

Abstract
Recent studies suggest that individual subunits of chromatin-remodeling complexes generate epigenetically specific signaling in tumorigenicity. The impact of environmental factors on the chromatin-remodeling factor has not been thoroughly elucidated to date. We detected the expression level of SMARCA6 (SWI/SNF2-Related, Matrix-Associated, Actin-Dependent Regulator of Chromatin, Subfamily A, Member 6) in NSCLC (Non-small-cell lung carcinoma) and measured it through quantitative real-time PCR (qRT-PCR) and immunohistochemistry. The effects of BaP on proliferation and cell cycle progression were evaluated using MTT, colony formation and FACS analyses. Tumor growth in vivo was observed in a xenograft model. ChIP and qPCR were performed to validate that SMARCA6 was a potential target of AhR in NSCLC. As a result, BaP increased SMARCA6 expression. Smoking was linked with elevated SMARCA6 expression in NSCLC. BaP promoted cancer progression in vitro and in vivo. ChIP assay confirmed that BaP increases SMARCA6 expression via recruitment of AhR and induces SMARCA6 expression by facilitating AhR translocation to the nucleus. Furthermore, inhibition of AhR expression decreases SMARCA6 expression in NSCLC. Finally, knockdown of SMARCA6 attenuates BaP-induced cancer progression. This study demonstrates that BaP promotes proliferation by activation of AhR, which promotes SMARCA6 expression, and may identify new diagnostic and therapeutic targets in lung cancer.
Keywords: SMARCA6, BaP, AhR, smoking, lung cancer
Introduction
Lung cancer is the leading cause of cancer-related deaths, and the most common cancer worldwide, including China. Lung cancer is classified into small cell lung cancer and non-small cell lung cancer (NSCLC), including adenocarcinoma (ADC) and squamous cell carcinoma (SCC), which accounts for 80 to 85% of all cases of lung cancer [1,2]. Lung cancer is associated with environmental factors such as cigarette smoke and air pollution. Smoke and air pollution contain chemicals which calls polycyclic aromatic hydrocarbons (PAHs). PAHs are reported as major carcinogens in regions where air is polluted by PM2.5/PM10 particulate [3]. These chemicals cause epigenetic changes in the lung, which are major risk for developing inflammation, oncogene senescence, and lung cancer progression [4,5]. Combination epigenetic therapy induces depletion of MYC and can increase responsiveness to interferon, potentiate T-cell attraction, and exert direct antiproliferative activity [6]. However, the link between PAHs, epigenetics and gene expression in lung cancer is unclear.
The SWI/SNF-related, matrix-associated, actin-dependent regulators of chromatin (SMARC), also called BRG1-associated factors, are components of human SWI/SNF-like chromatin-remodeling protein complexes. SMARCA6, also called lymphoid-specific helicase (LSH), is critical for the normal development of plants and mammals, because it maintains optimum levels of DNA methylation and acts as a critical chromatin modifier [7-11]. SMARCA6 maintains genome stability in mammalian somatic cells [12]. SMARCA6 also contributes to the malignant progression of prostate cancer, melanoma, nasopharyngeal carcinoma, and glioma [13-16]; however, the correlation between PAHs and SMARCA6 has not been thoroughly elucidated to date.
The aryl hydrocarbon receptor (AhR), a ligand-operated transcription factor, is a xenosensor traditionally associated with xenobiotic metabolism [17]. AhR facilitates tumor progression, disease tolerance defense, intestinal immunity and B-cell proliferation [18-21]. Interestingly, AhR influences the major stages of tumorigenesis, and studies of aggressive tumors and tumor cell lines have shown increased levels of AhR protein and constitutive nuclear localization in cancer tissue, whereas in normal tissues, AhR is primarily inactive and resides in the cytoplasm [22,23].
In this study, we show that BaP promotes proliferation by activation of AhR, which promotes SMARCA6 expression, and may identify new diagnostic and therapeutic targets in lung cancer.
Materials and methods
Animal study
Four-week-old male BALB/C-NU mice were purchased from Hunan SJA Laboratory Animal Co., Ltd. (Changsha, China). The animal study protocol was approved by the Institutional Animal Care and Use Committee of the Central South University of Xiangya School of Medicine and conformed to the legal mandates and federal guidelines for the care and maintenance of laboratory animals. For protein overexpression or knockdown, cells were transfected with lentivirus in 10 mg/ml polybrene, for 24 hours. Media was changed, and 2 mg/ml puromycin was supplemented for 7 days to promote selection. Cells were counted, and 5 × 106 cells were subcutaneously injected into the bilateral regions of the nude mice. Tumor volumes were later measured every 3 days until sacrifice at 31 days. Tumors were weighed and fixed in 10% formalin, followed by paraffin embedding or extraction of RNA with Trizol reagent.
Ethics approval and consent to participate
This study was conducted at the Cancer Research Institute, Central South University, Hunan, China. All of the protocols were reviewed and approved by the Joint Ethics Committee of the Central South University Health Authority and performed in accordance with national guidelines.
Cell lines and plasmids
The human lung bronchus cell line HBE (CRL-2741) and lung carcinomatous cell lines H358 (CRL-5807) and A549 (CCL-185) were obtained from American Type Culture Collection (University Boulevard, Manassas, VA, USA), The human lung carcinomatous cell lines SPC-A-1 and PC9 was stored in the Department of Molecular Biology, Cancer Research Institute (Central South University, Hunan, China). The cell lines were characterized by mycoplasma detection, DNA fingerprinting, isozyme detection, and cell viability by the provider; no further authentication of the cell lines was conducted. HBE, H358, PC9 and SPC-A-1 cells were cultured in Roswell Park Memorial Institute 1640 Media (Gibco) supplemented with 10% FBS, penicillin, and streptomycin. A549 cells were cultured in Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 (Gibco) supplemented with 10% FBS, penicillin, and streptomycin.
AhR shRNA vectors and control (GV248) were purchased from GeneChem Co., Ltd., Shanghai, China. Transfection of plasmids was performed using Lipofectamine® 2000 according to the manufacturer’s protocol. Stable expressing colonies were selected using 2 mg/ml puromycin.
Cell proliferation and colony formation
Cell proliferation assay was performed with MTT according to the manufacturer’s protocol. First, 500 cells were cultured in a 96-well plate. OD450 was measured 1 hour after adding MTS. For the cell colony formation assay, approximately 300 cells were seeded into each well of 6-well plates and cultured in media. After 2 weeks, cells were treated with methanol and stained with 0.1% crystal violet. The number of visible colonies with > 0.05 mm diameter were scored.
Cell cycle analysis
For cell cycle analysis assay, cells were washed twice with 1 × PBS, trypsinized, centrifuged at 4°C for 5 min, and washed with 1 × PBS twice. Cells were later stained with FxCycle PI/RNase Staining Solution in the dark for 30 min at room temperature. The events of cell cycle progression were analyzed using the BD FACSCanto Flow Cytometer (BD Biosciences). Flow cytometry data were analyzed using FlowJo to calculate cell-cycle distribution.
RT-qPCR
Total RNA was isolated using Trizol reagent. cDNA was generated using the PrimeScriptTM RT reagent kit with gDNA Eraser (Perfect Real Time). Real-Time PCR was performed using ABI 7500 with FastStart Universal SYBR Green Master (ROX). All gene expression levels were normalized by subtracting the Ct of the ACTB. The RT-qPCR primer sequences are listed below: SMARCA6-Forward: GATTTTGGATCGAATGCTGCCAG, SMARCA6-Reverse: ATGGACCCATCAAGCCTGCTGA; AhR-Forward: GTCGTCTAAGGTGTCTGCTGGA, AhR-Reverse: CGCAAACAAAGCCAACTGAGGTG.
Subcellular fractionation
To determine the cellular localization of AhR, cell cytoplasm and nuclear fractions were isolated and collected with Nuclear and Cytoplasmic Extraction Reagents Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Histology and immunohistochemistry
Lung and related diseases biopsies were validated and obtained from Department of Pathology in Xiangya Hospital. IHC analysis of paraffin sections, from lung cancer tissues was previously described. The images were analyzed and captured using a CX41 microscope (OLYMPUS, Tokyo, Japan) equipped with a Microscope Digital Camera System DP-72 (OLYMPUS, Tokyo, Japan), and differentially quantified by two pathologists from the Xiangya Hospital, Changsha, China.
Immunoblotting
Cells were harvested, washed with PBS, and lysed in IP lysis buffer with protease inhibitors cocktail. Cell lysates were separated on an SDS-polyacrylamide gel, transferred to a polyvinylidene fluoride membrane, and immunoblotted using the following primary antibodies. Protein detection was performed using mouse monoclonal anti-β-actin (Sigma Aldrich, A5441), mouse monoclonal anti-α-tubulin (Santa Cruz, sc-5286), rabbit polyclonal anti-histone-H3 (Proteintech, 17168-1-AP), rabbit polyclonal anti-AhR (Santa Cruz, sc-5579), mouse monoclonal anti-Flag (Santa Cruz, sc-166355), and mouse monoclonal anti-SMARCA6 (Santa Cruz, sc-46665).
Statistical analysis
The data in the figure legends are presented as the means ± SD. The exact values of the sample size (n) are presented in the figure legends and reflect either the number of experimental replications with cells, biochemical model systems, or the number of animals used in in vivo experiments. For measurements of human samples, n represents the number of patients. Statistical analyses were performed by either Student’s t test or one-way ANOVA for normally distributed data, and by Kruskal-Wallis one-way ANOVA on the ranks for data that were not normally distributed. When the overall ANOVA revealed a significant effect, the data were further analyzed with the Tukey/Dunn post hoc test to determine specific group differences. The statistical significance of differences was set at *P < 0.05, **P < 0.01, ***P<0.001.
Results
BaP increases SMARCA6 expression
PAHs were reported to be the major carcinogens in PM2.5/PM10 particles [3]. To test the effects of PAHs on SMARC genes, bronchial epithelial cell line HBE and lung adenocarcinoma cell line A549 were exposed to a representative PAH compound, benzo (a) pyrene (BaP), at 1 mM for 3 days. PCR arrays showed that expression of SMARCA6 was significantly up-regulated among the 18 SMARC genes with BaP treatment (Figure 1A). We further confirmed that BaP up-regulated SMARCA6 at mRNA levels in dose- and time-dependent manners in HBE and A549 cells (Figure 1B, 1C). Finally, Western blot analysis confirmed that BaP up-regulated SMARCA6 protein levels with BaP treatment (Figure 1D).
Figure 1.
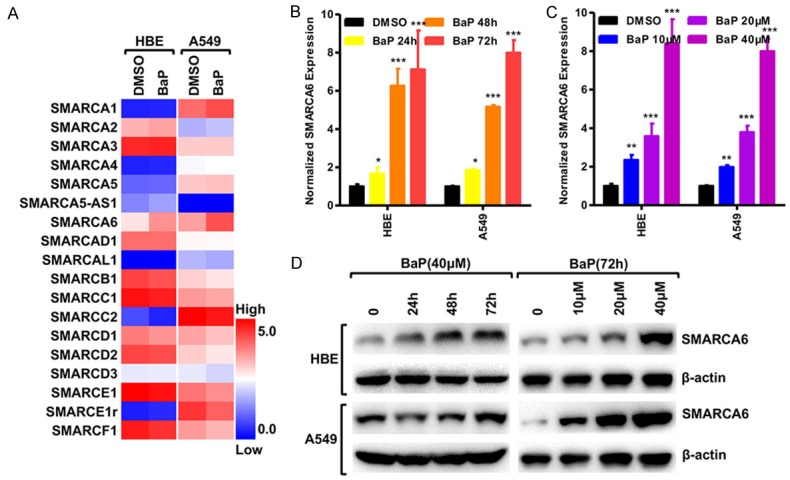
BaP increased SMARCA6 expression. (A) PCR array assessing the expression level of 18 SMARC genes in HBE and A549 cells after treatment with BaP. Log2-fold-change in RNA expression over the DMSO control; blue to red color gradation is based on the ranking of each condition from minimum (blue) to maximum (red). (B, C) qRT-PCR was used to detect SMARCA6 mRNA levels in HBE and A549 cells, in a time- (B) and dose-dependent manner (C) Treated with BaP. (D) Western blot shows the levels of SMARCA6 protein in HBE and A549 cells in a time- and dose-dependent manner in response to BaP. *P < 0.05, **P < 0.01, ***P < 0.001.
Smoking promoted SMARCA6 expression in NSCLC
To explore the expression of SMARCA6 in NSCLC, we examined the mRNA level of SMARCA6 in NSCLC tissues compared with adjacent normal tissues (n = 70). We determined that SMARCA6 expression was significantly increased in NSCLC samples compared with the adjacent normal tissues. Patient characteristics and correlations with SMARCA6 expression level are summarized in Table 1. SMARCA6 was significantly correlated with smoking history (P < 0.01), tumor type (P < 0.01), tumor differentiation (P < 0.05), classification (P < 0.05), clinical stage (TNM, P < 0.05), depth of invasion (P < 0.05) and relapse (P < 0.01). No statistically significant correlations were obtained between SMARCA6 and other clinicopathological characteristics, such as age or gender (P > 0.05) (Table 1). Importantly, the expression of SMARCA6 was significantly higher in smokers than in non-smokers in both SCC and ADC, suggesting a potential association between smoking and SMARCA6 expression level (Figure 2A, 2B). A total of 56 archived paraffin-embedded NSCLC specimens with extensive clinical follow-up, including 30 cases of ADC and 26 cases of SCC were analyzed by IHC staining with antibody against human SMARCA6. We found that SMARCA6 was up-regulated in all smoking tissues of NSCLC specimens (15 for ADC, 13 for SCC) compared with that in non-smoking tissues (Figure 2C-E). A Kaplan-Meier analysis was used to evaluate the relationship between SMARCA6 expression level and patient survival in NSCLC, and the results showed that high SMARCA6 expression was also associated with poor survival. The survival time of patients with low SMARCA6 expression (n = 45) was longer than that in patients with high SMARCA6 expression (n = 25) (P < 0.05, Figure 2F). Among patients who were smokers, the survival time of patients with low SMARCA6 (n = 11) was longer than in patients with high SMARCA6 (n = 20) (P < 0.05, Figure 2G), whereas the survival time of patients showed no significant changes among patients who were not smokers (P > 0.05, Figure 2H), indicating that smoking and SMARCA6 expression were associated with overall survival in lung cancer.
Table 1.
Correlation between SMARCA6 expression and clinicopathological parameters of NSCLC (n = 70)
| Clinical parameters | Factors | n | Relative high | Relative low | P values |
|---|---|---|---|---|---|
| Ages | ≤ 60 | 36 | 12 | 24 | 0.4973 |
| > 60 | 34 | 14 | 20 | ||
| Gender | Male | 51 | 22 | 29 | 0.3800 |
| Female | 19 | 6 | 13 | ||
| Smoking history | Yes | 31 | 20 | 11 | 0.0049 |
| No | 39 | 12 | 27 | ||
| Type | ADC | 35 | 9 | 26 | 0.0076 |
| SCC | 35 | 20 | 15 | ||
| Differentiation | Poor | 31 | 10 | 21 | 0.0261 |
| Well or Moderate | 39 | 23 | 16 | ||
| T-classification | T1-T2 | 44 | 14 | 30 | 0.0337 |
| T3-T4 | 26 | 15 | 11 | ||
| N- classification | N0 | 31 | 15 | 16 | 0.0485 |
| N1-N3 | 39 | 10 | 29 | ||
| Clinical Stage | I-II | 38 | 14 | 24 | 0.0324 |
| III-IV | 32 | 20 | 12 |
Chi-square test. *P < 0.05.
Figure 2.

Smoking links to elevated SMARCA6 expression in NSCLC. (A, B) qRT-PCR detected SMARCA6 mRNA level in lung ADC (A) and SCC (B) of smokers versus non-smokers. (C-E) IHC was performed using antibodies against SMARCA6 in human lung ADC and SCC of smokers and non-smokers (C); the mean values of quantitative immunohistochemistry are shown (D for ADC; E for SCC). (F-H) Kaplan-Meier curve, showing the overall survival rates associated with samples measured for the expression of SMARCA6, in the NSCLC tissues of smokers in lung cancer (F), smoking (G), and non-smoking (H). *P < 0.05, **P < 0.01, ***P < 0.001.
BaP increases cancer progression in vitro and in vivo
To further uncover the physiological role of BaP in cancer, we treated HBE and A549 cells with 1 mM of BaP. We determined that BaP significantly promoted the cell growth ability (Figure 3A, 3B). Moreover, BaP induced G0-G1 cell cycle progression, resulting in a considerable decrease of cell percentage in G1/S phases and an increase of G2/M-phase compared with the DMSO group (Figure 3C, 3D). Furthermore, the number of colonies formed was markedly increased in the BaP treatment of HBE and A549 cells (Figure 3E-G).
Figure 3.

BaP promoted cancer progression in NSCLC in vitro. (A, B) MTT assay was applied to assess cell viability in HBE (A) and A549 (B) treated with BaP, and the results indicate that BaP promotes cell growth. (C, D) BaP promotes the transition of G1/S to G2/M cell cycle phases in HBE (C) and A549 (D) that were treated with BaP, the representative images are shown in the left. (E-G) Growth in the plate was assessed with colony formation assay in HBE (E) and A549 (F) cells; the representative images are shown (G), and the results show that BaP promotes colony formation. *P < 0.05, **P < 0.01, ***P < 0.001.
Next, we assessed the phenotype changes induced by BaP in vivo using xenograft model. Using A549 cells, treated with BaP for 90 days, significantly generated larger tumors after 1 month of tumor growth compared with those generated by treatment with DMSO (Figure 4A); BaP increased tumor formation and weight (Figure 4B, 4C), while whole-body weight remained unchanged (Figure 4D). Finally, we determined that SMARCA6 was significantly up-regulated in the tumor samples with BaP treatment compared with their counterparts at both the protein and mRNA levels in the xenograft tumor (Figure 4E, 4F).
Figure 4.
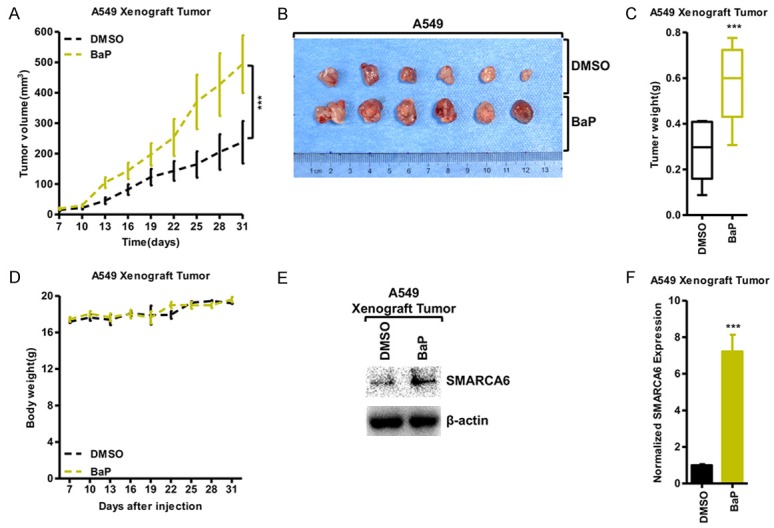
BaP promoted cancer progression in vivo. (A-D) Tumor volumes in nude mice after injection of A549 cells treated with BaP for 90 days at the indicated times (A); tumor formation (B) and weights (C) were recorded. (D) Nude mice after injection of A549 cells treated with BaP for 90 days; the weights of the mice were recorded at the indicated time points (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001. (E, F) Western blot (E) and qRT-PCR (F) demonstrated that BaP treatment in the A549 xenograft tumor increased the expression of SMARCA6.
BaP increased SMARCA6 expression via recruitment of AhR
Next, we screened for transcription factors that can regulate SMARCA6, and located potential aryl hydrocarbon receptor (AhR)-binding sites in the SMARCA6 promoter region (Figure 5C). AhR is a ligand-activated transcription factor that binds the xenobiotic-responsive element (XRE), or aryl hydrocarbon response element (AHRE) to regulate genes which respond to BaP [24]. Using qRT-PCR and Western blotting, we confirmed that the expression levels of AhR were significantly up-regulated in lung cancer cell lines compared with those of HBE. Consistent with our previous findings, SMARCA6 was up-regulated in a panel of lung cancer cells (Figure 5A, 5B). This finding indicates a strong correlation between AhR and SMARCA6. The potential interaction between AhR and SMARCA6 was analyzed by ChIP assay. The results showed that AhR can bind to the SMARCA6 promoter region in HBE and A549 cells, and BaP treatment promoted the enrichment of AhR binding in a dose- and time-dependent manner (Figure 5D-G), indicating that BaP increases SMARCA6 expression via recruitment of AhR to its promoter.
Figure 5.

BaP increases SMARCA6 expression via recruitment of AhR. (A, B) qPCR analysis (A) and Western blot (B) were used to detect the expression of AhR and SMARCA6 in a panel of lung cancer cells. (C) The AhR binding site is located at ~0.9 kb upstream of the SMARCA6 transcription start site (TSS). (D-G) ChIP assay was performed using BaP-treated or untreated HBE (D, F) and A549 (E, G) cells. The SMARCA6 promoter, enriched for AhR, was detected by qPCR in a dose- (D, F) and time-dependent (E, G) manner. *P < 0.05, **P < 0.01, ***P < 0.001.
BaP induced SMARCA6 expression by facilitating AhR translocation to the nucleus
AhR influences the major stages of tumorigenesis, and studies of aggressive tumors and tumor cell lines have shown increased levels of AhR protein and constitutive nuclear localization in cancer tissue, whereas in normal tissues AhR is mainly inactive and resides in the cytoplasm [22,23]. We found that BaP did not change AhR expression (Figure 6A). Interestingly, BaP induced AHR translocation from the cytoplasm to the nucleus in a dose and time-dependent manner indicating that BaP triggered the activation of AhR via nuclear relocation of AhR (Figure 6B). To further examine the regulation of AhR in SMARCA6, we knocked down AhR with two separate short hairpin RNAs (shRNAs) in A549 cell lines. The AhR expression was detected to confirm the efficiency of silencing by qRT-PCR and Western blot analysis (Figure 6C, 6D). The results demonstrated that the shRNA could efficiently knock down AhR expression by at least 35%. Knockdown of AhR in A549 cells decreased SMARCA6 expression at protein and mRNA levels (Figure 6E, 6F). Taken together, our results indicate that BaP increases SMARCA6 expression via the recruitment of AhR.
Figure 6.

BaP induced SMARCA6 expression by facilitating AhR translocation to the nucleus. (A) Western blot shows the protein level of AhR does not change in HBE and A549 after treatment with BaP in a time- and dose-dependent manner. (B) Activation of the AhR level in the nuclear and cytosolic fractions derived from HBE cells (Top) and A549 cells (Bottom) after treatment with BaP in a time- (left) and dose-dependent manner (right). (C, D) qPCR analysis (C) and Western blot (D) were used to detect the expression of AhR levels in A549 cells after depletion of AhR. (E, F) qPCR analysis (E) and Western blot (F) were used to detect the expression of SMARCA6 level in A549 cells after depletion of AhR. *P < 0.05, **P < 0.01, ***P < 0.001.
Knockdown of SMARCA6 attenuates BaP-induced lung cancer
To further uncover the critical role of SMARCA6 in BaP promoted lung cancer, we knocked down SMARCA6 expression in A549 cells using two separate shRNA sequences; we did not find the changes of AhR expression in the depletion of SMARCA6, but knockdown of SMARCA6 impaired SMARCA6 expression in response to BaP (Figure 7A, 7B). Moreover, knockdown of SMARCA6 decreased cell growth ability whereas BaP rescued the cell growth after knockdown of SMARCA6 in A549 cells (Figure 7C). Moreover, the number of colonies formed was markedly rescued after the BaP treatment in A549 cells after knockdown of SMARCA6 (Figure 7D, 7E). Taken together, SMARCA6 plays critical role of BaP-induced lung cancer progression.
Figure 7.
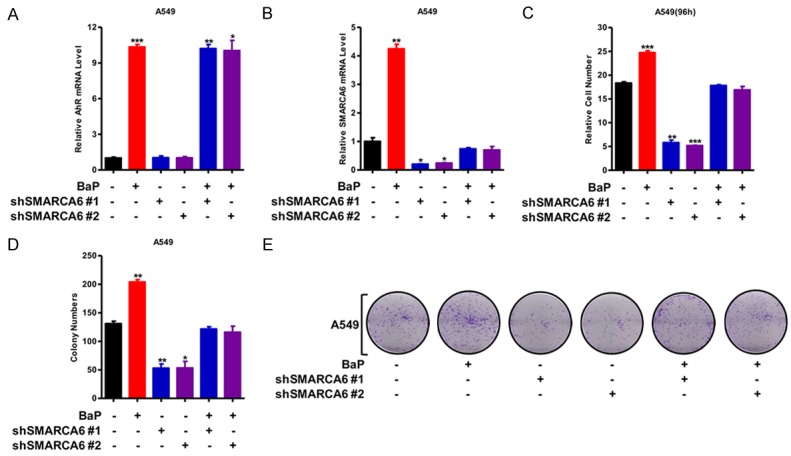
Knockdown of SMARCA6 attenuated BaP-induced lung cancer. (A, B) qRT-PCR detected AhR (A) and SMARCA6 (B) expression in A549 with BaP treatment and SMARCA6 knockdown. (C) MTT assay was applied to assess cell viability in A549 cells with BaP treatment and SMARCA6 knockdown, and the results indicate that knockdown of SMARCA6 impaired cell growth by BaP. (D, E) Growth in the plate was assessed with colony formation assay in A549 with BaP treatment and SMARCA6 knockdown (D), the representative images are shown (E), and the results indicate show that knockdown of SMARCA6 impaired colony formation and BaP rescued it. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Transcriptional regulation is a key regulatory mechanism determining cell fate; this regulation includes cis-regulatory elements and transacting factors. Among transacting factors, regulators of chromatin remodeling play a pivotal role in gene transcription and self-renewal [25-27]; these processes also rely on ATP-dependent chromatin remodeling complexes. The SWI/SNF complex is composed of 12-15 subunits, containing one of the two catalytic ATPase subunits, SMARCA4/BRG1 or SMARCA2/BRM, and several core components such as SMARCB1/SNF5, BAF170, and BAF155 [28,29]. SMARCA6/LSH is critical for the normal development of plants and mammals because it establishes correct levels of DNA methylation and patterns as a critical chromatin modifier [7,11]. SMARCA6/LSH maintains genome stability in mammalian somatic cells [12,30]. SMARCA6/LSH, as an oncogene, contributes to the malignant progression of prostate cancer, melanoma, nasopharyngeal carcinoma, and glioma via specific functions of LSH in DNA damage repair, ferroptosis, epithelial-mesenchymal transition (EMT), and metastasis [13-16,30-33]. Recent evidence indicates that SWI/SNF complexes, including SMARC genes, play a central role in human tumorigenesis.
SWI/SNF complexes are critical epigenetic regulators of tumorigenesis because of their pleiotropic roles in the regulation of the cell cycle, oncogenic pathways, and metabolism [29,34-36]. However, the interplay of gene expression between environmental factors and SMARC genes remains unknown. In this study, we show that smoking and BaP affect the expression of SMARCA6, which promotes tumorigenesis, and AhR, a critical gene that responds to environmental factors, induces SMARCA6 expression.
AhR, a widely expressed nuclear receptor that senses environmental stimuli and modulates target gene expression, plays a critical role in such cells as embryonic stem cells [37], hematopoietic stem and progenitor cells [38], and breast cancer stem cells [39,40]. AhR influences the major stages of tumorigenesis and chemoresistance [41,43], and studies of aggressive tumors and tumor cell lines have shown increased levels of AhR protein and constitutive nuclear localization in cancer tissue, whereas in normal tissues AhR is mainly inactive and resides in the cytoplasm [22,23]. In this study, we found an elevated AhR level in the nucleus in response to BaP, indicating that environmental factors might induce the activation of AhR that contributes to cancer progression and tumorigenesis. Continued expression of endogenous AhR promotes centrosome amplification in breast cancer cells in a pathway that involves cyclin E [44], which is consistent with BaP promoting cell cycle progression.
AhR signaling plays a pivotal role in self-renewal of hematopoietic stem cells, differentiation, immunity and antibacterial defense [20,45-47]. Aberrant activation of AhR signaling is implicated in breast cancer and leukemia [48,49]. In this study, we have shown that AhR signaling is impaired after knockdown of SMARCA6, and depression of SMARCA6 inhibits the progression of lung cancer and tumor-formation capacity in response to BaP. This result suggests that BaP-mediated AhR signaling is critical for promoting cancer progression and tumorigenicity in lung cells. Therefore, we consider that elevated SMARCA6 expression was activated by the signaling pathway of AhR in lung cancer cells. However, the specific mechanisms underlying the overexpression of SMARCA6 have not been thoroughly elucidated to date, and further investigations are needed.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities [2018zzts829 (M.Wang), 2015zzts099 (C.Mao)], the National Natural Science Foundation of China [81672787 and 81372427 (Y.Tao), 81772927 (D.Xiao), 81672991 (S.Liu), 81672307 (X.Wang)]; and the National Basic Research Program of China [2015CB553903 (Y.Tao)].
Disclosure of conflict of interest
None.
References
- 1.Reck M, Heigener DF, Mok T, Soria JC, Rabe KF. Management of non-small-cell lung cancer: recent developments. Lancet. 2013;382:709–19. doi: 10.1016/S0140-6736(13)61502-0. [DOI] [PubMed] [Google Scholar]
- 2.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in china, 2015. CA Cancer J Clin. 2016;66:115–32. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 3.Huang RJ, Zhang Y, Bozzetti C, Ho KF, Cao JJ, Han Y, Daellenbach KR, Slowik JG, Platt SM, Canonaco F, Zotter P, Wolf R, Pieber SM, Bruns EA, Crippa M, Ciarelli G, Piazzalunga A, Schwikowski M, Abbaszade G, Schnelle-Kreis J, Zimmermann R, An Z, Szidat S, Baltensperger U, El Haddad I, Prévôt AS. High secondary aerosol contribution to particulate pollution during haze events in china. Nature. 2014;514:218–222. doi: 10.1038/nature13774. [DOI] [PubMed] [Google Scholar]
- 4.Wang GZ, Cheng X, Zhou B, Ho KF, Cao JJ, Han Y, Daellenbach KR, Slowik JG, Platt SM, Canonaco F, Zotter P, Wolf R, Pieber SM, Bruns EA, Crippa M, Ciarelli G, Piazzalunga A, Schwikowski M, Abbaszade G, Schnelle-Kreis J, Zimmermann R, An Z, Szidat S, Baltensperger U, El Haddad I, Prévôt AS. The chemokine cxcl13 in lung cancers associated with environmental polycyclic aromatic hydrocarbons pollution. Nature. 2014;514:218–22. [Google Scholar]
- 5.Vaz M, Hwang SY, Kagiampakis I, Phallen J, Patil A, O’Hagan HM, Murphy L, Zahnow CA, Gabrielson E, Velculescu VE, Easwaran HP, Baylin SB. Chronic cigarette smoke-induced epigenomic changes precede sensitization of bronchial epithelial cells to single-step transformation by kras mutations. Cancer Cell. 2017;32:360–376. doi: 10.1016/j.ccell.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, Wenzel A, Hicks J, Ballew M, Stone M, Tran PT, Zahnow CA, Hellmann MD, Anagnostou V, Strissel PL, Strick R, Velculescu VE, Baylin SB. Epigenetic therapy ties mycdepletion to reversing immune evasion and treating lung cancer. Cell. 2017;171:1284–300. doi: 10.1016/j.cell.2017.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zemach A, Kim MY, Hsieh PH, Coleman-Derr D, Eshed-Williams L, Thao K, Harmer SL, Zilberman D. The arabidopsis nucleosome remodeler ddm1 allows DNA methyltransferases to access h1-containing heterochromatin. Cell. 2013;153:193–205. doi: 10.1016/j.cell.2013.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu W, McIntosh C, Lister R, Zhu I, Han Y, Ren J, Landsman D, Lee E, Briones V, Terashima M, Leighty R, Ecker JR, Muegge K. Genome-wide DNA methylation patterns in lsh mutant reveals de-repression of repeat elements and redundant epigenetic silencing pathways. Genome Res. 2014;24:1613–23. doi: 10.1101/gr.172015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tao Y, Xi S, Shan J, Maunakea A, Che A, Briones V, Lee EY, Geiman T, Huang J, Stephens R, Leighty RM, Zhao K, Muegge K. Lsh, chromatin remodeling family member, modulates genome-wide cytosine methylation patterns at nonrepeat sequences. Proc Natl Acad Sci U S A. 2011;108:5626–31. doi: 10.1073/pnas.1017000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myant K, Termanis A, Sundaram AY, Boe T, Li C, Merusi C, Burrage J, de Las Heras JI, Stancheva I. Lsh and g9a/glp complex are required for developmentally programmed DNA methylation. Genome Res. 2011;21:83–94. doi: 10.1101/gr.108498.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mao C, Wang X, Liu Y, Wang M, Yan B, Jiang Y, Shi Y, Shen Y, Liu X, Liai W, Yang R, Xiao D, Cheng Y, Liu S, Zhou H, Cao Y, Yu W, Muegge K, Yu H, Tao Y. A g3bp1-interacting lncrna promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res. 2018;78:3484–3496. doi: 10.1158/0008-5472.CAN-17-3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burrage J, Termanis A, Geissner A, Myant K, Gordon K, Stancheva I. The snf2 family atpase lsh promotes phosphorylation of h2ax and efficient repair of DNA double-strand breaks in mammalian cells. J Cell Sci. 2012;125:5524–34. doi: 10.1242/jcs.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Eyss B, Maaskola J, Memczak S, Möllmann K, Schuetz A, Loddenkemper C, Tanh MD, Otto A, Muegge K, Heinemann U, Rajewsky N, Ziebold U. The snf2-like helicase hells mediates e2f3-dependent transcription and cellular transformation. EMBO J. 2012;31:972–85. doi: 10.1038/emboj.2011.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao D, Huang J, Pan Y, Li H, Fu C, Mao C, Cheng Y, Shi Y, Chen L, Jiang Y, Yang R, Liu Y, Zhou J, Cao Y, Liu S, Tao Y. Chromatin remodeling factor lsh is upregulated by the lrp6-gsk3beta-e2f1 axis linking reversely with survival in gliomas. Theranostics. 2017;7:132–43. doi: 10.7150/thno.17032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keyes WM, Pecoraro M, Aranda V, Vernersson-Lindahl E, Li W, Vogel H, Guo X, Garcia EL, Michurina TV, Enikolopov G, Muthuswamy SK, Mills AA. ΔNp63α is an oncogene that targets chromatin remodeler lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell. 2011;8:164–76. doi: 10.1016/j.stem.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He X, Yan B, Liu S, Jia J, Lai W, Xin X, Tang CE, Luo D, Tan T, Jiang Y, Shi Y, Liu Y, Xiao D, Chen L, Liu S, Mao C, Yin G, Cheng Y, Fan J, Cao Y, Muegge K, Tao Y. Chromatin remodeling factor lsh drives cancer progression by suppressing the activity of fumarate hydratase. Cancer Res. 2016;76:5743–55. doi: 10.1158/0008-5472.CAN-16-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stejskalova L, Dvorak Z, Pavek P. Endogenous and exogenous ligands of aryl hydrocarbon receptor: current state of art. Cell Stem Cell. 2011;12:198–212. doi: 10.2174/138920011795016818. [DOI] [PubMed] [Google Scholar]
- 18.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, Jugold M, Guillemin GJ, Miller CL, Lutz C, Radlwimmer B, Lehmann I, von Deimling A, Wick W, Platten M. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 19.Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, Bicciato S, Mazza EM, Macchiarulo A, Vacca C, Iannitti R, Tissi L, Volpi C, Belladonna ML, Orabona C, Bianchi R, Lanz TV, Platten M, Della Fazia MA, Piobbico D, Zelante T, Funakoshi H, Nakamura T, Gilot D, Denison MS, Guillemin GJ, DuHadaway JB, Prendergast GC, Metz R, Geffard M, Boon L, Pirro M, Iorio A, Veyret B, Romani L, Grohmann U, Fallarino F, Puccetti P. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature. 2014;511:184–90. doi: 10.1038/nature13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schiering C, Wincent E, Metidji A, Iseppon A, Li Y, Potocnik AJ, Omenetti S, Henderson CJ, Wolf CR, Nebert DW, Stockinger B. Feedback control of ahr signalling regulates intestinal immunity. Nature. 2017;542:242–5. doi: 10.1038/nature21080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villa M, Gialitakis M, Tolaini M, Ahlfors H, Henderson CJ, Wolf CR, Brink R, Stockinger B. Aryl hydrocarbon receptor is required for optimal b-cell proliferation. EMBO J. 2017;36:116–28. doi: 10.15252/embj.201695027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer. 2014;14:801–14. doi: 10.1038/nrc3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan B, Liu S, Shi Y, Liu N, Chen L, Wang X, Xiao D, Liu X, Mao C, Jiang Y, Lai W, Xin X, Tang C, Luo D, Tan T, Jia J, Liu Y, Yang R, Huang J, Zhou H, Cheng Y, Cao Y, Yu W, Muegge K, Tao Y. Activation of AhR with nuclear IKKα regulates cancer stem-like properties in the occurrence of radioresistance. Cell Death Dis. 2018;9:490. doi: 10.1038/s41419-018-0542-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, Fujii-Kuriyama Y, Ishikawa T. Benzo[a] pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2000;97:779–82. doi: 10.1073/pnas.97.2.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. Mechanisms and functions of atp-dependent chromatin-remodeling enzymes. Cell. 2013;154:490–503. doi: 10.1016/j.cell.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clapier CR, Iwasa J, Cairns BR, Peterson CL. Mechanisms of action and regulation of atp-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol. 2017;18:407–22. doi: 10.1038/nrm.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Dieuleveult M, Yen K, Hmitou I, Depaux A, Boussouar F, Bou Dargham D, Jounier S, Humbertclaude H, Ribierre F, Baulard C, Farrell NP, Park B, Keime C, Carrière L, Berlivet S, Gut M, Gut I, Werner M, Deleuze JF, Olaso R, Aude JC, Chantalat S, Pugh BF, Gérard M. Genome-wide nucleosome specificity and function of chromatin remodellers in es cells. Nature. 2016;530:113–6. doi: 10.1038/nature16505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Helming KC, Wang X, Roberts CW. Vulnerabilities of mutant swi/snf complexes in cancer. Cancer cell. 2014;26:309–17. doi: 10.1016/j.ccr.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mathur R, Alver BH, San Roman AK, Wilson BG, Wang X, Agoston AT, Park PJ, Shivdasani RA, Roberts CW. Arid1a loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat Genet. 2017;49:296–302. doi: 10.1038/ng.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia J, Shi Y, Chen L, Lai W, Yan B, Jiang Y, Xiao D, Xi S, Cao Y, Liu S, Cheng Y, Tao Y. Decrease in lymphoid specific helicase and 5-hydroxymethylcytosine is associated with metastasis and genome instability. Theranostics. 2017;7:3920–32. doi: 10.7150/thno.21389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu S, Tao YG. Chromatin remodeling factor lsh affects fumarate hydratase as a cancer driver. Chin J Cancer. 2016;35:72. doi: 10.1186/s40880-016-0138-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang Y, Mao C, Yang R, Yan B, Shi Y, Liu X, Lai W, Liu Y, Wang X, Xiao D, Zhou H, Cheng Y, Yu F, Cao Y, Liu S, Yan Q, Tao Y. Egln1/c-myc induced lymphoid-specific helicase inhibits ferroptosis through lipid metabolic gene expression changes. Theranostics. 2017;7:3293–305. doi: 10.7150/thno.19988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang Y, He Y, Liu S, Tao Y. Chromatin remodeling factor lymphoid-specific helicase inhibits ferroptosis through lipid metabolic genes in lung cancer progression. Chin J Cancer. 2017;36:82. doi: 10.1186/s40880-017-0248-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masliah-Planchon J, Bièche I, Guinebretière JM, Bourdeaut F, Delattre O. Swi/snf chromatin remodeling and human malignancies. Annu Rev Pathol. 2015;10:145–71. doi: 10.1146/annurev-pathol-012414-040445. [DOI] [PubMed] [Google Scholar]
- 35.Dubey R, Lebensohn AM, Bahrami-Nejad Z, Marceau C, Champion M, Gevaert O, Sikic BI, Carette JE, Rohatgi R. Chromatin-remodeling complex swi/snf controls multidrug resistance by transcriptionally regulating the drug efflux pump abcb1. Cancer Res. 2016;76:5810–21. doi: 10.1158/0008-5472.CAN-16-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sokol ES, Feng YX, Jin DX, Tizabi MD, Miller DH, Cohen MA, Sanduja S, Reinhardt F, Pandey J, Superville DA, Jaenisch R, Gupta PB. Smarce1 is required for the invasive progression of in situ cancers. Proc Natl Acad Sci U S A. 2017;114:4153–8. doi: 10.1073/pnas.1703931114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gialitakis M, Tolaini M, Li Y, Pardo M, Yu L, Toribio A, Choudhary JS, Niakan K, Papayannopoulos V, Stockinger B. Activation of the aryl hydrocarbon receptor interferes with early embryonic development. Stem Cell Reports. 2017;9:1377–86. doi: 10.1016/j.stemcr.2017.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gazit R, Garrison BS, Rao TN, Shay T, Costello J, Ericson J, Kim F, Collins JJ, Regev A, Wagers AJ, Rossi DJ Immunological Genome Project Consortium. Transcriptome analysis identifies regulators of hematopoietic stem and progenitor cells. Stem Cell Reports. 2013;1:266–80. doi: 10.1016/j.stemcr.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prud’homme GJ, Glinka Y, Toulina A, Ace O, Subramaniam V, Jothy S. Breast cancer stem-like cells are inhibited by a non-toxic aryl hydrocarbon receptor agonist. PLoS One. 2010;5:e13831. doi: 10.1371/journal.pone.0013831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stanford EA, Wang Z, Novikov O, Mulas F, Landesman-Bollag E, Monti S, Smith BW, Seldin DC, Murphy GJ, Sherr DH. The role of the aryl hydrocarbon receptor in the development of cells with the molecular and functional characteristics of cancer stem-like cells. BMC Biol. 2016;14:20. doi: 10.1186/s12915-016-0240-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Dhfyan A, Alhoshani A, Korashy HM. Aryl hydrocarbon receptor/cytochrome p450 1a1 pathway mediates breast cancer stem cells expansion through pten inhibition and beta-catenin and akt activation. Mol Cancer. 2017;16:14. doi: 10.1186/s12943-016-0570-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stanford EA, Ramirez-Cardenas A, Wang Z, Novikov O, Alamoud K, Koutrakis P, Mizgerd JP, Genco CA, Kukuruzinska M, Monti S, Bais MV, Sherr DH. Role for the aryl hydrocarbon receptor and diverse ligands in oral squamous cell carcinoma migration and tumorigenesis. Mol Cancer Res. 2016;14:696–706. doi: 10.1158/1541-7786.MCR-16-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan KP, Wang B, Yang M, Boutros PC, Macaulay J, Xu H, Chuang AI, Kosuge K, Yamamoto M, Takahashi S, Wu AM, Ross DD, Harper PA, Ito S. Aryl hydrocarbon receptor is a transcriptional activator of the human breast cancer resistance protein (bcrp/abcg2) Mol Pharmacol. 2010;78:175–85. doi: 10.1124/mol.110.065078. [DOI] [PubMed] [Google Scholar]
- 44.Korzeniewski N, Wheeler S, Chatterjee P, Duensing A, Duensing S. A novel role of the aryl hydrocarbon receptor (ahr) in centrosome amplification-implications for chemoprevention. Mol Cancer. 2010;9:153. doi: 10.1186/1476-4598-9-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, Siddiqui S, Basso AS, Otterbein LE, Pardoll DM, Pan F, Priel A, Clish CB, Robson SC, Quintana FJ. Metabolic control of type 1 regulatory t cell differentiation by ahr and hif1-alpha. Nat Med. 2015;21:638–46. doi: 10.1038/nm.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moura-Alves P, Fae K, Houthuys E, Dorhoi A, Kreuchwig A, Furkert J, Barison N, Diehl A, Munder A, Constant P, Skrahina T, Guhlich-Bornhof U, Klemm M, Koehler AB, Bandermann S, Goosmann C, Mollenkopf HJ, Hurwitz R, Brinkmann V, Fillatreau S, Daffe M, Tümmler B, Kolbe M, Oschkinat H, Krause G, Kaufmann SH. Ahr sensing of bacterial pigments regulates antibacterial defence. Nature. 2014;512:387–92. doi: 10.1038/nature13684. [DOI] [PubMed] [Google Scholar]
- 47.Rentas S, Holzapfel N, Belew MS, Pratt G, Voisin V, Wilhelm BT, Bader GD, Yeo GW, Hope KJ. Musashi-2 attenuates ahr signalling to expand human haematopoietic stem cells. Nature. 2016;532:508–11. doi: 10.1038/nature17665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bunaciu RP, Yen A. Activation of the aryl hydrocarbon receptor ahr promotes retinoic acid-induced differentiation of myeloblastic leukemia cells by restricting expression of the stem cell transcription factor oct4. Cancer Res. 2011;71:2371–80. doi: 10.1158/0008-5472.CAN-10-2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.D’Amato NC, Rogers TJ, Gordon MA, Greene LI, Cochrane DR, Spoelstra NS, Nemkov TG, D’Alessandro A, Hansen KC, Richer JK. A tdo2-ahr signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015;75:4651–64. doi: 10.1158/0008-5472.CAN-15-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]