

Abstract
Low serum CC16 levels are associated with accelerated lung function decline in human population studies, but it is not known whether low serum CC16 levels contribute to lung function decline, or are an epiphenomenon. We tested the hypothesis that unchallenged Cc16 −/− mice develop accelerated rates of pulmonary function test abnormalities and pulmonary pathologies over time compared with unchallenged WT mice. Respiratory mechanics, airspace enlargement, and small airway fibrosis were measured in unchallenged wild‐type (WT) versus Cc16 −/− mice over 6–18 months of age. Lung leukocyte counts and lung levels of metalloproteinases (Mmps), cytokines, oxidative stress, cellular senescence markers (p19 and p21), and lung cell apoptosis, and serum C‐reactive protein (CRP) levels were measured in age‐matched WT versus Cc16 −/− mice. Unchallenged Cc16 −/− mice developed greater increases in lung compliance, airspace enlargement, and small airway fibrosis than age‐matched WT mice over 6–18 months of age. Cc16 −/− mice had greater: (1) lung leukocyte counts; (2) lung levels of Ccl2, Ccl‐5, interleukin‐10, Mmp‐2, and Mmp‐9; (3) pulmonary oxidative stress levels, (4) alveolar septal cell apoptosis and staining for p16 and p21; and (5) serum CRP levels. Unchallenged Cc16 −/− mice had greater nuclear factor‐κB (NF‐κB) activation in their lungs than age‐matched WT mice, but similar lung levels of secretory phospholipase‐A2 activity. Cc16 deficiency in mice leads spontaneously to an accelerated lung aging phenotype with exaggerated pulmonary inflammation and COPD‐like lung pathologies associated with increased activation of NF‐ κB in the lung. CC16 augmentation strategies may reduce lung aging in CC16‐deficient individuals.
Keywords: Club cell protein‐16, emphysema, inflammation, nuclear factor‐κB, small airway fibrosis
Introduction
Physiological aging in humans, in the absence of cigarette smoke or other exposures, induces changes in the lungs that are similar to those that occur in patients with chronic obstructive pulmonary disease (COPD) including airspace enlargement, small airway disease, and a decline in forced expiratory volume in 1 sec (FEV1) (Janssens et al. 1999; Vaz Fragoso and Gill 2010; Sorino et al. 2012). The pulmonary pathologies that contribute to age‐related decline in lung function over time in humans have not been well studied. In addition, some individuals have low FEV1 values by early adulthood, and others have accelerated lung function decline after 40 years of age (Lange et al. 2015), but the factors that contribute to these processes are not well understood. As‐yet‐unidentified circulating soluble factors reverse signs of aging in several tissues in elderly mice (Conboy et al. 2005), but circulating factors that regulate lung aging have not been identified in humans.
Club cell protein 16 (CC16; also known as uteroglobin, CC10, secretoglobin 1A1, Club cell secretory protein [CCSP]) is a circulating protein, and a member of the secretoglobin superfamily. CC16 is mainly expressed by Club cells in the airway epithelium, and is secreted by Club cells into the epithelial lining fluid and it readily diffuses into the circulation. CC16 has anti‐inflammatory and cytoprotective activities on various cell types including epithelial cells and leukocytes, and protects lungs from developing lung inflammation and injury mediated by allergens, viruses, bleomycin, and cigarette smoke (Chen et al. 2001; Wang et al. 2003; Lee et al. 2006; Snyder et al. 2010; Laucho‐Contreras et al. 2015a; Zhu et al. 2015). Low serum and lung levels of CC16 have been linked to the development of COPD, rapid decline in FEV1 in several human COPD studies (Bernard et al. 1992; Pilette et al. 2001; Lomas et al. 2008; Park et al. 2013; Laucho‐Contreras et al. 2015a; Petersen et al. 2015).
Recent human population studies have shown that, across the human adult lifespan, individuals with low serum CC16 levels have accelerated lung function decline compared with subjects with higher serum CC16 levels after correcting for covariates. Guerra et al. analyzed circulating CC16 levels measured at baseline in three population cohorts of adult subjects without COPD at baseline (>1600 adults aged 18–70 years in the United States and Europe) that had been followed longitudinally for 8–14 years (Celli and Owen 2013; Guerra et al. 2015). This study linked low baseline circulating CC16 levels to accelerated rates of FEV1 decline, the subsequent development of airflow obstruction (FEV1/forced vital capacity (FVC) < 70% predicted), and incident GOLD stage II COPD after adjusting for sex, age, height, smoking status, pack‐years smoking history, the presence of asthma, and FEV1 at baseline (Celli and Owen 2013; Guerra et al. 2015). However, it is not clear whether low circulating CC16 levels contribute directly to accelerate lung function decline in human, or whether they are an epiphenomenon. Also, the nature of the pulmonary pathologies underlying the lung function abnormalities were not evaluated in these human population studies.
Cc16‐deficient (Cc16 −/−) mice have normal lung development, and we reported previously that unchallenged young adult Cc16 −/− mice (~12 weeks of age) have normal lung architecture (Laucho‐Contreras et al. 2015a). However, unchallenged 12‐week‐old Cc16 −/− mice have higher lung leukocyte counts than age‐matched unchallenged WT mice indicating Cc16 deficiency is sufficient to induce low‐grade pulmonary inflammation in young adult mice (Laucho‐Contreras et al. 2015a; Zhu et al. 2015). Thus, we hypothesized that Cc16 deficiency (in the absence of any challenge) induces low‐grade chronic pulmonary inflammation which increases over time and contributes to accelerated lung aging characterized by COPD‐like lung pathologies to explain the accelerated lung function decline that is associated with low serum CC16 levels across the human lifespan (Guerra et al. 2015). To test this hypothesis, we compared the pulmonary phenotypes, and a readout of systemic inflammation (serum C‐reactive protein [CRP] levels), in WT and Cc16 −/− mice that were housed in the absence of any challenge until they were 6, 12, or 18 months of age to model young adulthood, middle age, and late middle age, respectively, in humans (Demetrius 2006; Dutta and Sengupta 2016). Our results show that Cc16 deficiency is sufficient to induce a low‐grade pulmonary inflammatory response in the lung which is associated with the development of airspace enlargement and small airway fibrosis, higher lung compliance, increased expression of markers of senescence in the lungs, and increased serum CRP levels in mice. In addition, we link exaggerated pulmonary lesions in unchallenged Cc16 −/− mice to increased activation of NF‐κB in the lung.
Materials and Methods
Animals
The Brigham and Women's Hospital Institutional Animal Care and Use Committee approved all procedures performed on mice. Animal experiments were conducted following ARRIVE guidelines. C57BL/6 Cc16 −/− mice were generated as described previously (Stripp et al. 2002). Unchallenged Cc16 −/− mice have a normal lifespan, fertility, and lung development (Stripp et al. 2002; Laucho‐Contreras et al. 2015a). Breeding pairs of C57BL/6 strain wild‐type (WT) mice were obtained from The Jackson Laboratory (Bar Harbor, ME), and housed in the same room in a barrier facility under specific pathogen‐free conditions as Cc16 −/− breeding pairs. The WT and Cc16 −/− progeny were used in all experiments. After weaning, male and female WT and Cc16 −/− pups were housed in the same room and fed the same diet in the barrier facility for 6, 12, or 18 months in the absence of any challenge. The genotypes of the Cc16 −/− mice were confirmed using PCR‐based protocols performed on genomic DNA extracted from tail biopsies.
Respiratory mechanic in mice
Mice were anesthetized with 100 mg/kg ketamine, 10 mg/kg xylazine, and 3 mg/kg acepromazine, and a tracheostomy was performed. Respiratory mechanics were measured on the mice using a tracheal cannula connected to a digitally controlled mechanical ventilator (FlexiVent device; Scireq Inc., Montreal, QC, Canada) using the following settings: f = 150/min; FiO2 = 0.21; tidal volume = 10 mL/kg body weight; and positive end‐expiratory pressure (PEEP) of 3 cm H20, as described previously (Laucho‐Contreras et al. 2015b). Briefly, the lungs were inflated to total lung capacity (TLC; 25 cm H2O) three times, and then tissue and peripheral airway resistance (G), tissue and peripheral airway elastance (H), and peripheral airway dynamic compliance were measured using 3 cm H2O PEEP. The animals were then euthanized, and lungs were inflated to 25 cm H2O pressure and fixed for 18 h in 10% buffered formalin, or removed and frozen to −80°C.
Airspace size
To quantify the distal airspace size in mice, alveolar chord lengths were measured on Gill's‐stained lung sections as described previously (Laucho‐Contreras et al. 2015b). Briefly, images of all well‐inflated microscopic fields of sections of both lung fields in each animal (~10 fields per animal) were captured (at ×100 magnification) using a Leica epi‐fluorescence microscope, a 3‐CCD color vision camera module, and Leica Qwin V3 software (Leica Microsystems Inc., Buffalo Grove, IL). Scion Image software (Scion Corp., Frederick, MD, USA) was used to measure alveolar chord length in microns (Laucho‐Contreras et al. 2015b).
Small airway fibrosis
Small airway fibrosis was measured in inflated lung sections from unchallenged WT and Cc16 −/− mice that were 6, 12, or 18 months of age. Formalin‐fixed paraffin‐embedded lung sections were stained with Masson's Trichrome stain using a commercial kit (Sigma‐Aldrich, St. Louis, MO). Images of both lung fields containing all airways having a diameter of 300–699 μm in each mouse (10–20 airways/mouse) were acquired using microscope interfaced with a digital camera a camera. The thickness of the extracellular matrix protein layer (stained blue) around small airways having a mean diameter of 300–699 microns was measured using MetaMorph software (Molecular Devices, San Jose, CA), as described previously (Laucho‐Contreras et al. 2015b).
Bronchoalveolar lavage leukocyte counts
Bronchoalveolar lavage (BAL) was performed on unchallenged WT and Cc16 −/− and WT mice at 6, 12, or 18 months of age as described previously (Craig et al. 2013). BAL total leukocytes were counted, differential leukocyte counts were performed on modified Wright's‐stained cytocentrifuge preparations, and absolute numbers of leukocyte subsets were calculated.
Lung levels of cytokines and growth factors
Levels of interleukin‐10 (Il‐10), Il‐6, Ccl5, Ccl2, and active transforming growth factor‐β 1 (Tgf‐β 1) were measured in homogenates of lung samples using ELISA kits (Peprotech, Rocky Hill, NJ; R&D Systems, Minneapolis, MN; and e‐Bioscience, San Diego, CA). All of the results were corrected for lung protein levels using a total protein assay kit (Thermo Fisher Scientific, Waltham, MA).
Lung levels of matrix metalloproteinases
Levels of pro and active Mmp‐9 protein were measured in homogenates of lung samples obtained from unchallenged WT and Cc16 −/− mice that were 6, 12, or 18 months of age using an ELISA kit (R&D Systems). The results were corrected for lung protein levels, using a total protein assay kit (Thermo Fisher Scientific, Waltham, MA).
Levels of active Mmp‐2 were measured in homogenates of lung samples using Western blotting after loading equal amounts of protein onto 12% SDS‐PAGE gels, separating the proteins by electrophoresis, and transferring the proteins to polyvinylidene difluoride membranes. Duplicate membranes were probed with either rabbit antimurine Mmp‐2 IgG diluted 1:1000 (Abcam, Cambridge, MA) or mouse antimurine alpha tubulin IgG (as a loading control) diluted 1:1000 (Abcam, Cambridge, MA) overnight at 4°C. After washing the membranes with PBS containing 0.1% Tween‐20, the membranes were incubated with horseradish peroxidase (HRP)‐conjugated goat antirabbit IgG or HRP‐conjugated goat antimurine IgG diluted 1:3000 (BioRad, Hercules, CA) for 1 h at room temperature. Signals were developed using a chemiluminescence kit (Thermo Scientific, Waltham, MA) following the manufacturer's instructions. Images of the signals were captured using a CCD camera, a ChemiDoc™ XRS+ System, and Image Lab software (Bio‐Rad Laboratories, Hercules, CA). The bands corresponding to active Mmp‐2 were analyzed using densitometry with ImageJ software (National Institutes of Health, Bethesda, MD) and signals for proMMP‐2 were normalized to alpha tubulin signals.
Lung cell apoptosis
Cytoplasmic and nuclear fractions were extracted from frozen lung tissue obtained from unchallenged WT and Cc16 −/− mice that were 6, 12, or 18 months of age using a commercial kit (Thermo Fisher Scientific, Waltham, MA). Cytoplasmic fractions and known standards of recombinant active caspase‐3 (BD Bioscience, San Jose, CA) were incubated in duplicate with a quenched fluorogenic substrate that is specific for active caspase‐3 (BD Bioscience, San Jose, CA) for 30 min at 37°C, protected from light. Cleavage of the substrate by active caspase‐3 in the samples was measured using fluorometry (with a F2500 fluorescence spectrophotometer; Hitachi, Tokyo, Japan; Ex λ 400 and Em λ 490). Concentrations of active caspase‐3 levels were calculated in fluorescence units by interpolation from the standard curve of active caspase‐3 exactly as described previously (Knolle et al. 2013).
Staining of lungs for 4‐hydroxy‐2‐nonenal (4‐HNE)
Formalin‐fixed sections of lung obtained from unchallenged WT and Cc16 −/− mice that were 6, 12, or 18 months of age (n = 3–4 mice/group) were incubated with blocking buffer followed by rabbit IgG to 4‐HNE (a marker of oxidative stress; Bioss, Woburn, MA) diluted 1:50 or nonimmune rabbit IgG at the same concentration for 1 h at 37°C. After washing the lung sections with PBS, the lung sections were incubated at 37°C for 1 h with Alexa 488‐conjugated goat antirabbit F(ab’)2 diluted 1:100. Sections were then washed in PBS, and nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI). Images of the immunostained lung sections were captured using an epi‐fluorescence microscope and a digital camera. The number of 4‐HNE‐positive cells in the alveolar walls was counted and normalized to alveolar wall areas measured using MetaMorph software.
Secretory phospholipase A2 (sPLA2) activity levels:
sPLA2 activity was measured in homogenates of whole lung samples obtained from unchallenged WT and Cc16 −/− mice that were 6, 12, or 18 months of age using a commercially available kit (Enzo Life Science Inc., Farmingdale, NY). The results were normalized to total protein levels measured using a commercial total protein assay kit (Thermo Fisher Scientific, Waltham, MA.
P16 and p21 staining in alveolar septal cells
Formalin‐fixed inflated lung sections obtained from unchallenged Cc16 −/− and WT mice that were 6, 12, or 18 months of age were immunostained for two cyclin‐dependent kinases inhibitors (CDKI), p16 and p21, which are markers of cellular senescence (Wu et al. 2016). The sections were incubated with blocking buffer followed by rabbit anti‐p16 IgG, rat anti‐p21 IgG (Abcam, Cambridge, MA), nonimmune rabbit IgG (Dako North America Inc, Carpinteria, CA), or nonimmune rat IgG (Sigma‐Aldrich, St. Louis, MO). The sections were washed in PBS and then incubated with Alexa‐488‐conjugated goat antirabbit F(ab’)2 or goat antirat F(ab’)2 (Life Technology, Grand Island, NY). Images of the lung sections were captured using an epifluorescence microscope and a digital camera (Leica DFC300, Allendale, NJ). The numbers of alveolar septal cells that were positively stained for p16 or p21 were counted, and normalized to unit area of alveolar wall measured in pixels (Laucho‐Contreras et al. 2015aa) using MetaMorph software (Molecular Devices, Sunnyvale, CA).
Nuclear Factor kappa B (NF‐κB) activation in lung samples
NF‐κB activation was measured in nuclear extracts prepared from whole lung samples obtained from unchallenged Cc16 −/− and WT mice that were 6, 12, or 18 months of age using electrophoretic mobility shift assays (EMSA; Thermo Fisher Scientific, Waltham, MA) as described previously (Laucho‐Contreras et al. 2015a). Nuclear fractions were extracted from frozen lung tissue obtained from unchallenged mice using a commercial kit (Thermo Fisher Scientific, Waltham, MA). Protein levels in the cytoplasmic fractions were quantified using a commercial kit (Thermo Fisher Scientific, Waltham, MA). Equal amounts of protein (2 μg) measured in the nuclear extracts were incubated with a biotin‐labeled probe having the consensus sequence to which NFκB binds (sense strand: 5′ AGT TGA GGG GAC TTT CC CAG GC) with or without excess unlabeled probe as a control, and subjected to gel electrophoresis for 3 h at 100 Volts. DNA:NFκB complexes were transferred onto positive nylon membranes, probed with streptavidin–HRP, and signals were developed using a chemiluminescence kit (Thermo Fisher Scientific, Waltham, MA). Images of the blots were captured using a ChemiDoc™ XRS+ System, and Image Lab software (Bio‐Rad laboratories, Hercules, CA). Signals were quantified using densitometry with Image J software (National Institutes of Health, Bethesda, MD). It was not possible to run all of the samples (6 experimental conditions with 5 mice per experimental condition and controls) on one gel. Multiple gels were run on different days and samples from WT and Cc16 −/− mice at the same time point were always analyzed on the same gels. As signal development time varied between gels, signals from Cc16 −/− samples were normalized to those of WT samples obtained from mice at the same time point analyzed on the same gel.
C‐reactive protein (CRP) levels in serum samples
Unchallenged WT and Cc16 −/− mice that were 6, 12, or 18 months of age were euthanized, and blood samples were obtained via right ventricular puncture, and serum samples were frozen to −80°C. CRP levels were quantified in the serum samples using a commercially available ELISA kit (R&D Systems, Minneapolis, MN).
Statistics
Statistical analyses were performed using SigmaPlot™ software (SSPS Inc, San Jose, USA). The Shapiro–Wilks test was performed to determine whether or not the data were normally distributed. Data were analyzed using one‐way analysis of variance and Holm–Sidak or Dunn post hoc methods for multiple group comparisons. Normally distributed data are presented as mean and SEM and pair‐wise testing was performed using Student's t tests. Data that were not normally distributed are presented as box‐plots showing medians and interquartile ranges (IQR), and error bars representing the 10th and 90th percentiles and pair‐wise testing was performed using Mann–Whitney U tests.
Results
Cc16 −/− mice spontaneously develop greater airspace enlargement, lower lung resistance, and higher lung compliance than WT mice over 18 months of age
None of the WT or Cc16 −/− mice died during the 18 month of study period. Unchallenged WT and Cc16 −/− mice that were 10 weeks of age had similar distal airspace size (Laucho‐Contreras et al. 2015a;). Unchallenged WT and Cc16 −/− mice that were 12 or 18 months old had higher alveolar airspace size (measured as alveolar cord length) than 6‐month‐old mice belonging to the same genotype (Fig. 1). Cc16 −/− mice had greater airspace enlargement than age‐matched WT mice at all time points tested. From 6 to 18 months of age, mean alveolar chord lengths increased to a greater extent in Cc16 −/− mice (1.7‐fold) than WT mice (1.4‐fold). There were no differences in emphysema development in males versus females belonging to the same genotype at the same time point (data not shown).
Figure 1.
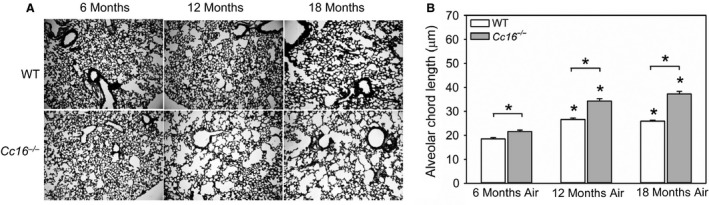
Greater airspace enlargement develops spontaneously in unchallenged Cc16 −/− versus WT mice. (A) Gill's‐stained inflated lungs sections from unchallenged WT versus Cc16 −/− mice at 6, 12, and 18 months of age that are representative of 8–12 mice per group. (B) Images of all well‐inflated microscopic fields of sections of both lung fields (~ten 100× microscopic fields per animal) were captured. Distal airspace size was measured as mean alveolar chord length in WT versus Cc16 −/− mice at 6, 12, or 18 months of age using Scion image software, as described in Methods. The bars show the means and error bars show the SEM values. Data were analyzed using a one‐way ANOVA and pairwise testing was performed using Student’s t‐test. Asterisks indicate P ≤ 0.05 versus air‐exposed mice belonging to the same genotype at 6 months of age, or the group indicated; n = 9–14 mice/group.
Unchallenged 18‐month‐old WT mice had lower tissue and peripheral airway elastance values; lower tissue and peripheral airway resistance values; and higher peripheral airway compliance values than unchallenged 6‐month‐old WT mice (Table 1). Unchallenged 18‐month‐old Cc16 −/− mice had lower tissue and peripheral airway elastance values; lower airway peripheral resistance values; and higher peripheral airway compliance values than unchallenged 6‐month‐old Cc16 −/− mice (Table 1). Unchallenged Cc16 −/− mice that were 6 months of age had lower tissue and peripheral airway elastance; lower peripheral airway resistance; and higher peripheral airway compliance than unchallenged WT mice that were 6 months old. Unchallenged Cc16 −/− mice that were 18 months of age had lower tissue and peripheral airway resistance; and higher peripheral airway compliance than unchallenged WT mice that were 18 months old (Table 1). Thus, from 6 to 18 months of age, WT and Cc16 −/− mice both developed airspace enlargement associated with increased lung compliance consistent with loss of elastic recoil in the lung. However, at 6 and 18 months of age, Cc16 −/− mice developed greater airspace enlargement and had more compliant lungs than age‐matched WT mice.
Table 1.
Respiratory mechanics in 6 and 18‐month‐old WT and Cc16 −/− mice
| Parameter measured | 6 Months of age | 18 Months of age | ||
|---|---|---|---|---|
| WT n = 11 | Cc16 −/− n = 11 | WT n = 12 | Cc16 −/− n = 13 | |
| Mean (SEM) or Median (IQR) | Mean (SEM) or Median (IQR) | Mean (SEM) or Median (IQR) | Mean (SEM) or Median (IQR) | |
| Tissue elastance (cmH2O/mL) | 19.3 (0.6) | 17.1 (0.6)a | 15.6 (0.7)b | 15.1 (0.5)c |
| Peripheral elastance (cmH2O/mL) | 21.0 (18.7–22.5) | 18.1 (16.9–19.4)a | 17.1 (15.4–19.0)b | 16.3 (15.7–17.9)c |
| Tissue resistance (cmH2O/mL) | 3.7 (0.3) | 2.9 (0.2)a | 3.0 (0.2)b | 2.3 (0.07)d |
| Peripheral resistance (cmH2O/s/mL) | 0.5 (0.02) | 0.40 (0.01) | 0.39 (0.02)b | 0.31 (0.01)c , d |
| Peripheral compliance (mL/cmH2O) | 0.047 (0.002) | 0.054 (0.002)a | 0.057 (0.003)b | 0.061 (0.002)c , d |
Respiratory mechanics were measured in unchallenged WT and Cc16 −/− mice at 6 and 18 months of age in the using a FlexiVent device.
Data are mean ± SEM for data that are normally distributed or medians and interquartile ranges (IQR) for data that are not normally distributed (peripheral elastance values). Data were analyzed with ANOVAs (P < 0.001 for all parameters measured). Pair‐wise comparisons were performed using Student's t‐tests for data that are normally distributed or the Mann–Whitney U tests for data that are not normally distributed.
P < 0.05 between 6‐month‐old WT and Cc16 −/− mice.
P < 0.05 between 6‐ and 18‐month‐old WT mice.
P < 0.05 between 6‐ and 18‐month‐old Cc16 −/− mice.
P < 0.05 between 18‐month‐old WT and Cc16 −/− mice.
Unlike WT mice, Cc16 −/− mice spontaneously develop small airway fibrosis over 18 months of age
Unchallenged WT mice that were 12 or 18 months of age did not have greater small airway fibrosis (measured as deposition of extracellular matrix (ECM) around their small airways) compared with unchallenged WT mice that were 6 months of age (Fig. 2). In contrast, unchallenged 12‐ and 18‐month‐old Cc16 −/− mice had greater small airway fibrosis than unchallenged 6‐month‐old Cc16 −/− mice, but small airway fibrosis did not differ between 12‐ and 18‐month‐old Cc16 −/− mice. Cc16 −/− mice had greater small airway fibrosis both at 12 and 18 months of age (but not at 6 months of age) when compared with age‐matched WT mice (Fig. 2). From 6 to 18 months of age, Cc16 −/− mice had a 1.3‐fold increase in the median value for small airway remodeling. There were no differences in small airway remodeling in males versus females belonging to the same genotype at the same time point (data not shown).
Figure 2.
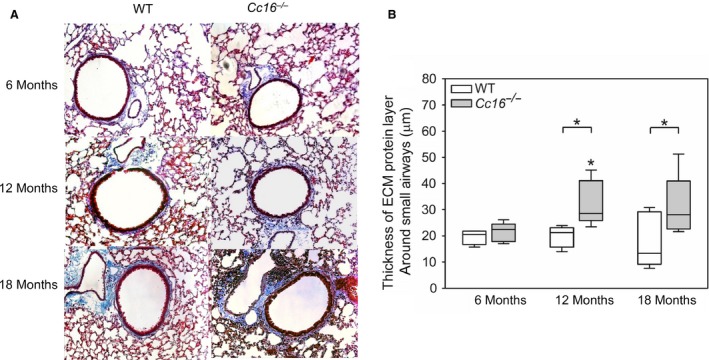
Small airway fibrosis develops over 18 months of age in unchallenged Cc16 −/− mice but not in unchallenged WT mice. Deposition of extracellular matrix (ECM) proteins around small airways having a diameter of 300–699 microns was measured on Masson's Trichrome‐stained sections of lungs from unchallenged Cc16 −/− versus WT mice at 6, 12, or 18 months of age. (A) Images of lung sections from mice that are representative of 5–9 mice/group at each time point. (B) Images of both lung fields containing all small airways having a diameter of 300–699 μm in each mouse (10–20 airways per mouse) were acquired using a microscope interfaced with a digital camera a camera. The thickness of the ECM protein layer deposited around the small airways measured at 12 equally spaced points around each airway using MetaMorph software. The boxes in the box‐plots show the median values and 25th and 75th percentiles for the thickness of the ECM layer, and the whiskers show the 10th and 90th percentiles (5–9 mice/group). Data were analyzed using a one‐way ANOVA and pair‐wise testing was performed using Mann–Whitney U tests. Asterisk indicates P < 0.05 compared with mice belonging to the same genotype at 6 months of age, or the group indicated.
Cc16 −/− mice spontaneously develop greater lung inflammation than WT mice over 18 months of age
Unchallenged WT mice and Cc16 −/− mice at 12 and 18 months of age had higher BAL total leukocyte counts than mice belonging to the same genotype at 6 months of age, and BAL leukocyte counts were higher in 18‐month‐old versus 12‐month‐old Cc16 −/− mice (Fig. 3A). BAL total leukocytes were higher in Cc16 −/− mice than age‐matched WT mice at all time points tested. From 6 to 18 months of age, the median BAL total leukocyte counts increased 1.8‐fold in WT mice and 1.6‐fold in Cc16 −/− mice. There were no differences in BAL total leukocyte counts at any time point in males versus females belonging to the same genotype at the same time point (data not shown).
Figure 3.
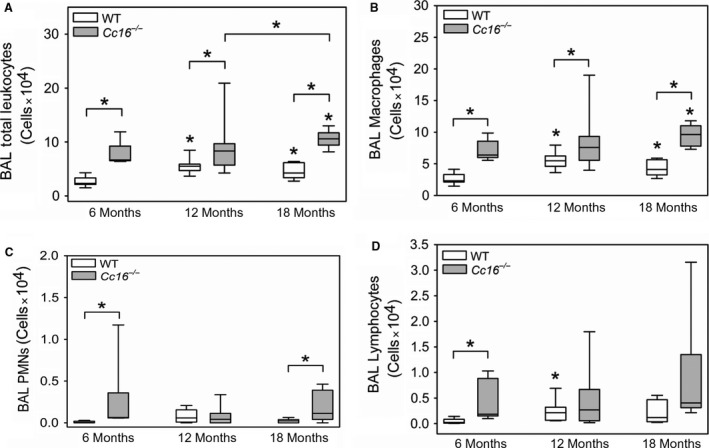
Greater pulmonary inflammation develops in unchallenged Cc16 −/− versus WT mice. WT and Cc16 −/− mice were housed for 6, 12, or 18 months in the absence of challenge. Bronchaolveolar lavage (BAL) was performed on each mouse, total leukocytes were counted with a hemocytometer, and differential leukocyte counts were performed on cytocentrifugre preparations that were stained with modified Wright's stain. Absolute numbers of leukocytes (A), macrophages (B), lymphocytes (C) and PMNs (D) were calculated. The boxes in the box‐plots show the median values and 25th and 75th percentiles, and the whiskers show the 10th and 90th percentiles (6–11 mice/group). Data were analyzed using one‐way ANOVAs and pairwise testing was performed using Mann–Whitney U tests. Asterisk indicates P < 0.05 compared with mice belonging to the same genotype at 6 months of age, or the group indicated.
Most of the leukocytes detected in BAL samples both WT and Cc16 −/− mice were macrophages, and BAL macrophage counts were significantly higher in Cc16 −/− mice than age‐matched WT mice at all time points tested (Fig. 3B). BAL PMN counts were higher in Cc16 −/− mice at 6 and 18 months of age than age‐matched WT mice (Fig. 3C). BAL lymphocyte counts were higher in Cc16 −/− mice 6 months of age than age‐matched WT mice, but not at later time points (Fig. 3D).
Cc16 −/− mice spontaneously develop higher lung levels of some cytokines than WT mice
Lung levels of two chemokines for monocytes and lymphocytes, Ccl2 and Ccl5, and proinflammatory (interleukin‐6) and anti‐inflammatory (interleukin‐10) cytokines were measured in WT and Cc16 −/− mice. Lung levels of Ccl2 protein were higher in 12‐ and 18‐month‐old WT mice than 6 month of WT mice, but similar in Cc16 −/− mice aged 6–18 months (Fig. 4A). Lung levels of Ccl2 protein were higher in 6‐month‐old Cc16 −/− mice than 6‐month‐old WT mice, but similar in 12‐ and 18‐month‐old Cc16 −/− mice and age‐matched WT mice. Lung levels of Ccl5 protein were higher in 18‐month‐old WT mice than 6‐month‐old WT mice (Fig. 4B), but similar in Cc16 −/− mice that were 6–18 months of age. Lung levels of Ccl5 protein were higher in 6‐month‐old Cc16 −/− mice than 6‐month‐old WT mice, but similar in 12‐ and 18‐month‐old Cc16 −/− mice and age‐matched WT mice. Lung levels of interleukin‐6 protein were higher in 18‐month‐old WT and Cc16 −/− mice than 6‐month‐old mice belonging to the same genotype (Fig. 4C). However, age‐matched WT and Cc16 −/− mice did not differ in lung levels of interleukin‐6 at any time point assessed. Lung levels of interleukin‐10 protein were similar in 6, 12, and 18 month old WT mice, but higher in 18‐month‐old Cc16 −/− mice than 6 month Cc16 −/− mice (Fig. 4D). Lung levels of interleukin‐10 were higher in 12‐month‐old Cc16 −/− mice than 12–month‐old WT mice, but were similar in WT and Cc16 −/− mice that were 18 months old.
Figure 4.
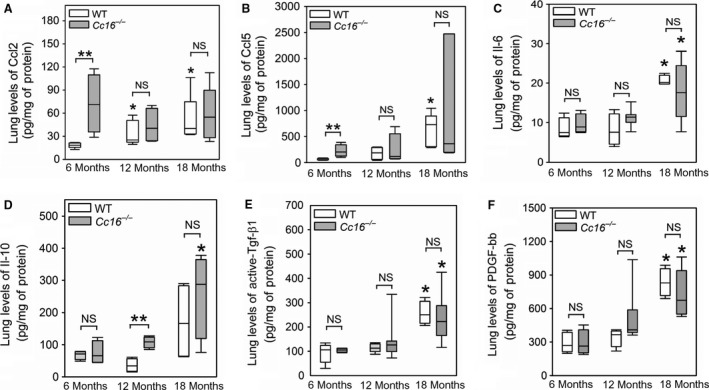
Higher levels of some proinflammatory chemokines and cytokines are detected in the lungs of unchallenged Cc16 −/− versus WT mice. Levels of Ccl2 (A), and Ccl5 (B), interleukin‐6 (il‐6) (C), interleukin‐10 (D), transforming growth factor‐β1 (Tgf‐β1) (E); and platelet‐derived growth factor bb (PDGF‐bb) (F) were measured in homogenates of lung samples obtained from unchallenged WT and Cc16 −/− mice at 6, 12, or 18 months of age using ELISAs, and values were normalized to lung total protein levels. In A–F, the boxes in the box‐plots show the median values and 25th and 75th percentiles, and the whiskers show the 10th and 90th percentiles (4–10 mice per group). Data were analyzed using one‐way ANOVAs and pair‐wise testing was performed using Mann–Whitney U tests. Asterisk indicates P < 0.05 compared with mice belonging to the same genotype at 6 months of age and **, P < 0.05 versus the groups indicated; NS, not significant.
Lung levels of a profibrotic mediator that have been linked to small airway fibrosis (active Tgf‐β1 and Pdgf‐bb (Churg et al. 2006; Dai et al. 1998)) were also measured. Lung levels of active Tgf‐β1 and Pdgf‐bb protein were higher in 18‐month‐old WT mice than 6‐ or 12‐month‐old WT mice (Fig. 4E–F). Lung levels of active Tgf‐β1 protein were higher in 18‐month‐old Cc16 −/− mice than 6‐ and 12‐month‐old Cc16 −/− mice. However, lung levels of either these growth factors did not differ in Cc16 −/− mice and age‐matched WT mice at any time point studied.
Cc16 −/− mice spontaneously develop higher lung levels of some matrix metallo‐proteinases than WT mice
Metalloproteinases (Mmps) are produced by leukocytes and lung parenchymal cells and contribute to emphysema development and small airway fibrosis in experimental animals exposed to cigarette smoke (Hautamaki et al. 1997; Churg et al. 2007). Lung levels of Mmp‐2 and ‐9 also increase as mice age (Sueblinvong et al. 2012). Lung levels of Mmp‐9 protein (pro and active forms) were higher in unchallenged 12‐ and 18‐month‐old WT and Cc16 −/− mice than unchallenged mice belonging to the same genotype at 6 months of age (Fig. 5A). Unchallenged Cc16 −/− mice that were 6‐ and 12‐months of age had higher lung levels of pro‐Mmp‐9 than age‐matched unchallenged WT mice. Lung levels of active Mmp‐2 protein were higher in 6‐month‐old Cc16 −/− mice than 6‐month‐old WT mice, but similar in 12‐month and 18‐month‐old Cc16 −/− mice and age‐matched WT mice (Fig. 5B).
Figure 5.
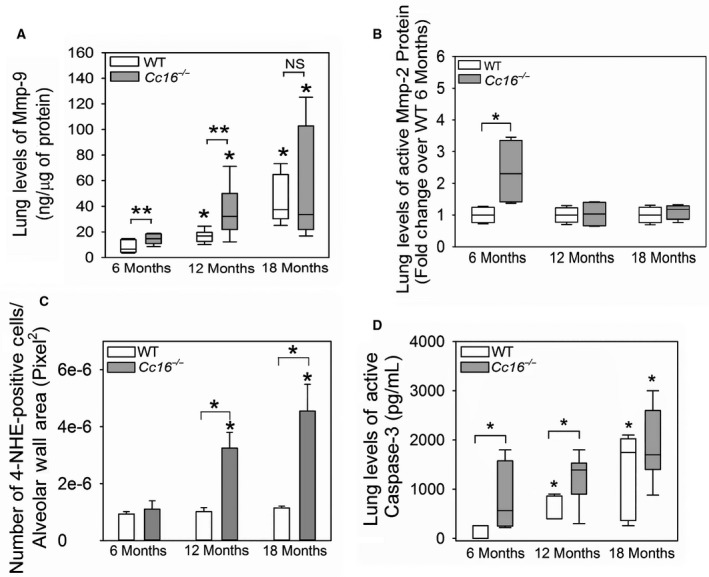
Greater lung levels of matrix metalloproteinases (Mmps) and oxidative stress and greater lung cell apoptosis develop in unchallenged Cc16 −/− versus WT mice. Lung levels of pro and active Mmp‐9 (A) and active Mmp‐2 (B) were measured in homogenates of lung samples from unchallenged WT and Cc16 −/− mice at 6, 12, or 18 months of age using ELISAs (5–9 mice/group) or western blotting (4 mice per group), respectively, and values were normalized to lung total protein levels. In (C), sections of inflated lungs harvested from 6‐, 12‐, or 18‐month‐old WT and Cc16 −/− mice (3 mice/group) were immunostained with a green fluorophore for 4‐hydroxy‐nonenol (4‐HNE) as a marker of oxidative stress and the number of 4‐HNE‐positively stained cells was counted per unit area of alveolar walls (measured in pixels2). In (D), active caspase‐3 levels cytoplasmic fractions of whole lung samples from unchallenged WT and Cc16 −/− mice at 6, 12, or 18 months of age (6–9 mice/group) using a quenched fluorogenic substrate that is specific for active capsase‐3, as described in Methods. In (A, B, and D), the boxes in the box‐plots show the median values and 25th and 75th percentiles, and the whiskers show the 10th and 90th percentiles. Data were analyzed using a one‐way ANOVA, and pairwise testing was performed using Mann–Whitney U tests. In (C), the bars represent the mean values and error bars represent SEM. Data were analyzed using a one‐way ANOVA, and pair‐wise testing was performed using Student's t‐tests. In (A–D), asterisk indicates P < 0.05 compared with mice belonging to the same genotype at 6 months of age, or the groups indicated; NS, not significant.
Cc16 −/− mice spontaneously develop greater lung oxidative stress levels than WT mice over 12–18 months of age
Pulmonary inflammation is associated with increases in lung oxidative stress levels (Owen 2005), and oxidative stress levels also increase in the aging lung and contribute to pulmonary inflammation and injury (Cannizzo et al. 2011; Sohal and Orr 2012). Lung oxidative stress levels [measured as staining for 4‐hydroxy‐2‐nonenal (4‐HNE) in the alveolar walls)] were similar in WT mice at 6, 12, and 18 months of age (Fig. 5C). However, 4‐NHE staining in the alveolar walls was higher in Cc16 −/− mice that were 12 and 18 months of age than in Cc16 −/− mice that were 6 months of age. Cc16 −/− mice that were 12 and 18 months old had greater staining for 4‐HNE in their alveolar walls than aged‐matched WT mice. From 6 to 18 months of age the mean 4‐HNE staining in the alveolar walls increased to a greater extent in Cc16 −/− mice (4.1‐fold) than WT mice (1.2‐fold).
Cc16 −/− mice spontaneously develop greater rates of lung cell apoptosis than WT mice over 18 months of age
Apoptosis of alveolar septal cells contributes to emphysema development in experimental murine models of COPD (Kasahara et al. 2000) and senile emphysema (Calvi et al. 2011). Lung levels of active caspase‐3 (a readout of lung cell apoptosis) were higher in 12‐ and 18‐month‐old WT mice than 6‐month‐old WT mice, and higher in 18‐month‐old Cc16 −/− mice than 6‐month‐old Cc16 −/− mice (Fig. 5D). Lung levels of active caspase‐3 were higher in Cc16 −/− mice that age‐matched WT mice at 6 and 12 (but not at 18) months of age.
Cc16 −/− mice have greater staining for cyclin‐dependent kinases (CDK) inhibitors in alveolar septal cells than WT mice over 18 months of age
CDK inhibitors (including p16 and p21) are the main inducers of cell senescence (Karrasch et al. 2008). P21 initiates cellular senescence, and p16 maintains this process (Stein et al. 1999). Staining for both p16 and p21 in the alveolar walls was higher in 12‐ and 18‐month‐old WT and Cc16 −/− mice than mice belonging to the same genotype at 6 months of age (Fig. 6). Cc16 −/− mice had greater staining for both p16 and p21 in their alveolar walls than age‐matched WT mice at all time points assessed. From 6 to 18 months of age, median P16 staining in the alveolar walls increased to a greater extent in Cc16 −/− mice (3.8‐fold) than WT mice (1.8‐fold). From 6 to 18 months of age, median p21 staining in the alveolar walls also increased to a greater extent in Cc16 −/− mice (6.4‐fold) than WT mice (2.3‐fold).
Figure 6.
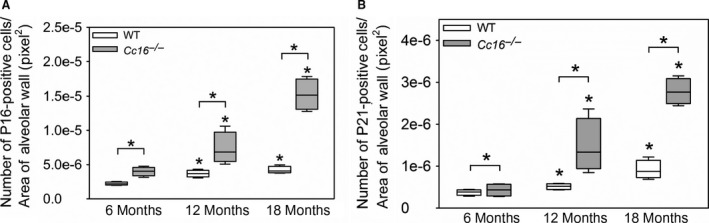
Greater expression of senescence markers (p16 and p21) in alveolar septal cells develops in unchallenged Cc16 −/− versus WT mice. WT and Cc16 −/− mice were housed in the absence of challenge for 6, 12, or 18 months. Fixed, inflated lungs sections from the animals were immunostained with a green fluorophore for p16 (A) or p21 (B), and the number of positively stained cells per unit of alveolar wall area (measured in pixels2) was quantified. The boxes in the box‐plots show the median values and 25th and 75th percentiles, and whiskers show the 10th and 90th percentiles; n = 4 mice/group. Data were analyzed using one‐way ANOVAs and pairwise testing was performed using Mann–Whitney U tests. Asterisk indicates P < 0.05 compared with mice belonging to the same genotype at 6 months of age, or the group indicated.
Cc16 −/− mice have greater serum C‐reactive protein (CRP) levels than WT mice at 18 months of age
Systemic inflammation increases during the aging process (Guarner and Rubio‐Ruiz 2015) and serum CRP levels are a measure of systemic inflammation (Roubenoff et al. 1998). Serum CRP levels were higher in unchallenged 18‐month‐old Cc16 −/− mice than unchallenged 6‐month‐old Cc16 −/− mice, but were similar in unchallenged WT mice at 6, 12, and 18 months of age (Fig. 7). Serum CRP levels were higher in 18‐month‐old unchallenged Cc16 −/− mice than 18‐month‐old unchallenged WT mice, but serum CRP levels did not differ between WT and Cc16 −/− mice that were 6 or 12 months of age (Fig. 7). From 6 to 18 months of age, there was a 1.4‐fold increase in the median serum CRP levels in Cc16 −/− mice.
Figure 7.
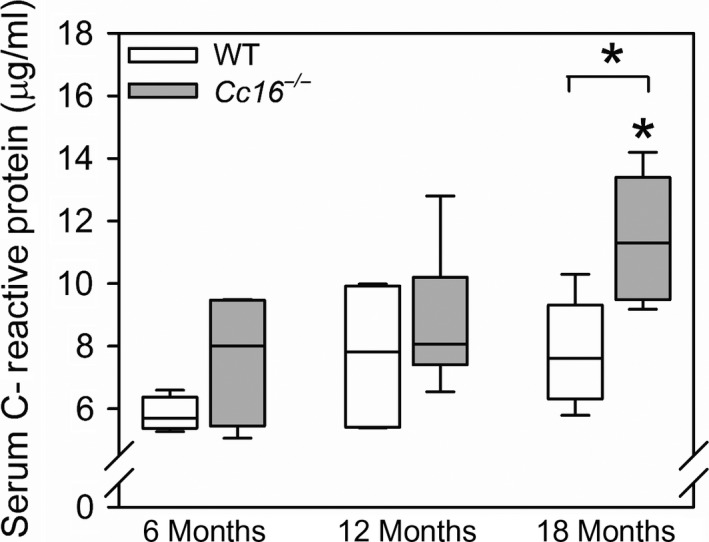
Serum C‐reactive protein (CRP) levels are higher in unchallenged Cc16 −/− than WT mice at 18 months of age: Serum samples were obtained from unchallenged WT and Cc16 −/− mice at 6 months of age (4 mice per group), 12 months of age (5–6 mice/group), or 18 months of age (5 mice/group) via cardiac puncture. Serum CRP levels were measured using an ELISA. The boxes in the box‐plots show the median values and 25th and 75th percentiles, and the whiskers show the 10th and 90th percentiles. Data were analyzed using a one‐way ANOVA followed by pairwise testing with Mann–Whitney U tests. Asterisk indicates 0.05 versus the group indicated or Cc16 −/− mice at 6 months of age.
Cc16 −/− mice have greater NF‐kB activation in their lungs than WT mice, but similar lung levels of sPLA2
Cc16 has been shown to mediate its anti‐inflammatory activities by inhibiting activation of NF‐kB or inhibiting the catalytic activity of secretory phospho‐lipase A2 (sPLA 2 , a proinflammatory pathway) (Laucho‐Contreras et al. 2016) in different model systems. NF‐kB activation (assessed using EMSA assays) was higher in nuclear extracts of whole lung samples from 18‐month‐old Cc16 −/− mice than 18‐month‐old WT mice (Fig. 8A–B), but 6‐ and 12‐month‐old WT mice did not differ in levels of NF‐kB activation in their lungs. Whole lung levels of active sPLA2 were similar in Cc16 −/− mice and aged‐matched WT mice at all time points assessed (Fig. 8C).
Figure 8.
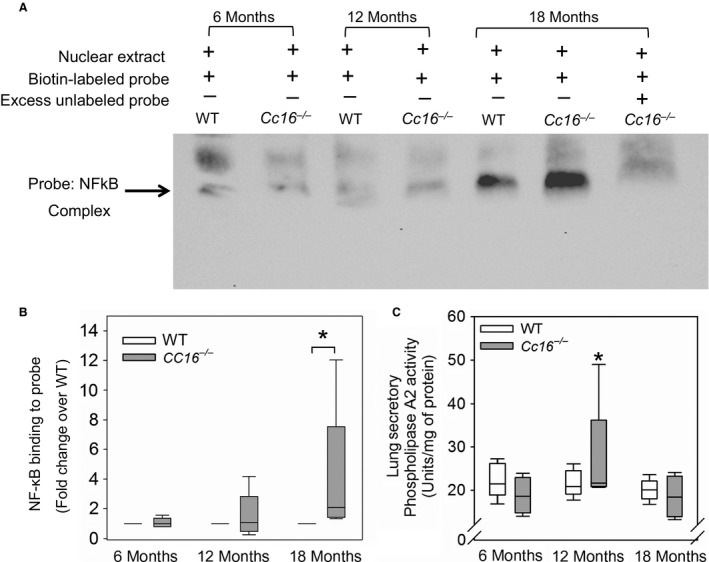
Greater NF‐kB activation occurs in whole lung samples from unchallenged Cc16 −/− mice versus WT mice at 18 months of age. NF κB activation was measured in nuclear extracts prepared from whole lung samples from unchallenged Cc16 −/− and WT mice at 6, 12, or 18 months of age using an electrophoretic mobility shift assay (EMSA) with a biotin‐labeled oligonucleotide probe having the consensus sequence to which the NF κB transcription factor binds, as described in Methods. (A) An EMSA gel is shown which is representative of five animals for each experimental condition. The last lane on the right of the gel shows a control in which an excess of the unlabeled oligonucleotide probe was added along with the biotin‐labeled probe to the nuclear extract from an aliquot of the sample prepared from the 18‐month‐old Cc16 −/− mouse, as a control. Loss of the signal corresponding to the NF κB‐oligonucleotide complex in the last lane indicates that the NF κB present in the nuclear extract binds specifically to the biotin‐labeled probe. (B) The intensities of the bands corresponding to NF‐ kB transcription factor bound to the oligonucleotide probe were quantified using densitometry (n = 5 mice per genotype per time point). As multiple gels had to be run to accommodate all of the samples, the band intensities for samples from Cc16 −/− mice normalized to the mean value of the WT sample at the same time point analyzed on the same gel, as described in Methods. The boxes in the box‐plots show the median values and 25th and 75th percentiles, and whiskers show the 10th and 90th percentiles. Data were analyzed using a one‐way ANOVA and pair‐wise testing was performed using Mann–Whitney U tests. Asterisk indicates P < 0.05 versus the group indicated. (C) Lung levels of secretory phospholipase A2 activity were measured in unchallenged Cc16 −/− and WT mice at 6, 12, or 18 months of age (n = 4–5 mice per group) using a kit. The boxes in the box‐plots show the median values and 25th and 75th percentiles, and the whiskers show the 10th and 90th percentiles. Data were analyzed using a one‐way ANOVA and pairwise testing was performed using Mann–Whitney U tests. Asterisk indicates P < 0.05 versus the Cc16 −/− mice that were 6 months of age.
Discussion
We report for the first time that unchallenged Cc16 −/− mice develop greater increases in lung compliance and greater airspace enlargement than WT mice before the onset of senescence. Unlike WT mice, unchallenged Cc16 −/− mice also developed small airway fibrosis by 12 months of age. The greater pulmonary inflammation and oxidative stress levels, and greater alveolar septal cell apoptosis and senescence that spontaneously developed in Cc16 −/− versus age‐matched WT mice likely contributed to their exaggerated pulmonary pathologies. In addition, the greater activation of NF‐kB in the lungs of Cc16 −/− mice may have contributed to the exaggerated pulmonary inflammation and alveolar septal cell perturbations detected in Cc16 −/− mice, as NF‐kB is a transcription factor that promotes inflammation and senescence and has been strongly linked to aging in many tissues in rodents and humans (Adler et al. 2007). Our data suggest that the low circulating levels of CC16 levels that occur in a subset of human subjects (Celli and Owen 2013; Guerra et al. 2015) may contribute directly to the accelerated lung function decline detected in these subjects by accelerating lung inflamm‐aging and inducing airspace enlargement and small airway fibrosis.
Physiological aging leads to changes in the lung similar to those occurring in COPD patients, including airspace enlargement (“senile emphysema”), a decline in the FEV1(Verbeken et al. 1992; Janssens et al. 1999; Vaz Fragoso and Gill 2010), and small airway disease, as assessed using computed tomography scans of the thorax, spirometry, and multiple breath washout methods (Janssens et al. 1999; Verbanck et al. 2012; Bommart et al. 2015). However, the mechanisms involved in these normal aging‐related pulmonary pathologies have not been well studied. In addition, the pulmonary pathologies and mechanisms underlying the accelerated decline in lung function that occurs in some individuals during middle age (Lange et al. 2015) have been even less well studied.
Mice are commonly used to model the human aging processes in organs of interest as they are less expensive to house than larger animals and have senescence pathways which are similar to those in humans (Demetrius 2006; Dutta and Sengupta 2016). Six months of age in mice is equivalent to young adulthood (~20 years of age) in humans; 12 months of age in mice is equivalent to middle age (~40 years of age in humans); 18 months is defined as the beginning of senescence in mice (~60 years of age in humans) (Dutta and Sengupta 2016). We studied time points at or before the normal onset of senescence in mice to test our hypothesis that Cc16 deficiency accelerates the lung aging process in mice, and leads to detectable pulmonary disease well before the onset of senescence. Furthermore, the period before middle age represents the optimal window in which novel therapies could be initiated to effectively prevent accelerated aging and preserve lung function.
Normal aging is associated with a low‐grade chronic inflammatory process or “inflamm‐aging” in the lungs and also in other organs including the kidney, cardiovascular and endocrine systems, and in skeletal muscle (Fougere et al. 2017; O'Sullivan et al. 2017). Herein, greater pulmonary inflammation along with higher serum levels of CRP were detected in Cc16 −/− mice versus age‐matched WT mice suggesting that Cc16 deficiency increases pulmonary and systemic inflammation. Future studies will assess whether deficiency of Cc16 leads to accelerated aging in organs other than the lung.
Human cohort studies have linked low serum levels of CC16 to increased rate of FEV1 decline, reduced FEV1/FVC ratios, increased airway hyper‐responsiveness, and the development of moderate airflow obstruction in adults after the adjusting for sex, age, height, baseline FEV1, smoking status, and pack/years smoking history (Guerra et al. 2015; Rava et al. 2015). However, the lung pathologies underlying the increased rate of FEV1 and airflow obstruction that are associated with low serum CC16 levels in humans have not been assessed. Our study adds to this literature by showing that Cc16 deficiency is sufficient to accelerate the spontaneous development of pulmonary inflammation and emphysema, and to induce small airway fibrosis over 18 months of age in mice. Thus, the increased rate of lung function decline that has been reported in humans with low serum CC16 levels (Guerra et al. 2015; Rava et al. 2015) may be due, in part, reduced CC16‐mediated protection from inflamm‐aging in the lung. Whether humans with low CC16 levels also have accelerated development of airspace enlargement and small airway fibrosis to explain their accelerated decline in FEV1 should be addressed by future lung imaging and/or studies of lung samples from humans with and without low serum CC16 levels.
Emphysema development
In cigarette smoke (CS)‐exposed mice, pulmonary inflammation, increases in lung proteinase and oxidative stress levels, and alveolar septal cell apoptosis promote loss of the alveolar walls and emphysema development (Kasahara et al. 2000; Rangasamy et al. 2004; Owen 2005, 2008). There have been fewer studies of senile emphysema in mice. However, the lung phenotypes of DBA/2 mice (an inbred strain which exhibits accelerated aging) have been examined over 20 months of age (Calvi et al. 2011). Between 8 and 12 months of age, DBA/2 mice developed greater lung oxidative stress levels, higher rates of alveolar septal cell apoptosis, and higher lung levels of Mmp‐9 (but not Mmp‐12), followed by increases in lung leukocyte counts and greater emphysema development than DBA/2 mice that were 2–4 months of age. Another study reported that C57BL/6 strain WT mice that were 9 months old did not have greater airspace enlargement or higher tissue PMN or macrophage counts, lung oxidative stress levels, or p21 or p16 gene expression levels than 18‐month‐old C57BL/6 WT mice (Zhou et al. 2013). However, the latter study did not evaluate C57BL/6 mice that were younger than 9 months of age as controls. Our study showed that increased BAL leukocyte counts and airspace enlargement were detected from 6 to 12 months of age in C57BL/6 WT mice but these changes did not progress further by 18 months of age.
The increased lung macrophage counts, and greater alveolar septal cell apoptosis, lung oxidative stress levels, and staining for two senescence markers (p21 and p16) detected in unchallenged Cc16 −/− mice likely contributed synergistically to the accelerated emphysema observed in these animals. Macrophages and their products (especially Mmp‐12) are required for the development of CS‐induced emphysema (Hautamaki et al. 1997; Ofulue and Ko 1999). Increases in lung macrophage counts (Takeda et al. 2008), Mmp expression by lung macrophages (Morris et al. 2003; Takeda et al. 2008; Xu et al. 2017), lung oxidative stress levels (Lao et al. 2016), alveolar septal cell apoptosis (Suga et al. 2000; Ishii et al. 2008), and the expression of senescence markers in the lung (Karrasch et al. 2008) have all been linked to the emphysema that develops spontaneously in gene‐targeted mice that develop an accelerated lung aging phenotype. It is possible that the increased lung macrophage counts detected in Cc16 −/− lungs contributed (in part) to the higher lung burden of some of the mediators detected in unchallenged Cc16 −/− mice (such as pro‐Mmp‐9 and ‐2, Ccl2, Ccl5, and interleukin‐10), as these mediators are produced by macrophages. The higher BAL lymphocyte counts detected in unchallenged Cc16 −/− versus WT mice at 6 months of age may be due to the higher lungs levels of Ccl‐2 and Ccl‐5 detected at this age.
The increased lung levels of Mmp‐9 and Mmp‐2 detected in Cc16 −/− mice likely contributed to increased lung ECM degradation and their increased loss of alveolar walls. The elevated lung levels of Ccl2 and Ccl5 may also have amplified the increased lung macrophage counts detected in Cc16 −/− mice aged 6–18 months as both of these mediators are chemokines for monocytes (Owen et al. 1994). Surprisingly, lung levels of interleukin‐10 (an anti‐inflammatory cytokine) were higher in Cc16 −/− mice than age‐matched WT mice, but were still insufficient to attenuate the increased lung inflammation detected in Cc16 −/− mice at 6–18 months of age.
Small airway fibrosis
Small airway fibrosis has been detected in older human subjects (Janssens et al. 1999; Verbanck et al. 2012; Bommart et al. 2015), and contributes significantly to airflow obstruction in COPD patients (Hogg et al. 2004). To our knowledge, there has been only one study of small airway fibrosis in unchallenged C57BL/6 WT mice older than 6 months of age. Zhou et al. reported that C57BL/6 WT mice aged 18 months did not have greater small airway fibrosis than C57BL/6 WT mice that were 9 months of age (Zhou et al. 2013). Our studies of this lung phenotype in WT mice agree with those of Zhou et al. Cc16 deficiency spontaneously led to the development of small airway fibrosis that was detectable by 12 months of age. Small airway fibrosis in CS‐exposed rodents has been linked to increased generation of profibrotic growth factors including Tgf‐β (Churg et al. 2006), and proinflammatory mediators (Churg et al. 2007, 2009, 2012). Lung levels of active Tgf‐β were similar in Cc16 −/− and WT mice at all time points assessed, but it is possible that Tgf‐β levels were higher in local microenvironments in Cc16 −/− small airways. Whether the greater lung inflammation detected in Cc16 −/− mice contributed to their exaggerated small airway fibrosis will be examined in future studies. Exogenous CC16 reduces the activation of fibroblast‐like cells in vitro (Antico et al. 2014). Whether the loss of Cc16‐mediated inhibition of fibroblast activation contributed to the accelerated development of small airway fibrosis in unchallenged Cc16 −/− mice will be examined in our future studies.
Cc16 has potent anti‐inflammatory activities in numerous models of acute and chronic lung disease (Laucho‐Contreras et al. 2015a, 2016). The anti‐inflammatory activities of Cc16 in other models have been linked to its capacity to inhibit the catalytic activity of secretory phospholipase A2 (sPLA2), a potent proinflammatory pathway by binding to co‐factors required for full catalytic activity of this enzyme (Levin et al. 1986; Lesur et al. 1995), or inhibition of activation of NFκB (Laucho‐Contreras et al. 2015a, 2016) depending on the model system studied. Herein, WT and Cc16 −/− mice did not differ in lung sPLA2 activity levels. The greater NF‐κB activation that was detected in the lungs of 18‐month‐old Cc16 −/− versus age‐matched WT mice likely contributed to the greater lung inflammation and pulmonary lesions detected in unchallenged Cc16 −/− mice. Greater (NFκB) activation was not detected in Cc16 −/− lungs at earlier time points despite the greater lung inflammation detected at 6–12 months of age in these mice. However, we measured NF‐κB activation only in whole lung samples, and signals due to increased NF‐κB activation in specific sub‐types of cells in the lung (e.g., inflammatory cells) at earlier time points may have been diluted out in EMSAs conducted on whole lung samples. How CC16 inhibits activation of NF‐kB is not known, and is currently an area of intense research investigations by a number of laboratories (Laucho‐Contreras et al. 2016).
Aging is thought to be induced by cumulative stochastic injury to proteins, DNA, mitochondria, and telomeres and other cellular components (Liu et al. 2002; Kirkwood 2005; Green et al. 2011) which is driven by oxidative stress (Packer and Fuehr 1977) generated by chronic inflammation (Franceschi et al. 2007; Green et al. 2011). Although cellular injury may cause aging by inducing loss of functional cells in tissues, the cellular response to injury may contribute significantly to the aging process. NF‐κB activation is a key component of the cellular response to injury (Hayden and Ghosh 2008). NF‐κB activation may drive cellular senescence in many tissues during normal and accelerating aging in mice and humans (Osorio et al. 2016). NF‐κB is the transcription factor that is most highly associated with mammalian aging (Adler et al. 2007), and NF‐κB activation is increased in most organs of old versus young rodents (Helenius et al. 1996; Korhonen et al. 1997). Chronic activation of NF‐κB occurs in numerous human age‐related diseases including sarcopenia, atherosclerosis, osteoarthritis, dementia, osteoporosis, and COPD (Yamamoto and Gaynor 2001; Tilstra et al. 2011; Mercado et al. 2015).
Soluble factors present in the circulation regulate aging in mammals. In a murine model of heterochronic parabiosis (in which old and young mice are joined together surgically to share a circulatory system), Ccl11 was present at elevated levels in the old mice, and was shown to induce accelerated aging of the nervous system in the young mice (Villeda et al. 2011). As‐yet‐unidentified circulating soluble factors in old mice that bind and activate Frizzled receptor converted muscle stem cells from a myogenic to a fibrogenic lineage with reduced regenerative potential by activating Wnt signaling (Villeda et al. 2011). Another study of murine heterochronic parabionts reported that as‐yet‐unidentified circulating factors in young mice reversed the aging process in the skeletal muscle and livers of the old mice (Conboy et al. 2005), but the lungs were not examined in this study. Whether CC16 is one such circulating protein that restrains inflamm‐aging in the lung and other organs will be evaluated in our future studies.
Limitations
Our sample sizes were relatively small which could have contributed to some of the negative findings for some of the readouts at some of the time points studied. In addition, we did not analyze correlations between the different parameters that were measured in individual mice as these analyses were not sufficiently powered to detect significant differences. We did not study mice beyond 18 months of age to determine whether the phenotype of the Cc16 −/− mice continues to progress after senescence is reached in mice. We also did not confirm that increased spontaneous activation of NF‐kB detected in the lungs of Cc16 −/− mice is required for their spontaneous development of greater pulmonary inflammation and COPD‐like lung lesions over time. These areas will be the focus of our future studies.
Conclusions
Our data suggest that CC16 is a circulating factor that protects lungs from aging by restraining lung inflamm‐aging as Cc16 deficiency accelerates pulmonary inflammation and COPD‐like pulmonary phenotypes in unchallenged mice. Thus, low serum CC16 levels may directly contribute to the impaired lung function that has been detected in human cohorts by increasing pulmonary inflamm‐aging to thereby accelerate the development of senile emphysema and small airway fibrosis which both contribute to airflow obstruction and FEV1 decline in humans. If our results are confirmed in human cohorts, our data suggest that CC16 augmentation approaches in young and middle‐aged adults with low serum CC16 levels may have potential as a novel therapeutic approach to preserve lung function in these subjects as they age.
Conflict of Interest
Dr. Owen is also an employee of Vertex Pharmaceuticals Inc., 50 Northern Avenue, Boston, MA, but has no disclosures relevant to the content of this manuscript.
Acknowledgments
The current institution for Maria Laucho‐Contreras is the University Hospital of Caracas, Caracas, Venezuela, 1040.
Laucho‐Contreras M. E., Polverino F., Rojas‐Quintero J., Wang X., Owen C. A.. Club cell protein 16 (Cc16) deficiency increases inflamm‐aging in the lungs of mice. Physiol Rep, 6 (15), 2018, e13797, 10.14814/phy2.13797
Funding Information
This work was supported by Public Health Service, NHLBI grant numbers HL063137, HL086814, HL111835, PO1 HL105339, and P01 HL114501, NIAID grant number AI111475–01, The Flight Attendants Medical Research Institute (#CIA123046 and YFEL141004), The Parker B. Francis Foundation Fellowship, the Brigham and Women's Hospital–Lovelace Respiratory Research Institute Consortium, and the Department of Defense (#PR152060).
References
- Adler, A. S. , Sinha S., Kawahara T. L., Zhang J. Y., Segal E., and Chang H. Y.. 2007. Motif module map reveals enforcement of aging by continual NF‐kappaB activity. Genes Dev. 21:3244–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antico, G. , Aloman M., Lakota K., Miele L., Fiore S., and Sodin‐Semrl S.. 2014. Uteroglobin, a possible ligand of the lipoxin receptor inhibits serum amyloid A‐driven inflammation. Mediators Inflamm. 2014:876395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard, A. , Marchandise F. X., Depelchin S., Lauwerys R., and Sibille Y.. 1992. Clara cell protein in serum and bronchoalveolar lavage. Eur. Respir. J. 5(1231–123):1992. [PubMed] [Google Scholar]
- Bommart, S. , Marin G., Bourdin A., Revel M. P., Klein F., Hayot M., et al. 2015. Computed tomography quantification of airway remodelling in normal ageing subjects: a cross‐sectional study. Eur. Respir. J. 45:1167–1170. [DOI] [PubMed] [Google Scholar]
- Calvi, C. L. , Podowski M., D'Alessio F. R., Metzger S. L., Misono K., Poonyagariyagorn H., et al. 2011. Critical transition in tissue homeostasis accompanies murine lung senescence. PLoS ONE 6:e20712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannizzo, E. S. , Clement C. C., Sahu R., Follo C., and Santambrogio L.. 2011. Oxidative stress, inflamm‐aging and immunosenescence. J. Proteomics. 74:2313–2323. [DOI] [PubMed] [Google Scholar]
- Celli, B. R. , and Owen C. A.. 2013. The club cell and its protein, CC16: time to shine. Lancet Respir. Med. 1:757–759. [DOI] [PubMed] [Google Scholar]
- Chen, L. C. , Zhang Z., Myers A. C., and Huang S. K.. 2001. Cutting edge: altered pulmonary eosinophilic inflammation in mice deficient for Clara cell secretory 10‐kDa protein. J. Immunol. 167:3025–3028. [DOI] [PubMed] [Google Scholar]
- Churg, A. , Tai H., Coulthard T., Wang R., and Wright J. L.. 2006. Cigarette smoke drives small airway remodeling by induction of growth factors in the airway wall. Am. J. Respir. Crit. Care Med. 174:1327–1334. [DOI] [PubMed] [Google Scholar]
- Churg, A. , Wang R., Wang X., Onnervik P. O., Thim K., and Wright J. L.. 2007. Effect of an MMP‐9/MMP‐12 inhibitor on smoke‐induced emphysema and airway remodelling in guinea pigs. Thorax 62:706–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churg, A. , Zhou S., Wang X., Wang R., and Wright J. L.. 2009. The role of interleukin‐1beta in murine cigarette smoke‐induced emphysema and small airway remodeling. Am. J. Respir. Cell Mol. Biol. 40:482–490. [DOI] [PubMed] [Google Scholar]
- Churg, A. , Marshall C. V., Sin D. D., Bolton S., Zhou S., Thain K., et al. 2012. Late intervention with a myeloperoxidase inhibitor stops progression of experimental chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 185:34–43. [DOI] [PubMed] [Google Scholar]
- Conboy, I. M. , Conboy M. J., Wagers A. J., Girma E. R., Weissman I. L., and Rando T. A.. 2005. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 433:760–764. [DOI] [PubMed] [Google Scholar]
- Craig, V. J. , Quintero P. A., Fyfe S. E., Patel A. S., Knolle M. D., Kobzik L., et al. 2013. Profibrotic activities for matrix metalloproteinase‐8 during bleomycin‐mediated lung injury. J. Immunol. 190:4283–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, J. , Gilks B., Price K., and Churg A.. 1998. Mineral dusts directly induce epithelial and interstitial fibrogenic mediators and matrix components in the airway wall. Am. J. Respir. Crit. Care Med. 158:1907–1913. [DOI] [PubMed] [Google Scholar]
- Demetrius, L. 2006. Aging in mouse and human systems. Ann. N.Y. Acad. Sci. 1067:66–82. [DOI] [PubMed] [Google Scholar]
- Dutta, S. , and Sengupta P.. 2016. Men and mice: relating their ages. Life Sci. 152:244–248. [DOI] [PubMed] [Google Scholar]
- Fougere, B. , Boulanger E., Nourhashemi F., Guyonnet S., and Cesari M.. 2017. Chronic inflammation: accelerator of biological aging. J. Gerontol. A Biol. Sci. Med. Sci. 72:1218–1225. [DOI] [PubMed] [Google Scholar]
- Franceschi, C. , Capri M., Monti D., Giunta S., Olivieri F., Sevini F., et al. 2007. Inflammaging and anti‐inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 128:92–105. [DOI] [PubMed] [Google Scholar]
- Green, D. R. , Galluzzi L., and Kroemer G.. 2011. Mitochondria and the autophagy‐inflammation‐cell death axis in organismal aging. Science 333:1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarner, V. , and Rubio‐Ruiz M. E.. 2015. Low‐grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip. Top. Gerontol. 40:99–106. [DOI] [PubMed] [Google Scholar]
- Guerra, S. , Halonen M., Vasquez M. M., Spangenberg A., Stern D. A., Morgan W. J., et al. 2015. Relation between circulating CC16 concentrations, lung function, and development of chronic obstructive pulmonary disease across the lifespan: a prospective study. Lancet Respir. Med. 3:613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hautamaki, R. D. , Kobayashi D. K., Senior R. M., and Shapiro S. D.. 1997. Requirement for macrophage elastase for cigarette smoke‐induced emphysema in mice. Science 277:2002–2004. [DOI] [PubMed] [Google Scholar]
- Hayden, M. S. , and Ghosh S.. 2008. Shared principles in NF‐κB signaling. Cell 132:344–362. [DOI] [PubMed] [Google Scholar]
- Helenius, M. , Hanninen M., Lehtinen S. K., and Salminen A.. 1996. Changes associated with aging and replicative senescence in the regulation of transcription factor nuclear factor‐kappa B. Biochem. J. 318(Pt 2):603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg, J. C. , Chu F., Utokaparch S., Woods R., Elliott W. M., Buzatu L., et al. 2004. The nature of small‐airway obstruction in chronic obstructive pulmonary disease. N.Engl . J. Med. 350:2645–2653. [DOI] [PubMed] [Google Scholar]
- Ishii, M. , Yamaguchi Y., Yamamoto H., Hanaoka Y., and Ouchi Y.. 2008. Airspace enlargement with airway cell apoptosis in klotho mice: a model of aging lung. J. Gerontol. A Biol. Sci. Med. Sci. 63:1289–1298. [DOI] [PubMed] [Google Scholar]
- Janssens, J. P. , Pache J. C., and Nicod L. P.. 1999. Physiological changes in respiratory function associated with ageing. Eur. Respir. J. 13:197–205. [DOI] [PubMed] [Google Scholar]
- Karrasch, S. , Holz O., and Jorres R. A.. 2008. Aging and induced senescence as factors in the pathogenesis of lung emphysema. Respir. Med. 102:1215–1230. [DOI] [PubMed] [Google Scholar]
- Kasahara, Y. , Tuder R. M., Taraseviciene‐Stewart L., Le Cras T. D., Abman S., Hirth P. K., et al. 2000. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Invest 106:1311–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood, T. B. 2005. Understanding the odd science of aging. Cell 120:437–447. [DOI] [PubMed] [Google Scholar]
- Knolle, M. D. , Nakajima T., Hergrueter A., Gupta K., Polverino F., Craig V. J., et al. 2013. Adam8 limits the development of allergic airway inflammation in mice. J. Immunol. 190:6434–6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen, P. , Helenius M., and Salminen A.. 1997. Age‐related changes in the regulation of transcription factor NF‐kappa B in rat brain. Neurosci. Lett. 225:61–64. [DOI] [PubMed] [Google Scholar]
- Lange, P. , Celli B., Agusti A., Jensen G. B., Divo M., Faner R., et al. ; for the COPD Progression Group . 2015. Function trajectories leading to chronic obstructive pulmonary disease. N. Engl. J. Med. 373(2):111–122. [DOI] [PubMed] [Google Scholar]
- Lao, T. , Jiang Z., Yun J., Qiu W., Guo F., Huang C., et al. 2016. Hhip haploinsufficiency sensitizes mice to age‐related emphysema. Proc. Natl Acad. Sci. USA 113:E4681–E4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laucho‐Contreras, M. E. , Polverino F., Gupta K., Taylor K. L., Kelly E., Pinto‐Plata V., et al. 2015a. Protective role for club cell secretory protein‐16 (CC16) in the development of chronic obstructive pulmonary disease. Eur. Respir. J. 45:1544–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laucho‐Contreras, M. E. , Taylor K. L., Mahadeva R., Boukedes S. S., and Owen C. A.. 2015b. Automated measurement of pulmonary emphysema and small airway remodeling in cigarette smoke‐exposed mice. JoVE 95:52236 10.3791/52236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laucho‐Contreras, M. E. , Polverino F., Tesfaigzi Y., Pilon A., Celli B. R., and Owen C. A.. 2016. Club cell protein 16 (CC16) augmentation: a potential disease‐modifying approach for chronic obstructive pulmonary disease (COPD). Expert. Opin. Ther. Targets. 20(869–883):2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y. C. , Zhang Z., and Mukherjee A. B.. 2006. Mice lacking uteroglobin are highly susceptible to developing pulmonary fibrosis. FEBS Lett. 580(4515–4520):2006. [DOI] [PubMed] [Google Scholar]
- Lesur, O. , Bernard A., Arsalane K., Lauwerys R., Begin R., Cantin A., et al. 1995. Clara cell protein (CC‐16) induces a phospholipase A2‐mediated inhibition of fibroblast migration in vitro. Am. J. Respir. Crit. Care Med. 152:290–297. [DOI] [PubMed] [Google Scholar]
- Levin, S. W. , Butler J. D., Schumacher U. K., Wightman P. D., and Mukherjee A. B.. 1986. Uteroglobin inhibits phospholipase A2 activity. Life Sci. 38:1813–1819. [DOI] [PubMed] [Google Scholar]
- Liu, L. , Trimarchi J. R., Smith P. J., and Keefe D. L.. 2002. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell 1:40–46. [DOI] [PubMed] [Google Scholar]
- Lomas, D. A. , Silverman E. K., Edwards D. L., Miller B. E., Coxson H. O., and Tal‐Singer R.. 2008. Evaluation of serum CC‐16 as a biomarker for COPD in the ECLIPSE cohort. Thorax 63:1058–1063. [DOI] [PubMed] [Google Scholar]
- Mercado, N. , Ito K., and Barnes P. J.. 2015. Accelerated ageing of the lung in COPD: new concepts. Thorax 70:482–489. [DOI] [PubMed] [Google Scholar]
- Morris, D. G. , Huang X., Kaminski V., Wang Y., Shapiro S. D., Dolganov G., et al. 2003. Loss of integrin alpha(v)beta6‐mediated TGF‐beta activation causes MMP12‐dependent emphysema. Nature 422:169–173. [DOI] [PubMed] [Google Scholar]
- Ofulue, A. F. , and Ko M.. 1999. Effects of depletion of neutrophils or macrophages on development of cigarette smoke‐induced emphysema. Am. J. Physiol. Lung Cell. Mol. Physiol. 277:L97–L105. [DOI] [PubMed] [Google Scholar]
- Osorio, F. G. , Soria‐Valles C., Santiago‐Fernandez O., Freije J. M., and Lopez‐Otin C.. 2016. NF‐kappaB signaling as a driver of ageing. Int. Rev. Cell Mol. Biol. 326:133–174. [DOI] [PubMed] [Google Scholar]
- O'Sullivan, E. D. , Hughes J., and Ferenbach D. A.. 2017. Renal aging: causes and consequences. J. Am. Soc. Nephrol. 28:407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen, C. A. 2005. Proteinases and oxidants as targets in the treatment of chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2:373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen, C. A. 2008. Roles for proteinases in the pathogenesis of chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 3:253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen, C. A. , Campbell M. A., Boukedes S. S., and Campbell E. J.. 1994. Monocytes recruited to sites of inflammation express a distinctive proinflammatory (P) phenotype. Am. J. Physiol. 267:L786–L796. [DOI] [PubMed] [Google Scholar]
- Packer, L. , and Fuehr K.. 1977. Low oxygen concentration extends the lifespan of cultured human diploid cells. Nature 267:423–425. [DOI] [PubMed] [Google Scholar]
- Park, H. Y. , Churg A., Wright J. L., Li Y., Tam S., Man S. F., et al. 2013. Club cell protein 16 and disease progression in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 188:1413–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, H. , Leng S., Belinsky S. A., Miller B. E., Tal‐Singer R., Owen C. A., et al. 2015. Low plasma CC16 levels in smokers are associated with a higher risk for chronic bronchitis. Eur. Respir. J. 46:1501–1503. [DOI] [PubMed] [Google Scholar]
- Pilette, C. , Godding V., Kiss R., Delos M., Verbeken E., Decaestecker C., et al. 2001. Reduced epithelial expression of secretory component in small airways correlates with airflow obstruction in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 163:185–194. [DOI] [PubMed] [Google Scholar]
- Rangasamy, T. , Cho C. Y., Thimmulappa R. K., Zhen L., Srisuma S. S., Kensler T. W., et al. 2004. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke‐induced emphysema in mice. J Clin Invest 114:1248–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rava, M. , Le M. N., Dumont X., Guerra S., Siroux V., Jacquemin B., et al. 2015. Serum club cell protein 16 is associated with asymptomatic airway responsiveness in adults: findings from the French epidemiological study on the genetics and environment of asthma. Respirology 20:1198–1205. [DOI] [PubMed] [Google Scholar]
- Roubenoff, R. , Harris T. B., Abad L. W., Wilson P. W., Dallal G. E., and Dinarello C. A.. 1998. Monocyte cytokine production in an elderly population: effect of age and inflammation. J. Gerontol. A Biol. Sci. Med. Sci. 53:M20–M26. [DOI] [PubMed] [Google Scholar]
- Snyder, J. C. , Reynolds S. D., Hollingsworth J. W., Li Z., Kaminski N., and Stripp B. R.. 2010. Clara cells attenuate the inflammatory response through regulation of macrophage behavior. Am. J. Respir. Cell Mol. Biol. 42:161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal, R. S. , and Orr W. C.. 2012. The redox stress hypothesis of aging. Free Radic. Biol. Med. 52:539–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorino, C. , Battaglia S., Scichilone N., Pedone C., Antonelli‐Incalzi R., Sherrill D., et al. 2012. Diagnosis of airway obstruction in the elderly: contribution of the SARA study. Int. J. Chron. Obstruct. Pulmon. Dis. 7:389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, G. H. , Drullinger L. F., Soulard A., and Dulic V.. 1999. Differential roles for cyclin‐dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol. 19:2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stripp, B. R. , Reynolds S. D., Boe I. M., Lund J., Power J. H., Coppens J. T., et al. 2002. Clara cell secretory protein deficiency alters clara cell secretory apparatus and the protein composition of airway lining fluid. Am. J. Respir. Cell Mol. Biol. 27:170–178. [DOI] [PubMed] [Google Scholar]
- Sueblinvong, V. , Neujahr D. C., Mills S. T., Roser‐Page S., Ritzenthaler J. D., Guidot D., et al. 2012. Predisposition for disrepair in the aged lung. Am. J. Med. Sci. 344:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga, T. , Kurabayashi M., Sando Y., Ohyama Y., Maeno T., Maeno Y., et al. 2000. Disruption of the klotho gene causes pulmonary emphysema in mice. Defect in maintenance of pulmonary integrity during postnatal life. Am. J. Respir. Cell Mol. Biol. 22:26–33. [DOI] [PubMed] [Google Scholar]
- Takeda, Y. , He P., Tachibana I., Zhou B., Miyado K., Kaneko H., et al. 2008. Double deficiency of tetraspanins CD9 and CD81 alters cell motility and protease production of macrophages and causes chronic obstructive pulmonary disease‐like phenotype in mice. J. Biol. Chem. 283:26089–26097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilstra, J. S. , Clauson C. L., Niedernhofer L. J., and Robbins P. D.. 2011. NF‐kappaB in aging and disease. Aging Dis. 2:449–465. [PMC free article] [PubMed] [Google Scholar]
- Vaz Fragoso, C. A. , and Gill T. M.. 2010. Defining chronic obstructive pulmonary disease in an aging population. J. Am. Geriatr. Soc. 58:2224–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbanck, S. , Thompson B. R., Schuermans D., Kalsi H., Biddiscombe M., Stuart‐Andrews C., et al. 2012. Ventilation heterogeneity in the acinar and conductive zones of the normal ageing lung. Thorax 67:789–795. [DOI] [PubMed] [Google Scholar]
- Verbeken, E. K. , Cauberghs M., Mertens I., Clement J., Lauweryns J. M., and Van de Woestijne K. P.. 1992. The senile lung. Comparison with normal and emphysematous lungs. 1 Structural aspects. Chest 101:793–799. [DOI] [PubMed] [Google Scholar]
- Villeda, S. A. , Luo J., Mosher K. I., Zou B., Britschgi M., Bieri G., et al. 2011. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. Z. , Rosenberger C. L., Bao Y. X., Stark J. M., and Harrod K. S.. 2003. Clara cell secretory protein modulates lung inflammatory and immune responses to respiratory syncytial virus infection. J. Immunol. 171:1051–1060. [DOI] [PubMed] [Google Scholar]
- Wu, Q. , Jiang D., Matsuda J. L., Ternyak K., Zhang B., and Chu H. W.. 2016. Cigarette smoke induces human airway epithelial senescence via growth differentiation factor 15 production. Am. J. Respir. Cell Mol. Biol. 55:429–438. [DOI] [PubMed] [Google Scholar]
- Xu, J. , Tao B., Guo X., Zhou S., Li Y., Zhang Y., et al. 2017. Macrophage‐restricted Shp2 tyrosine phosphatase acts as a rheostat for MMP12 through TGF‐beta activation in the prevention of age‐related emphysema in mice. J. Immunol. 199:2323–2332. [DOI] [PubMed] [Google Scholar]
- Yamamoto, Y. , and Gaynor R. B.. 2001. Role of the NF‐kappaB pathway in the pathogenesis of human disease states. Curr. Mol. Med. 1:287–296. [DOI] [PubMed] [Google Scholar]
- Zhou, S. , Wright J. L., Liu J., Sin D. D., and Churg A.. 2013. Aging does not enhance experimental cigarette smoke‐induced COPD in the mouse. PLoS ONE 8:e71410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, L. , Di P. Y., Wu R., Pinkerton K. E., and Chen Y.. 2015. Repression of CC16 by cigarette smoke (CS) exposure. PLoS ONE 10:e0116159. [DOI] [PMC free article] [PubMed] [Google Scholar]