

Abstract
The HIV-1 subtype B epidemic in French Guiana and Suriname is characterized by the co-circulation of the globally disseminated “BPANDEMIC” lineage and of non-pandemic subtype B lineages of Caribbean origin (BCAR). To reconstruct the spatiotemporal pattern of spread of those viral lineages circulating in these two countries, a total of 361 HIV-1 subtype B pol sequences recovered from treatment-naive adult patients from French Guiana and Suriname between 2006 and 2012 were combined with BPANDEMIC and BCAR reference sequences. Major Guianese/Surinamese BPANDEMIC and BCAR lineages were identified by Maximum Likelihood phylogenetic analysis and the spatiotemporal and demographic parameters estimated using a Bayesian coalescent-based method. We detected four BCAR and three BPANDEMIC transmission chains of large size that together comprise most pandemic and non-pandemic subtype B sequences from French Guiana (≥52%) and Suriname (≥70%) here analyzed. These major lineages were probably introduced into French Guiana and Suriname from the Caribbean (BCAR) and North/South America (BPANDEMIC) between the middle 1970s and the late 1980s and spread among populations from both countries with roughly comparable demographic growth rates. We detected a significant trend for higher viral loads and higher proportion of homosexual/bisexual men among subjects infected with BPANDEMIC relative to BCAR strains in French Guiana. These results show that the HIV subtype B epidemic in French Guiana and Suriname has been driven by multiple active BCAR and BPANDEMIC transmission chains that arose since the middle 1970s onward and operate in both countries simultaneously. Although no significant differences in the epidemic potential of major BCAR and BPANDEMIC lineages were observed, relevant associations between the infecting subtype B lineage and epidemiological and clinical characteristics were detected in French Guiana.
Keywords: HIV-1, subtype B, pandemic, non-pandemic, phylodynamics, French Guiana, Suriname
Introduction
The Guianas are a region located on the northeastern coast of South America, bordered by Brazil to the south and Venezuela to the west, which includes the French Guiana (an overseas department of France), and the sovereign states of Guyana (known as British Guiana until 1966) and Suriname (part of the Kingdom of the Netherlands until 1975). With a combined population of nearly 1.5 million inhabitants, the Guianas consists of a wide variety of ethnic groups due to historical colonization by Amerindians, Europeans, Africans, and Asians and recent migratory fluxes from neighboring South American and Caribbean countries (Hyles, 2014). In this singular geographic and demographic context, the HIV/AIDS epidemic is a major public health problem and HIV prevalence rates in adult populations from French Guiana, Guyana, and Suriname (1.0–1.5%) are among the highest in the American continent (Nacher et al., 2010; UNAIDS, 2013).
Subtype B is the predominant HIV-1 lineage circulating in French Guiana (Kazanji et al., 2001; Darcissac et al., 2016) and Suriname (Abdoel Wahid et al., 2016); but in sharp contrast to other continental American countries where the epidemic is mostly driven by the globally disseminated “BPANDEMIC” lineage, the subtype B epidemic in French Guiana and Suriname is driven by transmission of both BPANDEMIC and of non-pandemic subtype B lineages characteristic of the Caribbean region (“BCAR” lineages) (Cabello et al., 2015). This epidemiological pattern resembles that described in several Caribbean islands (Haiti, the Dominican Republic, Jamaica, The Bahamas and the Lesser Antilles) (Cabello et al., 2014) and in the Northern Brazilian state of Roraima (Divino et al., 2016). Previous phylogenetic analyses revealed that a substantial fraction (30–95%) of subtype B infections in Latin American and Caribbean countries resulted from the expansion of a few local (or regional) BPANDEMIC and BCAR founder strains (Delatorre and Bello, 2013; Cabello et al., 2014, 2015; Mendoza et al., 2014; Mir et al., 2015; Divino et al., 2016), thus supporting a great geographic compartmentalization of the HIV-1 subtype B epidemic in those regions.
Little is known about the spatiotemporal dynamics of dissemination, geographic compartmentalization, and demographic history of the BPANDEMIC and BCAR lineages circulating in French Guiana and Suriname. To answer these questions, we used Maximum Likelihood (ML) and Bayesian coalescent-based methods to analyze a comprehensive data set of 361 HIV-1 subtype B pol sequences from French Guiana and Suriname recently described (Abdoel Wahid et al., 2016; Darcissac et al., 2016). The sequences were compared with HIV-1 subtype B pol sequences from the Caribbean, South America, North America, and Europe to identify country-specific transmission clusters of the BPANDEMIC and BCAR lineages and to reconstruct their evolutionary and demographic dynamics. We also tested if individuals from French Guiana infected by the BPANDEMIC and BCAR lineages displayed or not comparable epidemiological and clinical characteristics.
Materials and Methods
Guianese and Surinamese HIV-1 Subtype B Sequences
HIV-1 subtype B pol sequences from treatment-naive adult patients from French Guiana (n = 271) and Suriname (n = 90) recently described (Abdoel Wahid et al., 2016; Darcissac et al., 2016) were included in the present study. HIV-1 sequences were sampled over a time period of seven years (2006–2012) and cover the complete protease (PR) and the first part of the reverse transcriptase (RT) regions (nucleotides 2,253–3,275 of reference strain HXB2). Only one sequence per subject was selected and the subtype of all sequences was confirmed using the REGA HIV subtyping tool v.2 (de Oliveira et al., 2005). HIV-1 pol sequences were aligned using the ClustalW program (Thompson et al., 1997) and codons associated with major antiretroviral (ARV) drug resistance positions in PR (n = 12) and RT (n = 21) were excluded. All patients were informed of the possible use of epidemiological and clinical data for research and provided written consent. The project was approved by the Comité de Recherche Clinique (CoRC) Pasteur Institute Paris Project number 2014–2016.
HIV-1 Subtype B Lineage Assignment and Identification of Guianese/Surinamese Subtype B Lineages
HIV-1 subtype B pol sequences from French Guiana and Suriname were first aligned with 500 subtype B sequences representative of the BPANDEMIC and the BCAR lineages described previously (Cabello et al., 2014; Mendoza et al., 2014) (Supplementary Table S1) and classified within corresponding lineages by using a ML phylogenetic approach. ML trees were inferred with the PhyML program (Guindon et al., 2010) using an online web server (Guindon et al., 2005) under the GTR+I+Γ nucleotide substitution model, as selected by the jModelTest program (Posada, 2008), and the SPR branch-swapping algorithm of heuristic tree search. The reliability of the obtained tree topology was estimated with the approximate likelihood-ratio test (aLRT) (Anisimova and Gascuel, 2006) based on the Shimodaira-Hasegawa-like procedure. Trees were rooted using subtype D sequences (the closets HIV-1 group M lineage relative to subtype B) taken from the Los Alamos HIV Database and visualized using the FigTree v1.4.0 program (Rambaut, 2009).
HIV-1 BCAR and BPANDEMIC pol sequences from French Guiana and Suriname were next aligned with BCAR sequences from the Caribbean and Brazil and with BPANDEMIC sequences from the United States (US), France, the Netherlands, and Northern Brazil characterized previously (Cabello et al., 2014, 2015, 2016; Divino et al., 2016) (Supplementary Table S2). Sequences were subjected to ML analyses as described above and Guianese and Surinamese BCAR and BPANDEMIC transmission clusters were defined as highly supported (aLRT ≥ 0.85) monophyletic clusters mostly (>90%) composed by sequences from these countries. For putative intra-subtype BCAR/BPANDEMIC recombinant sequences, similarity plots depicting the percentage identity to a panel of BPANDEMIC, BCAR, and subtype D reference sequences were generated using SimPlot v.3.5.1 (Lole et al., 1999) and Neighbor-Joining phylogenetic trees of different pol gene fragments were reconstructed under the Tamura-Nei model, in 500 bootstrapped datasets, using MEGA v6 (Tamura et al., 2013).
Spatiotemporal and Demographic Reconstructions
To reconstruct spatiotemporal dynamics and identify the most probable source location of major BCAR Guianese/Surinamese lineages here identified, we selected BCAR sequences from the major Caribbean islands with high prevalence of non-pandemic strains and BCAR sequences from neighboring South American countries, including: all BCAR sequences from Hispaniola and sequences of major BCAR lineages circulating in Trinidad and Tobago (BCAR-TT), Jamaica (BCAR-JM-I), Brazil (BCAR-BR-I and BCAR-BR-II), and Guyana (BCAR-GY) identified in previous studies (Cabello et al., 2014, 2015; Divino et al., 2016) (Supplementary Table S3). Similarly, to identify the most probable source location of major BPANDEMIC Guianese/Surinamese lineages we selected a subset of BPANDEMIC reference sequences from regions with the highest human flux from/to French Guiana and Suriname (the Caribbean, South America, Central America, North America, and Europe). A total of 40 BPANDEMIC reference sequences from each geographic region with known date of isolation and with the highest similarity to Guianese and Surinamese sequences were selected using the basic local alignment search tool (BLAST)1 (Supplementary Table S4).
The evolutionary rate, the age of the most recent common ancestor (TMRCA, years), the spatial diffusion pattern and the rate (r, years-1) of population growth of major HIV-1 Guianese/Surinamese subtype B lineages were jointly estimated using the Bayesian Markov Chain Monte Carlo (MCMC) approach as implemented in BEAST v1.8 (Drummond et al., 2002; Drummond and Rambaut, 2007) with BEAGLE (Suchard and Rambaut, 2009) to improve run-time. Because regression analyses using program TempEst (Rambaut et al., 2016) revealed that subtype B pol datasets here compiled does not contain sufficient temporal signal for reliable time-scale estimations (X-intercept [TMRCA] < 1,910), Bayesian MCMC analyses were performed using the GTR+I+Ã4 nucleotide substitution model and a relaxed uncorrelated lognormal molecular clock model (Drummond et al., 2006) with a uniform prior distribution on the substitution rate that encompass mean values previously estimated for the subtype B pol gene (2.0–3.0 × 10-3 subst./site/year) (Hue et al., 2005; Zehender et al., 2010; Chen et al., 2011; Mendoza et al., 2014). Migration events throughout the phylogenetic histories were reconstructed using a reversible discrete phylogeography model (Lemey et al., 2009) with a CTMC rate reference prior (Ferreira and Suchard, 2008). Changes in effective population size through time for each major HIV-1 Guianese/Surinamese subtype B lineages was independently estimated using a Bayesian Skyline coalescent tree prior (Drummond et al., 2005). Estimates of the population growth rate were obtained using the parametric model (logistic, exponential, or expansion) that provided the best fit to the demographic signal contained in datasets. Comparison between demographic models was performed using the log marginal likelihood estimation based on path sampling (PS) and stepping-stone sampling (SS) methods (Baele et al., 2012). MCMC chains were run for 50–200 × 106 generations. Convergence (Effective Sample Size > 200) and uncertainty (95% Highest Probability Density [HPD] values) in parameter estimates were assessed using the TRACER v1.6 program (Rambaut and Drummond, 2007). Maximum clade credibility (MCC) trees were summarized with TreeAnnotator v1.7.5 and visualized with FigTree v1.4.0.
Estimation of HIV Incidence Temporal Trend in French Guiana
To estimate the HIV incidence in French Guiana we used the Spectrum v5.51 package2. AIM (AIDS Impact Model) and CSAVER (Case Surveillance and Vital Registration) incidence fitting tools were used with a start in 1970 and projections until 2013. Historical HIV programmed data, treatment eligibility criteria for adults and children for different periods, the proportion of pregnant women with access to prevention of mother-to-child transmission of HIV, the number of patients receiving ARV therapy, the median CD4-count at ARV initiation, the proportion of virologically suppressed treated patients and the proportion of lost to follow-up patients each year were entered in Spectrum. The Epidemic was modeled as a concentrated epidemic. The yearly progression rate to the next CD4 category and the HIV mortality with and without ARV were selected from the options in Spectrum based on Latin America and the Caribbean. The default ratio of fertility of infected women versus uninfected ones was used.
Statistical Analyses
Epidemiological and demographic characteristics of the cohort included in the present study were compared using Fisher’s exact test or chi2 implemented in Stata 13 software. Statistical significance was defined as p < 0.05.
Results
Identification of Major Guianese/Surinamese Subtype B Lineages
HIV-1 subtype B sequences from French Guiana and Suriname (n = 361) were combined with viral strains representative of the BPANDEMIC (n = 300) and BCAR (n = 200) lineages (Supplementary Table S1). The ML analysis revealed that, as expected, a very large proportion of HIV-1 subtype B sequences from French Guiana (60%) and Suriname (50%) were intermixed among BCAR strains; whereas the others branched within the well-supported (aLRT = 0.90) BPANDEMIC lineage (Supplementary Figure S1 and Supplementary Table S5). A few sequences from French Guiana (4%) and Suriname (6%) remained unclassified as they branched basal to the BPANDEMIC lineage, but were not intermixed among BCAR sequences and, when included, greatly reduced the support of the BPANDEMIC lineage (aLRT < 0.80) (data not shown). Similarity plots revealed that most unclassified sequences displayed a higher similarity to BCAR reference strains in the initial portion of the pol fragment (covering the entire PR) compared to BPANDEMIC strains, and a higher similarity to BPANDEMIC sequences in the final portion of the pol fragment (Supplementary Figure S2). This suggests that they may represent intra-subtype BCAR/BPANDEMIC recombinant sequences. The phylogenetic branching pattern at the initial and final portions of the pol fragment also supports a BCAR/BPANDEMIC recombinant structure for half of unclassified sequences (Supplementary Figure S2). However, this result should be interpreted with caution due the overall low support (bootstrap < 60%) at nodes of phylogenetic trees.
The BCAR and BPANDEMIC sequences from French Guiana and Suriname were then combined with BCAR and BPANDEMIC sequences of countries from the Americas (Caribbean region and Brazil) and Europe (France and the Netherlands) that maintain historical intense migratory fluxes with French Guiana and Suriname. The ML phylogenetic analyses revealed that most BCAR and BPANDEMIC sequences from French Guiana (≥52%) and Suriname (≥70%) branched within a few monophyletic lineages of large size (n > 10 sequences) that were exclusively composed of sequences from both countries (BCAR-GF/SR-I, BCAR-GF/SR-II, BCAR-GF/SR-III, BPAN-GF/SR-I, and BPAN-GF/SR-II), from French Guiana (BPAN-GF-I), or that also include sequences from neighboring countries (BCAR-SA-I) (Figure 1 and Supplementary Table S6). Geographic distribution of major Guianese/Surinamese BCAR and BPANDEMIC lineages was not homogenous across countries (Supplementary Tables S7, S8). The remaining BCAR and BPANDEMIC sequences from French Guiana and Suriname branched in country-specific lineages of small size (≤10%) or appeared as non-clustered infections (≤41%) (Supplementary Table S6).
FIGURE 1.
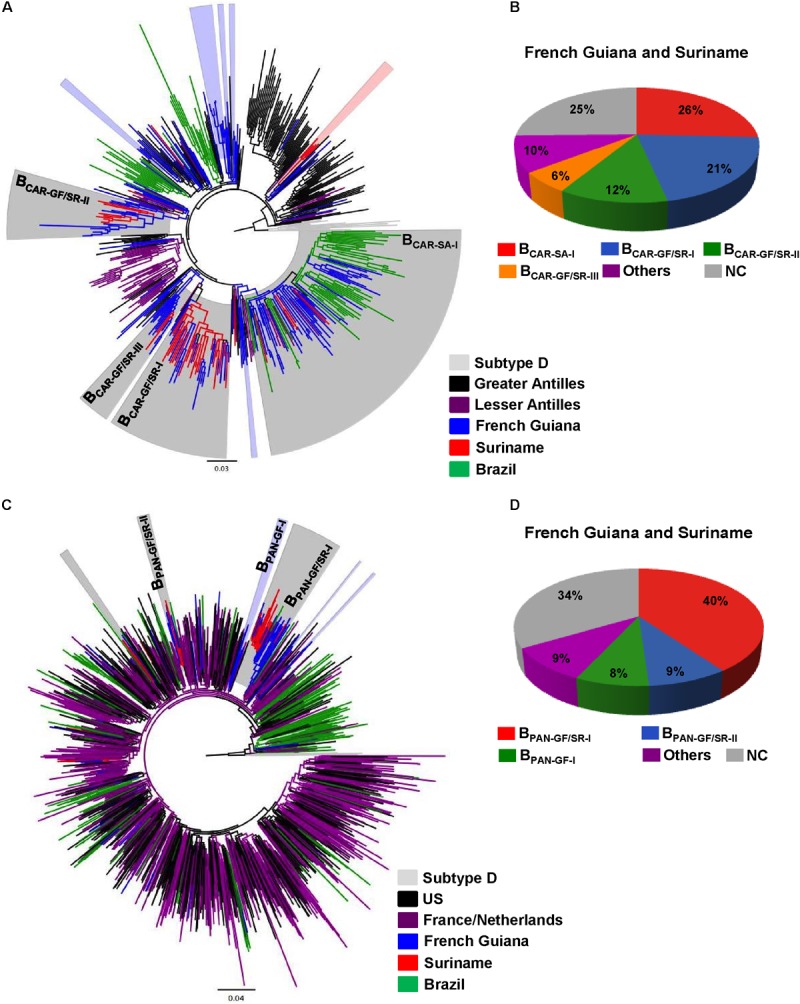
Identification of major Guianese/Surinamese subtype B lineages. (A,C) ML phylogenetic trees of HIV-1 BCAR and BPANDEMIC pol sequences circulating in French Guiana and Suriname together with representative BCAR sequences from the Caribbean and Brazil and BPANDEMIC sequences from the US, France, the Netherlands and Northern Brazil. Branches are colored according to the geographic origin of each sequence as indicated at the legend (bottom right). Shaded boxes highlight the position of BCAR and BPANDEMIC highly supported (SH-aLRT ≥ 0.85) clusters mostly/exclusively composed by Guianese and/or Surinamese sequences. Major (n ≥ 10) BCAR and BPANDEMIC lineages detected in French Guiana and Suriname are indicated with names. Trees were rooted using HIV-1 subtype D reference sequences. The branch lengths are drawn to scale with the bar at the bottom indicating nucleotide substitutions per site. (B,D) Estimated proportion of HIV-1 sequences branching in major and minor Guianese/Surinamese clusters as well of non-clustered (NC) sequences among BCAR and BPANDEMIC infected individuals from French Guiana and Suriname according to the ML analyses.
Spatiotemporal Dissemination of Major Guianese/Surinamese Subtype B Lineages
To identify the most probable source location of major BCAR and BPANDEMIC Guianese/Surinamese subtype B lineages, those sequences were combined with sequences of major BCAR lineages circulating in the Caribbean and Brazil and with BPANDEMIC sequences from the Americas and Europe with the highest BLAST search similarity score to Guianese/Surinamese sequences. Bayesian phylogeographic analyses support that lineage BCAR-SA-I arose after dissemination of a single variant strain from Trinidad and Tobago (Posterior State Probability [PSP] = 0.98) into French Guiana (PSP = 0.93) at around the middle 1970s, that subsequently passed the BCAR-SA-I lineage to Suriname, Brazil, and Guyana at multiples times, originating the sublineages BCAR-BR-I and BCAR-GY (Figure 2A and Supplementary Figure S3). The other Guianese/Surinamese BCAR lineages most probably arose after dissemination of viral strains from Hispaniola (PSP ≥ 0.97) into Suriname (lineage BCAR-GF/SR-I, PSP = 0.68) and French Guiana (lineages BCAR-GF/SR-II and BCAR-GF/SR-III, PSP ≥ 0.97) between the late 1970s and the middle 1980s, followed by multiple viral exchanges between those countries (Figure 2A and Table 1). The lineages BPAN-GF/SR-I and BPAN-GF/SR-II most probably arose after concurrent dissemination of BPANDEMIC strains from North America (PSP = 1) into French Guiana (PSP = 0.97) and Suriname (PSP = 0.75), respectively, at around the middle 1980s and were later disseminated between French Guiana and Suriname at multiple times (Figure 2B and Table 1). We detected only sporadic disseminations of the BPAN-GF/SR-I lineage from French Guiana into Northern Brazil (Amapá), North America (the US), and Europe (the Netherlands). The BPAN-GF-I lineage most probably arose after spreading of a BPANDEMIC strain from South America (PSP = 0.92) into French Guiana (PSP = 0.99) at around the early 1990s, with no evidence of dissemination outside this country (Figure 2B and Table 1).
FIGURE 2.
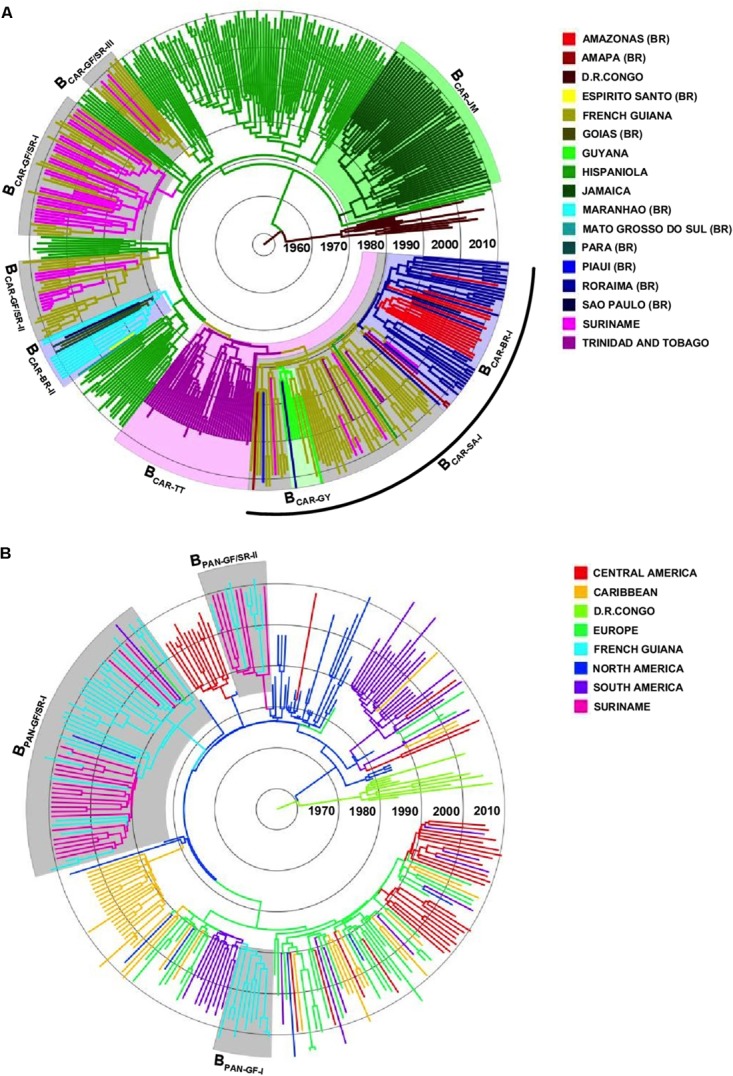
Spatiotemporal dissemination of major Guianese/Surinamese subtype B lineages. (A) Time-scaled Bayesian phylogenetic tree of HIV-1 BCAR pol sequences from French Guiana, Suriname, Brazil, Guyana, and the Caribbean combined with subtype D reference sequences. Shaded boxes highlight the position of major BCAR lineages from French Guiana/Suriname, Trinidad and Tobago, Jamaica, Brazil, and Guyana. The arc indicates the position of the BCAR-SA-I lineage. (B) Time-scaled Bayesian phylogenetic tree of HIV-1 BPANDEMIC pol sequences from the French Guiana, Suriname and closely related BPANDEMIC sequences from the Caribbean, South/Central/North America, and Europe, combined with subtype D reference sequences. Shaded boxes highlight the position of major BPANDEMIC lineages circulating in French Guiana and Suriname. Branches are colored according to the most probable location state of their descendent nodes as indicated in the legend at right. Branch lengths are drawn to a scale of years. The trees are automatically rooted under the assumption of a relaxed molecular clock.
Table 1.
Phylogeographic, evolutionary, and demographic parameters estimated for major HIV-1 BCAR and BPANDEMIC lineages circulating in French Guiana and Suriname.
| Clade | N | Sampling interval | Origin (PSP) | TMRCA (95% HPD) | Growth model | Growth rate (95% HPD) | R0 (95% HPD) |
|---|---|---|---|---|---|---|---|
| BCAR-SA-I | 54 | 2000–2012 | GF (0.93) | 1977 (1973–1981) | Logistic | 0.46 (0.30–0.64) | 4.7 (3.4–6.1) |
| BCAR-GF/SR-I | 45 | 2000–2012 | SR (0.68) | 1978 (1974–1982) | Logistic | 0.30 (0.21–0.40) | 3.4 (2.7–4.2) |
| BCAR-GF/SR-II | 25 | 2007–2012 | GF (0.97) | 1980 (1975–1985) | - | - | - |
| BCAR-GF/SR-III | 12 | 2007–2011 | GF (1.00) | 1984 (1979–1988) | - | - | - |
| BPAN-GF/SR-I | 55 | 2007–2012 | GF (0.97) | 1985 (1982–1988) | Logistic | 0.45 (0.27–0.70) | 4.6 (3.2–6.6) |
| BPAN-GF/SR-II | 13 | 2007–2011 | SR (0.75) | 1987 (1983–1990) | - | - | - |
| BPAN-GF-I | 11 | 2006–2011 | GF (0.99) | 1990 (1987–1992) | - | - | - |
GF, French Guiana; SR, Suriname.
Demographic History of Major Guianese/Surinamese Subtype B Lineages
Bayesian coalescent inference was next used to reconstruct the population dynamics of major HIV-1 subtype B Guianese/Surinamese lineages with more than 30 sequences. The Bayesian skyline plot (BSP) analyses suggest that lineages BCAR-SA-I, BCAR-GF/SR-I, and BPAN-GF/SR-I experienced an initial phase of slow growth until 1985–1990, followed by a phase of exponential growth during the 1990s and subsequent decline in growth rate from the middle 1990s and the middle 2000s (Figures 3A–C). The estimated temporal change of the HIV incidence rate in French Guiana also supports an epidemic stabilization around the early 2000s; but points to a continuous reduction of the HIV incidence afterward that was not captured by our coalescent-based demographic inference (Figure 3D). The logistic demographic model fit the data better than the other models tested (log BF > 3) for lineages BCAR-SA-I and BCAR-GF/SR-I; but was modestly supported (log BF = 1.9) over the exponential one for lineage BPAN-GF/SR-I, probably due to the very recent stabilization phase (Supplementary Table S9). According to the logistic growth model, the mean estimated epidemic growth rates of lineages BCAR-SA-I (0.46 year-1), BPAN-GF/SR-I (0.45 year-1), and BCAR-GF/SR-I (0.30 year-1) were roughly comparable and corresponded to mean R0 of between 3.4 and 4.7 (Table 1).
FIGURE 3.

(A–C) Demographic history of major HIV-1 BCAR and BPANDEMIC lineages (n > 30 sequences) circulating in French Guiana and Suriname. Mean estimates of the effective number of infections (Ne) (solid line) are shown together with the 95% HPD intervals (dashed curves) of the Bayesian skyline plot for lineages BCAR-SA-I (A), BCAR-GF/SR-I (B), and BPAN-GF/SR-I (C). The vertical axes represent the estimated Ne on a logarithmic scale. Time scales are in calendar years. (D) Spectrum-estimated national HIV incidence temporal trend in French Guiana.
Association Between Epidemiological Characteristics and HIV-1 Subtype B Lineages
Analysis of the epidemiological characteristics of Guianese subjects infected with BPANDEMIC and BCAR lineages revealed that both viral lineages circulated among males and females of different age groups. Most subjects infected with both BPANDEMIC and BCAR lineages were mainly followed-up at clinics from the capital city Cayenne and born outside French Guiana (Table 2). Although heterosexual sex was the main mode of HIV transmission for both lineages, the proportion of homosexual/bisexual men among those infected by the BPANDEMIC lineage (10%) was five-times higher than among those infected by BCAR lineages (2%). Among clinical characteristics, significant differences were observed in subject distribution according to plasma RNA viral load. The proportion of subjects with high viral loads (>10,000 copies/ml) among those infected with the BPANDEMIC lineage (64%) was higher that among those infected with BCAR lineages (46%).
Table 2.
Epidemiological information of subjects from French Guiana infected by HIV-1 BCAR and BPANDEMIC clades.
| Characteristic | Total (n = 260) | BCAR (n = 162) | BPANDEMIC (n = 98) | P |
|---|---|---|---|---|
| Sampling interval (years) | 2007–2012 | 2007–2012 | 2007–2012 | - |
| HIV diagnosis∗ | ||||
| 1980–1999 | 17 (7%) | 10 (6%) | 7 (7%) | 0.67 |
| 2000–2005 | 35 (13%) | 25 (15%) | 10 (10%) | |
| 2006–2012 | 202 (78%) | 123 (76%) | 79 (81%) | |
| Unknown | 6 (3%) | 4 (2%) | 2 (2%) | |
| Age group (years)∗∗ | ||||
| 18–24 | 23 (9%) | 14 (9%) | 9 (9%) | 0.55 |
| 25–34 | 89 (34%) | 56 (35%) | 33 (34%) | |
| 35–44 | 72 (28%) | 49 (30%) | 23 (23%) | |
| >44 | 76 (29%) | 43 (26%) | 33 (34%) | |
| Sex∗∗ | ||||
| Male | 113 (43%) | 65 (40%) | 48 (49%) | 0.16 |
| Female | 147 (57%) | 97 (60%) | 50 (51%) | |
| Mode of transmission∗ | ||||
| Homosexual/Bisexual | 14 (5%) | 4 (2%) | 10 (10%) | 0.009 |
| Heterosexual | 229 (88%) | 150 (93%) | 79 (81%) | |
| Others | 1 (<1%) | 0 | 1 (1%) | |
| Unknown | 16 (6%) | 8 (5%) | 8 (8%) | |
| Geographic location∗ | ||||
| Cayenne | 193 (74%) | 123 (76%) | 70 (71%) | 0.76 |
| Saint Laurent du Maroni | 54 (21%) | 31 (19%) | 23 (23%) | |
| Others | 4 (2%) | 3 (2%) | 1 (1%) | |
| Unknown | 9 (3%) | 5 (3%) | 4 (4%) | |
| Country of birth∗∗ | ||||
| French Guiana | 31 (12%) | 18 (11%) | 13 (13%) | 0.25 |
| Haiti | 56 (22%) | 32 (20%) | 24 (24%) | |
| Suriname | 52 (20%) | 32 (20%) | 20 (20%) | |
| Guyana | 33 (13%) | 28 (17%) | 5 (5%) | |
| France | 33 (13%) | 21 (13%) | 12 (12%) | |
| Brazil | 27 (10%) | 15 (9%) | 12 (12%) | |
| Others | 13 (5%) | 7 (4%) | 6 (6%) | |
| Unknown | 15 (6%) | 9 (6%) | 6 (6%) | |
| Clinical condition∗ | ||||
| Asymptomatic (A) | 194 (75%) | 119 (73%) | 75 (76%) | 0.57 |
| Symptomatic (B) | 24 (9%) | 18 (11%) | 6 (6%) | |
| AIDS (C) | 33 (13%) | 20 (12%) | 13 (13%) | |
| Unknown | 9 (3%) | 5 (3%) | 4 (4%) | |
| Viral load (copies/ml)∗ | ||||
| <LD | 10 (4%) | 7 (4%) | 3 (3%) | 0.03 |
| 51–2,000 | 42 (16%) | 32 (20%) | 10 (10%) | |
| 2,001–10,000 | 70 (27%) | 48 (30%) | 22 (22%) | |
| >10,000 | 138 (53%) | 75 (46%) | 63 (64%) | |
| CD4 count (cells/ml)∗∗ | ||||
| 350–500 | 133 (51%) | 85 (52%) | 48 (49%) | 0.58 |
| >500 | 127 (49%) | 77 (48%) | 50 (51%) | |
∗Fisher’s exact test. ∗∗Pearson’s chi2. P values < 0.05 are in bold.
Significant differences in epidemiological and clinical variables were also observed among major Guianese/Surinamese subtype B transmission chains (Supplementary Table S10). In contrast to the overall pattern, most (65%) subjects infected with the lineage BCAR-GF/SR-I were followed-up at clinics from Saint Laurent du Maroni, the second most populous city of French Guiana located at the border with Suriname. Significant associations were observed between infecting strain and the country of origin, including: born in Guyana and lineage BCAR-SA-I, born in Suriname and lineage BCAR-GF/SR-I, and born in mainland France and lineage BCAR-GF/SR-II. Notably, individuals of Haiti origin represent a large proportion (20%) of subjects from French Guiana infected with BCAR strains, but a low fraction (5%) of those infected with major Guianese/Surinamese BCAR lineages. We also observed that the proportion of subjects with high viral loads among those infected with lineage BPAN-GF/SR-I (82%) was higher than among those infected with lineages BCAR-SA-I (43%) and BCAR-GF/SR-II (25%).
Discussion
This study revealed that the HIV-1 subtype B epidemic in French Guiana and Suriname has been driven by multiple active BCAR and BPANDEMIC transmission chains that arose from several independent sources and operate in both countries simultaneously. Most BCAR (≥59%) and BPANDEMIC (≥40%) sequences from French Guiana and Suriname here analyzed branched within regional-specific monophyletic lineages that comprise sequences from both countries; while only a minor fraction of BCAR (≤10%) and BPANDEMIC (≤18%) sequences branched within country-specific clusters containing only sequences from a single country. This high degree of phylogenetic intermixing of HIV sequences is fully consistent with the intense cross-border population mobility between French Guiana and Suriname (Collomb and Fouck, 2016; Jaries et al., 2017) and supports the need to develop a coordinated bi-national healthcare response.
Our analyses identify four major BCAR (designated as BCAR-SA-I and BCAR-GF/SR-I to BCAR-GF/SR-III) and three major BPANDEMIC (designated as BPAN-GF/SR-I, BPAN-GF/SR-II, and BPAN-GF-I) transmission chains that together accounted for 54 and 73% of all HIV-1 subtype B sequences from French Guiana and Suriname here analyzed, respectively. This resembles the estimated proportion of HIV-1 subtype B infections that resulted from the expansion of major local (country- and regional-specific) BPANDEMIC and/or BCAR lineages in Argentina (31%), Brazil (31%), Mexico (37%), El Salvador (41%), Peru (51%), Jamaica (53%), Cuba (70%), Panama (77%), Honduras (91%), and Trinidad and Tobago (94%) (Delatorre and Bello, 2013; Cabello et al., 2014; Mendoza et al., 2014; Mir et al., 2015). This level of geographic compartmentalization is much higher than that observed for the subtype B epidemic in Europe where countries are highly interconnected and transcontinental migration is a significant driving force of virus dispersal (Paraskevis et al., 2009; Magiorkinis et al., 2016) and supports that subtype B transmissions in Latin America and the Caribbean are mainly occurring between individuals from the same country or neighboring countries (Junqueira et al., 2016).
The lineage BCAR-SA-I was the most widely disseminated BCAR lineage in French Guiana and the northern South America region. According to our Bayesian phylogeographic analysis, this regional lineage arose after dissemination of a BCAR-TT strain from Trinidad and Tobago into French Guiana at around the middle 1970s and later spread to Suriname, Guyana, and Northern Brazilian region. Nevertheless, a significant proportion (10%) of immigrants residing in Trinidad and Tobago is from Guyana (Borland et al., 2004). In addition, a very high proportion (45%) of subjects infected with the lineage BCAR-SA-I in French Guiana where from Guyana. At last, the intense human mobility across the Roraima border with Guyana (Pereira, 2006; Corbin, 2007) coincides with a high prevalence of the lineage BCAR-SA-I in that Northern Brazilian state (Divino et al., 2016). These epidemiological data therefore suggest that lineage BCAR-SA-I arose in Guyana. Nevertheless, the scarcity of HIV-1 sequences from that country seriously constrained our phylogeographic inference. The other major Guianese/Surinamese BCAR lineages most probably arose in the island of Hispaniola and were introduced into French Guiana (BCAR-GF/SR-II and BCAR-GF/SR-III) or Suriname (BCAR-GF/SR-I) between the late 1970s and the middle 1980s. It is interesting to note that a significant proportion (20%) of BCAR-infected subjects living in French Guiana were born in Haiti but individuals of Haitian origin only represent a minor fraction (5%) of lineages BCAR-GF/SR-I (5%), BCAR-GF/SR-II (0%), and BCAR-GF/SR-III (10%). Thus, Haitian migrants may provide an epidemiological link for sporadic BCAR transmissions from Haiti into French Guiana but only a minor fraction of those migrants seems to be actively participating in sustaining the local BCAR transmission networks.
Our phylogeographic analysis supports that major BPANDEMIC lineages most probably arose after dissemination of viral strains from North America into French Guiana (BPAN-GF/SR-I) and Suriname (BPAN-GF/SR-II) at around the middle 1980s and from South America into French Guiana (BPAN-GF-I) at around the early 1990s. Despite the historical ties and intense human mobility between French Guiana/mainland France and Suriname/Netherlands (Cordova Alcaraz, 2012), our analyses only support a few sporadic disseminations of BPANDEMIC strains between these regions. This is consistent with very restricted HIV transmissions between Surinamese and Dutch individuals living in the Netherlands (Kramer et al., 2011) and with the extremely low prevalence of BCAR strains in mainland France and the Netherlands (Cabello et al., 2016). Similarly, no intense BPANDEMIC fluxes were detected between French Guiana and Northern Brazil despite the intense human mobility and the favorable social conditions for the spread of HIV in the border region between Amapá (Northern Brazil) and French Guiana (Bourdier, 2005; Soares et al., 2011; Parriault et al., 2015; Collomb and Fouck, 2016).
Although the high population mobility promote a frequent HIV flux between French Guiana and Suriname, our results support some level of geographic subdivision for some major subtype B lineages. We found that lineages BCAR-SA-I and BPAN-GF-I comprise a much larger proportion of sequences from French Guiana than from Suriname, have their origin traced to French Guiana and mostly comprise Guianese subjects followed-up at the capital city, Cayenne. Lineages BCAR-GF/SR-I and BPAN-GF/SR-II, by contrast, were much more prevalent in Suriname than in French Guiana, had their origin traced to Suriname and mostly comprise subjects followed-up at Saint Laurent du Maroni, a French Guianese city located at the border with Suriname that attend a large number of patients from that country (Nacher et al., 2010; Jaries et al., 2017). We also detected a larger proportion of non-clustered BCAR and BPANDEMIC infections in French Guiana relative to Suriname, perhaps reflecting the greater influence of migrations and/or the larger number of sequences analyzed from French Guiana.
The study of the epidemiological characteristics of HIV-infected subjects from French Guiana revealed that BCAR and BPANDEMIC viruses have been disseminated between both MSM and heterosexual individuals. The MSM group has a much greater proportion of BPANDEMIC viruses (71%) than the heterosexual one (34%), but the role of MSM individuals in local spread of BPANDEMIC viruses greatly varied across transmission chains. While an important fraction of subjects infected with lineages BPAN-GF/SR-II (14%) and BPAN-GF-I (27%) were MSM; no MSM individuals were detected within the largest local BPAN-GF/SR-I transmission network. Overall, a large proportion of HIV-1 infections among heterosexuals (57%) and MSM (50%) in French Guiana can be ascribed to the seven major subtype B transmission chains here detected. More importantly, the proportion of individuals within these large cluster networks increased from 40% among those with HIV diagnosis between 1990 and 2005 to 62% among those diagnosed between 2006 and 2012. These data clearly support that large transmission clusters are a major driving force sustaining the recent dissemination of BCAR and BPANDEMIC epidemics in French Guiana. These results also emphasize that early detection and treatment as well as Pre Exposure Prophylaxis targeting people being part of large transmission chains should have a significant impact on reducing the spread of HIV-1 in French Guiana.
The analysis of clinical characteristics of individuals from French Guiana revealed a significant trend for higher RNA viral loads among BPANDEMIC-infected relative to BCAR-infected subjects, despite no significant differences in clinical condition or CD4+ T cell counts between groups. Previous studies suggested that viral genetic factors contribute to the heritability and variation of viral load set point (Bonhoeffer et al., 2015; Bertels et al., 2018) and may also modulate viral replication (Ronsard et al., 2017a,b). Whether differences in viral load here observed between subjects reflect selective advantages for BPANDEMIC strains over BCAR ones cannot be answered by the present study. Future in vitro studies comparing the replication dynamics of BCAR and BPANDEMIC strains and studies of carefully controlled retrospective cohorts could further explore this possibility. Characterization of full-length viral genomes should also be done in future studies to detect potential intra-subtype BCAR/BPANDEMIC recombinant lineages.
Although major lineages BPAN-GF/SR-I, BCAR-SA-I, and BCAR-GF/SR-I showed significant differences regarding city of residence, country of origin and RNA viral load, they exhibited roughly comparable mean epidemic growth rates (0.30–0.46 years-1). Transactional sex and concurrent sexual partnerships were pointed as important drivers of the HIV epidemic in French Guiana (Nacher et al., 2010) and most subjects (>90%) infected by major Guianese/Surinamese lineages were heterosexuals. Interestingly, the epidemic growth rates here obtained were similar to those estimated for BCAR and BPANDEMIC lineages spreading in countries from the Caribbean and Central America with generalized heterosexual epidemics (0.35–0.45 years-1) (Cabello et al., 2014; Mendoza et al., 2014); but lower than those estimated for BPANDEMIC lineages mainly transmitted among MSM networks (0.75–1.55 years-1) (Hue et al., 2005; Zehender et al., 2010; Chen et al., 2011; Delatorre and Bello, 2013; Worobey et al., 2016). This supports that the characteristics of the existing transmission network is a major driving force of the epidemic potential of different HIV-1 subtype B lineages.
In summary, this study highlights that HIV epidemics in French Guiana and Suriname are highly intermixed and that about 60% of HIV-1 subtype B infections in those countries resulted from the expansion of multiple BCAR and BPANDEMIC viral strains probably introduced between the middle 1970s and the early 1990s. Major BCAR and BPANDEMIC local lineages have independently spread among males and females of different age and risk groups from both countries and substantially contribute to the ongoing epidemic. Some associations between the infecting BCAR/BPANDEMIC lineage and epidemiological (geographic location and country of birth) and clinical (RNA viral load) variables were detected among individuals sampled in French Guiana, but no major differences in the epidemic potential of major BCAR and BPANDEMIC lineages were observed.
Author Contributions
GB and VL conceived and designed the study. MN, ED, and VL collected and analyzed the epidemiological data. MN performed the estimation of the HIV incidence temporal trend in French Guiana. FD performed the phylogenetic analyses. DM and GB performed the phylodynamics inferences and produced all figures. GB, MN, and VL wrote the manuscript. All authors discussed and reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding. This study has benefited from a European Commission “REGPOT-CT-2011-285837-STRonGer” grant within the FP7 and an “Investissement d’Avenir” grant managed by the Agence Nationale de la Recherche (CEBA, Ref. ANR-10-LABX-25-01). DM was funded by fellowships from “Agencia Nacional de Investigación e Innovación (ANII-Uruguay)” and “Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES-Brazil)”.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01738/full#supplementary-material
Lineage assignment of HIV-1 subtype B pol sequences from French Guiana and Suriname. (A,C) ML phylogenetic trees of HIV-1 subtype B pol PR/RT sequences (∼1,000 nt) from French Guiana (n = 271) and Suriname (n = 90) together with representative sequences of the BPANDEMIC (US = 165 and France = 135) and the BCAR (Caribbean = 200) lineages. Branches are colored according to the geographic origin of each sequence as indicated at the legend (bottom right). Asterisks point to highly supported (SH-aLRT > 0.90) key nodes of subtype B and the BPANDEMIC lineage. Trees were rooted using HIV-1 subtype D reference sequences. The branch lengths are drawn to scale with the bar at the bottom indicating nucleotide substitutions per site. (B,D) Estimated proportion of BCAR and BPANDEMIC lineages among HIV-1 subtype B infected individuals from French Guiana and Suriname according to the ML analyses. ND, not determined.
Analysis of HIV-1 subtype B pol sequences with no lineage assignment. Similarity plots generated using SimPlot are shown on the left. Plots displays the percent genetic similarity (y axis) of each subtype B query sequence from French Guiana/Suriname to the reference BCAR (blue line), BPANDEMIC (red line), and subtype D (black line) strains along the pol gene fragment (x axis; nucleotides 2,253–3,275 of reference strain HXB2). The analysis was performed with a window size of 250 nucleotides (nt) and a step size of 10 nt. Dashed vertical lines delimitate the pol gene fragments were most query sequences displayed highest similarity to BCAR references (fragment I), near equal similarity to both BCAR and BPANDEMIC references (fragment II), and highest similarity to BPANDEMIC references (fragment III). The GenBank accession number of each sequence is indicated in the upper left. NJ phylogenetic trees of subtype B query sequences from French Guiana/Suriname combined with BCAR (blue circles), BPANDEMIC (red circles), and subtype D (black circles) strains at pol fragments I and III are shown on the left. The subtype/lineage assignment of each query sequence at each fragment is indicated in the upper middle. Subtype D sequences were used as outgroups and the branch lengths are drawn to scale with the bar at the bottom indicating nucleotide substitutions per site. Low bootstrap support values were obtained for both subtype B (<70%) and BPANDEMIC (<30%) monophyletic groups in all phylogenetic trees.
Map summarizing viral migration events of major HIV-1 BCAR lineages circulating in French Guiana and Suriname. Arrows between locations represent branches in the Bayesian phylogenetic tree along, which location’s transitions occur. Map was created with Quantum GIS (QGIS) software from templates obtained from d-maps.com (Caribbean: http://d-maps.com/pays.php?num_pay=118&lang=es; South America: http://d-maps.com/carte.php?num_car=28522&lang=en).
HIV-1 subtype B pol (PR/RT) reference sequences of BPANDEMIC and BCAR clades used for subtype B clade assignment.
HIV-1 subtype B pol (PR/RT) reference sequences of BPANDEMIC and BCAR clades used for identification of Guianese/Surinamese subtype B clades.
HIV-1 BCAR pol (PR/RT) sequences used for Bayesian phylogeographic analysis.
HIV-1 BPANDEMIC pol (PR/RT) sequences used for Bayesian phylogeographic analysis.
Clade assignment of HIV-1 subtype B subtype pol sequences from French Guiana and Suriname.
Phylogenetic clustering of HIV-1 BCAR and BPANDEMIC pol sequences from French Guiana and Suriname.
Prevalence of major HIV-1 BCAR lineages circulating in French Guiana and Suriname.
Prevalence of major HIV-1 BPANDEMIC lineages circulating in French Guiana and Suriname.
Best fit demographic models for major HIV-1 subtype B lineages circulating in French Guiana and Suriname.
Epidemiological information of subjects from French Guiana infected by major HIV-1 BCAR and BPANDEMIC lineages.
References
- Abdoel Wahid F., Sno R., Darcissac E., Lavergne A., Adhin M. R., Lacoste V. (2016). HIV-1 genetic diversity and drug resistance mutations among treatment-naive adult patients in suriname. AIDS Res. Hum. Retroviruses 32 1223–1228. 10.1089/AID.2016.0161 [DOI] [PubMed] [Google Scholar]
- Anisimova M., Gascuel O. (2006). Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 55 539–552. 10.1080/10635150600755453 [DOI] [PubMed] [Google Scholar]
- Baele G., Lemey P., Bedford T., Rambaut A., Suchard M. A., Alekseyenko A. V. (2012). Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 29 2157–2167. 10.1093/molbev/mss084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertels F., Marzel A., Leventhal G., Mitov V., Fellay J., Gunthard H. F., et al. (2018). Dissecting HIV virulence: heritability of setpoint viral load, CD4+ T-cell decline, and per-parasite pathogenicity. Mol. Biol. Evol. 35 27–37. 10.1093/molbev/msx246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoeffer S., Fraser C., Leventhal G. E. (2015). High heritability is compatible with the broad distribution of set point viral load in HIV carriers. PLoS Pathog. 11:e1004634. 10.1371/journal.ppat.1004634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borland R., Faas L., Marshall D., Mclean R., Schroen M., Smit M., et al. (2004). HIV/AIDS and Mobile Populations in the Caribbean: A Baseline Assessment. International Organization for Migration (IOM). Available at: https://publications.iom.int/books/hivaids-and-mobile-populations-caribbean-baseline-assessment [Google Scholar]
- Bourdier F. (2005). L’avancée du sida dans les zones frontalières guyano-brésiliennes [in French]. Hommes Migr. 1256 116–129. 10.3406/homig.2005.4376 [DOI] [Google Scholar]
- Cabello M., Junqueira D. M., Bello G. (2015). Dissemination of nonpandemic Caribbean HIV-1 subtype B clades in Latin America. AIDS 29 483–492. 10.1097/QAD.0000000000000552 [DOI] [PubMed] [Google Scholar]
- Cabello M., Mendoza Y., Bello G. (2014). Spatiotemporal dynamics of dissemination of non-pandemic HIV-1 subtype B clades in the Caribbean region. PLoS One 9:e106045. 10.1371/journal.pone.0106045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabello M., Romero H., Bello G. (2016). Multiple introductions and onward transmission of non-pandemic HIV-1 subtype B strains in North America and Europe. Sci. Rep. 6:33971. 10.1038/srep33971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. H., Wong K. H., Chan K. C., To S. W., Chen Z., Yam W. C. (2011). Phylodynamics of HIV-1 subtype B among the men-having-sex-with-men (MSM) population in Hong Kong. PLoS One 6:e25286. 10.1371/journal.pone.0025286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collomb G., Fouck S. M. L. (2016). Mobilités, Ethnicités, Diversité Culturelle: La Guyane Entre Surinam et Brésil Eléments de Compréhension de la Situation Guyanaise [in French]. Matoury: Ibis Rouge Editions. [Google Scholar]
- Corbin H. P. (2007). Brazilian Migration to Guyana as a Livelihood Strategy: A Case Study Approach. Available at: http://www.repositorio.ufpa.br/jspui/bitstream/2011/1966/1/Dissertacao_BrazilianMigrationGuyana.pdf [Google Scholar]
- Cordova Alcaraz R. (2012). Migratory Routes and Dynamics Between Latin American and Caribbean (LAC) Countries and Between LAC and the European Union. International Organization for Migration (IOM). Available at: https://publications.iom.int/system/files/pdf/migration_routes_digital.pdf [Google Scholar]
- Darcissac E., Nacher M., Adriouch L., Berlioz-Arthaud A., Boukhari R., Couppie P., et al. (2016). HIV-1 Pol gene polymorphism and antiretroviral resistance mutations in treatment-naive adult patients in French Guiana between 2006 and 2012. AIDS Res. Hum. Retroviruses 32 801–811. 10.1089/AID.2016.0048 [DOI] [PubMed] [Google Scholar]
- de Oliveira T., Deforche K., Cassol S., Salminen M., Paraskevis D., Seebregts C., et al. (2005). An automated genotyping system for analysis of HIV-1 and other microbial sequences. Bioinformatics 21 3797–3800. 10.1093/bioinformatics/bti607 [DOI] [PubMed] [Google Scholar]
- Delatorre E., Bello G. (2013). Phylodynamics of the HIV-1 epidemic in Cuba. PLoS One 8:e72448. 10.1371/journal.pone.0072448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divino F., de Lima Guerra Corado A., Gomes Naveca F., Stefani M. M., Bello G. (2016). High prevalence and onward transmission of non-pandemic HIV-1 subtype B clades in Northern and Northeastern Brazilian regions. PLoS One 11:e0162112. 10.1371/journal.pone.0162112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J., Ho S. Y., Phillips M. J., Rambaut A. (2006). Relaxed phylogenetics and dating with confidence. PLoS Biol. 4:e88. 10.1371/journal.pbio.0040088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J., Nicholls G. K., Rodrigo A. G., Solomon W. (2002). Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161 1307–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J., Rambaut A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J., Rambaut A., Shapiro B., Pybus O. G. (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22 1185–1192. 10.1093/molbev/msi103 [DOI] [PubMed] [Google Scholar]
- Ferreira M. A. R., Suchard M. A. (2008). Bayesian analysis of elapsed times in continuous-time Markov chains. Can. J. Stat. 36 355–368. 10.1002/cjs.5550360302 [DOI] [Google Scholar]
- Guindon S., Dufayard J. F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59 307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- Guindon S., Lethiec F., Duroux P., Gascuel O. (2005). PHYML Online–a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33 W557–W559. 10.1093/nar/gki352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hue S., Pillay D., Clewley J. P., Pybus O. G. (2005). Genetic analysis reveals the complex structure of HIV-1 transmission within defined risk groups. Proc. Natl. Acad. Sci. U.S.A. 102 4425–4429. 10.1073/pnas.0407534102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyles J. R. (2014). Guiana and the Shadows of Empire: Colonial and Cultural Negotiations at the Edge of the World. Lanham, MD: Lexington Books. [Google Scholar]
- Jaries R., Vantilcke V., Clevenbergh P., Adoissi J., Boukhari R., Misslin C., et al. (2017). Population movements and the HIV cascade in recently diagnosed patients at the French Guiana -Suriname border. AIDS Care 29 1448–1452. 10.1080/09540121.2017.1291899 [DOI] [PubMed] [Google Scholar]
- Junqueira D. M., De Medeiros R. M., Graf T., Almeida S. E. (2016). Short-term dynamic and local epidemiological trends in the South American HIV-1B epidemic. PLoS One 11:e0156712. 10.1371/journal.pone.0156712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanji M., Lavergne A., Pouliquen J. F., Magnien C., Bissuel F., Marty C., et al. (2001). Genetic diversity and phylogenetic analysis of human immunodeficiency virus type 1 subtypes circulating in French Guiana. AIDS Res. Hum. Retroviruses 17 857–861. 10.1089/088922201750252052 [DOI] [PubMed] [Google Scholar]
- Kramer M. A., Cornelissen M., Paraskevis D., Prins M., Coutinho R. A., Van Sighem A. I., et al. (2011). HIV transmission patterns among The Netherlands, Suriname, and The Netherlands Antilles: a molecular epidemiological study. AIDS Res. Hum. Retroviruses 27 123–130. 10.1089/aid.2010.0115 [DOI] [PubMed] [Google Scholar]
- Lemey P., Rambaut A., Drummond A. J., Suchard M. A. (2009). Bayesian phylogeography finds its roots. PLoS Comput. Biol. 5:e1000520. 10.1371/journal.pcbi.1000520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lole K. S., Bollinger R. C., Paranjape R. S., Gadkari D., Kulkarni S. S., Novak N. G., et al. (1999). Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiorkinis G., Angelis K., Mamais I., Katzourakis A., Hatzakis A., Albert J., et al. (2016). The global spread of HIV-1 subtype B epidemic. Infect. Genet. Evol. 46 169–179. 10.1016/j.meegid.2016.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza Y., Martinez A. A., Castillo Mewa J., Gonzalez C., Garcia-Morales C., Avila-Rios S., et al. (2014). Human immunodeficiency virus type 1 (HIV-1) subtype B epidemic in panama is mainly driven by dissemination of country-specific clades. PLoS One 9:e95360. 10.1371/journal.pone.0095360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir D., Cabello M., Romero H., Bello G. (2015). Phylodynamics of major HIV-1 subtype B pandemic clades circulating in Latin America. AIDS 29 1863–1869. 10.1097/QAD.0000000000000770 [DOI] [PubMed] [Google Scholar]
- Nacher M., Vantilcke V., Parriault M. C., Van Melle A., Hanf M., Labadie G., et al. (2010). What is driving the HIV epidemic in French Guiana? Int. J. STD AIDS 21 359–361. 10.1258/ijsa.2010.009570 [DOI] [PubMed] [Google Scholar]
- Paraskevis D., Pybus O., Magiorkinis G., Hatzakis A., Wensing A. M., Van De Vijver D. A., et al. (2009). Tracing the HIV-1 subtype B mobility in Europe: a phylogeographic approach. Retrovirology 6:49. 10.1186/1742-4690-6-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parriault M. C., Van Melle A., Basurko C., Gaubert-Marechal E., Macena R. H., Rogier S., et al. (2015). HIV-testing among female sex workers on the border between Brazil and French Guiana: the need for targeted interventions. Cad. Saude Publica 31 1615–1622. 10.1590/0102-311X00138514 [DOI] [PubMed] [Google Scholar]
- Pereira M. C. (2006). Processos migratoìrios na fronteira Brasil-Guiana. Estud. Avançados 20 209–219. 10.1590/S0103-40142006000200016 [DOI] [Google Scholar]
- Posada D. (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25 1253–1256. 10.1093/molbev/msn083 [DOI] [PubMed] [Google Scholar]
- Rambaut A. (2009). FigTree v1.4: Tree Figure Drawing Tool. Available at: http://tree.bio.ed.ac.uk/software/figtree/ [Google Scholar]
- Rambaut A., Drummond A. (2007). Tracer v1.6. Available at: http://tree.bio.ed.ac.uk/software/tracer/ [Google Scholar]
- Rambaut A., Lam T. T., Carvalho L. M., Pybus O. G. (2016). Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2:vew007. 10.1093/ve/vew007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronsard L., Ganguli N., Singh V. K., Mohankumar K., Rai T., Sridharan S., et al. (2017a). Impact of genetic variations in HIV-1 Tat on LTR-mediated transcription via TAR RNA interaction. Front. Microbiol. 8:706. 10.3389/fmicb.2017.00706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronsard L., Rai T., Rai D., Ramachandran V. G., Banerjea A. C. (2017b). In silico analyses of subtype specific HIV-1 Tat-TAR RNA interaction reveals the structural determinants for viral activity. Front. Microbiol. 8:1467. 10.3389/fmicb.2017.01467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares C. L., De Souza Oliveira B., De Souza Pinto M. J. (2011). Trabalhadores brasileiros na Guiana Francesa: entre a invisibilidade e o desemprego. PRACS Rev. Hum. Curso Ciênc. Soc. 4 129–142. [Google Scholar]
- Suchard M. A., Rambaut A. (2009). Many-core algorithms for statistical phylogenetics. Bioinformatics 25 1370–1376. 10.1093/bioinformatics/btp244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25 4876–4882. 10.1093/nar/25.24.4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UNAIDS (2013). Report on the Global AIDS Epidemic. Available at: http://www.unaids.org/en/media/unaids/contentassets/documents/epidemiology/2013/gr2013/UNAIDS_Global_Report_2013_en.pdf [Google Scholar]
- Worobey M., Watts T. D., Mckay R. A., Suchard M. A., Granade T., Teuwen D. E., et al. (2016). 1970s and ’Patient 0’ HIV-1 genomes illuminate early HIV/AIDS history in North America. Nature 539 98–101. 10.1038/nature19827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehender G., Ebranati E., Lai A., Santoro M. M., Alteri C., Giuliani M., et al. (2010). Population dynamics of HIV-1 subtype B in a cohort of men-having-sex-with-men in Rome, Italy. J. Acquir. Immune Defic. Syndr. 55 156–160. 10.1097/QAI.0b013e3181eb3002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Lineage assignment of HIV-1 subtype B pol sequences from French Guiana and Suriname. (A,C) ML phylogenetic trees of HIV-1 subtype B pol PR/RT sequences (∼1,000 nt) from French Guiana (n = 271) and Suriname (n = 90) together with representative sequences of the BPANDEMIC (US = 165 and France = 135) and the BCAR (Caribbean = 200) lineages. Branches are colored according to the geographic origin of each sequence as indicated at the legend (bottom right). Asterisks point to highly supported (SH-aLRT > 0.90) key nodes of subtype B and the BPANDEMIC lineage. Trees were rooted using HIV-1 subtype D reference sequences. The branch lengths are drawn to scale with the bar at the bottom indicating nucleotide substitutions per site. (B,D) Estimated proportion of BCAR and BPANDEMIC lineages among HIV-1 subtype B infected individuals from French Guiana and Suriname according to the ML analyses. ND, not determined.
Analysis of HIV-1 subtype B pol sequences with no lineage assignment. Similarity plots generated using SimPlot are shown on the left. Plots displays the percent genetic similarity (y axis) of each subtype B query sequence from French Guiana/Suriname to the reference BCAR (blue line), BPANDEMIC (red line), and subtype D (black line) strains along the pol gene fragment (x axis; nucleotides 2,253–3,275 of reference strain HXB2). The analysis was performed with a window size of 250 nucleotides (nt) and a step size of 10 nt. Dashed vertical lines delimitate the pol gene fragments were most query sequences displayed highest similarity to BCAR references (fragment I), near equal similarity to both BCAR and BPANDEMIC references (fragment II), and highest similarity to BPANDEMIC references (fragment III). The GenBank accession number of each sequence is indicated in the upper left. NJ phylogenetic trees of subtype B query sequences from French Guiana/Suriname combined with BCAR (blue circles), BPANDEMIC (red circles), and subtype D (black circles) strains at pol fragments I and III are shown on the left. The subtype/lineage assignment of each query sequence at each fragment is indicated in the upper middle. Subtype D sequences were used as outgroups and the branch lengths are drawn to scale with the bar at the bottom indicating nucleotide substitutions per site. Low bootstrap support values were obtained for both subtype B (<70%) and BPANDEMIC (<30%) monophyletic groups in all phylogenetic trees.
Map summarizing viral migration events of major HIV-1 BCAR lineages circulating in French Guiana and Suriname. Arrows between locations represent branches in the Bayesian phylogenetic tree along, which location’s transitions occur. Map was created with Quantum GIS (QGIS) software from templates obtained from d-maps.com (Caribbean: http://d-maps.com/pays.php?num_pay=118&lang=es; South America: http://d-maps.com/carte.php?num_car=28522&lang=en).
HIV-1 subtype B pol (PR/RT) reference sequences of BPANDEMIC and BCAR clades used for subtype B clade assignment.
HIV-1 subtype B pol (PR/RT) reference sequences of BPANDEMIC and BCAR clades used for identification of Guianese/Surinamese subtype B clades.
HIV-1 BCAR pol (PR/RT) sequences used for Bayesian phylogeographic analysis.
HIV-1 BPANDEMIC pol (PR/RT) sequences used for Bayesian phylogeographic analysis.
Clade assignment of HIV-1 subtype B subtype pol sequences from French Guiana and Suriname.
Phylogenetic clustering of HIV-1 BCAR and BPANDEMIC pol sequences from French Guiana and Suriname.
Prevalence of major HIV-1 BCAR lineages circulating in French Guiana and Suriname.
Prevalence of major HIV-1 BPANDEMIC lineages circulating in French Guiana and Suriname.
Best fit demographic models for major HIV-1 subtype B lineages circulating in French Guiana and Suriname.
Epidemiological information of subjects from French Guiana infected by major HIV-1 BCAR and BPANDEMIC lineages.