin, and 10% (v/v) FBS. Cells (approximately 1×106 cells/ml) were seeded in 6-well plates before being subjected to treatments. RSV at 50 µM was added 2 h before LPS (from Escherichia coli, Sigma) stimulation.
Histologic examination and grading
After the animals were killed, lung tissues embedded in paraffin and cut in 3 μm thick sections. Sections were stained with hematoxylin and eosin stain. The total score was calculated by adding up the individual scores of each category [34].
Pulmonary MPO activity
MPO activity, an indicator of polymorphonuclear leukocytes accumulation, was determined using an MPO kit produced by Jiancheng Bioengineering Institute (Nanjing, China) according to the manufacturer’s instruction.
Cell-viability assay
The cell viability in the various treatment groups was assessed with an MTT assay. In this assay, MH-S cells were rested in 96-well plates (5,000 cells/well). Following incubation with RSV (10 µM) for 2 h, cells were subsequently treated with different concentrations of LPS in complete medium for 24 h. Subsequently, 10 µl MTT was added to each well, and the plates were incubated for an additional four hours at 37°C. Subsequently, media was carefully discarded and 100 μl of dimethyl sulfoxide were added to dissolve the formazan crystals, then the 96-well plates were put on a horizontal oscillator for ten minutes. The absorbance values were measured by the enzyme mark instrument at a wavelength of 490 and a reference wavelength of 570 nm.
Measurement of apoptosis by flow cytometry analysis using the PI/Annexin V double-staining method
Cells were harvested and washed three times with cold PBS, then stained with 5 μL Annexin V-FITC and 10 μL propidium iodide (PI) for 15 min at room temperature in 500 mL of the binding buffer. Surface exposure of phosphatidylserine in apoptotic cells was measured by an Annexin V-FITC apoptosis detection kit according to the manufacturer’s instructions, and then the cells were analyzed with a FACSCalibur flow cytometer (BD Biosciences).
Immunofluorescence staining
After drug treatment, cells were blocked with 10% normal fetal bovine serum for 15 min at room temperature. Subsequently, cells were incubated with mouse anti-C5aR (Hycult biotech) antibody for 1 h at 4°C. After a washing with PBS, cells were incubated with the donkey anti-rat IgG (Abcam) for a further 30 min on ice in the dark. Cells were washed twice with PBS and analyzed using FACSCalibur (BD Biosciences). Mean fluorescence intensity (MFI) was calculated using CellQuest ProTM software (BD Biosciences).
RNA interference
Independent siRNA sequences were used to silence VEGF-B expression. The sequences used were as follows: sense, 5’-GGUGCCAUGGAUAGACGUUTT (dTdT)-3’ and anti-sense, 5’-AACGUCUAUCCAUGGCACCTT (dTdT)-3’. MH-S cells were transfected with control siRNA or VEGF-B siRNA (Shanghai GenePharma) by using the supplied transfection reagents according to the manufacturer’s instructions. At 24 hr after transfection, the cells were incubated with RSV followed by LPS treatment.
RNA extraction and quantitative real-time polymerase chain reaction
Total RNA was isolated using trizol reagent (Takara Bio, Japan) according to the manufacturer’s protocol. Reverse transcription reactions were performed using a 1 μg total RNA with TransGen Reverse Transcription Kit from Applied Biosystems. Quantitative real-time PCR detection of gene expression was performed using SYBR Green Master Mix (Promega, China) in a Bio-Rad IQ5 detection system and the cycle threshold (CT) values were automatically determined in triplicates and averaged. The following primers were used for qPCR: TNF-α, forward primer, 5’-CGTCAGCCGATTTGCTATCT-3’, and reverse primer, 5’-CGGACTCCGCAAAGTCTAAG-3’; IL-6, forward primer, 5’-AGTTGCCTTCTTGGGACTGA-3’, and reverse primer, 5’- TCCACGATTTCCCAGAGAAC-3’; IL-1β, forward primer, 5’-CTATGTCTTGCCCGTGGAG-3’, and reverse primer, 5’-CATCATCCCACGAGTCACA-3’; VEGF-B, forward primer, 5’-TTCACAGGGAGAAGAGTGGAGC-3’, and reverse primer, 5’-TCCCGTTATTGGTAGAAGTTTGG-3’; C5aR, forward primer, 5’-ACTCCCTGTGCGTGTCCC-3’, and reverse primer, 5’-CCCTGCCCACTGAATCCTC-3’; C5L2, forward primer, 5’-GGCATCTCAGACACCATTTCG-3’, and reverse primer, 5’-TTTCATTTGCTGGACCCCTTAT-3’; GADPH, forward primer, 5’-GCCTCGTCTCATAGACAAGATG-3’, and reverse primer, 5’-CAGTAGACTCCACGACATAC-3’.
Western blot analysis
Cells were lysed and incubated for 30 min in ice-cold RIPA buffer. Equal amounts of protein were subjected to SDS-PAGE for separation. After transferring onto the PVDF membrane, blocking with 5% fat-free milk. Expressions of Bax, Bcl-2, NF-κB, P-NF-κB, ERK1/2, P-ERK1/2, p38, P-P38 (Cell Signalling), LC3 (Novus) and GAPDH (Abcam) proteins were determined using respective specific antibodies (1/1000).
Statistical analysis
All the calculations were conducted using SPSS 17.0 software), and data charts were made using Prism 5.0 (GraphPad Software). All values are expressed as mean ± SEM. Values of P<0.05 were considered a statistically significant difference. Data sets were analyzed using one-way analysis of variance, and pairwise comparison was conducted using the Student-Newman-Keuls test.
Results
Treatment with RSV protected against CLP-induced acute lung injury
To evaluate the protective effect of RSV in CLP-induced acute lung injury, histological analyses of lung tissues were performed. As shown in Figure 1A and 1B, at 24 h after sepsis, histopathology of the lung sections demonstrated an acute inflammatory response including interstitial edema and thickening with inflammatory cell infiltration, microvascular congestion, and alveolar integrity destruction. Meanwhile, histological damage was visibly alleviated in the RSV group compared with the CLP group. As shown in Figure 1C and 1D, compared with the sham group, MPO activity of the lung tissues was elevated in the CLP group, (P<0.01), while RSV decreased MPO activity of lung tissues in CLP mice. In addition, the albumin level in BAL fluids was elevated in the CLP group; however, RSV significantly reduced albumin content in the BAL fluids from CLP mice (P<0.01).
Figure 1.
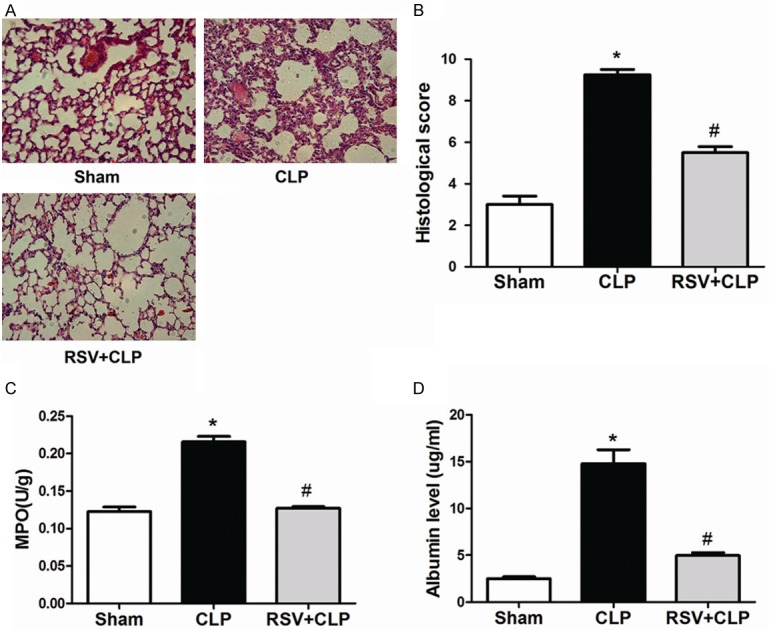
Treatment with RSV protected against CLP-induced acute lung injury. RSV alleviated sepsis-induced acute lung injury. A. Lung tissue sections stained with hematoxylin and eosin (original magnification 200×; scale bar 50 μm). B. Semi-quantitative analysis of lung tissues by lung injury score. C. MPO activity in lung tissues. D. Albumin level in BAL fluids was determined 24 h after CLP administration. The values presented are means ± SEM. (n = 6 in each group).*P<0.01 compared with the SHAM group, #P<0.01 compared with the CLP group.
Treatment with RSV suppressed production of proinflammatory cytokines by upregulating the expression of VEGF-B both in vivo and in vitro
To evaluate the protective effect of RSV on CLP-induced lung inflammatory response, proinflammatory cytokines were examined in BAL fluids. As shown in Figure 2A , compared with the sham group, levels of TNF-α, IL-1β, and IL-6 were significantly elevated in the CLP group, (P<0.01); meanwhile, this increase was significantly attenuated by RSV treatment. Macrophages are important cells involved in inflammation by secreting various inflammatory mediators. Therefore, we sought to determine the effects of RSV treatment on the secretion of proinflammatory mediators from LPS-stimulated alveolar macrophages. As shown in Figure 2B, levels of TNF-α, IL-6, and IL-1β mRNA markedly increased in MH-S cells, an alveolar macrophage cell line, after 1 h of LPS exposure. Results were acquired by real-time PCR. This increase in TNF-α, IL-6, and IL-1β mRNA was significantly attenuated by RSV treatment. As shown in Figure 2C, cytokine secretions in MH-S cells were evaluated by enzyme-linked immunosorbent assay (ELISA), which showed that levels of TNF-α, IL-6, and IL-1β were also noticeably attenuated by RSV treatment.
Figure 2.
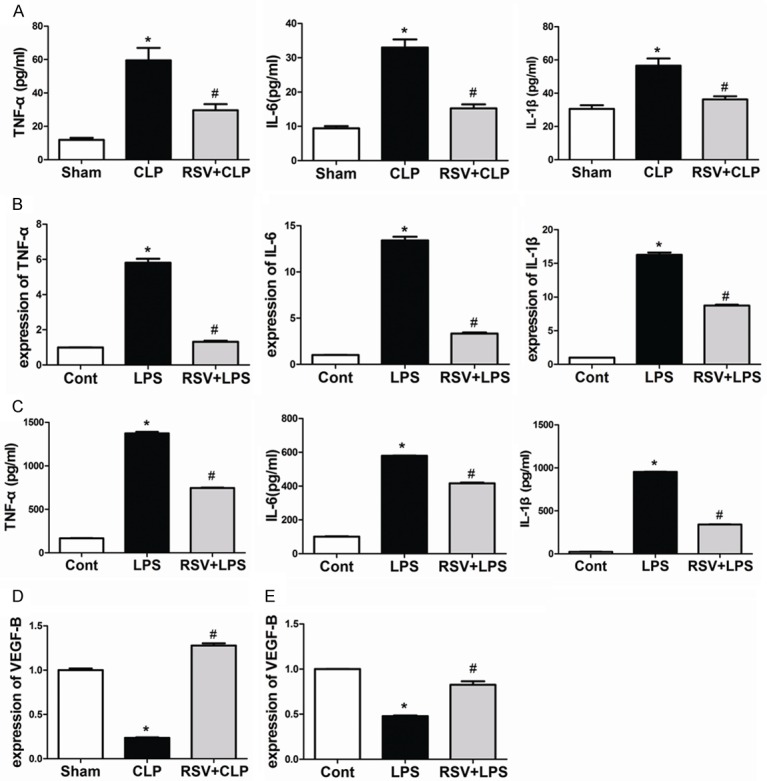
Treatment with RSV suppressed production of proinflammatory cytokines by upregulating the expression of VEGF-B both in vivo and in vitro. RSV inhibited the inflammatory response both in vivo and in vitro (A) Bronchoalveolar lavage fluid collected at 24 h after surgery to analyze inflammatory cytokines. *P<0.05 compared with the SHAM group, #P<0.05 compared with the CLP group. (B) The mRNA expression levels of TNF-α, IL-1β, and IL-6 (n = 4). Cells were exposed to 100 ng/mL LPS for 1 h. (C) Cell culture media were collected and the concentrations of TNF-α, IL-1β, and IL-6 were measured by ELISA (n = 4) (D) Lungs were harvested 24 h after surgery, and mRNA levels of VEGF-B were quantified by real-time PCR. (D) The mRNA expression levels of VEGF-B in Cells were quantified by real-time PCR (n = 3). *P<0.01 compared with the control group, #P<0.01 compared with the LPS group.
VEGF-B is abundantly expressed in tissues with highly active energy metabolism, such as in the heart, lung, endothelial cells, and macrophages. As shown in Figure 2D, with 24 h of CLP-induced sepsis, the mRNA expression of VEGF-B was significantly decreased in the lung compared with saline-treated controls. In addition, we showed that VEGF-B expression was efficiently up-regulated by RSV treatment. After treatment with LPS, the mRNA expression of VEGF-B was significantly decreased in MH-S cells, while RSV treatment significantly increased its expression in MH-S cells (Figure 2E).
VEGF-B siRNA abolished inhibitory effect of RSV on inflammation
We performed further experiments to determine whether VEGF-B was responsible for RSV-mediated secretion of proinflammatory mediators in MH-S cells. As shown in Figure 3A, VEGF-B expression was efficiently down-regulated by siRNA specific for VEGF-B in MH-S cells. These results were determined using real-time PCR. As shown in Figure 3B and 3C, the expression and secretion of TNF-α, IL-6, and IL-1β were all significantly induced from LPS-stimulated MH-S cells. Moreover, silencing VEGF-B led to a significant increase in LPS-induced secretion of TNF-α, IL-6, and IL-1β from MH-S cells when compared with control siRNA-treated cells. Importantly, knockdown of VEGF-B abolished RSV-mediated expression and secretion of proinflammatory mediators (Figure 3D, 3E).
Figure 3.
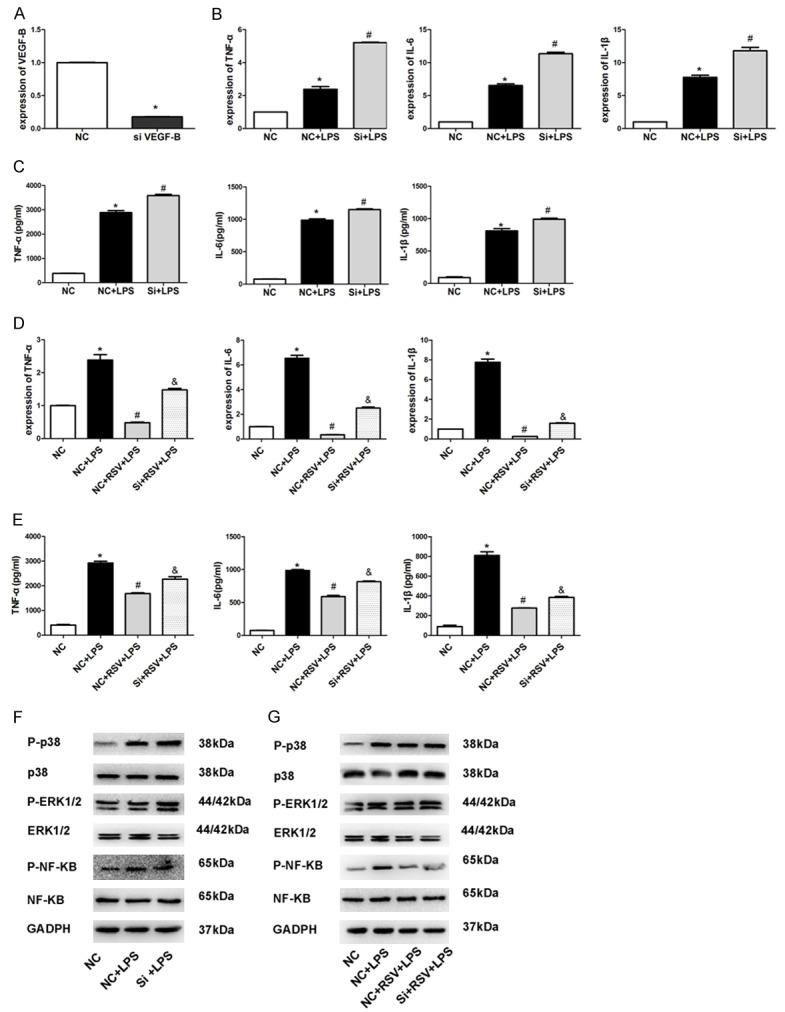
VEGF-B siRNA abolished inhibitory effect of RSV on inflammation. Efficiency of VEGF-B knockdown by using siRNA in MH-S cells. A. Levels of VEGF-B in MH-S cells after siRNA treatment. B. The mRNA expression levels of TNF-α, IL-6, and IL-1β. C. Concentrations of TNF-α, IL-6, and IL-1β measured by ELISA (n = 3). D. The mRNA expression levels of TNF-α, IL-6, and IL-1β (n = 3). E. Concentrations of TNF-α, IL-6, and IL-1β measured by ELISA (n = 3). F. NF-κB/P-NF-κB, ERK1/2/P-ERK1/2 and p38/P-P38 protein levels after siRNA treatment in MH-S cells. G. NF-κB/P-NF-κB, ERK1/2/P-ERK1/2, and p38/P-P38 protein levels after siRNA treatment in MH-S cells. *P<0.01 compared with the NC group, #P<0.01 compared with the NC+LPS group, &P<0.05 compared with the NC+RSV+LPS group.
The NF-κB and mitogen-activated protein kinase (MAPK) are 2 key pathways for LPS-stimulated signaling events [26]. Hence, we tested whether the suppression of proinflammatory cytokines was regulated through these pathways. As shown in Figure 3F, LPS strongly activated NF-κB, ERK1/2, and p38 in MH-S cells, while RSV prevented LPS-induced in-crease in levels of phosphorylated NF-κB, ERK1/2, and p38. Furthermore, knockdown of VEGF-B abolished RSV-mediated inhibition of NF-κB, ERK1/2, and p38. Additional experiments showed that silencing VEGF-B led to a significant increase in LPS-induced activation of NF-κB, ERK1/2, and p38 in MH-S cells compared with control siRNA-treated cells (Figure 3G).
RSV promotes cell viability and protects LPS-induced apoptosis in MH-S cells
To determine the working concentration of LPS, MH-S cells were cultured in 1640 supplemented with different concentrations of LPS (0, 100, 150, 200, 250 and 300 μg/mL) for 24 h, in which cell viability was evaluated using the MTT assay. LPS decreased the cell viability with dose increasing (Figure 4A). Based on this result, we used 150 μg/mL LPS in the subsequent experiments. Next, we investigated whether RSV exerted a protective effect on MH-S cells exposed to LPS. We pre-treated MH-S cells with 10 μM RSV (2 h) prior to LPS (150 μg/mL) exposure for 24 h, in which RSV augmented the decline of cell viability as a result of LPS insult (Figure 4B). To quantify the protective action of RSV against cytotoxic effects of LPS, we pre-treated MH-S cells with RSV (2 h) and then incubated them with LPS (150 μg/mL) for 24 h. Apoptosis was assessed by Annexin V-FITC and PI double staining. The sum of the early apoptotic cells (quadrant 4, Q4) and late apoptotic cells (quadrant 2, Q2). The apoptotic rate was defined as the number of apoptotic cells to the number of all cells. The apoptosis rate was increased significantly when cells were treated with 150 mg/mL LPS compared with the controls. However, RSV caused a significant reduction of early apoptotic cells as compared with the LPS (150 mg/mL) only groups (Figure 4C, 4D). Together, these results demonstrate that RSV promotes cell viability and protects LPS-induced apoptosis in MH-S cells. Cell survival is dependent on the ratio of Bcl-2 (antiapoptotic) to Bax (proapoptotic). To further investigate the molecular mechanism of RSV protection in apoptosis of MH-S cells after exposed to LPS, the expressions of Bcl-2 and Bax proteins were determined by western blotting. The treatment of MH-S cells with LPS (150 mg/mL) for 4 h caused a significant increase in the level of Bax (Figure 4E), while reduced Bcl-2 expression resulting in a decrease in the Bcl-2/Bax ratio (Figure 4F). In contrast, RSV increased the Bcl-2/Bax ratio by decreasing Bax expression and simultaneously increasing Bcl-2.
Figure 4.
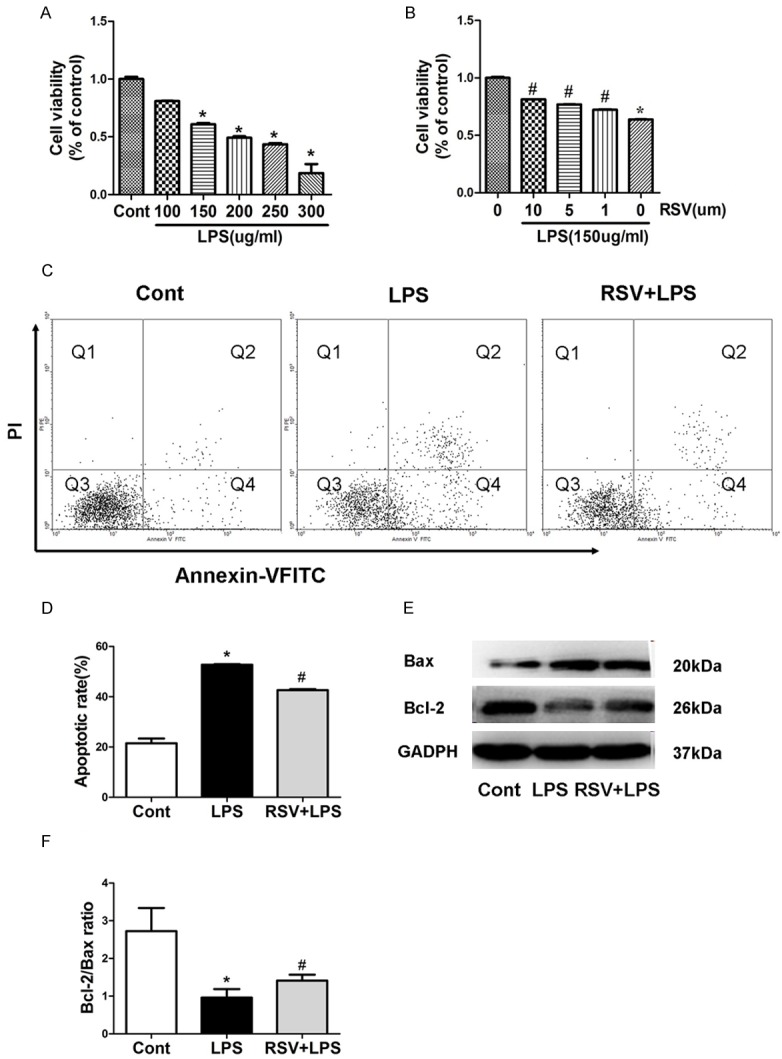
RSV promotes cell viability and protects LPS-induced apoptosis in MH-S cells. RSV decreased LPS-induced apoptosis in MH-S cells. A, B. Viability of MH-S cells was estimated using the MTT assay. Results are representative of three independent experiments, mean ± SD, *P<0.05 compared with control. C. Apoptosis of MH-S cells were stained with Annexin-VFITC and PI and analyzed by flow cytometry. D. Quantitative analysis of cell apoptosis in MH-S cells. E. Representative western blots show LPS-induced expression of Bax and Bcl-2 in MH-S cells with and without pre-treatment with RSV. F. Densitometric analysis shows that treatment with LPS reduced the Bcl-2/Bax ratio, and that this effect could be inhibited by RSV pre-treatment. *P<0.05 compared with control; #P<0.05 compared with LPS treated alone.
RSV inhibits LPS-induced apoptosis in macrophages in part via VEGF-B signaling
To investigate the role of VEGF-B in RSV mediating anti-apoptotic effect, we inhibited expression of VEGF-B by using siRNA in MH-S cells. After treatment with VEGF-B siRNA, increased apoptosis cells were found in comparison with the group receiving only RSV treatment, indicating that RSV attenuated LPS-induced apoptosis via the VEGF-B regulation (Figure 5A, 5B). Importantly, knockdown of VEGF-B blocked RSV-mediated Bcl-2 and Bax expression Figure 5C), suggesting that RSV regulates Bcl-2/Bax expression through the VEGF-B signaling pathway.
Figure 5.
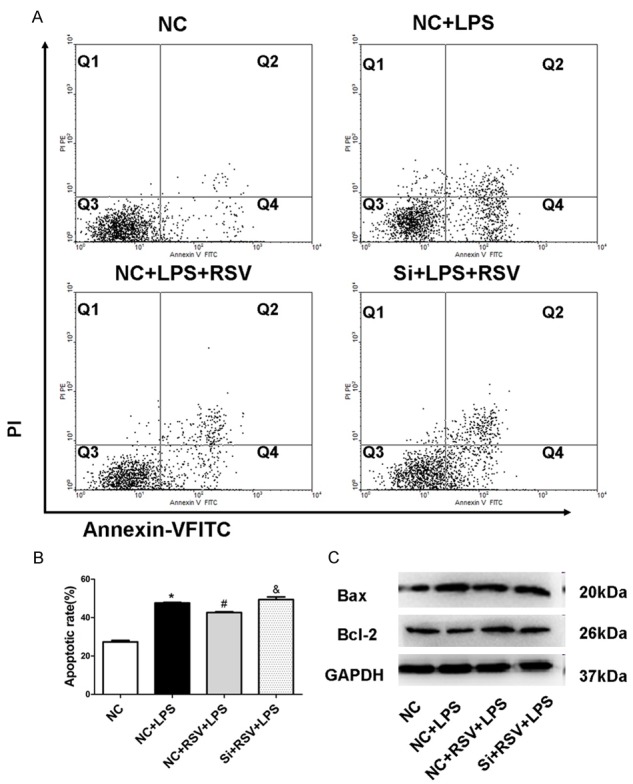
RSV inhibits LPS-induced apoptosis in macrophages in part via VEGF-B signaling. A. Apoptosis of MH-S cells were stained with Annexin-VFITC and PI and analyzed by flow cytometry. B. Quantitative analysis of cell apoptosis in MH-S cells. C. Bax, Bcl-2 protein levels after siRNA treatment in MH-S cells. *P<0.01 compared with the NC group, #P<0.01 compared with the NC+LPS group, &P<0.05 compared with the NC+RSV+LPS group.
RSV protects AMs against LPS-induced apoptosis in part via inhibiting LPS-induced autophagy
To determine whether RSV afected autophagy, we used monodansylcadaverine (MDC) staining to detect autophagic vacuoles. As shown in Figure 6A, LPS induced the accumulation of MDC-labeled vacuoles in the cytoplasm in MH-S cells, while RSV significantly lessened the accumulation of MDC-labeled vacuoles. Moreover, we determined the effect of RSV on the expression of autophagy marker LC3 by western blot. The ratio of LC3-II/I was decreased in RSV group in comparison with LPS group (Figure 6B). However, VEGF-B siRNA treatment markedly reversed RSV lessened expression LC3-II/I (Figure 6C), suggesting that RSV regulates autophagy through the VEGF-B signaling pathway.
Figure 6.
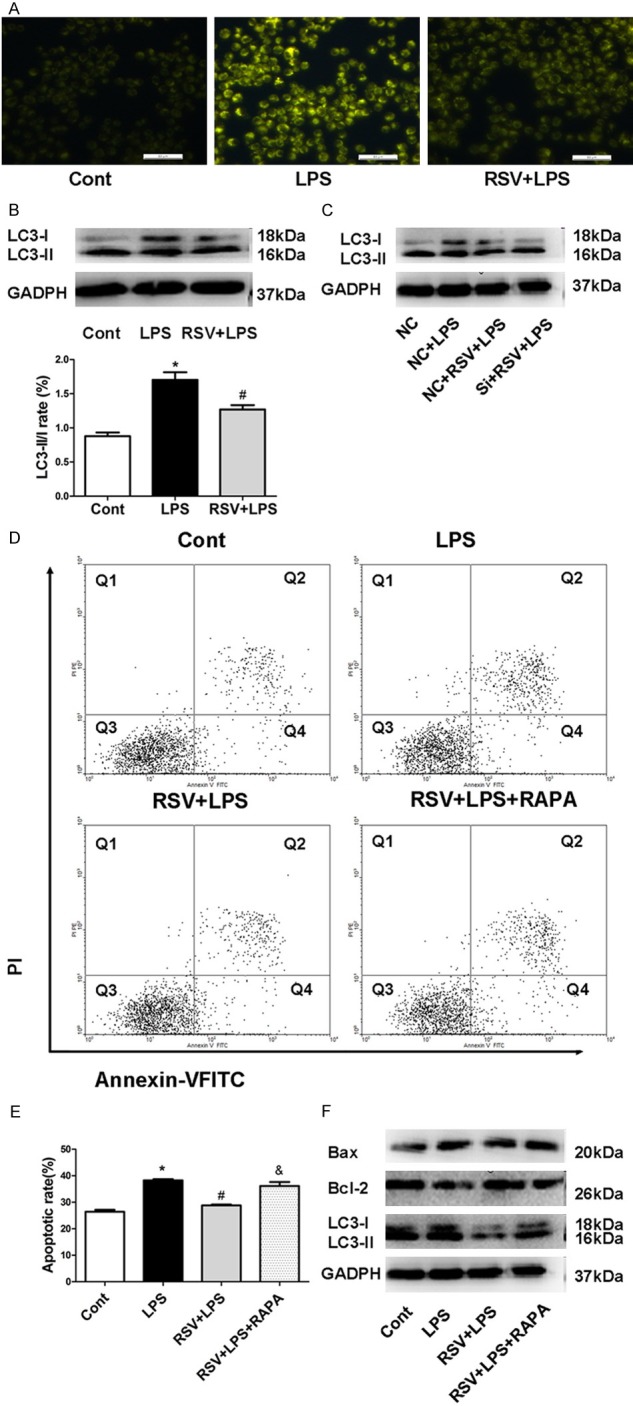
RSV protects AMs against LPS-induced apoptosis in part via inhibiting LPS-induced autophagy. RSV decreased LPS-induced autophagy in MH-S cells. (A) Autophagy level in MH-S cells was detected by MDC staining (B) RSV decreased protein level of LC3-II/I in MH-S cells (C) LC3-II/I protein levels after siRNA treatment in MH-S cells. (D) Cell apoptosis was detected using Annexin V/PI staining and analyzed by flow cytometry. (E) Quantitative analysis of cell apoptosis in MH-S cells. Quantitative analysis of cell apoptosis in MH-S cells, *P<0.01 compared with control. #P<0.05 compared with LPS treated alone &P<0.05 compared with the RSV+LPS group. (F) LC3-II/I expression was detected using western blot after rapamycin treatment in MH-S cells.
To further explore the involvement of RSV in MH-S cell apoptosis in the LPS-induced autophagy model, the autophagy inducer rapamycin was used to determine whether RSV’s effect on autophagy involved MH-S cell apoptosis. As shown in Figure 6D and 6E, rapamycin reversed the RSV-mediated anti-apoptosis effects and the expression of LC3 in the LPS-induced autophagy model (Figure 6F), suggesting that RSV could inhibit LPS-induced cell apoptosis by blocking autophagy.
Treatment with RSV affected both mRNA and protein levels of C5aR and C5L2 in MH-S cells
The complement anaphylatoxin, C5a, is a key factor for the regulation of inflammatory responses during sepsis, and has been shown to play a critical role in survival after CLP [35]. C5a binds to receptors C5aR and C5L2, both of which are critical factors during polymicrobial sepsis after CLP [36]. We thus examined the effects of RSV on the expression of C5aR and C5L2 in MH-S cells. Real-time PCR analysis showed that the LPS-dependent induce in C5aR gene expression at 1 h was significantly decreased by RSV treatment (Figure 7A), and the reduction in C5L2 gene expression was significantly increased by RSV treatment (Figure 7B). We tested whether RSV affected the cell surface expression of C5aR and C5L2. As shown in Figure 7C and 7D, LPS significantly increased C5aR expression and decreased C5L2 expression compared to the control group, while RSV treatment abrogated these effects. In summary, RSV affected both gene and protein expression of C5aR and C5L2 in MH-S cells.
Figure 7.
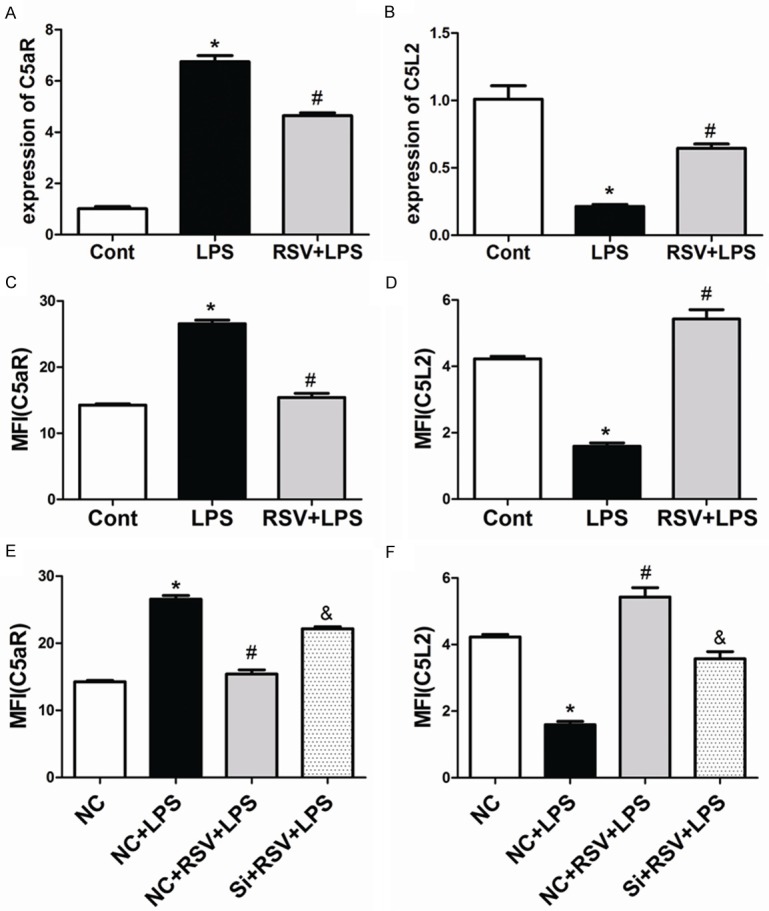
Treatment with RSV affected both mRNA and protein levels of C5aR and C5L2 in MH-S cells. RSV affected the expression of C5aR and C5L2 in MH-S Cells. A. The mRNA expression levels of C5aR. B. The mRNA expression levels of C5L2. C and D. Cell surface expression of C5aR and C5L2 in MH-S cells. P<0.01 compared with the control group, #P<0.01 compared with the LPS group. E and F. Cell surface expression levels of C5aR and C5L2 after siRNA treatment in MH-S cells. *P<0.01 compared with the NC group, #P<0.01 compared with the NC+LPS group, &P<0.05 compared with the NC+RSV+LPS group.
One mechanism by which RSV exerts effects in macrophages is via VEGF-B signaling. Therefore, we tested whether the effect of RSV on C5aR and C5L2 expression was VEGF-B-dependent. As shown in Figure 7E and 7F, RSV-mediated change in C5L2 and C5aR expressions in the presence of LPS were abrogated by the knockdown of VEGF-B.
Discussion
RSV, a polyphenolic compound in red wine, was well known for its anti-inflammation and anti-apoptotic properties [37,38]. However, its protective properties and underlying mechanisms are largely unknown in the occurrence of sepsis-induced ALI. In this study, we found that RSV pretreatment ameliorated LPS-induced ALI by inhibiting inflammation in vivo and in vitro. MPO content in mice lungs and albumin levels in BAL fluids were both significantly increased after CLP [39]. Our resulted showed MPO activity and albumin level were significantly reduced by RSV treatment. These results revealed that the effects of RSV on CLP-induced ALI are associated with a decrease of capillary permeability in lung tissues. Furthermore, we found that when RSV was given to septic mice, the elevated levels of proinflammatory cytokines in BAL fluids were greatly inhibited, and histologic damage in lungs, including inflammatory infiltration and parenchyma disorganization, were significantly mitigated.
In this study, we demonstrated the protective effects of RSV on CLP-induced ALI in relation to the VEGF-B signaling pathway. VEGF can regulate inflammatory cytokine production in murine polymicrobial sepsis via regulation of C/EBPβ [40]. VEGF-C significantly attenuated proinflammatory cytokine production by regulating the PI3-kinase-Akt signaling pathway and SOCS1 expression in macrophages [41]. VEGF-B mRNA is down-regulated 6-96 h post-wounding during the acute inflammatory phase of murine excisional wounds [42]. Thus, VEGF-B might participate in the network involving acute inflammation. To further clarify the role of VEGF-B during acute inflammation. We performed in vitro experiments with MH-S cells. We found that silencing VEGF-B led to a significant increase in LPS-induced expression and secretion of TNF-α, IL-6, and IL-1β from MH-S cells by activation of NF-κB (P65), ERK1/2, and P38. We then tested whether VEGF-B was involved in the anti-inflammatory effects by RSV in LPS-stimulated MH-S cells. We found that VEGF-B significantly increased in the cultured macrophages upon RSV treatment. Importantly, we showed that after the knockdown of VEGF-B expression in MH-S cells, RSV was unable to attenuate LPS-induced proinflammatory cytokine production, suggesting that VEGF-B indeed plays a critical role in the anti-inflammatory effects of RSV. Previous research showed that RSV exerts anti-inflammatory effects by modulating the NF-κB and Janus kinase/STAT signaling pathways in LPS-stimulated RAW264.7 cells in a dose-dependent manner [43], and the NF-κB and MAPK pathways play central roles in the transcriptional regulation of the expression of proinflammatory genes [44]. We found that RSV inhibited activation of NF-κB (P65), ERK1/2, and P38 by enhancing the expression of VEGF-B, which was abolished by VEGF-B siRNA. This suggests that the inhibitory effect of RSV on the overproduction of proinflammatory cytokines is at least in part due to activating the VEGF-B signaling pathway.
Moreover, in our study we found that LPS reduced cell viability in dose-dependent manners by MTT assays, and a reduction in death rate of MH-S cells was observed when cells were pretreated with RSV. Furthermore, the pre-incubation of MH-S cells with RSV inhibited LPS-induced apoptosis. An increase of Bax/Bcl-2 ratio was observed at 7 days post-sepsis, illustrating the induction of apoptosis in lung tissues [45]. Our findings showed the Bax/Bcl-2 ratio was increased by LPS insult, whereas RSV treatment completely restored Bax/Bcl-2 ratio to normal levels by enhancing the expression of VEGF-B. A recent study shows that autophagy and apoptosis might play distinct roles at different stages of LPS-induced ALI. Autophagy reached a peak at 2 hr; however, apoptosis reached its maximal level at later stages (6 hr) [46]. We found that LPS can also induce autophagy in MH-S cells and RSV reversed the LPS-induced increase in LC3 levels. Li GH, Luo B, et al. reported that VEGF-B inhibited I/R-induced autophagy in rat cardiomyocytes [47]. We then tested whether VEGF-B was involved in anti- autophagy by RSV in LPS-stimulated MH-S cells. We showed in our study that knockdown of VEGF-B expression in the MH-S cells made RSV no longer able to attenuate LPS-induced increase in LC3 levels, suggesting that VEGF-B indeed plays a critical role in the anti- autophagy of RSV. VEGF-B can inhibit H9c2 cell apoptosis by inhibiting H/R-induced autophagy and autophagy may increase LPS-induced apoptosis via the intrinsic apoptotic pathway in alveolar macrophages of human silicosis [47,48]. In the present study, we found that RSV inhibited MH-S cell apoptosis and abolished by rapamycin. Furthermore, RSV significantly up-regulated Bcl-2 and down-regulated Bax in MH-S cells by enhancing the expression of VEGF-B and was reversed by rapamycin. Therefore, the inhibitory effects of RSV on LPS-induced apoptosis in macrophages may partially via inhibiting LPS-induced autophagy.
Previous researched showed that RSV virtually completely attenuated the effects of C5a on vascular permeability, neutrophil migration, and release of proinflammatory cytokine in a C5a-induced model of acute peritonitis [49].Acute lung injury (ALI) results in the formation of complement fragment 5a (C5a). The complement activation product, C5a interacts with its two receptors, C5aR and C5L2 [50]. C5L2 plays an important anti-inflammatory role and suppresses lipopolysaccharide-induced acute lung injury [51]. We found that RSV affected the expression of C5L2 by enhancing the expression of VEGF-B, which was abolished by VEGF-B siRNA. Our results thus indicated that suppression of LPS-induced production of proinflammatory cytokines in MH-S cells by RSV in part via regulating C5L2 expression. Furethermore, interaction of C5a with C5aR can also lead to many pathophysiological changes as seen in acute lung injury. C5a produced during lung injury binds to C5aR can lead to apoptosis of alveolar macrophages through C5aR-mediated degradation of bcl-2 [52]. Our previous work with lung epithelial cells revealed that Silencing of C5aR reduces the severity of ALI [53]. In this study, we also found RSV inhibited the expression of C5aR by enhancing the expression of VEGF-B and was abolished by VEGF-B siRNA. Our results thus indicate that RSV protects alveolar macrophages against LPS-induced apoptosis in part via inhibiting the expression of C5aR.
In summary, we found that prophylactic treatment with RSV significantly alleviated sepsis-induced acute lung injury in mice, which was associated with anti- inflammation and anti-apoptotic effects of RSV depends on the VEGF-B signaling pathway. Therefore, RSV may be used as a potential adjuvant during sepsis-induced acute lung injury.
Conclusions
RSV could significantly improve sepsis-induced ARDS via decreasing the release of proinflammatory factors and the ratio of alveolar macrophages apoptosis. Therefore, RSV may be a promising pharmacological therapy for acute lung injury.
Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 81470263) and Tianjin science and technology project, China (No. 13RCGFSY19300) for their great supports in financing these researches.
Disclosure of conflict of interest
None.
References
- 1.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 2.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang C, Zheng H, He W, Lu G, Li X, Deng Y, Zeng M. Ghrelin ameliorates the human alveolar epithelial A549 cell apoptosis induced by lipopolysaccharide. Biochem Biophys Res Commun. 2016;474:83–90. doi: 10.1016/j.bbrc.2016.04.074. [DOI] [PubMed] [Google Scholar]
- 4.Gill SE, Rohan M, Mehta S. Role of pulmonary microvascular endothelial cell apoptosis in murine sepsis-induced lung injury in vivo. Respir Res. 2015;16:109. doi: 10.1186/s12931-015-0266-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li B, Zeng M, He W, Huang X, Luo L, Zhang H, Deng DY. Ghrelin protects alveolar macrophages against lipopolysaccharide-induced apoptosis through growth hormone secretagogue receptor 1a-dependent c-Jun N-terminal kinase and Wnt/beta-catenin signaling and suppresses lung inflammation. Endocrinology. 2015;156:203–217. doi: 10.1210/en.2014-1539. [DOI] [PubMed] [Google Scholar]
- 6.Niesler U, Palmer A, Radermacher P, Huber-Lang MS. Role of alveolar macrophages in the inflammatory response after trauma. Shock. 2014;42:3–10. doi: 10.1097/SHK.0000000000000167. [DOI] [PubMed] [Google Scholar]
- 7.Lu MC, Liu TA, Lee MR, Lin L, Chang WC. Apoptosis contributes to the decrement in numbers of alveolar macrophages from rats with polymicrobial sepsis. J Microbiol Immunol Infect. 2002;35:71–77. [PubMed] [Google Scholar]
- 8.Machado-Aranda D, V Suresh M, Yu B, Dolgachev V, Hemmila MR, Raghavendran K. Alveolar macrophage depletion increases the severity of acute inflammation following nonlethal unilateral lung contusion in mice. J Trauma Acute Care Surg. 2014;76:982–990. doi: 10.1097/TA.0000000000000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones HD, Crother TR, Gonzalez-Villalobos RA, Jupelli M, Chen S, Dagvadorj J, Arditi M, Shimada K. The NLRP3 inflammasome is required for the development of hypoxemia in LPS/mechanical ventilation acute lung injury. Am J Respir Cell Mol Biol. 2014;50:270–280. doi: 10.1165/rcmb.2013-0087OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pahuja M, Tran C, Wang H, Yin K. Alveolar macrophage suppression in sepsis is associated with high mobility group box 1 transmigration. Shock. 2008;29:754–760. doi: 10.1097/shk.0b013e31815d0c8f. [DOI] [PubMed] [Google Scholar]
- 11.Arunachalam G, Yao H, Sundar IK, Caito S, Rahman I. SIRT1 regulates oxidant- and cigarette smoke-induced eNOS acetylation in endothelial cells: role of resveratrol. Biochem Biophys Res Commun. 2010;393:66–72. doi: 10.1016/j.bbrc.2010.01.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 13.Kumerz M, Heiss EH, Schachner D, Atanasov AG, Dirsch VM. Resveratrol inhibits migration and Rac1 activation in EGF- but not PDGF-activated vascular smooth muscle cells. Mol Nutr Food Res. 2011;55:1230–1236. doi: 10.1002/mnfr.201100309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oh YC, Kang OH, Choi JG, Chae HS, Lee YS, Brice OO, Jung HJ, Hong SH, Lee YM, Kwon DY. Anti-inflammatory effect of resveratrol by inhibition of IL-8 production in LPS-induced THP-1 cells. Am J Chin Med. 2009;37:1203–1214. doi: 10.1142/S0192415X09007600. [DOI] [PubMed] [Google Scholar]
- 15.Sin TK, Yu AP, Yung BY, Yip SP, Chan LW, Wong CS, Ying M, Rudd JA, Siu PM. Modulating effect of SIRT1 activation induced by resveratrol on Foxo1-associated apoptotic signalling in senescent heart. J Physiol. 2014;592:2535–2548. doi: 10.1113/jphysiol.2014.271387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holthoff JH, Wang Z, Seely KA, Gokden N, Mayeux PR. Resveratrol improves renal microcirculation, protects the tubular epithelium, and prolongs survival in a mouse model of sepsis-induced acute kidney injury. Kidney Int. 2012;81:370–378. doi: 10.1038/ki.2011.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li T, Zhang J, Feng J, Li Q, Wu L, Ye Q, Sun J, Lin Y, Zhang M, Huang R, Cheng J, Cao Y, Xiang G, Zhang J, Wu Q. Resveratrol reduces acute lung injury in a LPS induced sepsis mouse model via activation of Sirt1. Mol Med Rep. 2013;7:1889–1895. doi: 10.3892/mmr.2013.1444. [DOI] [PubMed] [Google Scholar]
- 18.Bai X, Fan L, He T, Jia W, Yang L, Zhang J, Liu Y, Shi J, Su L, Hu D. SIRT1 protects rat lung tissue against severe burn-induced remote ALI by attenuating the apoptosis of PMVECs via p38 MAPK signaling. Sci Rep. 2015;5:10277. doi: 10.1038/srep10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo R, Su Y, Liu B, Li S, Zhou S, Xu Y. Resveratrol suppresses oxidised low-density lipoprotein-induced macrophage apoptosis through inhibition of intracellular reactive oxygen species generation, LOX-1, and the p38 MAPK pathway. Cell Physiol Biochem. 2014;34:603–616. doi: 10.1159/000363026. [DOI] [PubMed] [Google Scholar]
- 20.Grimmond S, Lagercrantz J, Drinkwater C, Silins G, Townson S, Pollock P, Gotley D, Carson E, Rakar S, Nordenskjold M, Ward L, Hayward N, Weber G. Cloning and characterization of a novel human gene related to vascular endothelial growth factor. Genome Res. 1996;6:124–131. doi: 10.1101/gr.6.2.124. [DOI] [PubMed] [Google Scholar]
- 21.Muhl L, Moessinger C, Adzemovic MZ, Dijkstra MH, Nilsson I, Zeitelhofer M, Hagberg CE, Huusko J, Falkevall A, Yla-Herttuala S, Eriksson U. Expression of vascular endothelial growth factor (VEGF)-B and its receptor (VEGFR1) in murine heart, lung and kidney. Cell Tissue Res. 2016;365:51–63. doi: 10.1007/s00441-016-2377-y. [DOI] [PubMed] [Google Scholar]
- 22.Granata F, Frattini A, Loffredo S, Staiano RI, Petraroli A, Ribatti D, Oslund R, Gelb MH, Lambeau G, Marone G, Triggiani M. Production of vascular endothelial growth factors from human lung macrophages induced by group IIA and group X secreted phospholipases A2. J Immunol. 2010;184:5232–5241. doi: 10.4049/jimmunol.0902501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagberg CE, Falkevall A, Wang X, Larsson E, Huusko J, Nilsson I, van Meeteren LA, Samen E, Lu L, Vanwildemeersch M, Klar J, Genove G, Pietras K, Stone-Elander S, Claesson-Welsh L, Yla-Herttuala S, Lindahl P, Eriksson U. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature. 2010;464:917–921. doi: 10.1038/nature08945. [DOI] [PubMed] [Google Scholar]
- 24.Hagberg CE, Mehlem A, Falkevall A, Muhl L, Fam BC, Ortsater H, Scotney P, Nyqvist D, Samen E, Lu L, Stone-Elander S, Proietto J, Andrikopoulos S, Sjoholm A, Nash A, Eriksson U. Targeting VEGF-B as a novel treatment for insulin resistance and type 2 diabetes. Nature. 2012;490:426–430. doi: 10.1038/nature11464. [DOI] [PubMed] [Google Scholar]
- 25.Hanrahan V, Currie MJ, Gunningham SP, Morrin HR, Scott PA, Robinson BA, Fox SB. The angiogenic switch for vascular endothelial growth factor (VEGF)-A, VEGF-B, VEGF-C, and VEGF-D in the adenoma-carcinoma sequence during colorectal cancer progression. J Pathol. 2003;200:183–194. doi: 10.1002/path.1339. [DOI] [PubMed] [Google Scholar]
- 26.Guaiquil VH, Pan Z, Karagianni N, Fukuoka S, Alegre G, Rosenblatt MI. VEGF-B selectively regenerates injured peripheral neurons and restores sensory and trophic functions. Proc Natl Acad Sci U S A. 2014;111:17272–17277. doi: 10.1073/pnas.1407227111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kivela R, Bry M, Robciuc MR, Rasanen M, Taavitsainen M, Silvola JM, Saraste A, Hulmi JJ, Anisimov A, Mayranpaa MI, Lindeman JH, Eklund L, Hellberg S, Hlushchuk R, Zhuang ZW, Simons M, Djonov V, Knuuti J, Mervaala E, Alitalo K. VEGF-B-induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO Mol Med. 2014;6:307–321. doi: 10.1002/emmm.201303147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sands M, Howell K, Costello CM, McLoughlin P. Placenta growth factor and vascular endothelial growth factor B expression in the hypoxic lung. Respir Res. 2011;12:17. doi: 10.1186/1465-9921-12-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Zhang F, Nagai N, Tang Z, Zhang S, Scotney P, Lennartsson J, Zhu C, Qu Y, Fang C, Hua J, Matsuo O, Fong GH, Ding H, Cao Y, Becker KG, Nash A, Heldin CH, Li X. VEGF-B inhibits apoptosis via VEGFR-1-mediated suppression of the expression of BH3-only protein genes in mice and rats. J Clin Invest. 2008;118:913–923. doi: 10.1172/JCI33673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bry M, Kivela R, Holopainen T, Anisimov A, Tammela T, Soronen J, Silvola J, Saraste A, Jeltsch M, Korpisalo P, Carmeliet P, Lemstrom KB, Shibuya M, Yla-Herttuala S, Alhonen L, Mervaala E, Andersson LC, Knuuti J, Alitalo K. Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation. 2010;122:1725–1733. doi: 10.1161/CIRCULATIONAHA.110.957332. [DOI] [PubMed] [Google Scholar]
- 31.Huang D, Zhao C, Ju R, Kumar A, Tian G, Huang L, Zheng L, Li X, Liu L, Wang S, Ren X, Ye Z, Chen W, Xing L, Chen Q, Gao Z, Mi J, Tang Z, Wang B, Zhang S, Lee C, Li X. VEGF-B inhibits hyperglycemia- and Macugen-induced retinal apoptosis. Sci Rep. 2016;6:26059. doi: 10.1038/srep26059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang L, Yang L, Tian W, Li J, Liu J, Zhu M, Zhang Y, Yang Y, Liu F, Zhang Q, Liu Q, Shen Y, Qi Z. Resveratrol plays dual roles in pancreatic cancer cells. J Cancer Res Clin Oncol. 2014;140:749–755. doi: 10.1007/s00432-014-1624-4. [DOI] [PubMed] [Google Scholar]
- 33.Yang L, Zhang Y, Zhu M, Zhang Q, Wang X, Wang Y, Zhang J, Li J, Yang L, Liu J, Liu F, Yang Y, Kang L, Shen Y, Qi Z. Resveratrol attenuates myocardial ischemia/reperfusion injury through up-regulation of vascular endothelial growth factor B. Free Radic Biol Med. 2016;101:1–9. doi: 10.1016/j.freeradbiomed.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 34.Zhong WT, Wu YC, Xie XX, Zhou X, Wei MM, Soromou LW, Ci XX, Wang DC. Phillyrin attenuates LPS-induced pulmonary inflammation via suppression of MAPK and NF-kappaB activation in acute lung injury mice. Fitoterapia. 2013;90:132–139. doi: 10.1016/j.fitote.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Czermak BJ, Sarma V, Pierson CL, Warner RL, Huber-Lang M, Bless NM, Schmal H, Friedl HP, Ward PA. Protective effects of C5a blockade in sepsis. Nat Med. 1999;5:788–792. doi: 10.1038/10512. [DOI] [PubMed] [Google Scholar]
- 36.Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber-Lang M, Mackay CR, Zetoune FS, Gerard NP, Cianflone K, Kohl J, Gerard C, Sarma JV, Ward PA. Functional roles for C5a receptors in sepsis. Nat Med. 2008;14:551–557. doi: 10.1038/nm1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trotta V, Lee WH, Loo CY, Young PM, Traini D, Scalia S. Co-spray dried resveratrol and budesonide inhalation formulation for reducing inflammation and oxidative stress in rat alveolar macrophages. Eur J Pharm Sci. 2016;86:20–28. doi: 10.1016/j.ejps.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 38.Liang Q, Wang XP, Chen TS. Resveratrol protects rabbit articular chondrocyte against sodium nitroprusside-induced apoptosis via scavenging ROS. Apoptosis. 2014;19:1354–1363. doi: 10.1007/s10495-014-1012-1. [DOI] [PubMed] [Google Scholar]
- 39.Hirano Y, Aziz M, Yang WL, Wang Z, Zhou M, Ochani M, Khader A, Wang P. Neutralization of osteopontin attenuates neutrophil migration in sepsis-induced acute lung injury. Crit Care. 2015;19:53. doi: 10.1186/s13054-015-0782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nolan A, Weiden MD, Thurston G, Gold JA. Vascular endothelial growth factor blockade reduces plasma cytokines in a murine model of polymicrobial sepsis. Inflammation. 2004;28:271–278. doi: 10.1007/s10753-004-6050-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Lu Y, Ma L, Cao X, Xiao J, Chen J, Jiao S, Gao Y, Liu C, Duan Z, Li D, He Y, Wei B, Wang H. Activation of vascular endothelial growth factor receptor-3 in macrophages restrains TLR4-NF-kappaB signaling and protects against endotoxin shock. Immunity. 2014;40:501–514. doi: 10.1016/j.immuni.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 42.Roy S, Khanna S, Rink C, Biswas S, Sen CK. Characterization of the acute temporal changes in excisional murine cutaneous wound inflammation by screening of the wound-edge transcriptome. Physiol Genomics. 2008;34:162–184. doi: 10.1152/physiolgenomics.00045.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma C, Wang Y, Dong L, Li M, Cai W. Anti-inflammatory effect of resveratrol through the suppression of NF-kappaB and JAK/STAT signaling pathways. Acta Biochim Biophys Sin (Shanghai) 2015;47:207–213. doi: 10.1093/abbs/gmu135. [DOI] [PubMed] [Google Scholar]
- 44.Campbell KJ, Perkins ND. Regulation of NF-kappaB function. Biochem Soc Symp. 2006:165–180. doi: 10.1042/bss0730165. [DOI] [PubMed] [Google Scholar]
- 45.Chopra M, Reuben JS, Sharma AC. Acute lung injury: apoptosis and signaling mechanisms. Exp Biol Med (Maywood) 2009;234:361–371. doi: 10.3181/0811-MR-318. [DOI] [PubMed] [Google Scholar]
- 46.Lin L, Zhang L, Yu L, Han L, Ji W, Shen H, Hu Z. Time-dependent changes of autophagy and apoptosis in lipopolysaccharide-induced rat acute lung injury. Iran J Basic Med Sci. 2016;19:632–637. [PMC free article] [PubMed] [Google Scholar]
- 47.Li GH, Luo B, Lv YX, Zheng F, Wang L, Wei MX, Li XY, Zhang L, Wang JN, Chen SY, Tang JM, He X. Dual effects of VEGF-B on activating cardiomyocytes and cardiac stem cells to protect the heart against short- and long-term ischemia-reperfusion injury. J Transl Med. 2016;14:116. doi: 10.1186/s12967-016-0847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen S, Yuan J, Yao S, Jin Y, Chen G, Tian W, Xi J, Xu Z, Weng D, Chen J. Lipopolysaccharides may aggravate apoptosis through accumulation of autophagosomes in alveolar macrophages of human silicosis. Autophagy. 2015;11:2346–2357. doi: 10.1080/15548627.2015.1109765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Issuree PD, Pushparaj PN, Pervaiz S, Melendez AJ. Resveratrol attenuates C5a-induced inflammatory responses in vitro and in vivo by inhibiting phospholipase D and sphingosine kinase activities. FASEB J. 2009;23:2412–2424. doi: 10.1096/fj.09-130542. [DOI] [PubMed] [Google Scholar]
- 50.Sarma JV, Ward PA. New developments in C5a receptor signaling. Cell Health Cytoskelet. 2012;4:73–82. doi: 10.2147/CHC.S27233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang R, Lu B, Gerard C, Gerard NP. C5L2, the second C5a anaphylatoxin receptor, suppresses LPS-induced acute lung injury. Am J Respir Cell Mol Biol. 2016;55:657–666. doi: 10.1165/rcmb.2016-0067OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun L, Guo RF, Gao H, Sarma JV, Zetoune FS, Ward PA. Attenuation of IgG immune complex-induced acute lung injury by silencing C5aR in lung epithelial cells. FASEB J. 2009;23:3808–3818. doi: 10.1096/fj.09-133694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu R, Chen ZF, Yan J, Li QF, Huang Y, Xu H, Zhang X, Jiang H. Complement C5a exacerbates acute lung injury induced through autophagy-mediated alveolar macrophage apoptosis. Cell Death Dis. 2014;5:e1330. doi: 10.1038/cddis.2014.274. [DOI] [PMC free article] [PubMed] [Google Scholar]