

Abstract
The prognosis of advanced hepatocellular carcinoma (HCC) patients remains extremely poor, partially due to the development of acquired resistance to sorafenib and chemotherapy. Cabazitaxel, a semisynthetic taxane, has been approved for the therapy of docetaxel-resistant prostate cancer. However, no studies have been performed on the effect of cabazitaxel on HCC, and whether cabazitaxel remains sensitive in chemotherapy-resistant and sorafenib-resistant HCC cells is not clear. Our results demonstrate that cabazitaxel is highly toxic to HCC cell lines in a time- and dose-dependent manner by inducing G2/M phase arrest and apoptosis in vitro. Cabazitaxel also significantly suppresses HCC tumor growth in vivo. In chemotherapy-resistant HCC cell Huh-TS-48 with P-gp-overexpression, cabazitaxel shows less cross-resistant to other chemotherapeutic agents. The resistance fold of cabazitaxel, doxorubicin, paclitaxel, docetaxel and vinorelbine is 1.53, 8.60, 38.58, 15.53 and 18.06 respectively. Furthermore, sorafenib-resistant HCC cell SK-sora-5 is still sensitive to cabazitaxel. The IC50 values of cabazitaxel after 72 h exposure for parental cell SK-hep-1 and resistant cell SK-sora-5 are 0.84 and 0.73 nM. The results indicate that cabazitaxel is a potential agent to treat HCC after developing chemotherapy resistance caused by overexpression of P-gp and acquired resistance to sorafenib, and might improve prognosis in advanced HCC patients.
Keywords: Hepatocellular carcinoma, cabazitaxel, acquired resistance, sorafenib, P-gp
Introduction
Globally, hepatocellular carcinoma (HCC), which accounts for 90% of primary liver cancers, is the fifth most common malignancy and the third leading cause of cancer-related mortality [1]. Curative treatments, such as surgical resection and liver transplantation, are suitable for early HCC. Nevertheless, the majority of HCC patients present with advanced stage at diagnosis, making chemotherapy and targeted agents the best option for controlling tumor progression in such patients [2-5].
Sorafenib, a multi-kinase inhibitor, is the only approved systemic treatment that prolongs overall survival in select advanced HCC patients [4,5]. Despite an evident survival benefit, the response rate is quite low, and the mean prolongation of survival is modest at about 2-3 months. Another limit to the efficacy of this medication is the acquired resistance. Although almost 40% of HCC tumors are initially controlled by sorafenib treatment, these patients acquire resistance over time and experience disease progression. Currently, there is no existing second-line therapy for patients who relapse following sorafenib therapy in clinic. Systematic chemotherapy has been applied for over 30 years, but still lacks definite evidence of improving prognosis [6]. Traditional therapeutic agents including doxorubicin, cisplatin and 5-FU have been limited by resistance, systemic toxicity and poor efficacy [7-9]. MDR1 (encoding P-gp) is one of the most vital reasons which cause multidrug resistance and leads to failure in HCC chemotherapy [10-13]. Partially due to the development of resistance to sorafenib and chemotherapy, the prognosis of advanced HCC patients remains extremely poor. This creates an unmet medical need for alternative therapies for advanced HCC. Novel therapeutic agents are urgently required to improve the situation.
Cabazitaxel, a semisynthetic taxane, has been approved by the American Food and Drug Agency (FDA) and European Medicines Agency for therapy of castration-resistant prostate cancer with disease progression after docetaxel treatment [14,15]. The TROPIC trial demonstrated that cabazitaxel offers an overall survival benefit of 2.4 months over mitoxantrone in such patients. Besides prostate cancer, clinical efficacy has been reported in metastatic breast cancer patients resistant to former taxanes [16,17]. The antitumor activity spectrum of cabazitaxel in mice bearing human xenografts is broad [18,19]. Same with other taxanes, cabazitaxel exerts cytotoxic effect through mitotic arrest by stabilizing microtubules, which leads to cell death [20]. Compared with paclitaxel and docetaxel, cabazitaxel possesses a relatively low affinity for P-gp, which may offer potential for a wider antitumor spectrum and more durable response. Although cabazitaxel has been used clinically in different cancers, its effect on HCC has not been researched before. Whether cabazitaxel remains sensitive in chemotherapy-resistant and sorafenib-resistant HCC cells is still unclear.
Herein, we demonstrate that cabazitaxel is highly toxic to HCC cell lines in a time- and dose-dependent manner by inducing G2/M phase arrest and apoptosis in vitro. Cabazitaxel substantially inhibits HCC tumor growth in vivo as well. To investigate whether chemotherapy-resistant and sorafenib-resistant HCC cells are sensitive to cabazitaxel, chemotherapy-resistant HCC cell line Huh-TS-48 with P-gp overexpression and sorafenib-resistant HCC cell line SK-sora-5 were established. Cabazitaxel is much less cross-resistant in Huh-TS-48 than doxorubicin, paclitaxel, docetaxel and vinorelbine. Furthermore, SK-sora-5 is still sensitive to cabazitaxel. These results indicate that cabazitaxel offers a potential approach for treating HCC after experiencing chemotherapy resistance caused by overexpression of P-gp and acquired resistance to sorafenib, and might improve prognosis in advanced HCC patients.
Materials and methods
Cell culture and mice
All cells were purchased from China Center for Type Culture Collection (CCTCC). Human hepatocellular carcinoma cell lines SK-hep-1, SMMC7721, Huh-7, HCC-LM3, Huh-TS-48 and SKsora-5 were maintained in minimum essential medium (Gibco-Invitrogen, USA) supplemented with 10% fetal bovine serum at 37°C in a humidified incubator with 5% CO2. Female aged 4-5 weeks of athymic nude (nu/nu) mice were purchased from Shanghai SLAC animal facility. All animals care and experiments were conducted according to the Guidelines of Zhejiang University Animal Care Committee.
Antitumor activity in tumor-bearing mice
HCC-LM3 cells (4 × 106 cells in 0.1 mL PBS) were implanted into the right flanks of the homozygous nude athymic mice (female, 5-6-weeks old). Three days after implantation, xenografts were randomly divided into 2 groups and treated with different regimens: (i) vehicle; (ii) cabazitaxel at 10 mg/kg, i.v. The same treatment regimens were repeated every 6 days for a total of 4 cycles. Two perpendicular diameters (width and length) of the tumors were measured every 3 days until the animals were terminated. After the animals were killed, the tumor tissues were removed and weighted. Cabazitaxel solution (1 mg/ml) was prepared by mixing 1 volume of DMSO, 1 volume of polysorbate 80, and 38 volumes of sterile water.
Establishment of drug resistant cell lines
Huh-TS-48 cells were produced based on a strategy of pulsed exposure to paclitaxel with time-stepwise increments. Briefly, Huh-7 cells were exposed to 50 nM paclitaxel in time increments ranging from 0.5 h to 24 h (0.5, 1, 2, 4, 12, 24 h) in the adaptation stage. The surviving cells were amplified in paclitaxel free medium. Each pulse treatment was repeated 3 times, followed by exposure to the next longer time-course. In the consolidation stage, previously selected cells were exposed to ten pulses of treatment with 50 nM paclitaxel for 48 h. The resulting cell line was named Huh-TS-48 and was maintained in paclitaxel-free medium.
SK-sora-5 cells were selected based on continuous exposure to sorafenib using a dose-stepwise incremental strategy from SK-hep-1. In the adaptation stage, SK-hep-1 cells were exposed to sorafenib for 72 h in stepwise increments of concentrations ranging from 1 μM to 2 μM (1, 2 μM). Following each dose-induced step, surviving cells were amplified in sorafenib-free medium. After repeating 3 times (for each dose), cells were exposed to the next higher concentration of sorafenib. In the consolidation stage, previously selected cells were continuously cultured in medium containing 5 μM sorafenib, until they multiplied normally in medium containing 5 μM sorafenib. The resulting resistant cell line (SK-sora-5) was maintained in medium containing 5 μM sorafenib.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (MTT assay)
Cells were harvested and resuspended to a final concentration of 10^4 cells/ml. Aliquots of the cell suspension were evenly distributed into 96-well tissue culture plates. After one night of incubation, the designated columns were treated with drug regimes. Four hours prior to the end of time point, MTT solution was added. The medium containing MTT was replaced with 150 μL of DMSO in each well to dissolve the formazan crystals after 4 h-incubation. The absorbance in individual wells was determined at 570 nM using a microplate reader (Bio-Rad, Sunnyvale, CA).
Colony-forming assay
SK-hep-1, Huh-7 cells were seeded into 6-well plates in triplicates at a density of 500 cells/well and were maintained with or without cabazitaxel for 10 days. SK-hep-1 and SK-sora-5 cells were seeded into 6-well plates in triplicates at a density of 500 cells/well, and were maintained with or without sorafenib for 12 days. The cell clones were stained with Giemsa.
Analysis of cell cycle
Cell cycle distribution was determined by flow cytometric analysis. Briefly, after drug exposure for 48 h, both detached and attached cells were harvested and washed twice with PBS, followed by fixation in 75% ethanol diluted in PBS. Cells were then incubated in PBS containing 100 μL/mL RNase and 40 μL/mL propidium iodide (PI) at room temperature for 0.5 h before flow cytometric analysis. Cell cycle distribution and DNA content were determined using a Coulter Epics V instrument (Beckman Coulter, Inc., Fullerton, CA).
Analysis of apoptosis
Annexin V/PI apoptosis detection kit (Beyotime, Haimen, China) was used to detect cell apoptosis according to the manufacturer’s instructions. Briefly, cells were harvested and washed twice with PBS after treatment for 48 h. Then the cells were suspended with 400 μL of binding buffer, 5 μL of Annexin V antibody conjugated with fluorescein isothiocyanate (FITC) and 5 μL of PI solution. After being incubated in the mixture for 15 min at room temperature in the dark, the percentage of apoptotic cells was determined by flow cytometry.
Western blotting
Equal amounts (30 μg/lane) of proteins were fractionated on 12% SDS-PAGE gels and transferred to polyvinylidene difluoride membranes. The membranes were incubated with anti-Cdc25c, anti-Cdc2, anti-pCdc2, anti-cyclin B1, anti-Bcl2, anti-PARP and anti-β-actin primary antibodies (Cell Signaling Technology, Danvers, MA, USA), respectively. After washing with TBS containing 0.1% (v/v) Tween 20, the membranes were incubated with peroxidase-conjugated goat anti-mouse or rabbit secondary antibodies. The western blot bands were visualized using ECL kits in accordance with the manufacturer’s instructions (Abcam, Cambridge, MA, USA). β-actin was used for normalization of protein loading.
Statistical analysis
Data are presented as mean ± standard error of three independent experiments. Two-sided Student’s t-test was used to determine the statistical difference between various experimental and control groups. Differences were considered statistically significant at a level of P < 0.05. Statistical analysis of the data presented was performed by SPSS version 22.0 (SPSS, Chicago, IL, USA).
Results
Cabazitaxel shows high cytotoxic effects on HCC cells
To test the cytotoxic effects of cabazitaxel on HCC cells, we examined the sensitivities of four HCC cell lines (SK-hep-1, SMMC7721, Huh-7, HCC-LM3) to cabazitaxel using MTT assay. As shown in Figure 1A, cabazitaxel treatment substantially inhibited HCC cell proliferation in a time- and dose-dependent manner. Colony formation assays demonstrated that when treated with 5 nM cabazitaxel, SK-hep-1 and Huh-7 cells both showed fewer and smaller colonies than the control group (Figure 1B). The IC50s of cabazitaxel after 24 h, 48 h and 72 h treatment were determined for each cell line (Figure 1C). IC50 values of 72 h cabazitaxel exposure for four HCC cell lines were all below 5 nM, ranging from 0.84 to 4.52 nM. Taken together, we demonstrate that cabazitaxel is effective in inhibiting cell viability of various HCC cell lines at nanomolar concentrations.
Figure 1.
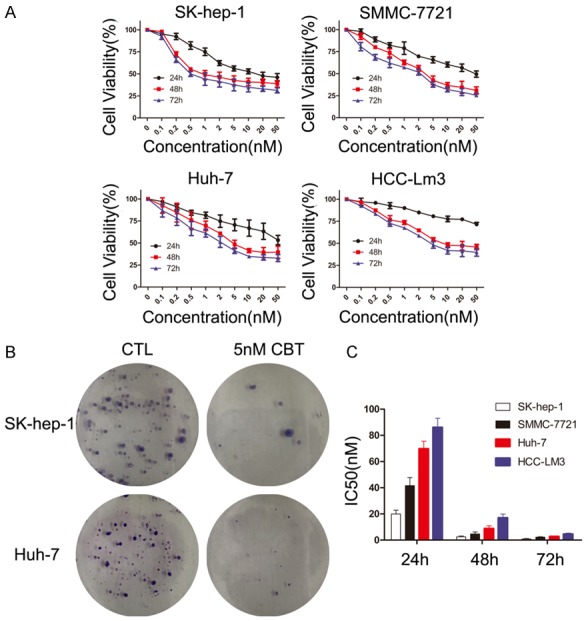
Effect of cabazitaxel on HCC cell lines in vitro. A. HCC cells were treated with increasing doses of cabazitaxel (0-50 nM) for 12 h, 24 h, 48 h, 72 h. Then, HCC cells proliferation was assessed by the MTT assay. B. SK-hep-1 and Huh-7 cells’ colony formation with or without 5 nM cabazitaxel treatment. C. The IC50s of cabazitaxel after 24 h, 48 h and 72 h treatment were determined for each cell line; Data are shown as mean ± SD. CTL, control; CBT, cabazitaxel.
Cabazitaxel suppresses HCC tumor growth in mouse xenograft models
The results presented above demonstrated that cabazitaxel treatment substantially inhibited HCC cell proliferation in vitro. Those findings had raised a clinically relevant concern about whether this phenomenon would also occur in vivo. As shown in Figure 2A-C, after four treatment cycles, the tumors of cabazitaxel treated mice were significantly smaller than those of the control group. The mean tumor volumes of control and cabazitaxel group were 539.9 ± 85.2, 121.7 ± 29.3 mm3 (n = 6, p < 0.01), and mean tumor weights were 0.29 ± 0.06, 0.06 ± 0.03 g (n = 6, p < 0.01). Furthermore, removed HCC-LM3 xenograft tumors were tested by immunohistochemistry staining with Ki-67. As shown in Figure 2D, Ki-67-positive cells significantly reduced in the cabazitaxel group, compared with the control group. Taken together, these results indicated that cabazitaxel treatment could significantly inhibit HCC tumor proliferation in vivo.
Figure 2.
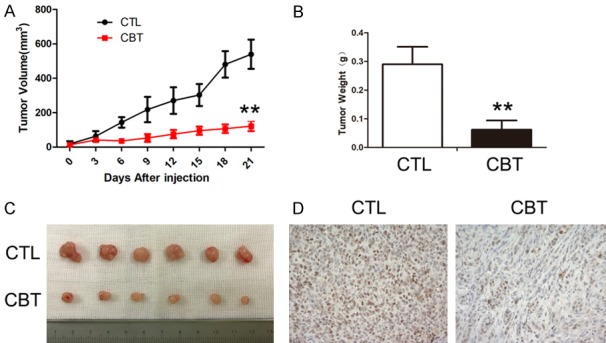
Effect of cabazitaxel on HCC tumor growth in vivo. The tumor volume (A) and tumor weight at the end of experiments (B) were compared in control and cabazitaxel group. (C) The tumor mass with or without cabazitaxel treatment. Data are shown as mean ± SD. **P < 0.01 vs. control group. Ki-67 immunohistochemistric staining (D, × 200) were performed. CTL, control; CBT, cabazitaxel.
Cabazitaxel triggers G2/M cell cycle arrest in HCC cells
The effect of cabazitaxel on cell cycle arrest in SK-hep-1 and Huh-7 cells was studied. As shown in Figure 3A, when treated with 5 nM cabazitaxel for 48 hours, the proportion of cells at G2-M phase significantly increased both in SK-hep-1 and Huh-7 from 5.14% to 57.77% and from 7.71% to 55.54%, respectively. Furthermore, we conducted a detailed analysis of several regulatory proteins associated with cabazitaxel-induced mitotic arrest. As depicted in Figure 3B, treatment of cabazitaxel resulted in a decrease in Cdc25c, Cdc2, pCdc2 and cyclin B1 protein levels. In a word, our data support that cabazitaxel induces G2/M phase arrest in HCC cells via Cdc25C/Cdc2/cyclin B1 pathway.
Figure 3.
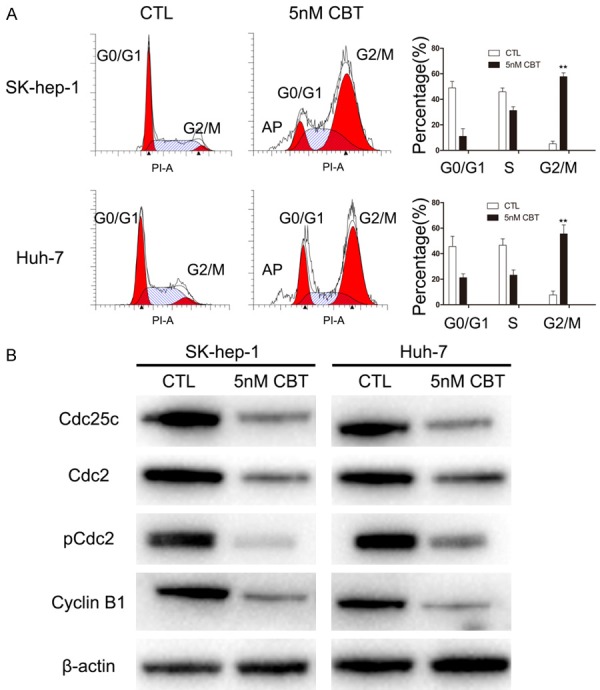
Cell cycle analysis of cabazitaxel-treated cells. A. Cabazitaxel treatment caused G2/M arrest. SK-hep-1 and Huh-7 were treated with or without 5 nM cabazitaxel for 48 h. B. Western blot analysis of Cdc25c, Cdc2, pCdc2 and Cyclin B1 proteins after cabazitaxel treatment. Data are shown as mean ± SD. *P < 0.05 vs. control group; **P < 0.01 vs. control group. CTL, control; CBT, cabazitaxel.
Cabazitaxel induces apoptosis in HCC cells
Annexin V and propidium iodide staining were used to assess apoptosis. The results indicated that in comparison with the control group, the ratio of apoptotic cells statistically increased when exposed to 5 nM cabazitaxel for 48 h in SK-hep-1 and Huh-7 (Figure 4A), increasing from 3.6% to 21% and from 4.7% to 20.3%, respectively. Then, we analyzed the molecules likely to be involved in cabazitaxel-induced apoptosis. As shown in Figure 4B, exposure to cabazitaxel led to a decline in anti-apoptotic protein Bcl2 and a stronger cleavage of poly ADP-ribose polymerase (PARP). Taken together, cabazitaxel promotes apoptosis in HCC cells through the Bcl2/PARP pathway.
Figure 4.
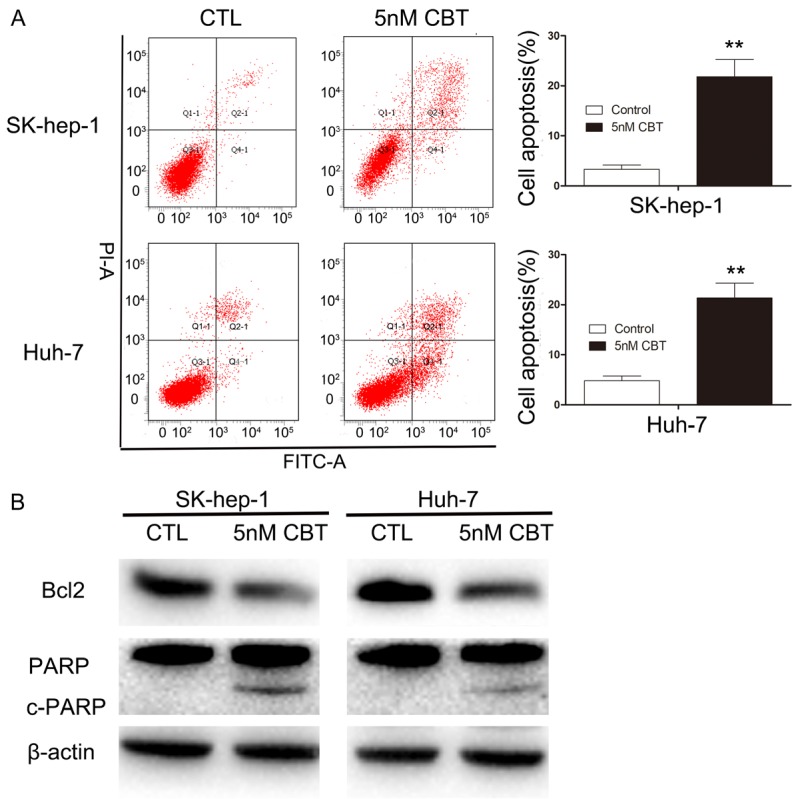
Effect of cabazitaxel on HCC cells apoptosis. A. Cabazitaxel promotes apoptosis in HCC cells. SK-hep-1 and Huh-7 were treated with or without 5 nM cabazitaxel for 48 h. FITC, Annexin V/PI staining was used to identify apoptosis. The data are shown as mean ± S.D. from three experiments. *P < 0.05 vs. control group; **P < 0.01 vs. control group. B. Bcl2, PARP and c-PARP proteins were measured by Western blot. CTL, control; CBT, cabazitaxel.
Cabazitaxel is much less cross-resistant in P-gp-overexpressed HCC cell
Elevated levels of P-gp plays a major role in acquired resistance to paclitaxel in HCC [21]. To explore whether cabazitaxel remains sensitive in P-gp-overexpressed HCC cell, we selected Huh-TS-48 based on pulsed exposure to paclitaxel with a time-stepwise increments strategy to build a P-gp-overexpressed HCC cell model. Compared to parent cell Huh-7, overexpression of P-gp was confirmed in Huh-TS-48 by western blot (Figure 5A). MTT assay showed cross-resistant of Huh-TS-48 to doxorubicin, paclitaxel, docetaxel and vinorelbine, all known to have a high affinity for P-gp (Table 1). The resistance fold was 8.60, 38.58, 15.53 and 18.06 respectively. Next, we tested the effect of cabazitaxel on Huh-7 and Huh-TS-48 (Figure 5B, 5C). The results showed that the resistance fold of cabazitaxel is 1.53 (Table 1), which is significantly smaller than the former four drugs (Figure 5D). Taken together, we demonstrated that cabazitaxel is much less cross-resistant in P-gp-overexpressed HCC cell compared to doxorubicin, paclitaxel, docetaxel and vinorelbine.
Figure 5.
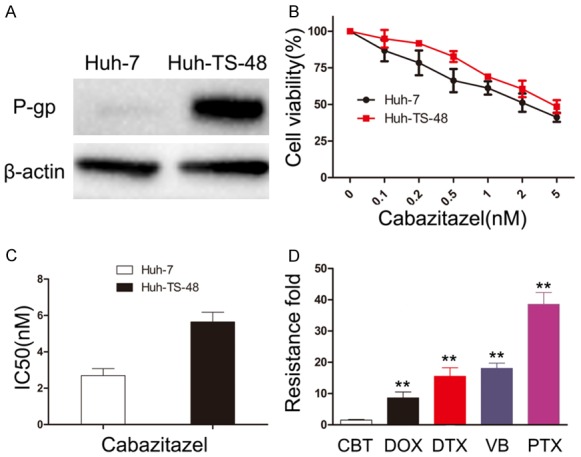
Effect of cabazitaxel on P-gp-overexpressed HCC cell. A. P-gp was measured by Western blot in Huh-7 and Huh-TS-48. B. Huh-7 and Huh-TS-48 were treated with increasing doses of cabazitaxel (0-5 nM) for 72 h. Then, cell proliferation was assessed by the MTT assay. C. Resistance fold was determined by dividing IC50 values of Huh-TS-48 by that of Huh-7 cells. D. Resistance fold of cabazitaxel, doxorubicin, paclitaxel, docetaxel and vinorelbine was calculated. The data are shown as mean ± S.D. from three experiments, *P < 0.05 vs. cabazitaxel, **P < 0.01 vs. cabazitaxel. CBT, cabazitaxel; DOX, doxorubicin; DTX, docetaxel; VB, vinorelbine; PTX, paclitaxel.
Table 1.
Drug sensitivity of Huh-7 and Huh-TS-48
| Drug (72 h) | Huh-7 IC50 (nM) | Huh-TS-48 | |
|---|---|---|---|
|
| |||
| IC50 (nM) | RF | ||
| Doxorubicin | 299.67 ± 44.81 | 2578.33 ± 301.22 | 8.60 |
| Paclitaxel | 11.70 ± 3.04 | 451.33 ± 36.47 | 38.58 |
| Docetaxel | 5.67 ± 0.96 | 88.05 ± 14.71 | 15.53 |
| Vinorelbine | 8.92 ± 2.22 | 161.09 ± 16.36 | 18.06 |
| Cabazitaxel | 2.69 ± 0.38 | 4.11 ± 0.53 | 1.53 |
Note: The IC50s of five chemotherapeutic agents after 72 h treatment were determined for Huh-7 and Huh-TS-48. Resistance fold (RF) was determined by dividing IC50 values of Huh-TS-48 by that of Huh-7 cells.
Cabazitaxel is sensitive in sorafenib-resistant HCC cell
Acquired resistance limits the efficacy of sorafenib treatment in clinic. To test whether cabazitaxel is still sensitive in sorafenib-resistant HCC cell, we selected SK-sora-5 based on continuous exposure to sorafenib using a dose-stepwise incremental strategy to establish a sorafenib-resistant HCC cell model. Colony formation assays demonstrated that while the number and size of colonies are comparable between SK-hep-1 and SK-sora-5 in the control group, SK-hep-1 showed much fewer and smaller colonies than SK-sora-5 when treated with 5 μM sorafenib (Figure 6A). We then examined the sensitivities of SK-hep-1 and SK-sora-5 to sorafenib using MTT assays. As shown in Figure 6B, the IC50 values of 72 h sorafenib exposure for SK-hep-1 and SK-sora-5 were 3.86 and 8.12 μM (P < 0.01). These two experiments validated that SK-sora-5 is resistant to sorafenib, compared with SK-hep-1. Next, we examined the effect of cabazitaxel on SK-hep-1 and SK-sora-5. As depicted in Figure 6C, the IC50 values of 72 h cabazitaxel exposure for SK-hep-1 and SK-sora-5 were 0.84 and 0.73 nM. Taken together, these results demonstrate that cabazitaxel is still sensitive in HCC cell resistant to sorafenib.
Figure 6.
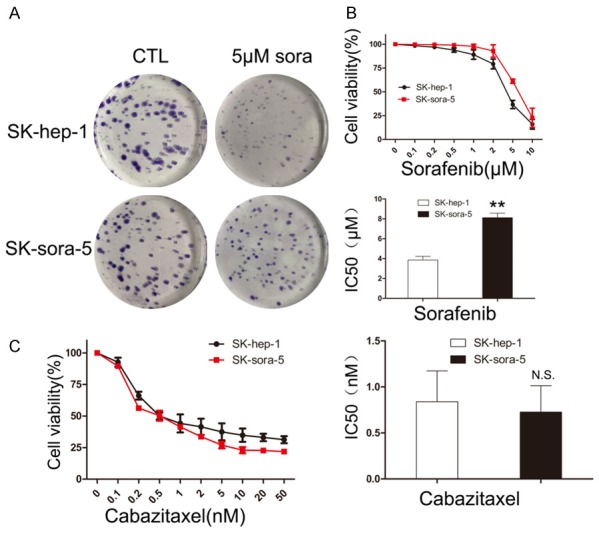
Effect of cabazitaxel on sorafenib-resistant HCC cell. A. SK-hep-1 and SK-sora-5 cells’ colony formation with or without 5 μM sorafenib treatment. B. SK-hep-1 and SK-sora-5 were treated with increasing doses of sorafenib (0-10 μM) for 72 h. Then, cell proliferation was assessed by the MTT assay. The IC50s of sorafenib after 72 h treatment were determined, Data are shown as mean ± S.D, *P < 0.05 vs. SK-hep-1, **P < 0.01 vs. SK-hep-1. C. SK-hep-1 and SK-sora-5 were treated with increasing doses of cabazitaxel (0-50 nM) for 72 h. Then, cell proliferation was assessed by the MTT assay. The IC50s of cabazitaxel after 72 h treatment were determined, Data are shown as mean ± S.D. **P < 0.01 vs. SK-hep-1, N.S vs. SK-hep-1. CTL, control; sora, sorafenib.
Discussion
HCC is a major global health issue, especially in China, with little improvement in prognosis during the past few decades. Hence, novel therapeutic agents are urgently required. Herein, we offer the most comprehensive preclinical assessment of a semisynthetic taxane, cabazitaxel, in HCC. Cabazitaxel shows high toxicity in HCC cell lines at nanomolar concentrations by inducing G2/M phase arrest and apoptosis in vitro, and significantly suppresses HCC tumor growth in vivo. Moreover, cabazitaxel is much less cross-resistant in chemotherapy-resistant HCC cell, and remains sensitive to sorafenib-resistant HCC cell.
Cabazitaxel is a second-generation semisynthetic taxane approved by American FDA for the therapy of patients with metastatic castration-resistant prostate cancer with disease progression after docetaxel-containing treatment [14]. In addition, clinical trials of cabazitaxel contained phase I/II studies in patients with various advanced solid tumors, including metastatic breast cancer, lung cancer and colorectal cancer, in which partial and complete responses were observed [16,22-24]. Besides, cabazitaxel showed antitumor activity in animals bearing glioblastoma xenografts [25]. However, the effect of cabazitaxel on HCC has not been previously researched. To our best knowledge, this is the only study to demonstrate the cytotoxic effect of cabazitaxel on HCC in vitro and in vivo. Our results revealed that cabazitaxel treatment inhibits HCC cell proliferation in a time- and dose-dependent manner. Cabazitaxel at nanomolar concentrations was sufficiently enough to affect HCC cells in MTT and colony formation assays. As shown in Table 1, cabazitaxel had dose advantage over the other chemotherapeutic agents in SK-hep-1 and Huh-7 cell, thus may avoid certain dose-related toxic effects and improve patient tolerance in clinic. Besides, we further confirmed that cabazitaxel treatment could generate anti-cancer effects on HCC in vivo. Mechanistically, cabazitaxel induced HCC cell G2/M phase arrest via Cdc25C/Cdc2/cyclin B1 pathway and apoptosis through the bcl2/PARP pathway, consistent with its effect on other cancer cells [20].
Previously, chemotherapy was not routinely used in terminal stage HCC, as it was considered chemo-refractory and because of adverse events (AEs) [26]. Numerous studies have reported low response rates of single or combined chemotherapeutic agents in HCC. Though when compared with best supportive care, doxorubicin was initially shown to improve survival [27], two subsequent clinic trials showed a low response rate when used as monotherapy or combination regimens [7]. The combination regimens PIAF (cisplatin, interferon, doxorubicin and 5-fluorouracil) showed positive outcomes with a median overall survival of 8.9 month [28]. However, a subsequent study comparing PIAF with doxorubicin failed to meet the primary endpoint (OS: 8.6 mo vs. 6.8 mo, P = 0.83), with no survival benefit [29]. To date, chemotherapy (monotherapy or combination regimens) has been researched in abundant clinical trials in HCC, but no significant persuasive efficacy in prolonging survival has been shown. Resistance is one of the major reasons leading to chemotherapy failure in HCC. The mechanisms of resistance include drug transport and metabolism, alterations in drug targets, DNA damage repair, activation of prosurvival pathways, ineffective induction of cell death and etc. MDR1 (encoding P-gp) is one of the most vital reasons which cause multidrug resistance and lead to chemotherapy failure in HCC [9,10]. The expression of P-gp is found in 80-90% of HCC patients and has been shown to negatively correlate with response to chemotherapy in one study [11]. In another study, P-gp expression is significantly higher both in HCC neoplastic and perineoplastic tissues, compared with cirrhotic and cholestatic liver tissues [12]. In this study, chemotherapy-resistant HCC cell line Huh-TS-48 with P-gp overexpression was successfully established to mimic the obstacles in chemotherapy of HCC in clinic. Huh-TS-48 exhibited a phenotype of MDR after continuous exposure to paclitaxel, as HCC is mostly chemotherapy-resistant and known to have innate and acquired MDR [30]. MTT assays show that cabazitaxel is much less cross-resistant in Huh-TS-48, indicating cabazitaxel as an effective agent for HCC with multidrug resistance.
Sorafenib was the first approved targeted agent which demonstrated survival benefits in advanced HCC patients. Two landmark phase III trials (SHARP and the Asia-Pacific studies) showed sorafenib as a prominent progress in the treatment of terminal HCC with prolonged overall survival (10.7 mo vs. 7.9 mo, 6.5 mo vs. 4.2 mo) [4,5]. Despite initial response to sorafenib, acquired resistance has been shown to limit the efficacy. Currently, no alternative second-line drugs are available after the failure of sorafenib therapy. Therefore, seeking for substitute therapy after sorafenib resistance is promising and worthwhile. In our study, sorafenib-resistant HCC cell SK-sora-5 was successfully established to imitate the setbacks in targeted therapies in HCC. SK-sora-5 became resistant to sorafenib as constantly cultured in drug-contained media, mimicking initial response and subsequent resistance to sorafenib therapy of HCC in clinic. MTT and colony formation assays show that SK-sora-5 remains sensitive to cabazitaxel. Our data demonstrated that cabazitaxel offers a potential approach for HCC with acquired resistance to sorafenib. Hence, combination or sequential therapy of cabazitaxel and sorafenib may be a novel effective regimen to treat advanced HCC. The mechanistic basis of acquired sorafenib resistance will be explored in further study.
Our study has two important implications. First, as far as we know, it is the only study demonstrating the effect of cabazitaxel on HCC both in vitro and in vivo. Second, we tested the effect of cabazitaxel on two novel drug-resistant cell models, which were built to imitate the setbacks in the systematic therapy of advanced HCC in clinic. The results indicated that cabazitaxel is a potential agent to treat HCC after experiencing chemotherapy resistance caused by overexpression of P-gp and acquired resistance to sorafenib, and might improve prognosis in advanced HCC patients. Nevertheless, additional studies involving HCC patients and patient-derived xenograft models will be needed to refine response prediction of cabazitaxel and optimize selection in clinic. Further studies are needed to identify the molecular basis of cabazitaxel resistance which may promote the development of effective drug combination strategies and maximize the anti-tumor effects of cabazitaxel. In conclusion, our findings provide evidence for future development of cabazitaxel as a novel chemotherapeutic agent against HCC in clinical settings.
Acknowledgements
This research was supported by Innovative Research Groups of National Natural Science Foundation of China (No. 81721091); National S&T Major Project (No. 2017ZX10203205); Zhejiang International Science and Technology Cooperation Project (NO. 2016C04003); Zhejiang Provincial Natural Science Foundation (LY18H160017).
Disclosure of conflict of interest
None.
References
- 1.Lindsey AT, Freddie B, Rebecca LS, Jacques F, Joannie LT, Ahmedin J. Global cancer statistics, 2012. Ca A Cancer Journal for Clinicians. 2015;61:69–90. [Google Scholar]
- 2.Schwartz M, Roayaie S, Konstadoulakis M. Strategies for the management of hepatocellular carcinoma. Nat Clin Pract Oncol. 2007;4:424–32. doi: 10.1038/ncponc0844. [DOI] [PubMed] [Google Scholar]
- 3.Forner A, Reig ME, de Lope CR, Bruix J. Current strategy for staging and treatment: the BCLC update and future prospects. Semin Liver Dis. 2010;30:61–74. doi: 10.1055/s-0030-1247133. [DOI] [PubMed] [Google Scholar]
- 4.Josep ML, Sergio R, Vincenzo M, Philip H, Edward G, Jean-Frédéric B, Andre Cosme de O, Armando S, Jean-Luc R, Alejandro F, Myron S, Camillo P, Stefan Z, Luigi B, Tim FG, Peter RG, Jean-François S, Ivan B, Dieter H, Tom G, Minghua S, Marius M, Dimitris V, Jordi B. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 5.Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, Liu J, Wang J, Tak WY, Pan H, Burock K, Zou J, Voliotis D, Guan Z. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phaseIII randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 6.Asghar U, Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J Hepatol. 2012;56:686–95. doi: 10.1016/j.jhep.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 7.Burroughs A, Hochhauser D, Meyer T. Systemic treatment and liver transplantation for hepatocellular carcinoma: two ends of the therapeutic spectrum. Lancet Oncol. 2004;5:409–18. doi: 10.1016/S1470-2045(04)01508-6. [DOI] [PubMed] [Google Scholar]
- 8.Ganne-Carrie N, Trinchet JC. Systemic treatment of hepatocellular carcinoma. Eur J Gastroenterol Hepatol. 2004;16:275–81. doi: 10.1097/00042737-200403000-00005. [DOI] [PubMed] [Google Scholar]
- 9.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–34. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 10.Ng IO, Liu CL, Fan ST, Ng M. Expression of P-glycoprotein in hepatocellular carcinoma. A determinant of chemotherapy response. Am J Clin Pathol. 2000;113:355–63. doi: 10.1309/AC1M-4TY4-U0TN-EN7T. [DOI] [PubMed] [Google Scholar]
- 11.Serena B, Lorella P, Lory SC, Giorgio S, Claudio T. Gene expression of ABC proteins in hepatocellular carcinoma, perineoplastic tissue, and liver diseases. Mol Med. 2002;8:318–25. [PMC free article] [PubMed] [Google Scholar]
- 12.Cox J, Weinman S. Mechanisms of doxorubicin resistance in hepatocellular carcinoma. Hepat Oncol. 2016;3:57–9. doi: 10.2217/hep.15.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park JG, Lee SK, Hong IG, Kim HS, Lim KH, Choe KJ, Kim WH, Kim YI, Tsuruo T, Gottesman MM. MDR1 gene expression: its effect on drug resistance to doxorubicin in human hepatocellular carcinoma cell lines. J Natl Cancer Inst. 1994;86:700–5. doi: 10.1093/jnci/86.9.700. [DOI] [PubMed] [Google Scholar]
- 14.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, Gravis G, Bodrogi I, Mackenzie MJ, Shen L. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147–54. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 15.Pean E, Demolis P, Moreau A, Hemmings RJ, O’Connor D, Brown D, Shepard T, Abadie E, Pignatti F. The European medicines agency review of cabazitaxel (Jevtana?) for the treatment of hormone-refractory metastatic prostate cancer: summary of the scientific assessment of the committee for medicinal products for human use. Oncologist. 2012;17:543–9. doi: 10.1634/theoncologist.2011-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villanueva C, Awada A, Campone M, Machiels JP, Besse T, Magherini E, Dubin F, Semiond D, Pivot X. A multicentre dose-escalating study of cabazitaxel (XRP6258) in combination with capecitabine in patients with metastatic breast cancer progressing after anthracycline and taxane treatment: a phase I/II study. Eur J Cancer. 2011;47:37–45. doi: 10.1016/j.ejca.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Vrignaud P, Sémiond D, Lejeune P, Bouchard H, Calvet L, Combeau C, Riou JF, Commerçon A, Lavelle F, Bissery MC. Preclinical antitumor activity of cabazitaxel, a semisynthetic taxane active in taxane-resistant tumors. Clin Cancer Res. 2013;19:2973–83. doi: 10.1158/1078-0432.CCR-12-3146. [DOI] [PubMed] [Google Scholar]
- 18.Issery MC, Bouchard H, Riou J, Vrignaud P, Combeau C, Bourzat JD. Preclinical evaluation of TXD258, a new taxoid. Proceedings of the American Association for Cancer Research 2000:41. Cancer Research, Incorporated, and American Association for Cancer Research, Incorporated. 2000 Abstract 1364. [Google Scholar]
- 19.Vrignaud P, Lejeune P, Chaplin D, Lavelle F, Bissery MC. In vivo efficacy of TXD258, a new taxoid, against human tumor xenografts. Proceedings of the American Association for Cancer Research 2000:41. Cancer Research, Incorporated, and American Association for Cancer Research, Incorporated. 2000 Abstract 1365. [Google Scholar]
- 20.Galsky MD, Dritselis A, Kirkpatrick P, Oh WK. Cabazitaxel. Nat Rev Drug Discov. 2010;9:677–8. doi: 10.1038/nrd3254. [DOI] [PubMed] [Google Scholar]
- 21.Meena AS, Sharma A, Kumari R, Mohammad N, Singh SV, Bhat MK. Inherent and acquired resistance to paclitaxel in hepatocellular carcinoma: molecular events involved. PLoS One. 2013;8:e61524–e61524. doi: 10.1371/journal.pone.0061524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pivot X, Koralewski P, Hidalgo JL, Chan A, Gonçalves A, Schwartsmann G, Assadourian S, Lotz JP. A multicenter phase II study of XRP6258 administered as a 1-h i. v. infusion every 3 weeks in taxane-resistant metastatic breast cancer patients. Ann Oncol. 2008;19:1547–52. doi: 10.1093/annonc/mdn171. [DOI] [PubMed] [Google Scholar]
- 23.Fumoleau P, Trigo JM, Isambert N, Sémiond D, Gupta S, Campone M. Phase I dose-finding study of cabazitaxel administered weekly in patients with advanced solid tumours. BMC Cancer. 2013;13:460. doi: 10.1186/1471-2407-13-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diéras V, Lortholary A, Laurence V, Delva R, Girre V, Livartowski A. Cabazitaxel in patients with advanced solid tumours: results of a phase I and pharmacokinetic study. Eur J Cancer. 2013;49:25–34. doi: 10.1016/j.ejca.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 25.Dykes DJ, Sarsat JP, Bissery MC. Efficacy evaluation of TXD258, a taxoid compound, against orthotopic and subcutaneous glioblastomas. Proc Am Assoc Cancer Res. 2000;41:301. [Google Scholar]
- 26.Yau T, Chan P, Epstein R, Poon RT. Management of advanced hepatocellular carcinoma in the era of targeted therapy. Liver Int. 2009;29:10–7. doi: 10.1111/j.1478-3231.2008.01916.x. [DOI] [PubMed] [Google Scholar]
- 27.Lai CL, Wu PC, Chan GC, Lok AS, Lin HJ. Doxorubicin versus no antitumor therapy in inoperable hepatocellular carcinoma. A prospective randomized trial. Cancer. 1988;62:479–83. doi: 10.1002/1097-0142(19880801)62:3<479::aid-cncr2820620306>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 28.Leung TW, Patt YZ, Lau WY, Ho SK, Yu SC, Chan AT, Mok TS, Yeo W, Liew CT, Leung NW, Tang AM, Johnson PJ. Complete pathological remission is possible with systemic combination chemotherapy for inoperable hepatocellular carcinoma. Clin Cancer Res. 1999;5:1676–81. [PubMed] [Google Scholar]
- 29.Yeo W, Mok TS, Zee B, Leung TW, Lai PB, Lau WY, Koh J, Mo FK, Yu SC, Chan AT, Hui P, Ma B, Lam KC, Ho WM, Wong HT, Tang A, Johnson PJ. A randomized phase III study of doxorubicin versus cisplatin/interferon alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst. 2005;97:1532–8. doi: 10.1093/jnci/dji315. [DOI] [PubMed] [Google Scholar]
- 30.Holohan C, Schaeybroeck SV, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]