

Abstract
Sepsis is a highly lethal and urgent unmet medical need. It is the result of a complex interplay of several pathways, including inflammation, immune activation, hypoxia, and metabolic reprogramming. Specifically, the regulation and the impact of the latter have become better understood in which the highly catabolic status during sepsis and its similarity with starvation responses appear to be essential in the poor prognosis in sepsis. It seems logical that new interventions based on the recognition of new therapeutic targets in the key metabolic pathways should be developed and may have a good chance to penetrate to the bedside. In this review, we concentrate on the pathological changes in metabolism, observed during sepsis, and the presumed underlying mechanisms, with a focus on the level of the organism and the interplay between different organ systems.
Keywords: hypoxia, inflammation, interventions, metabolic reprogramming, sepsis
Subject Categories: Immunology; Metabolism; Microbiology, Virology & Host Pathogen Interaction
Glossary
- Aerobic glycolysis
A.k.a. Warburg effect: first described by Otto Warburg as a characteristic of cancer cells. Warburg observed that cancer cells generate energy (ATP) through high rates of glycolysis, rather than through oxidative phosphorylation.
- Anorexia
Reduced food intake.
- Beta‐oxidation
Metabolic pathway in which fatty acids are metabolized to generate energy.
- Catabolism
Process that breaks down molecules into smaller units.
- Cecal ligation and puncture (CLP) model
One of the most stringent models of sepsis. It involves a combination of three insults: tissue trauma due to laparotomy, necrosis caused by ligation of the cecum, and infection due to the leakage of peritoneal microbial flora into the peritoneum.
- Cori cycle
A.k.a. glycose‐lactate cycle: metabolic pathway in which lactate produced by anaerobic glycolysis in the muscles moves to the liver and is converted to glucose, which in turn is metabolized back to lactate in the muscles.
- Disease tolerance
Defense strategy limiting the pathologic outcome of infections by preventing tissue damage or ameliorating tissue function without interfering with host's pathogen load.
- Electron transport chain
A system of molecules that pass electrons from one to another via redox reactions coupled with the transfer of protons across the inner membrane of mitochondria, creating a proton gradient that drives ATP synthesis.
- Epigenome
Heritable chemical changes of the genome, such as DNA methylation or histone modification, that modifies gene expression but does not change the DNA nucleotide sequence itself.
- Free fatty acid
A non‐esterified fatty acid, released by the hydrolysis of triglycerides within adipose tissue. Free fatty acids can be used as an immediate source of energy by many organs and can be converted by the liver into ketone bodies.
- Glasgow coma scale score
Provides a practical method for assessment of impairment of conscious level based on eye, verbal, and motor criteria.
- Gluconeogenesis
Metabolic process in which glucose is formed from smaller precursors, such as amino acids and glycerol.
- Glycolysis
Generation of ATP through degradation of glucose, usually associated with anaerobic conditions.
- Immunoparalysis
Persistence of a compensatory anti‐inflammatory innate immune response following an insult such as sepsis or trauma.
- Ketogenesis
Production of ketone bodies by breaking down fatty acids and ketogenic amino acids. This process supplies the needed energy of certain organs, especially the brain.
- Lipolysis
The process of breaking down of lipids into fatty acids and glycerol.
- Oxidative phosphorylation
A.k.a. mitochondrial respiration: generation of ATP through the mitochondrial inner membrane via the tricarboxylic acid cycle, whereby oxygen functions as terminal proton acceptor.
- Reactive oxygen species (ROS)
Derivatives of oxygen generated during mitochondrial oxidative metabolism. A buildup of ROS in cells may cause damage to DNA, RNA and proteins.
- Sepsis
A life‐threatening condition that arises when the body's response to an infection leads to tissue and organ damage.
- Sequential organ failure assessment (SOFA)
Assessment of a patients status, during the stay in an intensive care unit, used to determine the extent of organ dysfunction. The score is based on six different scores (respiratory, cardiovascular, hepatic, coagulation, renal and neurological systems). A higher SOFA score is associated with an increased risk of mortality.
- Starvation
Malnutrition following, for example, anorexia, gastrointestinal disease, cancer, and coma. The metabolic response to starvation is mobilization of energy via catabolism of body tissues (muscle, adipose tissue).
- Tricyclic acid cycle (TCA)
A.k.a. Krebs cycle, is the major energy‐producing pathway and occurs in mitochondria. Short carbon chains derived from sugars, fatty acids, or amino acids are metabolized to yield carbon dioxide, water, and high‐energy phosphate bonds (ATP).
Introduction: sepsis pathophysiology
Sepsis is a life‐threatening condition resulting from a dysregulated response to infection, with a global burden of at least 19 million cases annually (Singer et al, 2016). Classically, the acute phase of sepsis is characterized by an initial strong pro‐inflammatory and innate immune status aimed at eliminating the pathogen. During the later phase of sepsis, the immune system shifts toward an anti‐inflammatory, immune‐suppressive status, resulting in diminished inflammation, and initiation of tissue repair. Based on these insights, many immunomodulatory therapies have been tested in clinical trials in sepsis over the last decades, but no actual novel therapy based on inflammatory or immune pathways has demonstrated survival benefit. The only agent that has gained regulatory approval was activated protein C, but this was later withdrawn from the market due to safety and efficacy concerns (Ranieri et al, 2012). Current management of sepsis is supportive rather than curative and is aimed at eradication of the infection, fluid resuscitation to maintain organ perfusion, vasopressor administration to maintain an adequate blood pressure and mechanical support of failing organs. Even in the face of improved technical advances, mortality rates of 20–25% are commonly reported (Marshall, 2014). Increasing rates of antimicrobial resistance and aging of the human population add to the problem and underscore the need for innovative therapeutic strategies.
One of the explanations why so many clinical trials have been disappointing is that sepsis has been considered too much as an inflammatory disease, while recent research suggests an important contribution of coagulation, complement activation, microbiome composition, thermoregulation, circadian rhythm, and metabolism (Cohen et al, 2015). Indeed, the pathogenesis of sepsis is clearly influenced by profound changes in metabolic homeostasis. Typical features of sepsis, such as high fever, tachycardia, tachypnea, inflammation, immune activation, phagocytosis, and acute phase reactant production, all require supra‐physiological energy supplies. However, despite their increased nutritional requirements, patients are often unwilling or unable to eat, which creates a problem of energy deficit. Also, one recurrent feature in sepsis is a clear problem of mitochondrial respiration (Protti et al, 2007). Biopsies taken from skeletal muscle from deceased sepsis patients found a strong decrease in ATP/ADP ratio (Lee & Hüttemann, 2014).
Similarly, in mice and rats, sepsis led to decreased ATP levels in skeletal muscle and liver (Lee & Hüttemann, 2014). The immediate result is a high catabolic state leading to the breakdown of carbohydrates, lipid, and protein reserves (Englert & Rogers, 2016). Interestingly, a large proteomic and metabolic screen on plasma of sepsis patients identified glucose metabolism and fatty acid beta‐oxidation pathways as being significantly different between sepsis survivors and non‐survivors (Langley et al, 2013). In human patients, hyperglycemia is very often observed and occasionally hypoglycemia occurs. A clear increase in L‐lactate is usually seen directly related to poor outcome, besides signs of poor fatty acid oxidation and amino acid catabolism. It is believed that short‐term and mild changes in metabolism can positively modulate immune responses to eliminate pathogens and to protect the host via disease tolerance, that is, a defense strategy that limits the pathologic outcome of infections without interfering directly with the hosts pathogen load (Soares et al, 2014). Uncontrolled, severe disturbances of metabolic homeostasis are however detrimental (Balmer & Hess, 2017). Thus, changes in metabolism during sepsis can well be the master regulator pathways that aim to provide energy to overcome the critical situation, but at the same time save energy because of poor nutritional input. How the septic organism copes with this schism, and whether these insights can lead to new therapeutic options, is reviewed in this paper.
Sepsis and mitochondrial metabolism
A recurrent finding in sepsis is the obvious problem with mitochondrial metabolism (Park & Zmijewski, 2017). Even in septic conditions, glycolysis appears to be perfectly ongoing and pyruvate is abundantly generated in the cytoplasm of cells. Under well‐oxygenated conditions, pyruvate enters the mitochondria via a heterodimeric mitochondrial pyruvate carrier protein (MPC) complex. We are not aware of studies reporting a reduced expression or function of MPC in sepsis. In contrast, the enzyme complex responsible for the next necessary step of pyruvate breakdown is heavily compromised in sepsis: the transformation of pyruvate into acetyl‐CoA and CO2. The multiprotein enzyme pyruvate dehydrogenase complex (PDC) is a big, strongly conserved complex that is tightly regulated in activity and is an important sensor of oxygen and a regulator of TCA activity. In sepsis, PDC activity is clearly reduced (Vary, 1996; Nuzzo et al, 2015) and several explanations have been provided. PDC needs a co‐factor, thiamin (a.k.a. vitamin B1), and this molecule has been found to be reduced in sepsis samples, but clinical trials using thiamin were rather disappointing (Leite & de Lima, 2016). More likely, PDC loses activity due to phosphorylation by a set of kinases known as pyruvate dehydrogenase kinase, PDKs. There are four such kinases, PDK1 to PDK4. These kinases are regulated by transcription [e.g., by transcription factors such as hypoxia‐inducible factor 1α (HIF‐1α), glucocorticoid receptor (GR), peroxisome proliferator‐activated receptor (PPAR‐α), and others] as well as by other mechanisms (Jeoung, 2015). Researchers have also found other kinases that inhibit PDC activity, notably the mitogen‐activated protein kinase (MAPK) kinases (Park & Jeoung, 2016) and the kinase involved in necroptosis, receptor interacting protein kinase 1 (RIPK3; Yang et al, 2018). Obviously, there is a clear point of intersection of different potentially key pathways in sepsis, namely inflammation, hypoxia, and stress. Once inactivated, the PDC complex can return to an active status by means of de‐phosphorylating phosphatases. From an evolutionary point of view, PDC is closely related to α‐ketoglutarate dehydrogenase complex, which is similar in structure, also needs thiamin, and performs de‐carboxylation and addition of CoA to a substrate (forming succinyl CoA from α‐ketoglutarate in the TCA cycle). There are however no reports of changes of this enzyme in sepsis.
Other problems with mitochondria have been reported in relation to sepsis. Several groups have demonstrated that mitochondria display physical damage (Zang et al, 2014). Dysfunction of mitochondria in sepsis has been described as being a direct result of small, reactive molecules, produced in the first wave of inflammation, for example NO, CO, and reactive oxygen and nitrogen species, whereby mitochondrial DNA is damaged as well as proteins of the electron transport chain (ETC.; Svistunenko et al, 2006). The ETC. multiprotein complexes have also been described as being less abundant in cells from septic patients due to reduced expression (Lee & Hüttemann, 2014). The ETC. serves to transport the electrons generated in the TCA cycle and finally to reduce the terminal electron acceptor O2 to water, in coordination with flux of protons from the matrix into the intermembrane space and back to the mitochondrial matrix. In sepsis, complex I and complex II of the ETC. have been found to be less active, but also complex IV has been claimed as a major mitochondrial target (Hüttemann et al, 2012b). Phosphorylation of this complex [by inflammatory kinases such as c‐Jun‐N‐terminal kinase (JNK) that enter mitochondria] is thought to lead to physical problems with mitochondria, including leakage of matrix components into the cell, exposing cytochrome C which initiates apoptosis of the cells (Hüttemann et al, 2012a). This view is compatible with the observations that in sepsis, mitochondria are less involved in oxidative phosphorylation and more in the induction of apoptosis (Lee & Hüttemann, 2014).
Despite the strong evidence pointing toward mitochondrial dysfunction during sepsis, other studies have shown that mitochondrial function in sepsis is highly variable between organs and over the course of the disease. These statements open the discussion to whether mitochondrial impairment is the major driver of organ failure in septic shock and urges the field to investigate this issue in more detail.
The increased production of lactate in sepsis is thought to be the result of increased glycolysis activity (aerobic glycolysis a.k.a. Warburg effect) and increased reduction of pyruvate, and this is based on several changes, which may be problematic in sepsis. First, there is the reduction in PDC activity, and other issues with mitochondria, as just explained. Second, in sepsis there is cytopathic hypoxia. Several authors have shown that the oxygen levels in tissues during sepsis are rather normal (Fink, 2015) but that there is a reduced capacity of the tissues to utilize O2 from the blood (Suetrong & Walley, 2016). Other authors have demonstrated that during a decrease in oxygen delivery to cells (oxygen delivery = cardiac output × O2 carrying capacity of blood), normal cells will respond with a production of lactate when oxygen delivery reaches a lower limit of about 400 ml/min. Septic cells will however respond much more sensitive, that is, from a lower limit of about 600 ml/min (Suetrong & Walley, 2016). Third: despite there being no hypoxia senso stricto, septic cells will perform a hypoxic response and there is transcriptional increase and activation of HIF1‐α (further described below). This transcription factor will induce several genes, coding for proteins that are involved in glycolysis, namely hexokinase, phosphofructokinase‐1, and most importantly lactate dehydrogenase A (LDHA; Marín‐Hernández et al, 2009), which converts pyruvate into lactate. Lactate is then pumped into the extracellular space by increased expression of MCT4 (coded by the SLC16A3 gene). Fourth, lactate is transported in the blood and, as a reduced metabolite, can be used as an energy source by different tissues, such as cardiac muscle. Already four decades ago, it was shown that in sepsis, the oxidation of lactate to pyruvate is defective, most likely because due to aberrations of two mitochondrial shuttle systems which transport the protons (derived from the oxidation of lactate) into the mitochondria (Jones, 1974). Fifth, under physiological conditions and during physical activities, lactate released into the blood will be taken up by the liver and used as a substrate to form glucose by a process called gluconeogenesis. This cycle of glucose to lactate catabolism followed by new glucose production from lactate is called the Cori cycle, after Gerty and Carl Cori, rewarded by the Noble Award in 1947. As shown by Clemens et al, in a rat sepsis model (the cecal ligation and puncture (CLP) model; Dejager et al, 2011), this lactate‐based gluconeogenesis in liver is severely reduced (Clemens et al, 1983). The mechanism behind this defect in gluconeogenesis is unknown, but could in part be explained by the fact that, the first step that oxidizes lactate to pyruvate, again requires one of these defective mitochondrial shuttles. Moreover, even if pyruvate is formed, it will have to be transformed to phosphoenolpyruvate (PEP) via a step performed in mitochondria and again involving an oxaloacetate‐malate‐oxaloacetate shuttle, leading to oxaloacetate in the cytoplasm and its de‐carboxylation to PEP via the critical enzyme PEPCK (coded by PCK1). Several studies have shown that PCK1 expression is significantly decreased in the liver in sepsis (Deutschman et al, 1993).
In conclusion, based on these mechanisms, in sepsis, there is a clear increase in production of lactate, and the major pathways to remove lactate are blocked. Lactate is a typical biomarker of bad prognosis in sepsis, since lactate levels strongly correlate with disease severity, morbidity, and mortality in sepsis (Nichol et al, 2010; Rishu et al, 2013). The degree of lactate clearance in a septic patient, during the first 6 h of sepsis treatment, is highly associated with survival in sepsis (Nguyen et al, 2004). Lactate may lead to lactic acidosis (serum pH < 7.35), which results in reduced glucose uptake by the brain, leading to coma, and induction of heart arrhythmias and heart failure (Suetrong & Walley, 2016). A low Glasgow Coma Scale score and cardiovascular failure are both factors influencing the Sequential [Sepsis‐related] Organ Failure Assessment (SOFA) score, which is a score reflecting how severely compromised different organ systems are in sepsis (Singer et al, 2016). Severe lactic acidosis (pH < 7.2) is associated with a mortality rate of about 50%, and even no surviving patients has been reported with severe lactic acidosis with a pH under 7.0 (Kimmoun et al, 2015). Lactate has been reported not only as an important intermediate in metabolic processes and as a cause of pH decline, but also as a molecule actively influencing inflammation. For example, lactate boosts Toll‐like receptor‐4 (TLR4), that is, the major receptor of LPS, signaling, and NF‐κB‐mediated gene transcription (Samuvel et al, 2009) and promotes HMGB1 release (Yang et al, 2014) in macrophages, suggesting that lactate promotes inflammation.
The problems with mitochondrial function also impact fatty acid and protein catabolism, as described further.
Sepsis and carbohydrate metabolism
Hyperglycemia
The pathogenesis of sepsis is associated with a deregulation in glucose metabolism resulting in a metabolic and energetic failure (Fig 1). Hyperglycemia is one of the most prevalent metabolic derangements in sepsis patients, presumably resulting from altered glycogen metabolism and profound insulin resistance (Englert & Rogers, 2016). From an evolutionary perspective, hyperglycemia is thought to be desirable as it provides glucose to cells that do not rely on insulin for glucose uptake, such as neurons and leukocytes (Marik & Bellomo, 2013). Redirection of glucose to immune cells allows meeting their rapid division as well as their bio‐energetic demands during inflammation. Indeed, naïve cells rely mainly on oxidative phosphorylation and beta‐oxidation of free fatty acids as energy sources, whereas activated immune cells rely mainly on aerobic glycolysis, also referred to as the Warburg effect (Cheng et al, 2016; Fig 2). Although energetically less favorable in terms of ATP production, aerobic glycolysis is adopted by activated immune cells because it provides biosynthetic intermediates that are essential for the synthesis of nucleotides, lipids, and amino acids. These are important to support proliferation and the synthesis of inflammatory molecules. For instance, glucose 6‐phosphate (G6P), generated by the first step in glycolysis, can feed into the pentose phosphate pathway to support nucleotide synthesis. This pathway also generates NADPH which is essential for lipid synthesis. Furthermore, an adequate immune response requires rapid energy production and aerobic glycolysis provides ATP swiftly. Oxidative phosphorylation requires mitochondrial biogenesis, which is a complex and slow process, whereas glycolysis could be induced rapidly by inducing glycolytic enzymes (Loftus & Finlay, 2016; O'Neill et al, 2016). Conversion of glucose into lactate allows high rates of glycolysis providing rapid ATP production and lactate can also be used as a highly reduced (H‐rich) tri‐carbon fuel. Finally, because of suboptimal ETC. function, mitochondrial TCA‐mediated respiration in sepsis leads to the production of dangerous reactive oxygen species (ROS; Arulkumaran et al, 2016), a process which can also be prevented by switching to glycolysis, which generates ATP without ROS production and this might be a form of damage control.
Figure 1. Impact of sepsis on carbohydrate metabolism.
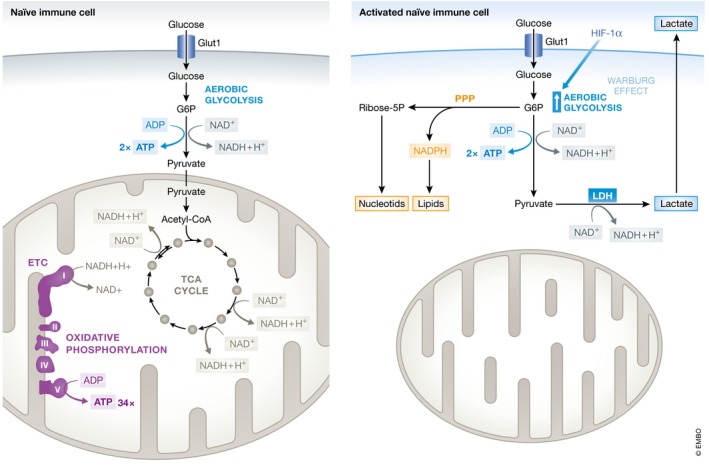
Naïve immune cells rely on glycolysis and oxidative phosphorylation as the main metabolic pathways to generate ATP. Glycolysis is the catabolic process in which glucose is converted into pyruvate. Extracellular glucose is imported into cells by GLUT1 (SLC2A1 gene). Subsequent intracellular processing of glucose yields two pyruvate molecules and two ATP molecules by a series of enzymatic reactions. At sufficient O2 tension, pyruvate is imported in mitochondria and converted into acetyl‐coA and enters the tricarboxylic acid cycle (TCA cycle, a.k.a. Kreb's cycle), after which each pyruvate yields 17 ATP molecules by the electron transport chain (ETC) and oxidative phosphorylation. Upon activation of immune cells, the glycolysis pathway is upregulated under the control of HIF‐1α. The metabolic pathway shifts from oxidative phosphorylation to aerobic glycolysis, also referred to as the Warburg effect, to meet the increased energy demand in activated immune cells. Despite the presence of abundant O2 in the environment, glucose is then directly metabolized into lactate. Although energetically less favorable (glycolysis generates 2 molecules of ATP out of 1 molecule glucose, whereas oxidative phosphorylation provides 36 ATP molecules), aerobic glycolysis allows higher velocity of glycolysis and thus faster ATP production, as well as provides important precursors for the synthesis of lipids, amino acids, and nucleotides required for proliferation. Interfering with aerobic glycolysis in immune cells has been shown to undermine their anti‐infectious activities, highlighting the importance of this altered metabolism in activated immune cells.
Figure 2. The pathogenesis of sepsis is associated with a deregulation in glucose metabolism.
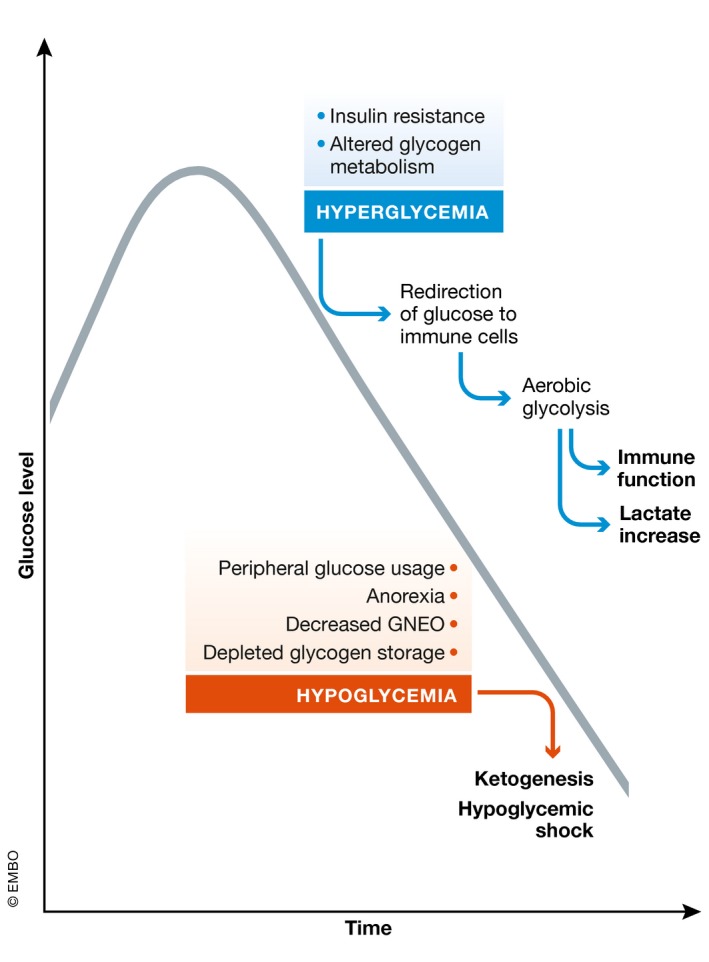
Both high and low glucose levels correlate with sepsis severity. Initially, a hyperglycemic response is observed in both animal models and in human patients, presumably resulting from insulin resistance and altered glycogen metabolism. This allows redirection of glucose to immune cells supporting aerobic glycolysis and thus immune function, and also leads to lactate production. In later stages, hypoglycemia could be observed as a consequence of several factors. In contrast to animal models, hypoglycemia is less frequently observed in human patients.
Glycolysis has been shown to be crucial for a number of immune cell functions in several activated immune cells in order to meet their increased energy needs. For example, glycolysis is specifically required for phagocytosis and cytokine production in pro‐inflammatory (M1) macrophages (Yang et al, 2014; Jha et al, 2015), for cytokine production in T cells (Chang et al, 2013), for antibody production in B cells (Caro‐Maldonado et al, 2014) and for antigen presentation in dendritic cells (Everts et al, 2012, 2014). The increased serum lactate levels during sepsis hence could also originate from activated immune cells undergoing aerobic glycolysis (Cheng et al, 2014b).
From the above‐mentioned studies, it is clear that there is a close link between metabolism and inflammatory responses. If it is true that high lactate levels would be derived from white blood cells, therapeutic modulation of leukocyte metabolism could potentially represent a novel therapeutic approach in sepsis. Appropriate inflammatory responses are essential for pathogen elimination, but excessive inflammation may result in overstimulation, organ damage, and even death.
One key mediator of aerobic glycolysis is the transcription factor HIF‐1α (Corcoran & O'Neill, 2016). HIF‐1α regulates the expression of aerobic glycolysis‐related genes (such as glucose transporter 1 (GLUT1, encoded by SLC2A1), LDHA, and PDK4, as well as inflammatory genes, notably interleukin‐1β (IL1B gene; Corcoran & O'Neill, 2016) and inducible NO synthase (NOS2 gene). Under hypoxic conditions, but also by stimulation by bacterial products [e.g., lipopolysaccharide, LPS, of Gram‐negative bacteria (Nishi et al, 2008)], and pro‐inflammatory cytokines [e.g., TNF (Regueira et al, 2009)] under normoxic conditions, HIF‐1α protein is stabilized and accumulates in cells. Conditional knock‐out of HIF‐1α in myeloid cells was found to decrease inflammatory cytokine production and to protect animals against endotoxemia, an experimental model of sepsis (Peyssonnaux et al, 2007). Functional work with HIF‐1α KO mice in real bacterial sepsis models has not been performed so far.
Another important mediator in aerobic glycolysis is pyruvate kinase isoenzyme M2 (PKM2), which acts as a co‐activator for HIF‐1α and is also involved in a classical metabolic step, namely the last step of glycolysis, that is, the de‐phosphorylation of phosphoenolpyruvate to pyruvate. It is activated in hypoxia but also induced after LPS stimulation. Knockdown of PKM2 decreases lactate production and cytokine release both in vitro and in vivo leading to protection of mice from lethal endotoxemia and sepsis (Yang et al, 2014). Impairing the transcriptional activity of the PKM2/HIF‐1α complex by use of small molecules polarizes M1 macrophages into an M2 phenotype, leading to reduced IL‐1β and induced IL‐10 production. However, counteracting the inflammatory response with these small molecules in an Salmonella typhimurium model interferes with the bacterial killing capacity leading to increased bacterial dissemination (Palsson‐Mcdermott et al, 2015).
Also mammalian target of rapamycin (mTOR) has been described as an important regulator of HIF‐1α activity. mTOR activity is induced by hypoxia, growth factors, and a number of ligand‐receptor systems, and leads to increased translation of a number of key mRNAs, including HIF‐1α mRNA (Cheng et al, 2014a). mTOR is negatively involved in the function of regulatory T cells (Treg), because increased glycolysis in peripheral Treg cells may impair their lineage and survival integrity (Wei et al, 2016). Inhibition of the mTOR pathway by metformin decreases cytokine production in a Candida albicans infection model, leading to increased fungal outgrowth and decreased survival of mice in this infection model (Cheng et al, 2016).
We thus see here a number of examples that show that inhibition of cytokine release, via manipulation of metabolic pathways, undermines the activities of essential immune cells, which blunts the proper clearage of the infection. So, it is important to limit exaggerated immune and inflammatory responses during sepsis, while ensuring a minimal amount of immunity for the clearing of the ongoing infection.
Besides targeting HIF‐1α, PKM2, mTOR, or other similar central coordinating factors, another strategy to target glycolysis is with 2‐deoxy‐D‐glucose (2‐DG). Hexokinase (HK) is the initial and rate‐limiting enzyme in glycolysis). HK converts 2‐DG to phosphorylated 2‐DG (2‐DG‐6p), which inhibits HK activity thereby inhibiting glycolysis. In vitro, 2‐DG suppresses HIF‐1α and its targets IL‐1β and HMGB1 in activated macrophages (Tannahill et al, 2013; Yang et al, 2014). In T cells, blocking glycolysis with 2‐DG leads to a differentiation of Th17 cells into Treg cells (Shi et al, 2011). In vivo, 2‐DG significantly improves survival in both the LPS (Wang et al, 2016) and CLP (Zheng et al, 2017) model of sepsis by downregulating lactate and inflammatory cytokine production. In contrast, 2‐DG conferred no protection in a poly(I:C)—induced systemic inflammation model (Wang et al, 2016).
Patients that survive the acute hyperinflammatory phase of sepsis remain at an increased risk for secondary infections, and consequently late stage mortality (Wang et al, 2014). Understanding the mechanisms that underlie immunoparalysis in sepsis is also crucial for the identification of novel therapeutic approaches in sepsis. Immunotolerant leukocytes show broad metabolic defects at the level of both glycolysis and oxidative metabolism. One group has demonstrated that restoring the ability of immunotolerant leukocytes to mount a glycolytic response by treatment with IFN‐γ leads to an increased cytokine production and might represent a promising novel therapeutic approach to revert the immunotolerant state of sepsis (Cheng et al, 2016). A phase III clinical trial evaluating the effect of IFN‐γ on sepsis‐induced immunoparalysis (NCT01649921) has been conducted, but results have not yet been reported. The same group showed that aerobic glycolysis is also important for trained immunity, that is, the memory characteristics of the innate immune system. Pretreatment of mice with the β‐glucan component of C. albicans induces a non‐specific protection against infections, such as with Staphylococcus aureus. However, mice with a myeloid‐cell‐specific defect in HIF‐1α are unable to mount a trained immunity against S. aureus and succumb to the infection (Cheng et al, 2014a).
Hypoglycemia
In later stages, sepsis can also be characterized by hypoglycemia. In preclinical animal models, for example, mouse endotoxemia or mouse CLP, hypoglycemia is inevitable and linked with severity (Drechsler et al, 2015). Also in human patients, spontaneous hypoglycemia can be observed (Miller et al, 1980; Romijn et al, 1990; Rattarasarn, 1997; Singanayagam et al, 2009) but occurs less frequently. The fact that hypoglycemia occurs in mouse sepsis models and is less observed in human sepsis, may be based on (i) the relatively big surface to volume ratio in mice compared to humans, by which mice therefore lose heat much faster than men, and have to compensate this by a higher heart rate and metabolic turnover, and (ii) because humans are fed with glucose and other nutrients in the ICU. The mechanisms leading to hypoglycemia are not well understood. Depleted glycogen storages, increased peripheral usage of glucose, anorexia (Wang et al, 2016; Weis et al, 2017), and/or decreased gluconeogenesis (Dendoncker & Libert, 2017; Weis et al, 2017) may all be contributing factors.
Acute infections are associated with the development of anorexia, that is, an aspect of sickness behavior characterized by reduced food intake and thereby reduced blood glucose levels. It has recently been shown that this fasting metabolic state is crucial for survival of bacterial sepsis, whereas it may be detrimental for viral inflammation (Wang et al, 2016). Prolonged fasting results in hypoglycemia accompanied by lipolysis and ketogenesis, that is, the generation of ketone bodies. Ketone bodies [there are three common such ketone bodies, namely acetone, acetoacetate, and β‐hydroxybutyrate (BHB)] have been shown to confer resistance to reactive oxygen species (ROS)‐mediated damage. By supplementing glucose during bacterial sepsis, ketogenesis is suppressed, leading to inhibition of these ketone body‐mediated ROS adaptation pathways. This mechanism seems to be required for preventing ROS‐induced neuronal damage in bacterial inflammation, whereas it is dispensable in case of viral inflammation (Wang et al, 2016). Next to ketone bodies, alternative pathways that control ROS production, the release of ROS into the cytoplasm, the interaction of ROS with specific substrates such as kinases, and the detoxification of ROS can play important roles during sepsis pathophysiology, as was recently reviewed comprehensively by Kozlov et al (2017). In contrast, the study of Weis et al has shown that liver glucose production in response to bacterial infection is essential to establish disease tolerance. The authors suggest that liver gluconeogenesis is required to prevent lethal hypoglycemia in response to acute infection. According to this study, gluconeogenesis in the liver is hampered during sepsis due to iron‐driven oxidative inhibition of glucose‐6‐phosphatase (Weis et al, 2017). Presumably, anorexia is important to establish disease tolerance to systemic bacterial infections, and this could explain why this behavior is conserved from humans to insects (Wang et al, 2016). However, the blood glucose levels need to be maintained within a physiological range compatible with host survival and might thus not fall below a certain threshold (Chervonsky, 2017).
Control of glucose levels in sepsis: controversy
Both high (Gunst & Van den Berghe, 2016) and low glucose (Singanayagam et al, 2009; Krinsley et al, 2011) levels correlate with poor outcome in sepsis patients. Severe hyperglycemia has been shown to induce, that is, mitochondrial damage, endothelial dysfunction, acute kidney injury, liver dysfunction, muscle weakness, and long‐term neurocognitive impairment (Gunst & Van den Berghe, 2016). Severe hypoglycemia, in contrast, has been demonstrated to induce, for example, sudden cardiac arrest by inducing ischemic or depolarization/repolarization changes, neurocognitive impairment (acute coma), endothelial dysfunction, blood coagulation abnormalities, and increased inflammation (Kalra et al, 2013).
Three pioneer randomized controlled trials on tight glucose control found that control of glucose levels with insulin improved the outcome in sepsis patients as compared to control groups (Van den Berghe et al, 2001, 2006; Vlasselaers et al, 2009). These studies have led to a change in standard of care and have implemented control of glycemic levels to prevent excessive hyperglycemia (Gunst & Van den Berghe, 2016). However, there have been some concerns about the risk of developing severe hypoglycemia, the difficulty of achieving normoglycemia in ICU patients, and the contradicting results of some follow‐up studies (Marik, 2016). Therefore, a new clinical trial to test the value of tight glycemic control was conducted, namely the Normoglycemia in Intensive Care Evaluation–Survival Using Glucose Algorithm Regulation (NICE‐SUGAR) trial (Riske et al, 2009). The NICE‐SUGAR trial found that intensive glucose control in fact increased mortality, suggesting that slightly elevated glucose levels are rather beneficial and that glycemic control in ICU patients should be abandoned. It should be noted that in the NICE‐SUGAR study, the control group was also treated with insulin to keep glycemic levels between 140 and 180 mg/dl, instead of the uncontrolled hyperglycemia (up to 215 mg/dl) in the first studies (Gunst & Van den Berghe, 2016). It thus remains unclear how to manage glucose levels in ICU patients, but it seems that safe, effective glucose control may be advantageous over tight glycemic control.
Sepsis and fatty acid metabolism
Substantial activation of the immune system during sepsis, combined with the inability of most patients to keep up adequate nutrition, induces a starvation response in which next to glycolysis, energy needs are also supplied by lipid mobilization and oxidation (Jorgen et al, 1982; Wolowczuk et al, 2008). The storage of lipids as triglycerides (TGs) in adipose tissue comprises the bodies’ largest endogenous energy supply, allowing the release of fatty acids to become crucial during states of acutely increased energy needs (Cahill, 1970; Rittig et al, 2016). When energy demands are elevated, the body responds by upregulating lipolysis in adipose tissue, converting TGs into glycerol and free fatty acids (FFAs), which are subsequently released into the bloodstream (Cahill, 1970). FFAs can be taken up by peripheral organs and converted into energy by means of the β‐oxidation pathway followed by the TCA cycle (Fig 3). The average fat storage in a young human adult is 10–20 kg, the variation depending on gender, age, and fitness. The human glycogen reserve, in contrast, consists only of approximately 400 g, of which 300 g is largely unavailable and sealed within the skeletal muscles, reinforcing the importance of fat as potential energy storage (Sloan et al, 1973).
Figure 3. An overview of lipolysis and lipid oxidation in healthy conditions.
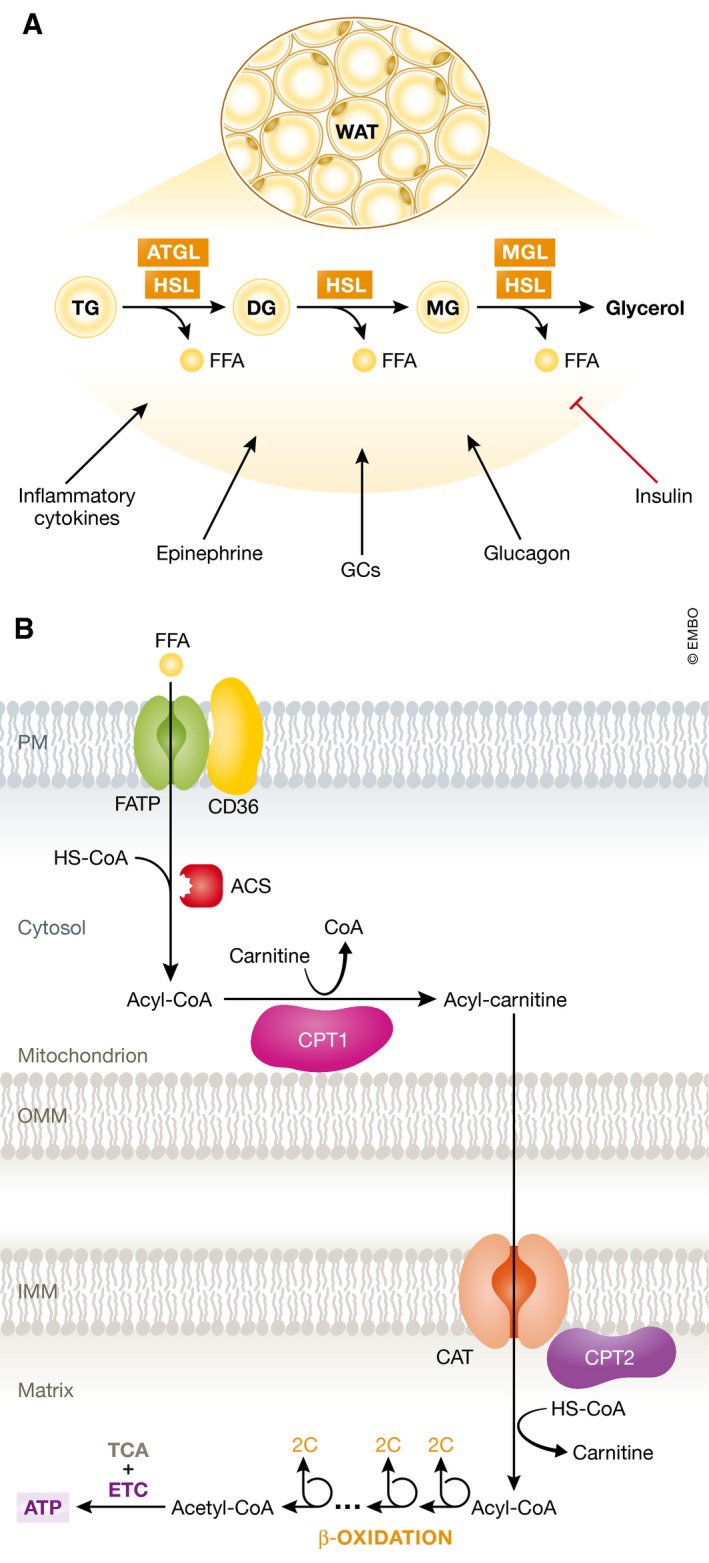
(A) Lipolysis is the hydrolytic conversion of triglycerides (TG) into glycerol and free fatty acids (FFAs) and is most abundant in white and brown adipose tissue. This process is tightly regulated by glucagon, (nor)epinephrine, and other hormones, but also by pro‐inflammatory cytokines. Hydrolysis of the ester bonds between long‐chain fatty acids and the glycerol backbone is executed by lipases. Up to now, three enzymes have been implicated in performing the complete hydrolysis of TG into FFAs and glycerol: adipose triglyceride lipase (ATGL), hormone sensitive lipase (HSL), and monoglyceride lipase (MGL). The FFAs are released into the blood stream and can be taken up by peripheral organs to produce energy via mitochondrial β‐oxidation. (B) Fatty acid β‐oxidation is a multistep process breaking down fatty acids in the mitochondria of the cell to produce acetyl‐CoA, which can be used by the tricarboxylic acid (TCA) cycle to produce ATP. In brief, FFAs are transported across the cell membrane by members of the FATP transporter family. Once inside the cytosol, the FFA is coupled to coenzyme A (CoA) by acyl‐CoA synthetase (ACS) and shuttled across the inner mitochondrial membrane by carnitine palmitoyltransferase II (CPT2) and the carnitine acyltransferase (CAT) after being coupled to carnitine by carnitine palmitoyltransferase I (CPT1). In the mitochondrial matrix, β‐oxidation is conducted by cleaving two carbon molecules in every oxidation cycle to form acetyl‐CoA. The cycle is repeated until the complete fatty acid has been reduced to acetyl‐CoA, which is subsequently enters the TCA cycle.
Lipolysis
Next to a deregulated glucose metabolism, the pathogenesis of sepsis is also known to influence fatty acid metabolism. Decades ago, it has already been described that lipolysis is upregulated in white adipose tissue (WAT) during the early phase of endotoxemia and sepsis (Forse et al, 1987), a finding that was more recently confirmed by several other studies (Wellhoener et al, 2011; Ilias et al, 2014; Rittig et al, 2016). Consequently, plasma TGs and FFA levels have been found to be increased up to 4‐fold in septic patients compared to healthy individuals (Scholl et al, 1984; Lanza‐Jacoby & Tabares, 1990; Nogueira et al, 2008). The molecular mechanisms behind this upregulation of lipolysis have hardly been addressed. One study showed that LPS infusion in human subjects led to an increased phosphorylation of hormone sensitive lipase (HSL) at Ser650, a modification known to activate the enzyme (Rittig et al, 2016). HSL is one of the best described lipases (encoded by the LIPE gene) and could, together with the other important enzyme adipose triglyceride lipase (ATGL, PNPLA2 gene), be involved in the (patho)physiological lipolysis in white adipose tissue (WAT). Also, cAMP‐dependent protein kinase A (PKA)‐dependent phosphorylation of perilipin 1 (PLIN1) is a decisive step in the activation of lipolysis (Choi et al, 2010) and was found to be elevated after LPS infusion (Rittig et al, 2016). This suggests that increased PKA activity might be one of the driving forces behind the increased lipolysis. Sepsis and endotoxemia are known to cause an acute increase in stress hormones such as (nor)epinephrine, growth hormone, glucagon, and cortisol, all of them being typical pro‐lipolytic hormones, which indeed stimulate lipolysis by direct stimulation of lipolysis in fat cells (Fong et al, 1990; Bloesch et al, 1993). Moreover, insulin resistance is a well‐established phenomenon in septic patients and insulin is a potent inhibitor of lipolysis (Choi et al, 2010). Eventually, all these mechanisms are likely to converge into an increase in the amounts of FFAs released by lipolysis into the bloodstream in sepsis.
β‐oxidation
Oxidation of FFAs provides a crucial source of energy during conditions of energy needs, starvation, or acute inflammation, including sepsis (Askanazi et al, 1980; Jorgen et al, 1982). However, the pro‐inflammatory status of this pathology is known to cause a dysregulation in the breakdown of FFAs in a wide variety of organs. A whole blood genome‐wide expression profiling in patients with septic shock showed that the expression of genes belonging to the PPAR‐α signaling pathway was significantly downregulated in children with a more severe disease status (Wong et al, 2009). PPAR‐α is a member of the nuclear receptor family and the most important transcription factor involved in the transcriptional regulation of genes coding for proteins involved in the β‐oxidation pathway (Hiukka et al, 2010).
Standage et al (2012) demonstrated more recently that the downregulated expression of PPAR‐α dependent genes was due to a reduced PPAR‐α expression in whole blood samples, a decrease which was associated with decreased survival and increased tissue bacterial load in an experimental polymicrobial septic mouse model. Furthermore, using bone marrow transplantation experiments, it was proven that disease severity is primarily determined by organ tissue PPAR‐α expression and not by hematopoietic immune cell PPAR‐α (Standage et al, 2012). Downregulation of PPAR‐α expression levels during infection has also been demonstrated in the liver, heart, and kidney (Beigneux et al, 2000; Feingold et al, 2008; Drosatos et al, 2011). In the heart, it was shown that LPS injection caused a decrease in PPAR‐α as well as in PPAR‐y co‐activator (PGC)‐1α. PGC‐1α is a major transcriptional co‐factor of PPAR‐α, and its downregulation thus leads to decreased expression of several PPAR‐α target genes and a reduction in the rate of FFA acid β‐oxidation in cardiomyocytes. As a consequence of the reduced oxidative breakdown of FFAs, severe organ damage may occur due to (i) energy deprivation, (ii) FFA accumulation and lipotoxicity, and (iii) mitochondrial dysfunction and endoplasmic reticulum (ER) stress (Fig 4).
Figure 4. Lipolysis, fatty acid oxidation, and ketogenesis in sepsis.

Sepsis is associated with the development of an anorectic response since patients are often unwilling or unable to eat. During a normal starvation response and during sepsis, lipolysis in white and brown adipose tissue is being upregulated by several pro‐lipolytic signals. The inhibitory effect on lipolysis of insulin, which is upregulated in sepsis due to high glucose levels, is however absent due to insulin resistance. Free fatty acids (FFAs) in the blood are upregulated in both conditions and can be taken up by peripheral organs to produce energy. The increased FFA levels activate and upregulate the expression of PPAR‐α, the main transcription factor responsible for the induction of genes involved in the β‐oxidation of fatty acids and the production of ketone bodies (KBs). During sepsis, PPAR‐α levels are downregulated and the breakdown of fatty acids through β‐oxidation is compromised, causing FFAs to accumulate in organs such as the liver, heart, and kidney, but also in the blood. Overall, the deficits in FFA breakdown during sepsis cause a shortage of energy and lipotoxicity and mitochondrial damage due to FFA accumulation. Green represents the normal starvation response, and red represents the response during sepsis.
Energy deprivation
Each of the previously specified organs is highly metabolically active in order to provide adequate energy to perform vital homeostatic functions within the body. When increased metabolic needs during acute inflammation are not met, in part due to mitochondrial dysfunction, life‐threatening organ dysfunction can develop, a recurrent feature of septic shock (Carré & Singer, 2008). It has been hypothesized that restoration of the FFA oxidation in specific organs could protect against organ dysfunction and potentially lead to increased survival in sepsis. In a zymosan‐induced (sterile) peritonitis model, it was shown that the upregulation of β‐oxidation of FFAs was associated with the resolution of inflammation (Fujieda et al, 2013). Furthermore, in macrophages and adipocytes, enhanced FFA oxidation led to reduced ER stress and ROS damage. It was also demonstrated that increased β‐oxidation improved insulin resistance by reducing the triglyceride accumulation and the expression of inflammatory cytokines, two factors that have been shown to reduce the sensitivity to insulin (Malandrino et al, 2015). Another study demonstrated that intervention in the LPS‐induced JNK signaling pathway restored PPAR‐α expression in the heart and prevented LPS‐induced heart dysfunction (Drosatos et al, 2011).
Lipotoxicity
The combination of increased lipolysis and deficits in the oxidation of FFAs can lead to the accumulation of these FFAs and subsequently to lipid‐induced toxicity. A proteomic and metabolic screen on plasma of septic patients identified several FFAs to be significantly upregulated in the blood of non‐survivors compared to survivors (Langley et al, 2013). Lipotoxicity is defined as a metabolic syndrome resulting from the accumulation of lipid intermediates in non‐adipose tissue and can lead to cellular dysfunction and cell death (Engin, 2017). Under normal conditions, lipids are stored primarily in adipocytes, with only a minimal presence in other tissues (Tontonoz & Spiegelman, 2008). When an overload of FFAs is present due to increased lipolysis and/or decreases in β‐oxidation, lipid deposits can be formed in peripheral organs. Presumably, this accumulation of fatty acids is not toxic at first; in fact, it can be seen as some sort of defense mechanism against the excess of circulating FFAs (Brindley et al, 2010). By taking up a part of the fatty acid overload, metabolically active organs such as the liver may protect more sensitive organs including the lungs from the toxic side effects of high FFA levels. Clinically, this phenomenon is addressed as steatosis and has been identified in liver, kidney, and heart after the onset of endotoxemia and sepsis (Zager et al, 2005; Rossi et al, 2007; Koskinas et al, 2008). When lipids continue to accumulate, certain lipid metabolites such as diacylglycerol (DAG), ceramide, and saturated fatty acids reach a critical level that could potentially be harmful to the tissue cells. It has been accepted that excess of lipids are ultimately steered toward non‐oxidative pathways which results in the formation of toxic lipid species that cause mitochondrial dysfunction (Bugger & Abel, 2008), modify cellular signaling(Yang & Barouch, 2007) and increase apoptosis (i.e., “lipoapoptosis”; Unger & Orci, 2002). More recently, toxic lipid species have been associated with the induction of ferroptosis, an alternative type of programmed cell death dependent on iron. The role of ferroptosis as a cell death mechanism contributing to organ damage in sepsis has been hardly explored. Nevertheless, ferroptosis has been implicated in acute kidney failure and could be an important alternative cell death pathway during sepsis (Linkermann et al, 2014; Müller et al, 2017; Wenzel et al, 2017). The precise contribution and impact of each of above‐mentioned altered cellular processes have not been properly delineated and seem to depend on lipid composition and cell type (Ghosh & Rodrigues, 2006). For a more detailed discussion on lipotoxicity, we refer to a number of excellent recent reviews (Wende & Abel, 2010; Ertunc & Hotamisligil, 2016; Engin, 2017).
Mitochondrial dysfunction
Mitochondrial dysfunction due to the fatty acid overload can be caused by the increased production of ROS (Schönfeld & Wojtczak, 2008), the uncoupling of the oxidative phosphorylation (Rial et al, 2010) and the permeabilization of the outer mitochondrial membrane (Listenberger & Schaffer, 2002). As stated before, it has been described that systemic inflammation can affect mitochondria by several mechanisms such as the generation of excess amounts of nitric oxide, carbon monoxide and ROS which directly inhibit mitochondrial function and damage the lipid membrane (Bauer & Pannen, 2009; Larsen et al, 2012). It seems plausible that the excess amount of fatty acids might contribute to the development of mitochondrial dysfunction during sepsis. Lipotoxicity has also been linked to ER stress and excessive activation of the unfolded protein response (UPR; Cunha et al, 2008; Baldwin et al, 2012). Many ER stress responses converge with inflammatory pathways via the activation of inflammatory kinases such as c‐Jun‐N‐terminal kinase (JNK) and the I‐kappa‐B‐kinase complex (IKK) and the activation of the inflammasome (Hu et al, 2006; Nakamura et al, 2010). Saturated fatty acids, including palmitate, can bind to TRL4 and induce a MyD88‐dependent signaling to eventually activate the inflammasome and NF‐κB signaling pathways (Lee et al, 2001). Moreover, ceramide, which is produced from long‐chain saturated fatty acids, has been shown to be directly associated with the induction of insulin resistance and lipoapoptosis (Kanety et al, 1996; Paz et al, 1997; Brookheart et al, 2009).
Therapeutic intervention
It is highly possible that the overload of FFAs, caused by aberrations in the fatty acid metabolism, contributes to the pathogenesis of sepsis and that interventions to prevent this accumulation could potentially ameliorate outcomes in sepsis. To avoid the accumulation and toxic effects of FFAs, one could target several pathways of the fatty acid metabolism: lipolysis, β‐oxidation, or lipogenesis.
Pharmacological inhibition of one of the major lipases, adipose triglyceride lipase (ATGL), was successful in correcting high‐fat diet‐induced insulin resistance and hepatosteatosis in mice (Schweiger et al, 2017). Several potent inhibitors that are directed toward the other major lipase, HSL, have recently been produced and might provide an attractive alternative to treat sepsis (Ogiyama et al, 2017).
Pharmacological prevention of PPAR‐α downregulation shows potential since inhibition of JNK restored PPAR‐α protein levels and was enough to prevent myocardial dysfunction after infection(Drosatos et al, 2011; Drosatos & Schulze, 2013). Also, interventions that target co‐factors or downstream targets of PPAR‐α could possibly contribute to improved oxidation of FFAs and enhanced energy production. However, extensive research into the mechanisms by which PPAR‐α is downregulated in different organs and other changes within this metabolic pathway are still needed to provide a complete view on possible druggable targets.
Another option could be to interfere before PPAR‐α is downregulated by actively stimulating PPAR‐α by the means of agonists. Indeed, mice on a fenofibrate‐loaded diet were found to show improved survival and repression of the inflammatory response in infection models (Tancevski et al, 2014). Such a therapy, of course, is purely preventive, and indeed rarely relevant in a clinical setting. One could argue that increasing the capacity of WAT to store lipids through adipogenesis by PPAR‐γ stimulation might decrease ectopic lipid overload (Gema et al, 2007). The PPAR‐γ agonist thiazolidinedione (TZD) was shown to improve peripheral insulin sensitivity and clearance of TGs in type 2 diabetes (Mayerson et al, 2002), while ameliorating murine polymicrobial sepsis by reducing the pro‐inflammatory signal transducer and activator of transcription‐1 (STAT‐1) signaling pathway (Ferreira et al, 2014). Moreover, several studies reported a role for PPAR‐γ in inducing mitochondrial biogenesis (Bolten et al, 2007; Pardo et al, 2011), a process that has been associated with survival during early sepsis (Carré et al, 2010). Combinatorial PPAR‐α and PPAR‐γ stimulation, while actively repressing lipolysis during the early stages of sepsis could represent an interesting alternative approach. Prospective clinical studies toward the use of fibrates and TZDs in infectious conditions have been conducted, but are however still insufficient and should be revisited (Fujita et al, 2006; Boggild et al, 2009).
In conclusion, an activation of lipolysis, associated with a break on FFA β‐oxidation, due to reduced activity of PPAR‐α and PGC‐1α and poor function of mitochondria, appears very critical in sepsis and is open to several key therapeutic interventions. The precise genes that suffer from reduced transcriptional activity of PPAR‐α and the impact on poor FFA oxidation have hardly been investigated, but the ones that coordinate cellular (CD36) and mitochondrial FFA import (SLC25A20, CPT1A, CPT1B), as well as those involved in the real oxidation pathway, appear logical priorities to study.
Sepsis and ketogenesis
As discussed above, sepsis leads to a fasting response accompanied by a pro‐lipolytic status with increased lipolysis and ketogenesis (Wang et al, 2016). Synthesis of the three known ketone bodies is tightly controlled by PPAR‐α and is mainly conducted by mitochondria of liver cells. Next to conferring resistance against ROS‐mediated damage, KBs are also an important source of energy for brain and skeletal muscle in times of high metabolic activity and limited glucose availability such as fasting and exercise (Randle et al, 1964). The production of KBs thus ensures that not all energy which is contained in FFAs is entirely degraded to acetyl‐CoA and further to CO2 and H2O in the liver TCA cycle, but that other organs, with much less FFA oxidation capacity can profit from the FFAs released by lipolysis. Aberrations in ketone metabolism during sepsis have been described a long time ago by several research groups (Felig et al, 1971; Flatt & Blackburn, 1974; Wannemacher et al, 1979). This suggests that the reduced production of KBs could lead to the increased breakdown of muscle protein, a universally accepted consequence of sepsis. Moreover, a recent study has shown that in PPAR‐α‐deficient mice LPS‐induced endotoxemia was lethal due to the absence of ketogenesis (Wang et al, 2016). Thus ketogenesis appears essential in the defense to sepsis but may be inadequately active in sepsis. Also, BHB was shown to block NLRP3‐inflammasome‐mediated inflammation not through the classical starvation‐regulated mechanisms such as AMP‐activated protein kinase (AMPK) or ROS, but by mechanically preventing the K+ efflux and reducing oligomerization of inflammasome components (Youm et al, 2015). Furthermore, BHB administration to mice exposed to high doses of fish oil was protective against acute liver failure and the lipotoxic effect of peroxidized fatty acids (Pawlak et al, 2015). In 1994, a study by Beylot et al demonstrated the inhibitory effect of BHB on lipolysis in septic patients, unfortunately, mortality rates were not considered during this trial (Beylot et al, 1994). Since NLRP3 and high levels of FFAs have been implicated in the pathogenesis of sepsis (Jin et al, 2017), administration of ketone bodies might thus be considered as a valuable novel therapeutic direction.
Sepsis and amino acid metabolism
In sepsis, a general catabolic status is observed. As we described above, a starvation‐like status leads to breakdown of carbohydrate and fat reserves, but also protein is degraded in several organs. Proteolysis, the trimming of proteins into smaller polypeptides and amino acids (AAs) has been best documented in skeletal muscle. The signals that lead to extensive muscle wasting or the proteases involved are poorly described in this condition. Similar to glycogen breakdown in liver and lipolysis in white adipose tissue, proteolysis is most likely the result of hormonal regulation, with glucagon and glucocorticoids being potential candidate regulators, as well as proteasomal proteases and inflammatory stimuli (Biolo et al, 1997). The precise reason for the increased proteolysis in sepsis fits into a general reshuffling of energy‐rich molecules, as well as an increased need of AAs in the liver to sustain the acute phase response (Hasselgren et al, 1988). Several amino acids are also known to play an important role in inflammatory cells. For example, glutamine is an important precursor for peptide and protein synthesis supporting cytokine production. Glutamine is also required for purine and pyrimidine and thus nucleic acid and nucleotide synthesis allowing proliferation of immune cells. Arginine can be converted into nitric oxide by nitric oxide synthase, which is then released by M1 macrophages.
All AAs are found to be increased in muscle cells, by proteolysis, and new amino acids seem to be formed due to catabolic chemical reactions (Su et al, 2015). Also in plasma, most amino acids are highly altered. Certain amino acids, especially taurine, are found to play an important role in predicting the severity and outcome in sepsis (Su et al, 2015). Supplementation of amino acids, such as glutamine, arginine, and taurine, during sepsis showed positive outcomes such as reduced length of stay and reduced number of secondary infections (Arts et al, 2016).
Specifically the branched‐chain amino acids (BCAAs), such as alanine, leucine, and isoleucine, are of interest. These AAs can function as acceptors of keto‐groups from pyruvate and glutamate, leading to glutamine and alanine. These AAs can enter the blood and can be used as energy‐rich AAs by other organs. Organs that are classically considered as targets are the kidney, intestine, and liver (Fig 5). In kidney and intestine, glutamine can be de‐aminated to glutamate, which can enter the TCA cycle via α‐ketoglutarate, potentially leading to the formation of alanine. Under normal conditions, the NH3 that is released during glutamine conversion is removed via urine or the intestinal lumen. Glutamine metabolism in the liver is low, especially because the liver has no big capacity to deal with NH3. Alanine, produced in the muscle, kidney, and intestine is transported to the liver, where it is oxidized and de‐aminated into pyruvate, which can subsequently enter the TCA cycle. Depending on the degree of hypoxia, pyruvate can be reduced to lactate or lead to glucose via gluconeogenesis. In septic patients, an increase in gluconeogenesis due to elevated alanine uptake in liver would be preferential, but, as stated above, gluconeogenesis is compromised in sepsis, which might therefore increase alanine levels in the blood of septic patients as observed by Langley et al (2013). Increases in ammonia in sepsis blood are observed in case of severe hepatic failure (Nesseler et al, 2012), a condition that is estimated to occur in about 20% of septic shock patients. Under such conditions, liver failure leads to a multitude of problems, such as bad hepatic capture and transport of bilirubin and bile salts, but also increased ammonia, resulting from a poor urea cycle. This latter, in essence a direct result of proteolysis, leads to hepatic encephalopathy, that is, mental disorientation of patients, a common finding, and coma (Felipo & Butterworth, 2002).
Figure 5. Overview of protein catabolism in sepsis.
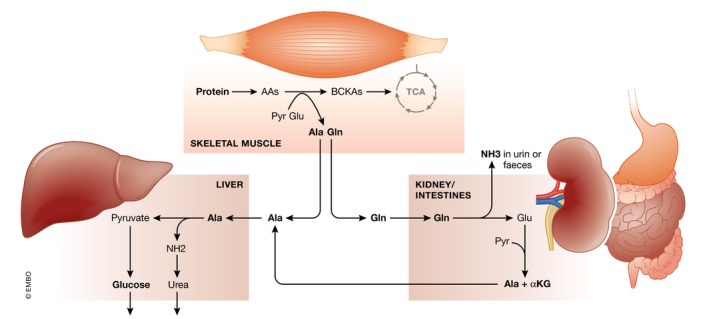
In sepsis conditions, catabolism of proteins in skeletal muscle is a recurrent feature, but the main regulators are still not identified. Branched‐chain amino acids (AAs) are oxidized to branched‐chain keto acids (BCKAs), which can be used in the TCA cycle. Glutamine (Gln) and alanine (Ala) find their way to kidney and intestine and liver. In the former two, Gln is de‐aminated to glutamate (Glu) and ammonia (NH3), which is removed. Glu and pyruvate can yield Ala and α‐ketoglutaric acid (αKG), which can enter the TCA cycle. Ala is mainly used as a gluconeogenic substrate and is transformed to pyruvate, whereby the NH2 group is removed via the urea cycle. During liver failure, ammonia may leak into the blood, leading to brain damage and coma.
Epigenetic‐metabolic link in sepsis
Epigenetics is defined as a “stably heritable phenotype that does not involve alterations in the DNA sequence, but chemical changes within the chromatin” (Berger et al, 2009). These changes include posttranslational modification of histone proteins (such as acetylation, methylation, phosphorylation, and ubiquitination), DNA methylation, and more recently also transient chromatin modifications and miRNAs (Natoli, 2010). Histone modifications are completed by specific sets of enzymes adding or removing different categories of posttranslational modifications, thereby directly modulating the chromatic structure and gene expression (Bannister & Kouzarides, 2011; Verdin & Ott, 2015). Interestingly, the activity of these different enzymes is depending on the presence of specific metabolites, causing chromatin modifications to be directly coupled to the metabolic state of the cell. It is now generally accepted that cells evolutionary acquired sensing mechanisms to detect changes in nutrients and accommodate the transcriptional program via epigenetic changes (Etchegaray & Mostoslavsky, 2016). On the other hand, key metabolic‐regulating genes are under the control of epigenetic machineries, creating a feedback loop that could potentially result into a vicious circle during conditions where metabolic pathways are severely altered.
Epigenetic changes during sepsis
Bomsztyk et al (2015) were the first to investigate changes in histone modifications on a whole‐organ level in sepsis‐related multiple organs dysfunction syndrome (MODS). They showed that 6 h after the onset of infection, major epigenetic changes were occurring in several organs. Moreover, the decrease in gene expression of key genes involved in endothelial function was associated with a decrease in permissive histone modifications (acetylation), while repressive marks (methylation) were unchanged, suggesting that intervention early into the disease to preserve these permissive marks could help to keep endothelial integrity intact. A major limitation of this study is the fact that within every organ, many different cell types are found, each with their own epigenome. More importantly, during sepsis, there is an extensive infiltration of immune cells, making a cell‐specific approach crucial.
Acetylation of lysine residues of histones and other proteins is one of the best known posttranslational modifications and is performed by KATs (lysine acetyl transferases). The activity of these enzymes relies on the availability of acetyl‐CoA, an intermediary metabolite produced mainly from β‐oxidation of fatty acids and the oxidative de‐carboxylation of pyruvate by PDC (Lee & Workman, 2007; Etchegaray & Mostoslavsky, 2016). In nutrient‐favorable conditions, high acetyl‐CoA levels trigger the regulation of specific genes involved in growth and proliferation through histone acetylation, resulting in a more permissive chromatin configuration (Wellen et al, 2009; Cai et al, 2011). As described above, both acetyl‐CoA‐producing pathways are reduced in sepsis with the oxidation of fatty acids being downregulated and pyruvate being shuttled toward aerobic glycolysis for the production of lactate. As a consequence, it is possible that less acetyl‐CoA is available for acetylation of proteins which could explain the reduction in permissive histone modifications observed by Bomsztyk et al (2015). Interestingly, a clear link between the reduction in FFA oxidation, consequent low acetyl‐CoA levels, and low expression of neovascular genes by suboptimal histone acetylation has been shown in mouse models in the recent past (Schoors et al, 2015).
Histone deacetylases in sepsis
Another group of enzymes, involved in the removal of acetylation groups, histone deacetylases (HDACs), might be of interest during the pathology of sepsis. Several histone deacetylase inhibitors (HDACi) have been tested as negative regulators of the expression of important immune receptors and antimicrobial pathways in immune cells. It was shown in multiple studies that HDACi can improve the outcome after septic shock by reducing the cytokine storm, confirming the hypothesis that these inhibitors have potential as sepsis therapeutics (Li et al, 2009; Zhang et al, 2010; Roger et al, 2011). However, broad HDACi have also been associated with impaired bacterial clearance and a more recent study suggested that specific HDAC6 inhibition might improve responses during sepsis, while preserving important microbicidal actions (Zhao et al, 2014). The catalytic activity of SIRT6, a member of the class III HDACs, was shown to be increased by certain FFAs up to 35‐fold in vitro (Feldman et al, 2013). It has been hypothesized that in nutrient conditions where FFAs are increased, SIRT6 activity could repress the transcription of HIF‐1α driven glycolytic genes by removing permissive acetylated histone marks (Zhong et al, 2010). During serious infections where the increase in lipolysis causes FFAs levels to rise, this mechanism could contribute to the reduction in histone acetylation and alter the glucose metabolism. On the other hand, SIRT6 acts as a tumor suppressor in cancer cells by inhibition of aerobic glycolysis (Zhong et al, 2010; Sebastián et al, 2012). This raises the question whether SIRT6 activation could also have potential as a druggable target during severe acute inflammatory conditions.
Another SIRT enzyme that provides a link between metabolism and inflammation is SIRT1. This deacetylase is activated by AMPK, a crucial metabolic sensor that is activated under conditions of elevated energy demand characterized by an increase in the AMP/ATP ratio (Lan et al, 2008). SIRT1 has been shown to directly interact with and deacetylate PGC‐1α, as discussed above, a potent co‐factor of many transcription factors involved in energy expenditure such as GR, PPAR‐α, and PPAR‐γ, thereby enhancing their transcriptional activity (Vega et al, 2000; Rodgers et al, 2005). Pharmacological activation of SIRT1 shows potential for the treatment of sepsis since PGC‐1α expression was found to be severely downregulated in muscle of septic mice, while SIRT1 activation proved to increase survival in the CLP sepsis model (Rocheteau et al, 2015; Opal et al, 2016). Whether this reduction in mortality is caused in part by improved PGC‐1α transcriptional activity is still unexplored.
It is clear that the impaired energy metabolism in sepsis can drive many epigenetic changes that could exacerbate the dysfunction of inflammatory and metabolic pathways. With more extensive research into the variable epigenetics during sepsis and the possible consequences, we could be paving the way to a new kind of drugs for care of septic patients.
Conclusion
In a healthy organism, energy expenditure and energy income are in balance. During extensive exercise, the consumption of O2 by the TCA cycle exceeds the available amounts, and the resulting hypoxia leads to a mainly HIF‐1α coordinated closure of the mitochondrial import of pyruvate, and an increased production of lactate, which can be consumed elsewhere in the body. The toxic lactate causes muscle cramps, enforcing the end of the exercise. During starvation, that is, when no or limited amounts of food enters the system, a prolonged imbalance of the energy homeostasis is created and in essence two systems are initiated to cause rearrangements of the metabolism. First, hormones (glucagon, epinephrine, norepinephrine, glucocorticoids), sensing low metabolic concentrations, coordinate a starvation response, leading to the release of energy‐rich molecules from the resources, for example, glucose from glycogen, AAs from protein and FFAs and glycerol from TGs. The AAs will lead to glucose via gluconeogenesis in the liver, a process strongly coordinated by glucocorticoids and the GR, while several of the FFAs act as ligands for another nuclear receptor PPAR‐α. These receptors are transcription factors and induce genes that are essential in the gluconeogenesis and β‐oxidation processes, respectively. This is a system of high efficiency and strongly stimulated by the transcriptional co‐factor PGC1‐α. Second, the amounts of mitochondria, well‐known as the organelles where TCA cycle and FFA β‐oxidation occur, are increased by a process of mitochondrial biogenesis, again by the stimulating action of PGC1‐α.
In sepsis, the energy balance is clearly disturbed. There is energy needed, but patients are unwilling or incapable to eat and a starvation response develops. Due to the infection, an inflammation and immune response develop, and as a consequence a HIF‐1α signature is seen (Fig 6). This leads to a limited mitochondrial function, which may serve several goals. First, inflammation leads to mitochondrial damage, so limiting the importance of these organelles in ATP production seems logical. Second, the metabolism of glucose shifts to aerobic glycolysis, which may be of interest for a more efficient function of white blood cells. Third, some authors suggest that the reduction in mitochondrial respiration under such conditions is a conserved pathway of limiting energy expenditure, leading to a metabolic reprogramming, as is observed during hibernation in several mammalian taxa. Of course, neither humans nor mice are hibernating species, so the importance of these pathways may be questioned. Nevertheless, the danger of a reduced mitochondrial activity is obvious: Most of the energy‐rich molecules produced from the energy stores need active mitochondria to be properly consumed by cells. It is likely that the systemic aspect of sepsis forms the major hurdle of this strategy, because mitochondrial function in sepsis seems to be failing in all tissues, and as a consequence, lactate, FFAs, and other catabolic products accumulate and cause tissue damage and death.
Figure 6. The toxic consequences of metabolic reprogramming in sepsis.
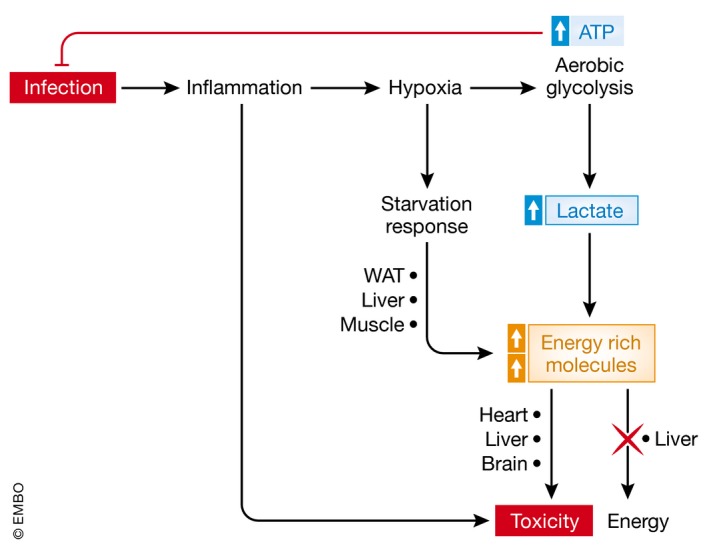
Infection is the start of sepsis. It leads to direct tissue damage and to inflammation, which in turn leads to hypoxia, which is essential to allow white blood cells (WBCs) to produce fast ATP from glucose and act fast on the infectious agents. The hypoxic response also leads to mobilization of energy‐rich molecules such as lactate and fatty acids, which however can also lead to toxicity, when over abundant.
Although it is clear that there is very significant metabolic reprogramming in sepsis, besides the activation of other complex systems (inflammation, coagulation, complement activation, hypoxia response), and given the fact that these pathways all influence one another, it is hard to conclude how, where, and when the metabolic pathways that are calling for therapeutic modulation have to be addressed in a safe and effective way. Based on this overview of the literature, it is our opinion that three early pathways deserve special attention, namely (i) the generation of lactate by the increased HIF‐stimulated glycolysis, (ii) the accumulation of free fatty acids in the blood, by the decreased ability of tissues to oxidize them via beta‐oxidation, and (iii) the decreased generation of ketone bodies by the liver. Since the liver also appears to be undergoing these metabolic rearrangements, and based on the availability of liver‐targeting approaches in today's pharmacology, this organ could be the best option to study in preclinical models of sepsis.
Conflict of interest
The authors declare that they have no conflict of interest.
For more information
(i) https://clinicaltrials.gov/ct2/show/NCT01649921?term=NCT01649921&rank=1
Pending issues.
-
(i)
Failure of all clinical sepsis trials is likely caused due to patient heterogeneity: stratification is the key.
-
(ii)
Studies directed toward the ideal sepsis animal model.
-
(iii)
Studies elucidating sepsis as an inflammatory versus metabolic disorder and identification of key target organs.
Acknowledgements
Research in the author's laboratory was funded by the Agency for Innovation of Science and Technology in Flanders (IWT), the Research Council of Ghent University (GOA Program), the Research Foundation Flanders (FWO Vlaanderen), COST Action BM1402, and the Interuniversity Attraction Poles Program of the Belgian Science Policy (IAP‐VI‐18). LV and JV are research fellows with the Research Foundation Flanders (FWO Vlaanderen).
EMBO Mol Med (2018) 10: e8712
See the Glossary for abbreviations used in this article.
References
- Arts RJW, Gresnigt MS, Joosten LAB, Netea MG (2016) Cellular metabolism of myeloid cells in sepsis. J Leukoc Biol 100: 1–14 [DOI] [PubMed] [Google Scholar]
- Arulkumaran N, Deutschman CS, Pinsky MR, Zuckerbraun B, Schumacker PT, Gomez H, Gomez A, Murray P, Kellum JA (2016) Mitochondrial function in sepsis. Shock 45: 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askanazi J, Carpentier Y, Elwyn D, Noenström J, Jeevanandam M, Rosenum S, Gump F, Kinney J (1980) Influence of total parenteral nutrition on fuel utilization in injury and sepsis. Ann Surg 191: 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AC, Green CD, Olson KL, Moxley MA, Corbett JA (2012) A role for aberrant protein palmitoylation in FFA‐induced ER stress and β‐cell death. Am J Physiol Endocrinol Metab 302: E1390–E1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer ML, Hess C (2017) Starving for survival—how catabolic metabolism fuels immune function. Curr Opin Immunol 46: 8–13 [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21: cr201122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer I, Pannen BHJ (2009) Bench‐to‐bedside review: carbon monoxide – from mitochondrial poisoning to therapeutic use. Crit Care 13: 220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigneux AP, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR (2000) The acute phase response is associated with retinoid X receptor repression in rodent liver. J Biol Chem 275: 16390–16399 [DOI] [PubMed] [Google Scholar]
- Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A (2009) An operational definition of epigenetics. Genes Dev 23: 781–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beylot M, Chassard D, Chambrier C, Guiraud M, Odeon M, Beaufrère B, Bouletreau P (1994) Metabolic effects of a D‐beta‐hydroxybutyrate infusion in septic patients: inhibition of lipolysis and glucose production but not leucine oxidation. Crit Care Med 22: 1091–1098 [DOI] [PubMed] [Google Scholar]
- Biolo G, Toigo G, Ciocchi B, Situlin R, Iscra F, Gullo A, Guarnieri G (1997) Metabolic response to injury and sepsis: changes in protein metabolism. Nutrition 13: 52–57 [DOI] [PubMed] [Google Scholar]
- Bloesch D, Keller U, Spinas GA, Küry D, Girard J, Stauffacher W (1993) Effects of endotoxin on leucine and glucose kinetics in man: contribution of prostaglandin E2 assessed by a cyclooxygenase inhibitor. J Clin Endocrinol Metab 77: 1156–1163 [DOI] [PubMed] [Google Scholar]
- Boggild AK, Krudsood S, Patel SN, Serghides L, Tangpukdee N, Katz K, Wilairatana P, Liles WC, Looareesuwan S, Kain KC (2009) Use of peroxisome proliferator‐activated receptor gamma agonists as adjunctive treatment for Plasmodium falciparum malaria: a randomized, double‐blind, placebo‐controlled trial. Clin Infect Dis 49: 841–849 [DOI] [PubMed] [Google Scholar]
- Bolten CW, Blanner PM, McDonald WG, Staten NR, Mazzarella RA, Arhancet GB, Meier MF, Weiss DJ, Sullivan PM, Hromockyj AE et al (2007) Insulin sensitizing pharmacology of thiazolidinediones correlates with mitochondrial gene expression rather than activation of PPARγ. Gene Regul Syst Bio 1: 73–82 [PMC free article] [PubMed] [Google Scholar]
- Bomsztyk K, Mar D, An D, Sharifian R, Mikula M, Gharib SA, Altemeier WA, Liles WC, Denisenko O (2015) Experimental acute lung injury induces multi‐organ epigenetic modifications in key angiogenic genes implicated in sepsis‐associated endothelial dysfunction. Crit Care 19: 225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindley DN, Kok BPC, Kienesberger PC, Lehner R, Dyck JRB (2010) Shedding light on the enigma of myocardial lipotoxicity: the involvement of known and putative regulators of fatty acid storage and mobilization. Am J Physiol Endocrinol Metab 298: E897–E908 [DOI] [PubMed] [Google Scholar]
- Brookheart RT, Michel CI, Schaffer JE (2009) As a matter of fat. Cell Metab 10: 9–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugger H, Abel ED (2008) Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci 114: 195–210 [DOI] [PubMed] [Google Scholar]
- Cahill GF (1970) Starvation in man. N Engl J Med 282: 668–675 [DOI] [PubMed] [Google Scholar]
- Cai L, Sutter BM, Li B, Tu BP (2011) Acetyl‐CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell 42: 426–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro‐Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, Gavin AL, Abel ED, Kelsoe G, Green DR et al (2014) Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF‐exposed B cells. J Immunol 192: 3626–3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carré JE, Singer M (2008) Cellular energetic metabolism in sepsis: the need for a systems approach. Biochim Biophys Acta 1777: 763–771 [DOI] [PubMed] [Google Scholar]
- Carré JE, Orban J‐C, Re L, Felsmann K, Iffert W, Bauer M, Suliman HB, Piantadosi CA, Mayhew TM, Breen P et al (2010) Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med 182: 745–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB, Faubert B, Villarino AV, O'Sullivan D, Huang SCC, Van Der Windt GJW, Blagih J, Qiu J et al (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153: 1239–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S‐C, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos‐Bourboulis EJ, Martens JHA, Rao NA, Aghajanirefah A et al (2014a) mTOR‐ and HIF‐1 ‐mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345: 1250684–1250684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SC, Joosten LAB, Netea MG (2014b) The interplay between central metabolism and innate immune responses. Cytokine Growth Factor Rev 25: 707–713 [DOI] [PubMed] [Google Scholar]
- Cheng S‐C, Scicluna BP, Arts RJW, Gresnigt MS, Lachmandas E, Giamarellos‐Bourboulis EJ, Kox M, Manjeri GR, Wagenaars JAL, Cremer OL et al (2016) Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol 17: 406–413 [DOI] [PubMed] [Google Scholar]
- Chervonsky AV (2017) Just a spoonful of sugar helps the tolerance go up. Cell 169: 1170–1172 [DOI] [PubMed] [Google Scholar]
- Choi SM, Tucker DF, Gross DN, Easton RM, DiPilato LM, Dean AS, Monks BR, Birnbaum MJ (2010) Insulin regulates adipocyte lipolysis via an Akt‐independent signaling pathway. Mol Cell Biol 30: 5009–5020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens MG, Chaudry IH, McDermott PH, Baue AE (1983) Regulation of glucose production from lactate in experimental sepsis. Am J Physiol 244: 794–800 [DOI] [PubMed] [Google Scholar]
- Cohen J, Vincent J‐L, Adhikari NKJ, Machado FR, Angus DC, Calandra T, Jaton K, Giulieri S, Delaloye J, Opal S et al (2015) Sepsis: a roadmap for future research. Lancet Infect Dis 15: 581–614 [DOI] [PubMed] [Google Scholar]
- Corcoran SE, O'Neill LAJ (2016) HIF1a and metabolic reprogramming in inflammation. J Clin Invest 126: 3699–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha DA, Hekerman P, Ladrière L, Angie B‐C, Ortis F, Wakeham MC, Moore F, Rasschaert J, Cardozo AK, Bellomo E et al (2008) Initiation and execution of lipotoxic ER stress in pancreatic β‐cells. J Cell Sci 121: 2308–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejager L, Pinheiro I, Dejonckheere E, Libert C (2011) Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol 19: 198–208 [DOI] [PubMed] [Google Scholar]
- Dendoncker K, Libert C (2017) Glucocorticoid resistance as a major drive in sepsis pathology. Cytokine Growth Factor Rev 35: 85–96 [DOI] [PubMed] [Google Scholar]
- Deutschman CS, De Maio A, Buchman TG, Clemens MG (1993) Sepsis‐induced alterations in phosphoenolpyruvate carboxykinase expression: the role of insulin and glucagon. Circ Shock 40: 295–302 [PubMed] [Google Scholar]
- Drechsler S, Weixelbaumer KM, Weidinger A, Raeven P, Khadem A, Redl H, Van Griensven M, Bahrami S, Remick D, Kozlov A et al (2015) Why do they die? Comparison of selected aspects of organ injury and dysfunction in mice surviving and dying in acute abdominal sepsis. Intensive Care Med Exp 3: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosatos K, Zoi D‐T, Khan R, Homma S, Schulze CP, Zannis VI, Goldberg IJ (2011) Inhibition of c‐Jun‐N‐terminal kinase increases cardiac peroxisome proliferator‐activated receptor α expression and fatty acid oxidation and prevents lipopolysaccharide‐induced heart dysfunction. J Biol Chem 286: 36331–36339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosatos K, Schulze PC (2013) Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr Heart Fail Rep 10: 109–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin A (2017) What is lipotoxicity? Adv Exp Med Biol 960: 197–220 [DOI] [PubMed] [Google Scholar]
- Englert J, Rogers A (2016) Metabolism, metabolomics, and nutritional support of patients with sepsis. Clin Chest Med 37: 321–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertunc ME, Hotamisligil GSS (2016) Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res 57: 2099–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchegaray JP, Mostoslavsky R (2016) Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol Cell 62: 695–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts B, Amiel E, Van Der Windt GJW, Freitas TC, Chott R, Yarasheski KE, Pearce EL, Pearce EJ (2012) Commitment to glycolysis sustains survival of NO‐producing inflammatory dendritic cells. Blood 120: 1422–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SC, Smith AM, Chang C, Lam WY, Redmann V, Freitas TC, Blagih J, Van Der Windt GJW et al (2014) TLR‐driven early glycolytic reprogramming via the kinases TBK1‐IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol 15: 323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold KR, Wang Y, Moser A, Shigenaga JK, Grunfeld C (2008) LPS decreases fatty acid oxidation and nuclear hormone receptors in the kidney. J Lipid Res 49: 2179–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Baeza J, Denu JM (2013) Activation of the protein deacetylase SIRT6 by long‐chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem 288: 31350–31356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felig P, Marliss EB, Cahill GF (1971) Metabolic response to human growth hormone during prolonged starvation. J Clin Invest 50: 411–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felipo V, Butterworth RF (2002) Neurobiology of ammonia. Prog Neurobiol 67: 259–279 [DOI] [PubMed] [Google Scholar]
- Ferreira AE, Sisti F, Sônego F, Wang S, Filgueiras LR, Brandt S, Serezani AP, Du H, Cunha FQ, Alves‐Filho JC et al (2014) PPAR‐γ/IL‐10 axis inhibits MyD88 expression and ameliorates murine polymicrobial sepsis. J Immunol 192: 2357–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink MP (2015) Cytopathic hypoxia and sepsis: Is mitochondrial dysfunction pathophysiologically important or just an epiphenomenon. Pediatr Crit Care Med 16: 89–91 [DOI] [PubMed] [Google Scholar]
- Flatt JP, Blackburn GL (1974) The metabolic fuel regulatory system: implications for protein‐sparing therapies during caloric deprivation and disease. Am J Clin Nutr 27: 175–187 [DOI] [PubMed] [Google Scholar]
- Fong YM, Marano MA, Moldawer LL, Wei H, Calvano SE, Kenney JS, Allison AC, Cerami A, Shires GT, Lowry SF (1990) The acute splanchnic and peripheral tissue metabolic response to endotoxin in humans. J Clin Invest 85: 1896–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forse RA, Leibel R, Askanazi J, Hirsch J, Kinney JM (1987) Adrenergic control of adipocyte lipolysis in trauma and sepsis. Ann Surg 206: 744–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujieda Y, Manno A, Hayashi Y, Rhodes N, Guo L, Arita M, Bamba T, Fukusaki E (2013) Inflammation and resolution are associated with upregulation of fatty acid β‐oxidation in zymosan‐induced peritonitis. PLoS One 8: e66270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Kaito M, Kai M, Sugimoto R, Tanaka H, Horiike S, Konishi M, Iwasa M, Watanabe S, Adachi Y (2006) Effects of bezafibrate in patients with chronic hepatitis C virus infection: combination with interferon and ribavirin. J Viral Hepat 13: 441–448 [DOI] [PubMed] [Google Scholar]
- Gema M‐G, Gray S, Antonio V‐P (2007) Adipogenesis and lipotoxicity: role of peroxisome proliferator‐activated receptor gamma (PPARgamma) and PPARgammacoactivator‐1 (PGC1). Public Health Nutr 10: 1132–1137 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Rodrigues B (2006) Cardiac cell death in early diabetes and its modulation by dietary fatty acids. Biochim Biophys Acta 1761: 1148–1162 [DOI] [PubMed] [Google Scholar]
- Gunst J, Van den Berghe G (2016) Blood glucose control in the ICU: don't throw out the baby with the bathwater!. Intensive Care Med 42: 1478–1481 [DOI] [PubMed] [Google Scholar]
- Hasselgren PO, Pedersen P, Sax HC, Warner BW, Fischer JE (1988) Current concepts of protein turnover and amino acid transport in liver and skeletal muscle during sepsis. Arch Surg 123: 992–999 [DOI] [PubMed] [Google Scholar]
- Hiukka A, Maranghi M, Matikainen N, Taskinen MR (2010) PPARα: an emerging therapeutic target in diabetic microvascular damage. Nat Rev Endocrinol 6: 454–463 [DOI] [PubMed] [Google Scholar]
- Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH (2006) Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α‐mediated NF‐κB activation and down‐regulation of TRAF2 expression. Mol Cell Biol 26: 3071–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttemann M, Helling S, Sanderson TH, Sinkler C, Samavati L, Mahapatra G, Varughese A, Lu G, Liu J, Ramzan R et al (2012a) Regulation of mitochondrial respiration and apoptosis through cell signaling: cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim Biophys Acta 1817: 598–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttemann M, Lee I, Grossman LI, Doan JW, Sanderson TH (2012b) Phosphorylation of mammalian cytochrome c and cytochrome c oxidase in the regulation of cell destiny: respiration, apoptosis, and human disease. Adv Exp Med Biol 748: 237–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilias I, Vassiliadi DA, Theodorakopoulou M, Boutati E, Maratou E, Mitrou P, Nikitas N, Apollonatou S, Dimitriadis G, Armaganidis A et al (2014) Adipose tissue lipolysis and circulating lipids in acute and subacute critical illness: effects of shock and treatment. J Crit Care 29: e5–e9 [DOI] [PubMed] [Google Scholar]
- Jeoung NH (2015) Pyruvate dehydrogenase kinases: therapeutic targets for diabetes and cancers. Diabetes Metab J 39: 188–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha AK, Huang SCC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B et al (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42: 419–430 [DOI] [PubMed] [Google Scholar]
- Jin L, Batra S, Jeyaseelan S (2017) Deletion of Nlrp3 augments survival during polymicrobial sepsis by decreasing autophagy and enhancing phagocytosis. J Immunol 198: 1253–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GRN (1974) Ionic shuttles in shock. Lancet 11: 905 [DOI] [PubMed] [Google Scholar]
- Jorgen N, Carpentier A, Jeffrey A, Robin A, Elwyn D, Hensle T, Kinney J (1982) Metabolic utilization of intravenous fat emulsion during total parenteral nutrition. Ann Surg 196: 221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra S, Mukherjee J, Ramachandran A, Saboo B, Shaikh S, Venkataraman S, Bantwal G, Das A (2013) Hypoglycemia: the neglected complication. Indian J Endocrinol Metab 17: 819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanety H, Hemi R, Papa MZ, Karasik A (1996) Sphingomyelinase and ceramide suppress insulin‐induced tyrosine phosphorylation of the insulin receptor substrate‐1. J Biol Chem 271: 9895–9897 [DOI] [PubMed] [Google Scholar]
- Kimmoun A, Novy E, Auchet T, Ducrocq N, Levy B (2015) Hemodynamic consequences of severe lactic acidosis in shock states: from bench to bedside. Crit Care 19: 175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskinas J, Gomatos IP, Tiniakos DG, Memos N, Boutsikou M, Garatzioti A, Archimandritis A, Betrosian A (2008) Liver histology in ICU patients dying from sepsis: a clinico‐pathological study. World J Gastroenterol 14: 1389–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov AV, Lancaster JR Jr, Meszaros AT, Weidinger A (2017) Mitochondria‐meditated pathways of organ failure upon inflammation. Redox Biol 13: 170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krinsley JS, Schultz MJ, Spronk PE, Harmsen RE, van Braam Houckgeest F, van der Sluijs JP, Mélot C, Preiser J (2011) Mild hypoglycemia is independently associated with increased mortality in the critically ill. Crit Care 15: R173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan F, Cacicedo JM, Ruderman N, Ido Y (2008) SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP‐activated protein kinase activation. J Biol Chem 283: 27628–27635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley RJ, Tsalik EL, van Velkinburgh JC, Glickman SW, Rice BJ, Wang C, Chen B, Carin L, Suarez A, Mohney RP et al (2013) An integrated clinico‐metabolomic model improves prediction of death in sepsis. Sci Transl Med 5: 195ra95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza‐Jacoby S, Tabares A (1990) Triglyceride kinetics, tissue lipoprotein lipase, and liver lipogenesis in septic rats. Am J Physiol 258: E678–85 [DOI] [PubMed] [Google Scholar]
- Larsen FJ, Schiffer TA, Weitzberg E, Lundberg JO (2012) Regulation of mitochondrial function and energetics by reactive nitrogen oxides. Free Radic Biol Med 53: 1919–1928 [DOI] [PubMed] [Google Scholar]
- Lee JY, Sohn KH, Rhee SH, Hwang D (2001) Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase‐2 mediated through toll‐like receptor 4. J Biol Chem 276: 16683–16689 [DOI] [PubMed] [Google Scholar]
- Lee KK, Workman JL (2007) Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol 8: 284–295 [DOI] [PubMed] [Google Scholar]
- Lee I, Hüttemann M (2014) Energy crisis: the role of oxidative phosphorylation in acute inflammation and sepsis. Biochim Biophys Acta 1842: 1579–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite HP, de Lima LFP (2016) Metabolic resuscitation in sepsis: a necessary step beyond the hemodynamic? J Thorac Dis 8: E552–E557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu B, Zhao H, Sailhamer EA, Fukudome EY, Zhang X, Kheirbek T, Finkelstein RA, Velmahos GC, DeMoya M et al (2009) Protective effect of suberoylanilide hydroxamic acid against LPS‐induced septic shock in rodents. Shock 32: 517–523 [DOI] [PubMed] [Google Scholar]
- Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz P‐S et al (2014) Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci 111: 16836–16841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listenberger LL, Schaffer JE (2002) Mechanisms of lipoapoptosis implications for human heart disease. Trends Cardiovasc Med 12: 134–138 [DOI] [PubMed] [Google Scholar]
- Loftus RM, Finlay DK (2016) Immunometabolism: cellular metabolism turns immune regulator. J Biol Chem 291: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malandrino M, Fucho R, Weber M, María C‐D, Mir J, Valcarcel L, Escoté X, María G‐S, Peral B, Salvadó L et al (2015) Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid‐induced triglyceride accumulation and inflammation. Am J Physiol Endocrinol Metab 308: E756–E769 [DOI] [PubMed] [Google Scholar]
- Marik PE, Bellomo R (2013) Stress hyperglycemia: an essential survival response!. Crit Care 17: 305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marik PE (2016) Tight glycemic control in acutely ill patients: low evidence of benefit, high evidence of harm!. Intensive Care Med 42: 1475–1477 [DOI] [PubMed] [Google Scholar]
- Marín‐Hernández A, Gallardo‐Pérez JC, Ralph SJ, Rodríguez‐Enríquez S, Moreno‐Sánchez R (2009) HIF‐1alpha modulates energy metabolism in cancer cells by inducing over‐expression of specific glycolytic isoforms. Mini Rev Med Chem 9: 1084–101 [DOI] [PubMed] [Google Scholar]
- Marshall JC (2014) Why have clinical trials in sepsis failed?. Trends Mol Med 20: 195–203 [DOI] [PubMed] [Google Scholar]
- Mayerson AB, Hundal RS, Dufour S, Lebon V, Befroy D, Cline GW, Enocksson S, Inzucchi SE, Shulman GI, Petersen KF (2002) The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 51: 797–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SI, Wallace RJ, Musher DM, Septimus EJ, Kohl S, Baughn RE (1980) Hypoglycemia as a manifestation of sepsis. Am J Med 68: 649–654 [DOI] [PubMed] [Google Scholar]
- Müller T, Dewitz C, Schmitz J, Schröder AS, Bräsen JH, Stockwell BR, Murphy JM, Kunzendorf U, Krautwald S (2017) Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci 74: 3631–3645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Furuhashi M, Li P, Cao H, Tuncman G, Sonenberg N, Gorgun CZ, Hotamisligil GS (2010) Double‐stranded RNA‐dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 140: 338–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli G (2010) Maintaining cell identity through global control of genomic organization. Immunity 33: 12–24 [DOI] [PubMed] [Google Scholar]
- Nesseler N, Launey Y, Aninat C, Morel F, Mallédant Y, Seguin P (2012) Clinical review: the liver in sepsis. Crit Care 16: 235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HB, Rivers EP, Knoblich BP, Jacobsen G, Muzzin A, Ressler JA, Tomlanovich MC (2004) Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 32: 1637–1642 [DOI] [PubMed] [Google Scholar]
- Nichol AD, Egi M, Pettila V, Bellomo R, French C, Hart G, Davies A, Stachowski E, Reade MC, Bailey M et al (2010) Relative hyperlactatemia and hospital mortality in critically ill patients: a retrospective multi‐centre study. Crit Care 14: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi K, Oda T, Takabuchi S, Oda S, Fukuda K, Adachi T, Semenza GL, Shingu K, Hirota K (2008) LPS induces hypoxia‐inducible factor 1 activation in macrophage‐differentiated cells in a reactive oxygen species‐dependent manner. Antioxid Redox Signal 10: 983–996 [DOI] [PubMed] [Google Scholar]
- Nogueira A, Kawabata V, Biselli P, Lins M, Valeri C, Seckler M, Hoshino W, Júnior L, Bernik M, de Machado JB et al (2008) Changes in plasma free fatty acid levels in septic patients are associated with cardiac damage and reduction in heart rate variability. Shock 29: 342–348 [DOI] [PubMed] [Google Scholar]
- Nuzzo E, Berg KM, Andersen LW, Balkema J, Montissol S, Cocchi MN, Liu X, Donnino MW (2015) Pyruvate dehydrogenase activity is decreased in the peripheral blood mononuclear cells of patients with sepsis: a prospective observational trial. Ann Am Thorac Soc 12: 1662–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiyama T, Yamaguchi M, Kurikawa N, Honzumi S, Terayama K, Nagaoka N, Yamamoto Y, Kimura T, Sugiyama D, Inoue S‐II (2017) Design, synthesis, and pharmacological evaluation of a novel series of hormone sensitive lipase inhibitor. Bioorg Med Chem 25: 4817–4828 [DOI] [PubMed] [Google Scholar]
- O'Neill LAJ, Kishton RJ, Rathmell J (2016) A guide to immunometabolism for immunologists. Nat Rev Immunol 16: 553–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opal SM, Ellis JL, Suri V, Freudenberg JM, Vlasuk GP, Li Y, Chahin AB, Palardy JE, Parejo N, Yamamoto M et al (2016) Pharmacological SIRT1 activation improves mortality and markedly alters transcriptional profiles that accompany experimental sepsis. Shock 45: 411–418 [DOI] [PubMed] [Google Scholar]
- Palsson‐Mcdermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, Van Den Bosch MWM, Quinn SR, Domingo‐Fernandez R, Johnson DGW et al (2015) Pyruvate kinase M2 regulates hif‐1α activity and il‐1β induction and is a critical determinant of the warburg effect in LPS‐activated macrophages. Cell Metab 21: 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo R, Enguix N, Lasheras J, Feliu JE, Kralli A, Villena JA (2011) Rosiglitazone‐induced mitochondrial biogenesis in white adipose tissue is independent of peroxisome proliferator‐activated receptor γ coactivator‐1α. PLoS One 6: e26989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Jeoung NH (2016) Inflammation increases pyruvate dehydrogenase kinase 4 (PDK4) expression via the Jun N‐Terminal Kinase (JNK) pathway in C2C12 cells. Biochem Biophys Res Commun 469: 1049–1054 [DOI] [PubMed] [Google Scholar]
- Park DW, Zmijewski JW (2017) Mitochondrial dysfunction and immune cell metabolism in sepsis. Infect Chemother 49: 10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak M, Lefebvre P, Staels B (2015) Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non‐alcoholic fatty liver disease. J Hepatol 62: 720–733 [DOI] [PubMed] [Google Scholar]
- Paz K, Hemi R, Derek L, Karasik A, Elhanany E, Kanety H, Zick Y (1997) A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS‐1 and IRS‐2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin‐induced tyrosine phosphorylation. J Biol Chem 272: 29911–29918 [DOI] [PubMed] [Google Scholar]
- Peyssonnaux C, Cejudo‐Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V (2007) Cutting edge: essential role of hypoxia inducible factor‐1 in development of lipopolysaccharide‐induced sepsis. J Immunol 178: 7516–7519 [DOI] [PubMed] [Google Scholar]
- Protti A, Carré J, Frost MT, Taylor V, Stidwill R, Rudiger A, Singer M (2007) Succinate recovers mitochondrial oxygen consumption in septic rat skeletal muscle. Crit Care Med 35: 2150–2155 [DOI] [PubMed] [Google Scholar]
- Randle PJ, Newsholme EA, Garland PB (1964) Regulation of glucose uptake by muscle. Effects of fatty acids, ketone bodies and pyruvate, and of alloxan‐diabetes and starvation, on the uptake and metabolic fate of glucose in rat heart and diaphragm muscles. Biochem J 93: 652–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranieri M, Thompson T, Barie P, Dhainaut J‐F, Douglas I, Finfer S, Garlund B, Marshall J, Rhodes A, Artigas A et al (2012) Drotrecogin Alfa (Activated) in adults with septic shock V. N Engl J Med 366: 2055–2064 [DOI] [PubMed] [Google Scholar]
- Rattarasarn C (1997) Hypoglycemia in sepsis: risk factors and clinical characteristics. J Med Assoc Thai 80: 760–766 [PubMed] [Google Scholar]
- Regueira T, Lepper PM, Brandt S, Ochs M, Vuda M, Takala J, Jakob SM, Djafarzadeh S (2009) Hypoxia inducible factor‐1 alpha induction by tumour necrosis factor‐alpha, but not by toll‐like receptor agonists, modulates cellular respiration in cultured human hepatocytes. Liver Int 29: 1582–92 [DOI] [PubMed] [Google Scholar]
- Rial E, Leonor R‐S, Eunate G‐V, Zaragoza P, Moyano E, González‐Barroso MM (2010) Lipotoxicity, fatty acid uncoupling and mitochondrial carrier function. Biochim Biophys Acta 1797: 800–806 [DOI] [PubMed] [Google Scholar]
- Rishu AH, Khan R, Al‐Dorzi HM, Tamim HM, Al‐Qahtani S, Al‐Ghamdi G, Arabi YM (2013) Even mild hyperlactatemia is associated with increased mortality in critically ill patients. Crit Care 17: R197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riske B, Hacker MR, Sc D, Kilcoyne R, Ingram JD, Manco‐johnson ML, Funk S, Sc B, Jacobson L, Valentino LA et al (2009) Intensive versus conventional glucose control in critically ill patients. N Engl J Med 360: 1283–1297 [DOI] [PubMed] [Google Scholar]
- Rittig N, Bach E, Thomsen HH, Pedersen SB, Nielsen TS, Jørgensen JO, Jessen N, Møller N (2016) Regulation of lipolysis and adipose tissue signaling during acute endotoxin‐induced inflammation: a human randomized crossover trial. PLoS One 11: e0162167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheteau P, Chatre L, Briand D, Mebarki M, Jouvion G, Bardon J, Crochemore C, Serrani P, Lecci PP, Latil M et al (2015) Sepsis induces long‐term metabolic and mitochondrial muscle stem cell dysfunction amenable by mesenchymal stem cell therapy. Nat Commun 6: 10145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC‐1alpha and SIRT1. Nature 434: 113–118 [DOI] [PubMed] [Google Scholar]
- Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, Ding XC, Chanson A‐LL, Reymond MK, Miconnet I et al (2011) Histone deacetylase inhibitors impair innate immune responses to Toll‐like receptor agonists and to infection. Blood 117: 1205–1217 [DOI] [PubMed] [Google Scholar]
- Romijn JA, Godfried MH, Wortel C, Sauerwein P (1990) Hypoglycemia, hormones and cytokines in fatal meningococcal septicemia. J Endocrinol Invest 13: 743–744 [DOI] [PubMed] [Google Scholar]
- Rossi MA, Celes M, Prado CM, Saggioro FP (2007) Myocardial structural changes in long‐term human severe sepsis/septic shock may be responsible for cardiac dysfunction. Shock 27: 10 [DOI] [PubMed] [Google Scholar]
- Samuvel DJ, Sundararaj KP, Nareika A, Lopes‐Virella MF, Huang Y (2009) Lactate boosts TLR4 signaling and NF‐κB pathway‐mediated gene transcription in macrophages via monocarboxylate transporters and MD‐2 up‐regulation. J Immunol 182: 2476–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl RA, Lang CH, Bagby GJ (1984) Hypertriglyceridemia and its relation to tissue lipoprotein lipase activity in endotoxemic, Escherichia coli bacteremic, and polymicrobial septic rats. J Surg Res 37: 394–401 [DOI] [PubMed] [Google Scholar]
- Schönfeld P, Wojtczak L (2008) Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic Biol Med 45: 231–241 [DOI] [PubMed] [Google Scholar]
- Schoors S, Bruning U, Missiaen R, Queiroz KCS, Borgers G, Elia I, Zecchin A, Cantelmo AR, Christen S, Goveia J et al (2015) Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520: 192–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger M, Romauch M, Schreiber R, Grabner GF, Hütter S, Kotzbeck P, Benedikt P, Eichmann TO, Yamada S, Knittelfelder O et al (2017) Pharmacological inhibition of adipose triglyceride lipase corrects high‐fat diet‐induced insulin resistance and hepatosteatosis in mice. Nat Commun 8: 14859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastián C, Zwaans BMM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D et al (2012) The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 151: 1185–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H (2011) HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of T H 17 and T reg cells. J Exp Med 208: 1367–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singanayagam A, Chalmers JD, Hill AT (2009) Admission hypoglycaemia is associated with adverse outcome in community‐acquired pneumonia. Eur Respir J 34: 932–939 [DOI] [PubMed] [Google Scholar]
- Singer M, Deutschman CS, Seymour C, Shankar‐Har M, Annane D, Angus DC (2016) The third international consensus definitions for sepsis and septic shock (Sepsis‐3). JAMA 315: 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan AW, Koeslag JH, Bredell GAG (1973) Body composition, work capacity, and work efficiency of active and inactive young men. Eur J Appl Physiol 32: 17–24 [Google Scholar]
- Soares MP, Gozzelino R, Weis S (2014) Tissue damage control in disease tolerance. Trends Immunol 35: 483–494 [DOI] [PubMed] [Google Scholar]
- Standage SW, Caldwell CC, Zingarelli B, Wong HR (2012) Reduced peroxisome proliferator‐activated receptor α expression is associated with decreased survival and increased tissue bacterial load in sepsis. Shock 37: 164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L, Li H, Xie A, Liu D, Rao W, Lan L, Li X, Li F, Xiao K, Wang H et al (2015) Dynamic changes in amino acid concentration profiles in patients with sepsis. PLoS One 10: e0121933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suetrong B, Walley KR (2016) Lactic acidosis in sepsis: It's Not All anaerobic: implications for diagnosis and management. Chest 149: 252–261 [DOI] [PubMed] [Google Scholar]
- Svistunenko DA, Davies N, Brealey D, Singer M, Cooper CE (2006) Mitochondrial dysfunction in patients with severe sepsis: an EPR interrogation of individual respiratory chain components. Biochim Biophys Acta 1757: 262–272 [DOI] [PubMed] [Google Scholar]
- Tancevski I, Nairz M, Duwensee K, Auer K, Schroll A, Heim C, Feistritzer C, Hoefer J, Gerner RR, Moschen AR et al (2014) Fibrates ameliorate the course of bacterial sepsis by promoting neutrophil recruitment via CXCR2. EMBO Mol Med 6: 810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson‐Mcdermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH et al (2013) Succinate is an inflammatory signal that induces IL‐1beta through HIF‐1alpha. Nature 496: 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM (2008) Fat and beyond: the diverse biology of PPARγ. Annu Rev Biochem 77: 289–312 [DOI] [PubMed] [Google Scholar]
- Unger RH, Orci L (2002) Lipoapoptosis: its mechanism and its diseases. Biochim Biophys Acta 1585: 202–212 [DOI] [PubMed] [Google Scholar]
- Van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R (2001) Intensive insulin therapy in critically ill patients. N Engl J Med 345: 1359–1367 [DOI] [PubMed] [Google Scholar]
- Van den Berghe G, Wilmer A, Hermans G, Meersseman W, Wouters PJ, Milants I, Van Wijngaerden E, Bobbaers H, Bouillon R (2006) Intensive insulin therapy in the medical ICU greet. N Engl J Med 354: 449–461 [DOI] [PubMed] [Google Scholar]
- Vary TC (1996) Sepsis‐induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 6: 89–94 [DOI] [PubMed] [Google Scholar]
- Vega RB, Huss JM, Kelly DP (2000) The coactivator PGC‐1 cooperates with peroxisome proliferator‐activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol 20: 1868–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E, Ott M (2015) 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol 16: 258–264 [DOI] [PubMed] [Google Scholar]
- Vlasselaers D, Milants I, Desmet L, Wouters PJ, Vanhorebeek I, van den Heuvel I, Mesotten D, Casaer MP, Meyfroidt G, Ingels C et al (2009) Intensive insulin therapy for patients in paediatric intensive care: a prospective, randomised controlled study. Lancet 373: 547–556 [DOI] [PubMed] [Google Scholar]
- Wang T, Derhovanessian A, De Cruz S, Belperio JA, Deng JC, Hoo GS (2014) Subsequent infections in survivors of sepsis: epidemiology and outcomes. J Intensive Care Med 29: 87–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Huen SC, Luan HH, Yu S, Zhang C, Gallezot JD, Booth CJ, Medzhitov R (2016) Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell 166: 1512–1525.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wannemacher RW, Pace JG, Beall RA, Dinterman RE, Petrella VJ, Neufeld HA (1979) Role of the liver in regulation of ketone body production during sepsis. J Clin Invest 64: 1565–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, Vogel P, Neale G, Green DR, Chi H (2016) Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol 17: 277–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis S, Carlos AR, Moita MR, Singh S, Blankenhaus B, Cardoso S, Larsen R, Rebelo S, Schäuble S, Del Barrio L et al (2017) Metabolic adaptation establishes disease tolerance to sepsis. Cell 169: 1263–1275.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB (2009) ATP‐citrate lyase links cellular metabolism to histone acetylation. Science 324: 1076–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellhoener P, Vietheer A, Sayk F, Schaaf B, Lehnert H, Dodt C (2011) Metabolic alterations in adipose tissue during the early phase of experimental endotoxemia in humans. Horm Metab Res 43: 754–759 [DOI] [PubMed] [Google Scholar]
- Wende AR, Abel DE (2010) Lipotoxicity in the heart. Biochim Biophys Acta 1801: 311–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel SE, Tyurina YY, Zhao J, St. Croix CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA, Amoscato AA et al (2017) PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171: 628–641.e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolowczuk I, Verwaerde C, Viltart O, Delanoye A, Delacre M, Pot B, Grangette C (2008) Feeding our immune system: impact on metabolism. Clin Dev Immunol 2008: 639803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HR, Cvijanovich N, Allen GL, Lin R, Anas N, Meyer K, Freishtat RJ, Monaco M, Odoms K, Sakthivel B et al (2009) Genomic expression profiling across the pediatric systemic inflammatory response syndrome, sepsis, and septic shock spectrum. Crit Care Med 37: 1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Barouch LA (2007) Leptin signaling and obesity: cardiovascular consequences. Circ Res 101: 545–559 [DOI] [PubMed] [Google Scholar]
- Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, Kang R, Lotze MT, Billiar TR, Wang H et al (2014) PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun 5: 4436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Wang Y, Zhang Y, He X, Hong C, Ni H, Chen X, Liang Y, Wu J, Zhao S et al (2018) RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF‐induced necroptosis. Nat Cell Biol 20: 186–197 [DOI] [PubMed] [Google Scholar]
- Youm Y‐HH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, Dominic D, Planavsky N, Lupfer C, Kanneganti TD et al (2015) The ketone metabolite β‐hydroxybutyrate blocks NLRP3 inflammasome‐mediated inflammatory disease. Nat Med 21: 263–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zager RA, Johnson ACM, Hanson SY (2005) Renal tubular triglyercide accumulation following endotoxic, toxic, and ischemic injury. Kidney Int 67: 111–121 [DOI] [PubMed] [Google Scholar]
- Zang QS, Wolf SE, Minei JP (2014) Sepsis‐induced cardiac mitochondrial damage and potential therapeutic interventions in the elderly. Aging Dis 5: 137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Jin S, Wang C, Jiang R, Wan J (2010) Histone deacetylase inhibitors attenuate acute lung injury during cecal ligation and puncture‐induced polymicrobial sepsis. World J Surg 34: 1676–1683 [DOI] [PubMed] [Google Scholar]
- Zhao T, Li Y, Liu B, Halaweish I, Mazitschek R, Alam HB (2014) Selective inhibition of histone deacetylase 6 alters the composition of circulating blood cells in a lethal septic model. J Surg Res 190: 647–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Ma H, Zhang X, Tu F, Wang X, Ha T, Fan M, Liu L, Xu J, Yu K et al (2017) Enhanced glycolytic metabolism contributes to cardiac dysfunction in polymicrobial sepsis. J Infect Dis 215: 1396–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Agustina D, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T et al (2010) The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 140: 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]